Introduction
Rheumatic heart disease (RHD) is an autoimmune
disease caused by rheumatic fever following group A hemolytic
streptococcal infection (1). RHD
remains a major global health problem and although primary (the use
of antibiotics for people suffering from acute streptococcal
pharyngitis to reduce the occurrence of rheumatic fever) and
secondary prevention (the use of antibiotic prophylactic measures
for people with a history of rheumatic fever or RHD to reduce the
recurrence of rheumatic fever) strategies have been clearly defined
(2), but their global
implementation is not perfect (3).
Moreover, the exact pathological mechanism of RHD-induced cardiac
valve damage remains unclear. RHD causes ~300,000 mortalities
worldwide every year (4,5) and the effective treatment at present
is surgery (6). While valve surgery
is expensive, has high technical requirements and requires
long-term medication afterwards (3), severely limiting the quality of life
of the patients. Therefore, it is important to study the pathogenic
mechanism and identify new therapeutic targets for RHD. To this
end, the present study investigated the most common RHD, identified
as mitral valve disease following review of the results of previous
histopathological analyses (7).
This finding led to the question: By which process is mitral valve
disease directly associated? The mitral valve is formed by two
layers of endothelial cells that tightly cover connective tissue
(3). When diseased, endothelial
cells form lesions, which involves connective tissue, leading to
scarring and causing permanent damage and chronic disease (8,9). Thus,
valvular disease is directly associated with endothelial
damage.
The endothelial-mesenchymal transition (EndMT) is
associated with endothelial damage and is a process by which
endothelial cells lose the endothelial phenotype and acquire the
characteristic phenotype of mesenchymal cells. In the heart, the
process of EndMT and activation of related signaling pathways are
primarily associated with cardiac development and a variety of
diseases processes (10). For
example, Wei et al (11)
showed that the EndMT serves an important role in cardiac fibrosis
and that inhibiting EndMT-related signaling pathways slows the
progression of heart disease caused by cardiac fibrosis.
Furthermore, Xiao et al (12) demonstrated that the TGF-β1/Smad
signaling pathway is involved in the RHD-mediated atrial fibrosis.
Smads are also key EndMT factors, as the phosphorylation of Smad2
and 3 regulates EndMT-related transcription factors to induce the
EndMT process (13,14), which also suggests that the EndMT
may be involved in RHD.
The EndMT is regulated by the TGF-β signaling
pathway and important factors in this regulatory process include
activin and Smad2 and 3 (15).
Activin, which is a member of the TGF-β subfamily, has the same
expression pattern as TGF-β during TGF-β-induced signal
transduction (16). Activin forms
complexes with related receptors (activin receptor I and II) on the
cell membrane and following phosphorylation it forms docking sites
that bind to the transcription factors Smad2 and Smad3 (17,18).
The Smad2/3 complex is phosphorylated at Ser residues in the
C-terminal domain, inducing transcription of key genes that are
associated with the EndMT. EndMT-related transcription factors that
are directly regulated by the Smad2/3 complex include lymphoid
enhancer factor-1 (LEF-1), Snail1, TWIST, zinc finger E-box-binding
homeobox (ZEB)1 and ZEB2 (13,14,16,19–21).
To date, there have been no studies investigating the association
between RHD and the EndMT to the best of the authors'
knowledge.
In summary, RHD is associated with endothelial
damage, endothelial damage is associated with EndMT and EndMT is
regulated by the activin/Smad2 and 3 signaling pathway. Therefore,
it was hypothesized that activin/Smad2 and 3 signaling is activated
during the development RHD-mediated valvular damage and that the
EndMT may be involved in RHD-induced cardiac valve damage.
The present study established an RHD rat model to
assess whether the activin/Smad2 and 3 signaling pathway is
activated during the development of RHD-induced cardiac valve
damage by evaluating differences in expression of activin/Smad2 and
3 signaling pathway-related factors [activin A, Smad2, Smad3,
phosphorylated (p-)Smad2 and p-Smad3] between RHD model rats and
normal rats. The present study also assessed whether the EndMT is
involved in RHD-induced cardiac valve damage by evaluating whether
there were differences in expression of EndMT-related factors that
are directly regulated by the activin/Smad2 and 3 signaling pathway
(LEF-1, Snail1, TWIST, ZEB1 and ZEB2) and mesenchymal markers [α
smooth muscle actin (α-SMA) and type I collagen α 1 (COL1A1)]
(22,23) between RHD model rats and normal
rats. The results of the present study may improve our
understanding of the potential association between EndMT and RHD as
well as provide new ideas for exploring the pathogenesis and
therapeutic targets of RHD.
Materials and methods
Antigen preparation
Group A streptococci (GAS, cat. no. ATCC19615;
American Type Culture Collection) were cultured in brain heart
infusion medium (Guangdong Huankai Microbial Sci. & Tech. Co.,
Ltd.) at 37°C for 24 h and then washed with normal saline (NS).
Following harvesting, the GAS were inactivated by incubating in 10%
neutral formalin for 12 h. Subsequently, the inactivated GAS were
washed in NS, resuspended in NS and the density was adjusted to
4.0×1011 CFU/ml. Finally, the suspensions were
emulsified by sonication (Sonics & Materials, Inc.) and the
antigen was prepared. Each sonication treatment lasted 2 sec, with
a 2.5 sec interval between each treatment. The entire sonication
treatment was performed at 10°C and lasted 10 min, and the
frequency of ultrasound was 20 kHz.
Animals and groups
A total of 16 healthy 8-week-old adult female Lewis
rats (160–180 g) were purchased from Beijing Vital River Animal
Technology Co., Ltd. Rats were acclimatized for 5 days. The rats
were housed in a specific pathogen-free animal laboratory at the
Center of Animal Experiments of Guangxi Medical University at
23±2°C with a 12-h light/dark cycle and were allowed unrestricted
cage activity and unlimited access to water and standard chow. All
animal experimental procedures performed in this study were
performed in accordance with the ethical guidelines for the care
and use of experimental animals and were approved by The Medical
Ethics Committee of the First Affiliated Hospital of Guangxi
Medical University (Nanning, China; approval no. 2019-KY-E-053).
All rats were randomly divided into two groups: i) The control
group (n=8); and ii) the RHD group (n=8). All rats were maintained
on soft bedding and not in wire-bottomed cages, as footpad
injection with complete Freund's adjuvant (CFA; Sigma-Aldrich,
Merck KGaA) was essential for establishing the RHD rat model. A
total of 9 weeks were required to establish the RHD rat model.
First, 100 µl of a solution comprising inactivated GAS
(4.0×1011 CFU/ml) and CFA at a 1:1 (v/v) ratio was
injected into one hindfoot pad of the rat. Subsequently, 1 week
later, 500 µl of the same solution was subcutaneously injected
subcutaneously into the abdomen at the same interval once a week
for 4 weeks. For the last 4 weeks, subcutaneous abdominal
injections were performed at the same intervals once a week, but
the injection was adjusted to 500 µl of inactivated GAS
(4.0×1011 CFU/ml). Prior to subcutaneous injection, the
rats were intraperitoneally anesthetized with ketamine and xylazine
at 10 mg and 0.2 mg per 250 g of body weight, respectively
(24–26). Rats in the control group were
injected using the same protocol as that described for the RHD
group, but the injected solution was an equivalent volume of
NS.
Animal sacrifice
Following all treatments, 1 ml of blood was
collected from the tail vein of each rat without anesthesia,
following which all rats were sacrificed with an intraperitoneal
injection of sodium pentobarbital (150 mg/kg). Mortality was
determined when >5 min passed without evidence of breathing or
heartbeat. A body weight loss of >15% with a decreased ability
to consume food and water was used as the humane endpoint.
Sample preparation
Once rats had been sacrificed, a sample of the heart
valve was rapidly collected using surgical instruments then quickly
frozen in liquid nitrogen and stored at −80°C for following
experiments. All samples were strictly stored in a refrigerator at
−80°C and were not removed until performing the experiments to
reduce the effect of temperature changes on the samples.
Histochemistry
The samples collected from valves in each group were
fixed with 4% paraformaldehyde at 4°C for 24 h and then dehydrated
by 70, 80, 90 and 95% alcohol and anhydrous alcohol in sequence.
After that, the samples were immersed in xylene to replace the
alcohol, then placed in the paraffin wax that has been melted in
the incubator (58-60°C) to replace the xylene. Finally, the samples
were placed in melted paraffin. After the melted paraffin was
solidified, the samples were embedded in paraffin. To perform
hematoxylin and eosin (H&E) staining and Sirius red staining,
5-µm-thick serial sections of each block were prepared. For H&E
staining, the sections were stained with H at room temperature for
4–10 min followed by E for 0.5–2 min at room temperature.
Subsequently, a BX43 light microscope (Olympus Corporation) was
used to capture images of each sample. For Sirius red staining,
sections were stained with a Sirius red solution at room
temperature for 1 h before being imaged with a BX43 confocal
microscope (magnification, 100×; Olympus Corporation). Then ImageJ
(version 1.51j; National Institutes of Health) was used to
calculate the area of the valve in the images of H&E staining
and the number of inflammatory cells on the valve area was
calculated. The ratio of the number of inflammatory cells to the
area of the valve was defined as inflammatory cell density to
quantify the H&E staining results.
Reverse transcription-quantitative
(RT-q) PCR
Total RNA was extracted from the valve tissues using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. The RNA
concentration was measured using a NanoDrop™ 2000 spectrophotometer
(NanoDrop Technologies; Thermo Fisher Scientific, Inc.) and 0.5 µg
of total RNA was then reverse transcribed into cDNA using a
PrimeScript RT reagent kit (Takara Bio, Inc.) according to the
manufacturer's protocol (incubation at 37°C for 15 min followed by
85°C for 5 sec). Finally, qPCR was performed using a TB Green
Premix Ex Taq II (Takara Bio, Inc.) with a StepOne system (Applied
Biosystems; Thermo Fisher Scientific, Inc.) under the following
thermal cycling conditions: 95°C for 30 min, followed by 40 cycles
of 95°C for 5 sec and 60°C for 30 sec. The primer sequences used in
the present study are listed in Table
I and β-actin was used as an internal control. The relative
changes in mRNA levels were determined as the fold difference
relative to β-actin for each gene using the 2−ΔΔCq
method (27).
 | Table I.Sequences of primers used in reverse
transcription-quantitative PCR. |
Table I.
Sequences of primers used in reverse
transcription-quantitative PCR.
| Gene | Primer sequence,
5′-3′ |
|---|
| Activin A | Forward:
TTGCTTGTGAACAGTGCCAGGAG |
|
| Reverse:
TCCCGTCTCCATCCACCTCTTTC |
| Smad2 | Forward:
TGTCGTCCATCTTGCCATTCACTC |
|
| Reverse:
TGTTCTCCACCACCTGCTCCTC |
| Smad3 | Forward:
CTTCACAGCCGTCCATGACAGTAG |
|
| Reverse:
CCAATGTAGTAGAGCCGCACACC |
| LEF-1 | Forward:
CCGAAGAGGAGGGCGACTTA |
|
| Reverse:
TGGGATGATTTCGGACTCGTT |
| Snail1 | Forward:
TTCTCTTCCACCTCGGCCTCATC |
|
| Reverse:
GGCTTCGGATGTGCATCTTCAGAG |
| TWIST | Forward:
CGACGACAGCCTGAGCAACAG |
|
| Reverse:
GCCGACTGCTGCGTCTCTTG |
| ZEB1 | Forward:
AGCCACCGAGAAGCCAGAGTC |
|
| Reverse:
CCAGCGGCAGGTTCACAGAATC |
| ZEB2 | Forward:
GAGATAAGGGAGAGCGTTGTG |
|
| Reverse:
AATTGTGGTCTGGATCGTGG |
| α-SMA | Forward:
GCGTGGCTATTCCTTCGTGACTAC |
|
| Reverse:
CCATCAGGCAGTTCGTAGCTCTTC |
| COL1A1 | Forward:
TGTTGGTCCTGCTGGCAAGAATG |
|
| Reverse:
GTCACCTTGTTCGCCTGTCTCAC |
| COL3A1 | Forward:
ACTTCTGGTCCTCCTGGTCTGC |
|
| Reverse:
CGCCTGGCTCACCCTTTTCAC |
| FSP1 | Forward:
TGGGGAGAAGGACAGACGAAGC |
|
| Reverse:
TGGCAATGCAGGACAGGAAGAC |
| β-actin | Forward:
GGAGATTACTGCCCTGGCTCCTA |
|
| Reverse:
GACTCATCGTACTCCTGCTTGCTG |
| BAX | Forward:
GATGCGTCCACCAAGAAGC |
|
| Reverse:
CCAGTTGAAGTTGCCGTCAG |
Western blot analysis
Total protein was extracted by treating the valve
tissues with RIPA lysis buffer (Sangon Biotech Co., Ltd.) according
to the manufacturer's protocol and the protein concentration was
measured using a bicinchoninic acid protein assay kit (Sangon
Biotech Co., Ltd.). Equal amounts of protein (30 µg) from each
sample were loaded per lane, separated using 10% SDS-PAGE at 80 V
for 30 min and 120 V for 60 min using a blotting system (Bio-Rad
Laboratories, Inc.) and then electrotransferred to 0.22-µm
polyvinylidene fluoride membranes (EMD Millipore) at a constant 80
V for 80 min. Subsequently, the membranes were blocked with 3% BSA
blocking solution (Sangon Biotech Co., Ltd.) for 1 h at room
temperature and then incubated for 12 h at 4°C with antibodies
against the following proteins: Activin A (1:1,000; cat. no.
ab128958; Abcam), Smad2 (1:1,000; cat. no. 5339; Cell Signaling
Technology, Inc.), Smad3 (1:1,000; cat. no. 9523; Cell Signaling
Technology, Inc.), p-Smad2 (1:1,000; cat. no. ab53100; Abcam),
p-Smad3 (1:2,000; cat. no. 9145; Cell Signaling Technology, Inc.),
LEF-1 (1:1,000; cat. no. ab137872; Abcam), Snail1 (1:500; cat. no.
13099-1-AP; ProteinTech Group, Inc.), TWIST (1:500; cat. no.
25465–1-AP; ProteinTech Group, Inc.), ZEB1 (1:500; cat. no.
21544-1-AP; ProteinTech Group, Inc.), ZEB2 (1:1,000; cat. no.
ab138222; Abcam), α-SMA (1:3,000; cat. no. ab32575; Abcam), COL1A1
(1:3,000; cat. no. ab34710; Abcam) and β-actin (1:3,000; cat. no.
10068-1-AP; ProteinTech Group, Inc.). Levels of NF-κB (1:2,000;
cat. no. ab16502; Abcam) and p-NF-κB (1:5,000; cat. no. ab86299;
Abcam) as valvular inflammation markers (28) were also examined. As apoptosis
serves a key role in the valvular damage (29), levels of the apoptosis-related
markers caspase-3 (1:1,000; cat. no. 9662; Cell Signaling
Technology, Inc.) and cleaved caspase-3 (1:1,000; cat. no. 9664;
Cell Signaling Technology, Inc.) were also detected (30). Following three washes in
tris-buffered saline with Tween-20, the membranes were incubated in
the dark for 1 h at room temperature with an HRP-conjugated
secondary antibody (1:10,000; cat. no. ab6721; Abcam). A
chemiluminescence imaging system (Alpha FluorChem FC3; Alpha, Inc.)
was used to visualize the protein bands and ImageJ was used to
quantify the expression levels of activin A, Smad2, Smad3, p-Smad2,
p-Smad3, LEF-1, Snail1, TWIST, ZEB1, ZEB2, α-SMA and COL1A1, which
were normalized to β-actin.
ELISA
ELISA kits (cat. nos. E04640r, E07451r, E11987r and
E13666r; Cusabio Technology LLC) were used to measure the serum
concentrations of rat IL-6, IL-17, TNF-α and rheumatoid factor (RF)
according to the manufacturer's protocols. After preparing all
reagents, working standards, samples (serum) and the assay plate,
100 µl of standard solution and sample was added to each well, the
assay plate covered with the adhesive strip and then the plate was
incubated for 2 h at 37°C. After removing the liquid in each well,
100 µl of biotin-antibody (1X) was added to each well, following
which the assay plate was covered with a new adhesive strip and
incubated for 1 h at 37°C. Subsequently, following removing the
liquid by aspiration, each well was washed with wash buffer
(provided by the ELISA kit) three times before the addition of 100
µl of HRP-avidin (1X) to each well and the assay plate was then
covered with a new adhesive strip and incubated for 1 h at 37°C.
After the aspiration/washing process aforementioned was performed
five times, 90 µl of TMB substrate was added to each well and the
plate was incubated at 37°C for 15–30 min away from the light.
Subsequently, after adding 50 µl of stop solution to each well and
gently tapping the plate to ensure adequate mixing, the absorbance
of each well at 450 nm was measured using a microplate reader
(Varioskan LUX; Thermo Fisher, Inc.).
Statistical analysis
All data are presented as the means ± standard
deviation of three independent experiments, unless otherwise shown.
Differences between two groups were analyzed using unpaired
Student's t-tests. Statistical analyses were performed using SPSS
16.0 (SPSS, Inc.) and P<0.05 was considered to indicate a
statistically significant difference.
Results
Histochemistry
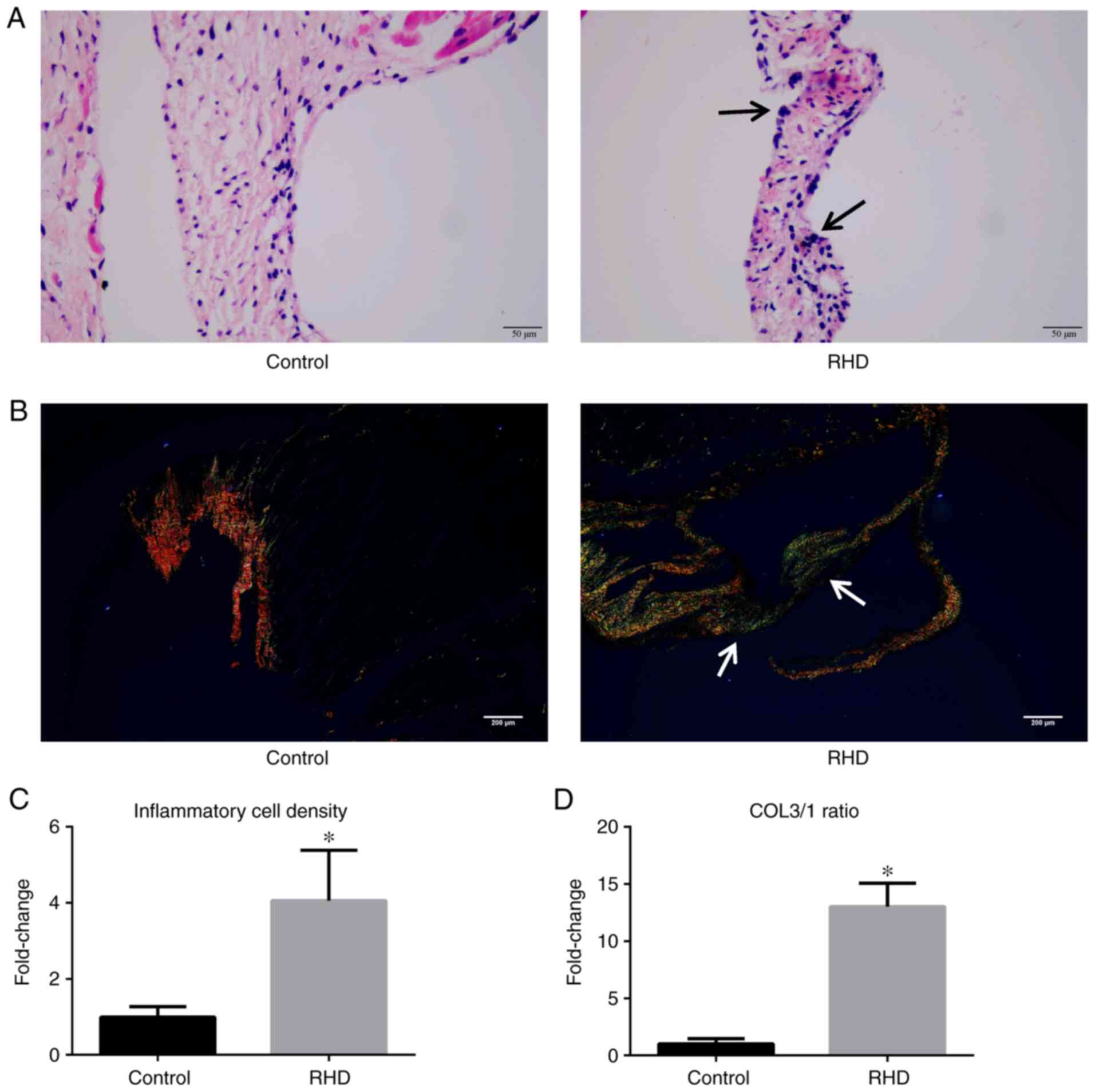
In the H&E staining experiment, H stained the
cell nucleus a vivid blue and E stained the cytoplasm pink.
Inflammatory cells have a large nucleus and a high ratio of
nucleus-to-cytoplasm (31).
Therefore, following H&E staining, inflammatory cells appear
blue because of the higher ratio of nucleus-to-cytoplasm, while
other cells appear pink because of the lower ratio of
nucleus-to-cytoplasm (32). H&E
staining results, as assessed by microscopy, showed myocarditis or
valvulitis in the valves of rats in the RHD group, which was not
observed in the control group (Fig.
1A). In this experiment, the inflammatory cell density was used
as a quantitative indicator of H&E staining results, which
showed that the inflammatory cell density of the valves in RHD
group was significantly higher compared with the control group
(P<0.05; Fig. 1C).
Sirius red staining was used to distinguish the
types of collagen fibers. A previous study showed that type 1
collagen (COL1) fibers are the primary type of collagen in
non-fibrotic valves (33). As the
ratio of type 3 collagen (COL3) fibers gradually increases with the
progression of fibrosis, a significant increase in the COL3/COL1
(COL3/1) ratio can be used to confirm the onset of valve and
myocardial fibrosis. Following Sirius red staining, COL1 fibers
appeared as closely packed yellow and red fibers, with obvious
birefringence, while COL3 fibers appeared as loosely arranged green
fibers, with weak birefringence. The results showed that the COL3/1
ratio in the valves from rats in the RHD group were significantly
increased compared with that observed in the control group
(P<0.05; Fig. 1B and D).
RT-qPCR
mRNA expression of activin/Smad2 and 3 signaling
pathway-related factors (activin A) in the RHD group was
significantly increased (P<0.05) compared with that observed in
the control group, while no differences in the mRNA levels of Smad2
and Smad3 were observed between these groups (Fig. 2A). In addition, mRNA expression of
EndMT-related factors (LEF-1, Snail1, TWIST, ZEB1, ZEB2, α-SMA and
COL1A1) in the RHD group was significantly increased (all
P<0.05) compared with that observed the control group (Fig. 2B and C). mRNA expression of collagen
type III α1 chain (COL3A1) and fibroblast-specific protein 1 (FSP1)
as fibrosis molecular markers (27)
and BAX as apoptosis molecular marker (30) was also examined, the levels of which
were significantly increased in the RHD group compared with that
observed in the control group (P<0.05; Fig. 2D and E).
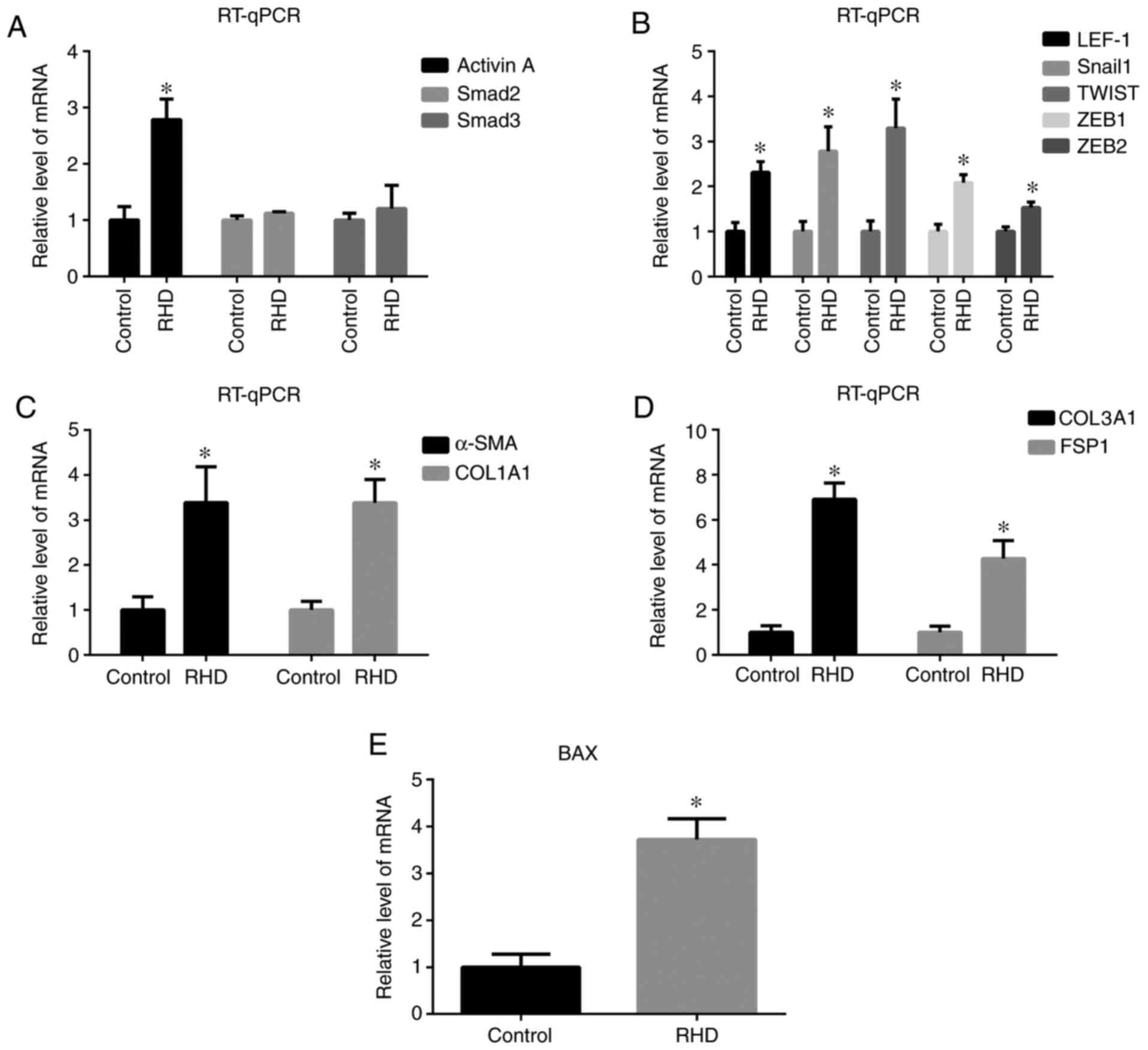 | Figure 2.RT-qPCR results for the relative
expression of (A) activin A, Smad2 and Smad3 in the two groups, (B)
LEF-1, Snail1, TWIST, ZEB1 and ZEB2, (C) α-SMA and COL1A1, (D)
COL3A1 and FSP1 and (E) BAX in the two groups. The results show
that expression of activin A, LEF-1, Snail1, TWIST, ZEB1, ZEB2,
α-SMA, COL1A1, COL3A1, FSP1 and BAX was increased in the RHD group.
Data are presented as the means ± standard deviation. *P<0.05
vs. respective control group. RT-qPCR, reverse
transcription-quantitative PCR; LEF-1, lymphoid enhancer factor-1;
ZEB, zinc finger E-box binding homeobox; α-SMA, α smooth muscle
actin; COL1A1, type I collagen; COL3A1, collagen type III α1 chain;
FSP1, fibroblast-specific protein 1; RHD, rheumatic heart
disease. |
Western blot analysis
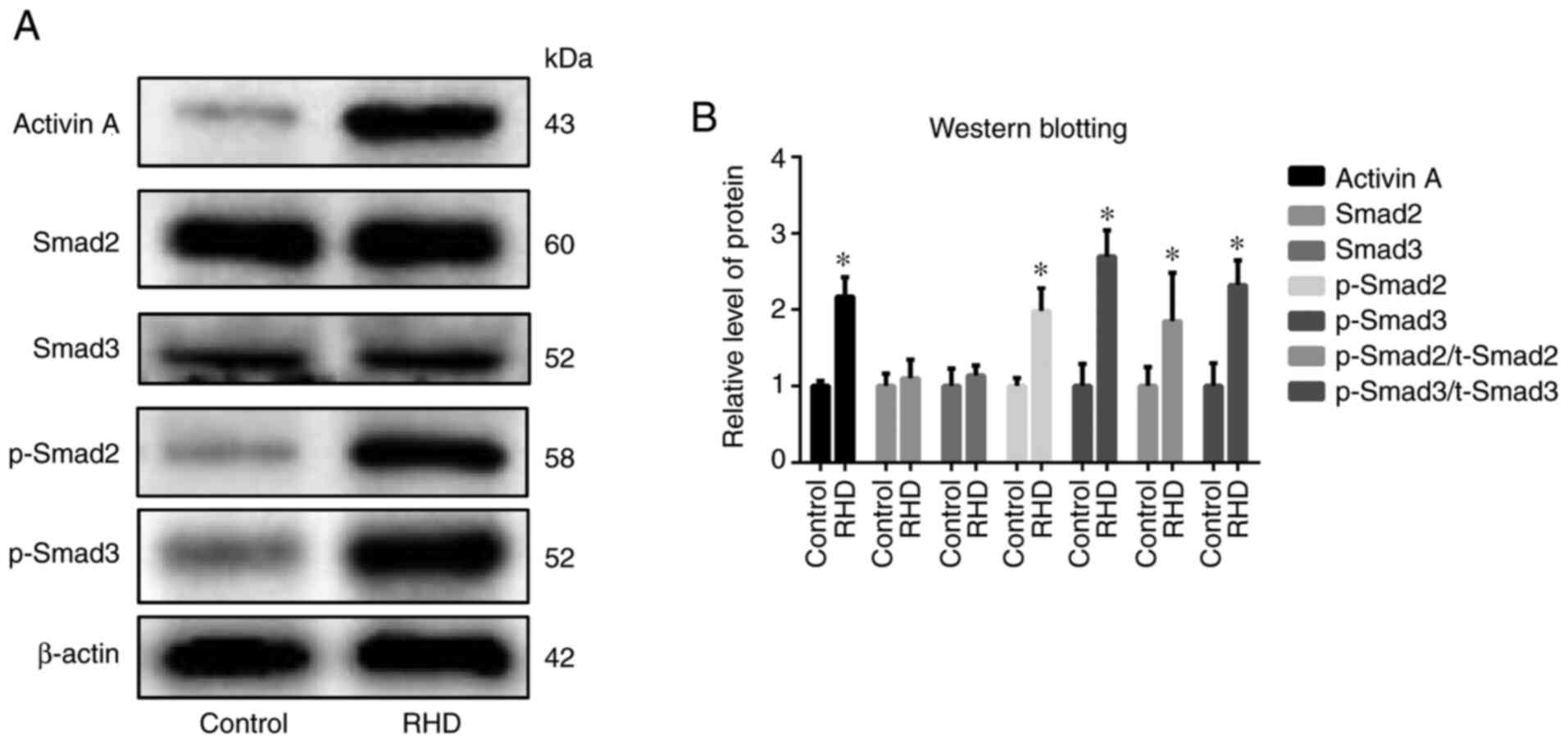
Protein expression of activin/Smad2 and 3 signaling
pathway-related factors (activin A, p-Smad2 and p-Smad3) in the RHD
group was significantly increased (all P<0.05) compared with the
control group, with no differences observed in the protein
expression of Smad2 and Smad3 between these groups (Fig. 3). Protein expression of
EndMT-related factors (LEF-1, Snail1, TWIST, ZEB1, ZEB2, α-SMA and
COL1A1) in the RHD group was significantly increased (all
P<0.05) compared with that in the control group (Fig. 4). In addition, protein expression of
cleaved caspase-3 and the ratio of p-NF-κB/total NF-κB in the RHD
group was significantly increased (all P<0.05) compared with
that in the control group (Fig.
5A-C).
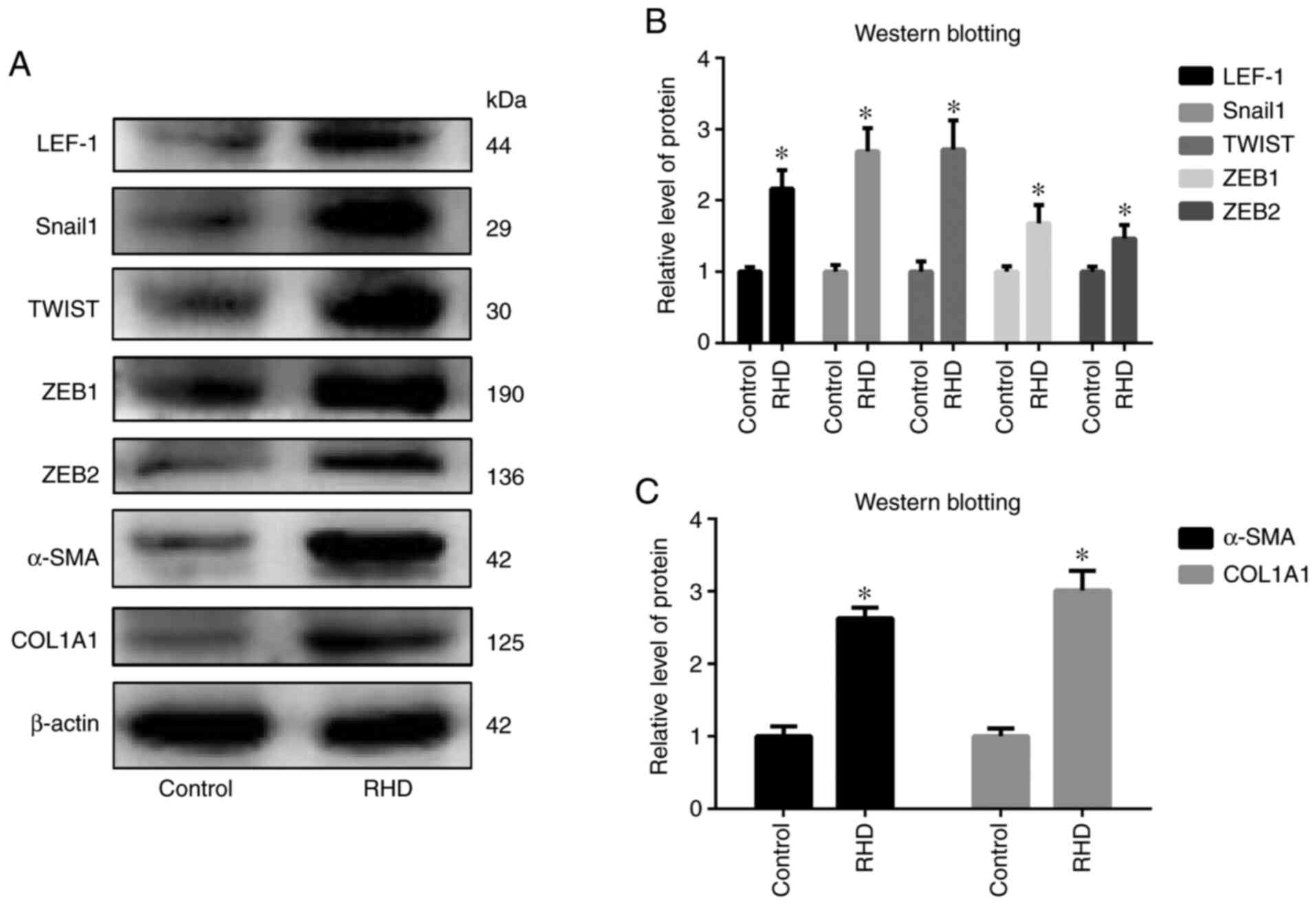 | Figure 4.Western blot analysis of (A) LEF-1,
Snail1, TWIST, ZEB1, ZEB2, α-SMA and COL1A1 levels in the two
groups and (B) quantified protein expression levels. (C) Relative
protein expression of α-SMA and COL1A1. Protein expression of
LEF-1, Snail1, TWIST, ZEB1, ZEB2, α-SMA and COL1A1 was increased in
RHD group. The data are presented as the means ± standard
deviation. *P<0.05 vs. respective control. LEF-1, lymphoid
enhancer factor-1; ZEB, zinc finger E-box binding homeobox; α-SMA,
α smooth muscle actin; COL1A1, type I collagen; RHD, rheumatic
heart disease. |
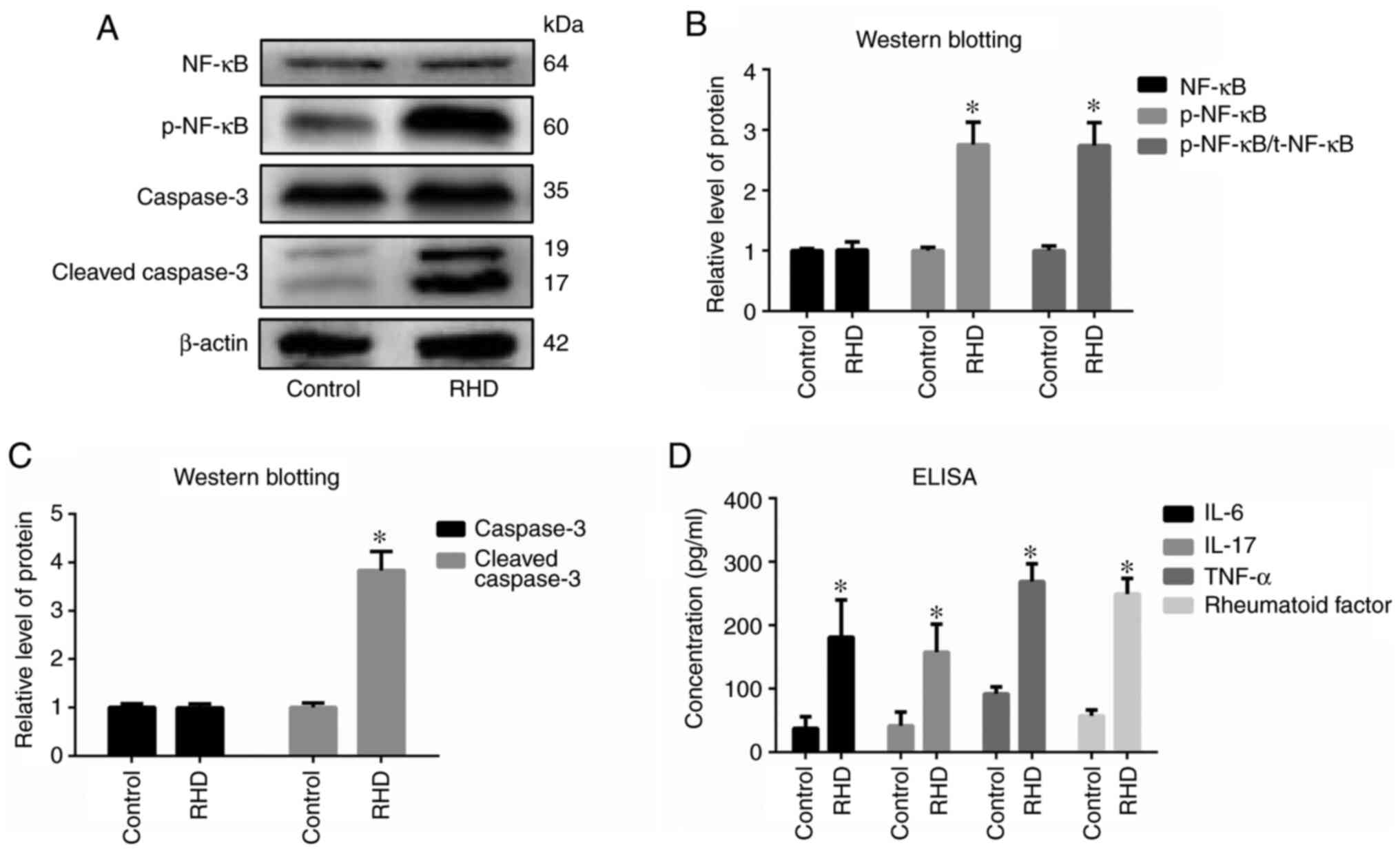 | Figure 5.Analysis of valvular inflammation
markers, apoptosis-related markers, cytokines and rheumatoid
factor. (A) Western blot analysis of NF-κB, p-NF-κB, caspase-3 and
cleaved caspase-3 levels in the two groups and (B) quantified
relative protein expression levels. (C) Relative protein expression
of caspase-3 and cleaved caspase-3. (D) Concentrations of IL-6,
IL-17, TNF-α and rheumatoid factor in serum detected using ELISA.
Results show that the ratio of p-NF-κB/t-NF-κB and the expression
of p-NF-κB, cleaved caspase-3, IL-6, IL-17, TNF-α and rheumatoid
factor was increased in the RHD group. Data are presented as the
means ± standard deviation. *P<0.05 vs. respective control. p-,
phosphorylated; RHD, rheumatic heart disease. |
ELISA
Levels of IL-6, IL-17, TNF-α and RF in the RHD group
were significantly increased compared with those observed in the
control group (P<0.05; Fig.
5D).
Discussion
RHD is an autoimmune disease caused by rheumatic
fever following group A hemolytic streptococcal infection and
threatens the health of a large number of patients every year
(34). RHD remains a global health
issue, especially in developing countries, being responsible for
the deaths of 250,000 young people worldwide each year, with >15
million people exhibiting evidence of rheumatic heart disease
(35). The results of previous
studies have shown that acute valvulitis is present in the valves
of RHD model rats and that valve damage in these animals is
associated with Th17 cell-related cytokines (27,36,37).
However, the exact pathological mechanism of RHD-induced cardiac
valve damage remains to be elucidated.
Mitral valve disease is the most common condition in
RHD, while isolated aortic valve disease occurs in ~2% of patients
with RHD in India (38). The mitral
valve is formed by two layers of endothelial cells that tightly
cover connective tissue and endothelial damage is directly
associated with valvular disease (3).
The EndMT is the process by which endothelial cells
lose the endothelial phenotype and acquire the characteristic
phenotype of mesenchymal cells, a process first discovered by
Elizabeth Hay in a study of chicken embryos (39). The transformation of endothelial
cells occurs throughout the life cycle, from the development of
heart valves during the embryonic stage to pathological changes in
valvular disease after birth (40).
The EndMT serves a key role in the occurrence and development of
various cardiovascular diseases. As early as 2003, Kuwahara et
al (41) observed that the
TGF-β signaling pathway is important in cardiac fibrosis and
diastolic heart failure in rats and that altering the activity of
this pathway inhibits the progression of cardiac fibrosis. Recent
studies have shown that the EndMT is associated with heart failure
(20,42–44)
and is also crucial in cardiac fibrosis following myocardial
infarction (45). Cardiac function
can be improved by inhibiting EndMT (46). Multiple studies have found that
attenuated EndMT can reduce cardiac fibrosis (11,20,47–51).
EndMT can also cause vascular endothelial dysfunction and serves an
important role in the process of atherosclerosis (52,53)
and pulmonary fibrosis (54–56).
The results of a study by Zhong et al (57) further elucidated the mechanism by
which Wnt1/β activates the EndMT in response to valve stiffening
and promotes myofibrogenesis. Wylie-Sears et al (58), by isolating and culturing sheep
mitral valve endothelial cells, reported that mitral valve
endothelial cells possess the potential to differentiate into
mesenchymal cells. Therefore, EndMT is widely involved in the
pathological process related to endothelial cells. The mitral valve
is formed by two layers of endothelial cells (3) and its pathology is probably related to
EndMT.
Mitral valve disease is closely associated with
mitral endothelial dysfunction. In patients with mitral valve
disease, the levels of microparticles in endothelial cells are
significantly increased compared with those observed in normal
individuals and the normal function of endothelial cells is
impaired (59). A previous study
observed the EndMT and activated myofibroblast-like interstitial
cells in mitral endothelial tissue of ischemic mitral regurgitation
in sheep, which showed that endothelial disease of the mitral valve
is related to EndMT (60). During
the EndMT, mitral valve endothelial cells secrete a bone-protecting
protein that is positively correlated with the severity of mitral
valve prolapse (9). In 2016,
Bischoff et al (61)
reported that the EndMT is the pathological basis of leaflet
fibrosis and leaflet dysfunction in the ischemic mitral
regurgitation model in Dorset hybrid sheep. These findings
demonstrated that endothelial cell dysfunction caused by the EndMT
is involved in the pathological process of mitral valve prolapse
and ischemic mitral valve reflux. Based on these results, it was
hypothesized that the EndMT may be involved in the pathological
process of RHD-induced mitral valve disease.
A number of studies have identified multiple
signaling pathways involved in regulating the EndMT, among which
the cardiac-specific signaling pathways include the TGF-β, BMP,
Notch, Wnt and Gata4 signaling pathways (15,62–66).
The TGF-β signaling pathway induces the EndMT through a variety of
intracellular messengers (67). The
TGF-β family includes at least 30 ligand molecules, which can be
divided into two subfamilies, TGF-β/activin/Nodal and BMP/GDF/MIS,
based on the similarities between the molecules and the specific
downstream signaling pathways that they activate (16). Activin, a member of the TGF-β
subfamily, has the same expression pattern as that of TGF-β during
TGF-β-induced signal transduction. Activin first binds activinRII
to form a complex on the cell membrane that subsequently binds
activinRI to form an activinRII-activin-activinRI complex and,
following phosphorylation, this complex forms docking sites that
bind the transcription factors Smad2 and Smad3 (16–18).
The Smad2/3 complex is phosphorylated and forms a complex with
SMAD4 (68–70). Once inside the nucleus, the Smad
complex induces the transcription of key genes associated with the
EndMT (such as snail1, smad2 and smad3) and directly regulates a
number of transcription factors that induce the EndMT (14,71). A
number of studies have demonstrated that EndMT is regulated by
Smad2/3. A recent study reported that using forkhead box M1 to
increase the expression of Smad2/3 can promote the EndMT (72). EndMT is involved in the cardiac
fibrosis of diabetic cardiomyopathy and is regulated by the Smad2/3
signaling pathway (51). Calcitriol
can attenuate TGF-β-induced EndMT by inhibiting the Smad2 pathway
(42). Sirtuin 1 activated by
resveratrol can regulate EndMT to alleviate isoproterenol-induced
cardiac fibrosis by regulating the TGF-β/Smad2/3 pathway (19). Activated TGF-β/Smad signaling,
elevated levels of EndMT and fibrosis were also observed in the
hearts of diabetic mice and cathelicidin-related antimicrobial
peptide can inhibit EndMT and cardiac fibrosis though TGF-β/Smad
signaling pathway (73). Twist1
overexpression and Smad2 phosphorylation can induce EndMT in human
pulmonary artery endothelial cells (74). MicroRNA-142-3p can attenuate EndMT
in human aortic endothelial cells induced by high glucose by
blocking the TGF-β1/Smad signaling pathway (75). Therefore, the EndMT has a close
association with the Smad2/3 signaling pathway and, since the EndMT
may be involved in the pathological process of RHD-induced mitral
valve disease, it was hypothesized that the activin/Smad2 and 3
signaling pathway is activated during RHD-induced valve damage.
The results of the present study supported this
hypothesis. The results of H&E staining, Sirius red staining,
ELISA, apoptosis-related (BAX and cleaved caspase-3), valvular
inflammation (NF-κB and TNF-α) and fibrosis molecular (COL3A1 and
FSP1) marker levels showed that valve damage was caused by RHD,
consistent with previous results obtained using RHD model rats
(27,36,37).
RT-qPCR, western blotting and ELISA results showed that the levels
of activin/Smad2 and 3 signaling pathway-related factors (activin
A, p-Smad2 and p-Smad3) in the RHD group were significantly higher
compared with those observed in the control group, suggesting that
the activin/Smad2 and 3 signaling pathway was activated during the
development of RHD-induced cardiac valve damage. Furthermore,
RT-qPCR and western blotting results revealed significantly higher
expression of EndMT-related factors (LEF-1, Snail1, TWIST, ZEB1,
ZEB2, α-SMA and COL1A1) in the RHD group compared with the control
group, suggesting that the EndMT is involved in RHD-induced cardiac
valve damage. The main purpose of the present study on
activin/Smad2 and 3 signaling pathway was to verify whether this
pathway was activated in RHD, so the main indicators detected were
activin, Smad2, Smad3, p-Smad2 and p-Smad3. Therefore, other
upstream and downstream signaling molecules in the activin/Smad2
and 3 signaling pathway were not tested in the present study and
should be investigated in future research.
The present study possessed some limitations. As the
results were obtained using an RHD rat model, the findings may not
accurately reflect the pathogenesis of RHD in human patients. Thus,
additional studies are required to validate the present findings in
an animal model that mimics human biology more closely. Relevant
cellular experiments are also needed to further corroborate the
results such as using genetic technology to intervene in the
expression of activin/Smad2 and 3 signaling pathway and then
detecting the expression of downstream molecules to further explore
the regulatory mechanism of activin/Smad2 and 3 signaling pathway,
as are studies on the specific mechanism by which the activin/Smad2
and 3 signaling pathway and the EndMT are involved in RHD-induced
cardiac valve damage. In addition, further clinical/physiological
examinations could improve the establishment of the RHD rat model,
which should be improved in future experiments.
RHD is a major disease worldwide that threatens the
health of ~300,000 individuals worldwide every year (1). However, the pathogenesis of RHD
remains poorly understood. Based on the results of previous
research on the EndMT, it was hypothesized that the EndMT is
involved in valvular damage due to RHD. Due to the association
between the activin/Smad2 and 3 signaling pathway and the EndMT, it
was hypothesized that this pathway is activated during the process
of valve damage caused by RHD. Therefore, the goal of the present
study was to verify the hypothesis by establishing an RHD rat model
and evaluating the differences in expression of activin/Smad2 and 3
signaling pathway-related factors and EndMT-related factors between
the RHD and control groups. The results suggested that the
activin/Smad2 and 3 signaling pathway was activated during the
development of valvular damage caused by RHD and that the EndMT is
involved in RHD-induced cardiac valve damage. Our findings can help
explore the pathogenesis of RHD and further our understanding to
inform improved treatments of RHD.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81960082), the
Guangxi Key Laboratory Base of Precision Medicine in
Cardio-cerebrovascular Disease Control and Prevention (grant no.
17-259-85), the Guangxi Clinical Research Center for
Cardio-cerebrovascular Diseases (grant no. AD17129014) and the
Guangxi Medical High-level Backbone Talents ‘139’ Program (grant
no. G201901006).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
ZZ and FH conceived and designed the study. SX, AC
and XW participated in the experimental design. SX, AC and YW
performed the experiments. SX, AC and CL analyzed the data. SX
wrote the manuscript and all authors contributed to the final
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Protocols involving animals were approved by The
Medical Ethics Committee of the First Affiliated Hospital of
Guangxi Medical University (Nanning, China; approval no.
2019-KY-E-053).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Watkins DA, Johnson CO, Colquhoun SM,
Karthikeyan G, Beaton A, Bukhman G, Forouzanfar MH, Longenecker CT,
Mayosi BM, Mensah GA, et al: Global, regional, and national burden
of rheumatic heart disease, 1990–2015. N Engl J Med. 377:713–722.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Remenyi B, Carapetis J, Wyber R, Taubert K
and Mayosi BM; World Heart Federation, : Position statement of the
World Heart Federation on the prevention and control of rheumatic
heart disease. Nat Rev Cardiol. 10:284–292. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Remenyi B, ElGuindy A, Smith SC Jr, Yacoub
M and Holmes DR Jr: Valvular aspects of rheumatic heart disease.
Lancet. 387:1335–1346. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Naghavi M, Abajobir AA, Abbafati C, Abbas
KM, Abd-Allah F, Abera SF, Aboyans V, Adetokunboh O, Afshin A,
Agrawal A, et al GBD 2016 Causes of Death Collaborators, : Global,
regional, and national age-sex specific mortality for 264 causes of
death, 1980–2016: A systematic analysis for the Global Burden of
Disease Study 2016. Lancet. 390:1151–1210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roth GA, Abate D, Abate KH, Abay SM,
Abbafati C, Abbasi N, Abbastabar H, Abd-Allah F, Abdela J,
Abdelalim A, et al GBD 2017 Causes of Death Collaborators, :
Global, regional, and national age-sex-specific mortality for 282
causes of death in 195 countries and territories, 1980–2017: A
systematic analysis for the Global Burden of Disease Study 2017.
Lancet. 392:1736–1788. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Salem A, Abdelgawad AME and Elshemy A:
Early and midterm outcomes of rheumatic mitral valve repair. Heart
Surg Forum. 21:E352–E358. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Elsayed AAA, Abdelaal KM, Abdelghaffar
AMM, Mohamed EEH, Mahran TMA, Ahmed MSM, Ibrahim AM and Mansour AA:
Poor outcome of surgical management of acute malfunctioning
mechanical mitral valve during pregnancy. Should centers with
limited resources find different options? Heart Surg Forum.
22:E405–E410. 2019.PubMed/NCBI
|
|
8
|
Pagnozzi LA and Butcher JT:
Mechanotransduction mechanisms in mitral valve physiology and
disease pathogenesis. Front Cardiovasc Med. 4:832017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Songia P, Branchetti E, Parolari A,
Myasoedova V, Ferrari G, Alamanni F, Tremoli E and Poggio P: Mitral
valve endothelial cells secrete osteoprotegerin during endothelial
mesenchymal transition. J Mol Cell Cardiol. 98:48–57. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hong L, Du X, Li W, Mao Y, Sun L and Li X:
EndMT: A promising and controversial field. Eur J Cell Biol.
97:493–500. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei WY, Zhang N, Li LL, Ma ZG, Xu M, Yuan
YP, Deng W and Tang QZ: Pioglitazone alleviates cardiac fibrosis
and inhibits endothelial to mesenchymal transition induced by
pressure overload. Cell Physiol Biochem. 45:26–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao M, Zhang M, Bie M, Wang X, Guo J and
Xiao H: Galectin-3 induces atrial fibrosis by activating the
TGF-β1/Smad pathway in patients with atrial fibrillation.
Cardiology. 145:446–455. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thuault S, Tan EJ, Peinado H, Cano A,
Heldin CH and Moustakas A: HMGA2 and Smads co-regulate SNAIL1
expression during induction of epithelial-to-mesenchymal
transition. J Biol Chem. 283:33437–33446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vincent T, Neve EP, Johnson JR, Kukalev A,
Rojo F, Albanell J, Pietras K, Virtanen I, Philipson L, Leopold PL,
et al: A SNAIL1-SMAD3/4 transcriptional repressor complex promotes
TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol.
11:943–950. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kovacic JC, Mercader N, Torres M, Boehm M
and Fuster V: Epithelial-to-mesenchymal and
endothelial-to-mesenchymal transition: From cardiovascular
development to disease. Circulation. 125:1795–1808. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Massagué J: How cells read TGF-beta
signals. Nat Rev Mol Cell Biol. 1:169–178. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Itoh S, Itoh F, Goumans MJ and Ten Dijke
P: Signaling of transforming growth factor-beta family members
through Smad proteins. Eur J Biochem. 267:6954–6967. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moustakas A, Souchelnytskyi S and Heldin
CH: Smad regulation in TGF-beta signal transduction. J Cell Sci.
114:4359–4369. 2001.PubMed/NCBI
|
|
19
|
Liu ZH, Zhang Y, Wang X, Fan XF, Zhang Y,
Li X, Gong YS and Han LP: SIRT1 activation attenuates cardiac
fibrosis by endothelial-to-mesenchymal transition. Biomed
Pharmacother. 118:1092272019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu L, Fu M, Chen D, Han W, Ostrowski MC,
Grossfeld P, Gao P and Ye M: Endothelial-specific deletion of Ets-1
attenuates Angiotensin II-induced cardiac fibrosis via suppression
of endothelial-to-mesenchymal transition. BMB Rep. 52:595–600.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maleki S, Cottrill KA, Poujade FA,
Bhattachariya A, Bergman O, Gådin JR, Simon N, Lundströmer K,
Franco-Cereceda A, Björck HM, et al: The mir-200 family regulates
key pathogenic events in ascending aortas of individuals with
bicuspid aortic valves. J Intern Med. 285:102–114. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang B, Niu W, Dong HY, Liu M-L, Luo Y
and Li ZC: Hypoxia induces endothelial mesenchymal transition in
pulmonary vascular remodeling. Int J Mol Med. 42:270–278.
2018.PubMed/NCBI
|
|
23
|
Fang S, Guo H, Cheng Y, Zhou Z, Zhang W,
Han B, Luo W, Wang J, Xie W and Chao J: circHECTD1 promotes the
silica-induced pulmonary endothelial-mesenchymal transition via
HECTD1. Cell Death Dis. 9:396. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gorton D, Govan B, Olive C and Ketheesan
N: B- and T-cell responses in group a streptococcus M-protein- or
Peptide-induced experimental carditis. Infect Immun. 77:2177–2183.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gorton D, Blyth S, Gorton JG, Govan B and
Ketheesan N: An alternative technique for the induction of
autoimmune valvulitis in a rat model of rheumatic heart disease. J
Immunol Methods. 355:80–85. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lymbury RS, Olive C, Powell KA, Good MF,
Hirst RG, LaBrooy JT and Ketheesan N: Induction of autoimmune
valvulitis in Lewis rats following immunization with peptides from
the conserved region of group A streptococcal M protein. J
Autoimmun. 20:211–217. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen A, Wen J, Lu C, Lin B, Xian S, Huang
F, Wu Y and Zeng Z: Inhibition of miR-155-5p attenuates the
valvular damage induced by rheumatic heart disease. Int J Mol Med.
45:429–440. 2020.PubMed/NCBI
|
|
28
|
Zhan Q, Zeng Q, Song R, Zhai Y, Xu D,
Fullerton DA, Dinarello CA and Meng X: IL-37 suppresses
MyD88-mediated inflammatory responses in human aortic valve
interstitial cells. Mol Med. 23:83–91. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Blake RR, Markby GR, Culshaw GJ,
Martinez-Pereira Y, Lu CC and Corcoran BM: Survival of activated
myofibroblasts in canine myxomatous mitral valve disease and the
role of apoptosis. Res Vet Sci. 128:99–106. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Pande S, Tewari P, Agarwal SK, Agarwal V,
Agrawal V, Chagtoo M, Majumdar G and Tewari S: Evidence of
apoptosis in right ventricular dysfunction in rheumatic mitral
valve stenosis. Indian J Med Res. 144:718–724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Trihia H, Siatra H, Gklisty H,
Diamantopoulos P, Arapantoni-Dadiotis P and Kalogerakos K:
Lymphoepithelioma-like carcinoma of the breast: Cytological and
histological features and review of the literature. Acta Cytol.
56:85–91. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cardiff RD, Miller CH and Munn RJ: Manual
hematoxylin and eosin staining of mouse tissue sections. Cold
Spring Harb Protoc. 2014:655–658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Purushothaman KR, Purushothaman M,
Turnbull IC, Adams DH, Anyanwu A, Krishnan P, Kini A, Sharma SK,
O'Connor WN and Moreno PR: Association of altered collagen content
and lysyl oxidase expression in degenerative mitral valve disease.
Cardiovasc Pathol. 29:11–18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sharma N and Toor D: Interleukin-10: Role
in increasing susceptibility and pathogenesis of rheumatic
fever/rheumatic heart disease. Cytokine. 90:169–176. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dass C and Kanmanthareddy A: Rheumatic
heart disease. StatPearls [Internet]. StatPearls Publishing;
Treasure Island, FL: 2020
|
|
36
|
Wu XD, Zeng ZY, Gong DP, Wen JL and Huang
F: Potential involvement of S1PR1/STAT3 signaling pathway in
cardiac valve damage due to rheumatic heart disease. Biotech
Histochem. 94:398–403. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wen Y, Zeng Z, Gui C, Li L and Li W:
Changes in the expression of Th17 cell-associated cytokines in the
development of rheumatic heart disease. Cardiovasc Pathol.
24:382–387. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chockalingam A, Gnanavelu G, Elangovan S
and Chockalingam V: Clinical spectrum of chronic rheumatic heart
disease in India. J Heart Valve Dis. 12:577–581. 2003.PubMed/NCBI
|
|
39
|
Trelstad RL, Hay ED and Revel JD: Cell
contact during early morphogenesis in the chick embryo. Dev Biol.
16:78–106. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bischoff J: Endothelial-to-mesenchymal
transition. Circ Res. 124:1163–1165. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kuwahara F, Kai H, Tokuda K, Kai M,
Takeshita A, Egashira K and Imaizumi T: Transforming growth
factor-beta function blocking prevents myocardial fibrosis and
diastolic dysfunction in pressure-overloaded rats. Circulation.
106:130–135. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tsai TH, Lin CJ, Hang CL and Chen WY:
Calcitriol attenuates doxorubicin-induced cardiac dysfunction and
inhibits endothelial-to-mesenchymal transition in mice. Cells.
8:82019. View Article : Google Scholar
|
|
43
|
Zheng G, Cai J, Chen X, Chen L, Ge W, Zhou
X and Zhou H: Relaxin ameliorates renal fibrosis and expression of
endothelial cell transition markers in rats of
isoproterenol-induced heart failure. Biol Pharm Bull. 40:960–966.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mai JT, Hu QS, Xie Y, Su SC, Qiu Q, Yuan
WL, Yang Y, Song YW, Chen YX and Wang JF: Dyssynchronous pacing
triggers endothelial-mesenchymal transition through heterogeneity
of mechanical stretch in a canine model. Circ J. 79:201–209. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen J, Jia J, Ma L, Li B, Qin Q, Qian J
and Ge J: Nur77 deficiency exacerbates cardiac fibrosis after
myocardial infarction by promoting endothelial-to-mesenchymal
transition. J Cell Physiol. Jun 15–2020.(Epub ahead of print).
|
|
46
|
Wu Y, Xu M, Bao H and Zhang JH:
Sitagliptin inhibits EndMT in vitro and improves cardiac function
of diabetic rats through the SDF-1α/PKA pathway. Eur Rev Med
Pharmacol Sci. 23:841–848. 2019.PubMed/NCBI
|
|
47
|
Song S, Liu L, Yu Y, Zhang R, Li Y, Cao W,
Xiao Y, Fang G, Li Z, Wang X, et al: Inhibition of BRD4 attenuates
transverse aortic constriction- and TGF-β-induced
endothelial-mesenchymal transition and cardiac fibrosis. J Mol Cell
Cardiol. 127:83–96. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu Y, Gao L, Zhao X, Guo S, Liu Y, Li R,
Liang C, Li L, Dong J, Li L, et al: Saikosaponin A protects from
pressure overload-induced cardiac fibrosis via inhibiting
fibroblast activation or endothelial cell EndMT. Int J Biol Sci.
14:1923–1934. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lai YJ, Chen IC, Li HH and Huang CC: EP4
Agonist L-902,688 suppresses EndMT and attenuates right ventricular
cardiac fibrosis in experimental pulmonary arterial hypertension.
Int J Mol Sci. 19:192018. View Article : Google Scholar
|
|
50
|
Wang Z, Wang Z, Gao L, Xiao L, Yao R, Du
B, Li Y, Wu L, Liang C, Huang Z, et al: miR-222 inhibits cardiac
fibrosis in diabetic mice heart via regulating
Wnt/β-catenin-mediated endothelium to mesenchymal transition. J
Cell Physiol. 235:2149–2160. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang B, Wu Y, Ge Z, Zhang X, Yan Y and Xie
Y: NLRC5 deficiency ameliorates cardiac fibrosis in diabetic
cardiomyopathy by regulating EndMT through Smad2/3 signaling
pathway. Biochem Biophys Res Commun. 528:545–553. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Souilhol C, Harmsen MC, Evans PC and
Krenning G: Endothelial-mesenchymal transition in atherosclerosis.
Cardiovasc Res. 114:565–577. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hao YM, Yuan HQ, Ren Z, Qu SL, Liu LS, Wei
DH, Yin K, Fu M and Jiang ZS: Endothelial to mesenchymal transition
in atherosclerotic vascular remodeling. Clin Chim Acta. 490:34–38.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Song S, Ji Y, Zhang G, Zhang X, Li B, Li D
and Jiang W: Protective effect of atazanavir sulphate against
pulmonary fibrosis in vivo and in vitro. Basic Clin Pharmacol
Toxicol. 122:199–207. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gaikwad AV, Eapen MS, McAlinden KD, Chia
C, Larby J, Myers S, Dey S, Haug G, Markos J, Glanville AR, et al:
Endothelial to mesenchymal transition (EndMT) and vascular
remodeling in pulmonary hypertension and idiopathic pulmonary
fibrosis. Expert Rev Respir Med. 14:1027–1043. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yu J, Deng Y and Han M: Blocking protein
phosphatase 2A with a peptide protects mice against
bleomycin-induced pulmonary fibrosis. Exp Lung Res. 46:234–242.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhong A, Mirzaei Z and Simmons CA: The
roles of matrix stiffness and β-catenin signaling in
endothelial-to-mesenchymal transition of aortic valve endothelial
cells. Cardiovasc Eng Technol. 9:158–167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wylie-Sears J, Aikawa E, Levine RA, Yang
JH and Bischoff J: Mitral valve endothelial cells with osteogenic
differentiation potential. Arterioscler Thromb Vasc Biol.
31:598–607. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Ci HB, Ou ZJ, Chang FJ, Liu DH, He GW, Xu
Z, Yuan HY, Wang ZP, Zhang X and Ou JS: Endothelial microparticles
increase in mitral valve disease and impair mitral valve
endothelial function. Am J Physiol Endocrinol Metab. 304:E695–E702.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Shapero K, Wylie-Sears J, Levine RA, Mayer
JE Jr and Bischoff J: Reciprocal interactions between mitral valve
endothelial and interstitial cells reduce
endothelial-to-mesenchymal transition and myofibroblastic
activation. J Mol Cell Cardiol. 80:175–185. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bischoff J, Casanovas G, Wylie-Sears J,
Kim DH, Bartko PE, Guerrero JL, Dal-Bianco JP, Beaudoin J, Garcia
ML, Sullivan SM, et al: CD45 expression in mitral valve endothelial
cells after myocardial infarction. Circ Res. 119:1215–1225. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lan Y, Liu B, Yao H, Li F, Weng T, Yang G,
Li W, Cheng X, Mao N and Yang X: Essential role of endothelial
Smad4 in vascular remodeling and integrity. Mol Cell Biol.
27:7683–7692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zeisberg M, Hanai J, Sugimoto H, Mammoto
T, Charytan D, Strutz F and Kalluri R: BMP-7 counteracts
TGF-beta1-induced epithelial-to-mesenchymal transition and reverses
chronic renal injury. Nat Med. 9:964–968. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Grieskamp T, Rudat C, Lüdtke TH, Norden J
and Kispert A: Notch signaling regulates smooth muscle
differentiation of epicardium-derived cells. Circ Res. 108:813–823.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li H, Zhao Q, Chang L, Wei C, Bei H, Yin
Y, Chen M, Wang H, Liang J and Wu Y: LncRNA MALAT1 modulates ox-LDL
induced EndMT through the Wnt/β-catenin signaling pathway. Lipids
Health Dis. 18:622019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Rivera-Feliciano J, Lee KH, Kong SW,
Rajagopal S, Ma Q, Springer Z, Izumo S, Tabin CJ and Pu WT:
Development of heart valves requires Gata4 expression in
endothelial-derived cells. Development. 133:3607–3618. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jiang Y, Zhou X, Hu R and Dai A:
TGF-β1-induced SMAD2/3/4 activation promotes RELM-β transcription
to modulate the endothelium-mesenchymal transition in human
endothelial cells. Int J Biochem Cell Biol. 105:52–60. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pickup MW, Owens P and Moses HL: TGF-β,
bone morphogenetic protein, and activin signaling and the tumor
microenvironment. Cold Spring Harb Perspect Biol. 9:92017.
View Article : Google Scholar
|
|
69
|
Morianos I, Papadopoulou G, Semitekolou M
and Xanthou G: Activin-A in the regulation of immunity in health
and disease. J Autoimmun. 104:1023142019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Goh BC, Singhal V, Herrera AJ, Tomlinson
RE, Kim S, Faugere MC, Germain-Lee EL, Clemens TL, Lee SJ and
DiGirolamo DJ: Activin receptor type 2A (ACVR2A) functions directly
in osteoblasts as a negative regulator of bone mass. J Biol Chem.
292:13809–13822. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Peinado H, Quintanilla M and Cano A:
Transforming growth factor beta-1 induces snail transcription
factor in epithelial cell lines: Mechanisms for epithelial
mesenchymal transitions. J Biol Chem. 278:21113–21123. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Song S, Zhang R, Cao W, Fang G, Yu Y, Wan
Y, Wang C, Li Y and Wang Q: Foxm1 is a critical driver of
TGF-β-induced EndMT in endothelial cells through Smad2/3 and binds
to the Snail promoter. J Cell Physiol. 234:9052–9064. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zheng X, Peng M, Li Y, Wang X, Lu W, Wang
X, Shan Y, Li R, Gao L and Qiu C: Cathelicidin-related
antimicrobial peptide protects against cardiac fibrosis in diabetic
mice heart by regulating endothelial-mesenchymal transition. Int J
Biol Sci. 15:2393–2407. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mammoto T, Muyleart M, Konduri GG and
Mammoto A: Twist1 in hypoxia-induced pulmonary hypertension through
transforming growth factor-β-Smad signaling. Am J Respir Cell Mol
Biol. 58:194–207. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhu GH, Li R, Zeng Y, Zhou T, Xiong F and
Zhu M: MicroRNA-142-3p inhibits high-glucose-induced
endothelial-to-mesenchymal transition through targeting TGF-β1/Smad
pathway in primary human aortic endothelial cells. Int J Clin Exp
Pathol. 11:1208–1217. 2018.PubMed/NCBI
|