Introduction
Cardiac disease is the most prevalent cause of
mortality worldwide (1). Damage to
the mammalian heart leads to cardiomyocyte (CM) loss, cardiac
fibrosis and hypertrophy followed by cardiac dysfunction, which
eventually leads to heart failure (2). Adult mammalian CMs become fully
differentiated, exit the cell cycle and are unable to regenerate
CMs following cardiac diseases (3).
Therefore, the induction of CM proliferation presents a promising
strategy to promote cardiac regeneration (4,5).
MicroRNAs (miRNAs/miRs) are a group of
~22-nucleotide, highly conserved, non-coding RNAs that regulate
gene expression via post-transcriptional modification in numerous
biological and pathological processes, such as cell proliferation
and apoptosis (6). Previous studies
have revealed that miRNA is a critical regulator in cardiac
conditions, including in cardiac hypertrophy, myocardial infarction
and heart failure (7,8). To date, multiple miRNAs have been
identified that control fetal CM proliferation and postnatal CM
cell cycle reentry (6,9,10). A
high-throughput functional screening identified that ~204 miRNAs
promoted neonatal CM proliferation, whereas 331 miRNAs decreased CM
proliferation (11). The miRNA
clusters miR-17-92 (12) and
miR-302-367 (9) are reported to be
involved in CM proliferation in mouse embryonic and postnatal
hearts. Moreover, miR-590-3p and miR-199a-3p increase cyclin A
(CCNA), cyclin E (CCNE) and cyclin B (CCNB) expression levels in
proliferating CMs (11). It has
also been shown that miR-204-dependent regulation of Jumonji And
AT-Rich Interaction Domain Containing 2 (Jarid2) in CM is essential
during embryo development and cardiogenesis (13).
miR-449a-5p has been reported to be a tumor
suppressor in numerous types of cancer including gastric cancer,
liver cancer and neuroblastoma (14–16).
Previous studies have demonstrated that miR-449a-5p inhibits cell
cycle progression, proliferation, growth and invasion in cancer
cells by targeting different target genes (17). A previous study also revealed that
miR-449a-5p regulated hypoxia/reoxygenation injury-induced CM
mortality (18). Furthermore, the
members of the miR-34/449 family share high sequence homology
(19). miR-34a has been shown to
inhibit CM proliferation in postnatal mice, whereas inhibition of
miR-34a promotes CM proliferation and improves cardiac function in
adult post-myocardial infarcted hearts (10). However, to the best of our knowledge
there is no direct evidence of miR-449a-5p modulation of CM
proliferation.
Integrated bioinformatics analyses have revealed
that CDK6 is a potential target of miR-449a-5p (16,20).
CDK6 is a cyclin D activated kinase that phosphorylates the
retinoblastoma protein (Rb) in the G1 phase, and
associates with E2 transcription factor (E2F) to regulate the
transition from G1 to S in the cell cycle (21). Additionally, previous studies have
reported that miR-449a-5p directly targets CDK6 to inhibit cell
proliferation (16,20).
Based on the relationship between miR-449a-5p and
CDK6, it was hypothesized that miR-449a-5p may suppress CM
proliferation by inhibiting CDK6. The aims of the current study
were to elucidate whether loss of miR-449a-5p promotes CM
proliferation and to identify the molecular mechanism via which
miR-449a-5p regulates CM proliferation. The present study provides
a novel insight into miR-449a-5p, which could potentially be used
as an effective therapeutic approach in cardiac diseases.
Materials and methods
Reagents and antibodies
Trypsin, collagenase type II, FBS and Opti-MEM were
purchased from Gibco, Thermo Fisher Scientific, Inc. DMEM/F12 1:1
medium was obtained from HyClone, Cytiva. An E.Z.N.A. Total RNA kit
II was purchased from Omega Bio-Tek, Inc. An RNeasy Midi kit was
acquired from Qiagen, Inc. DNase I and Lipofectamine®
2000 were purchased from Invitrogen, Thermo Fisher Scientific, Inc.
A PrimeScript™ RT Master mix and a SYBRs Premix Ex Taq™ kit were
obtained from Takara Biotechnology Co., Ltd. A miRNA First Strand
cDNA Synthesis and miRNAs Quantitation PCR kit were purchased from
Sangon Biotech Co., Ltd. Paraformaldehyde (PFA) was acquired from
Leagene. DAPI was purchased from Bestbio, while Hoechst 33342 was
purchased from Bioworld Technology, Inc. Tris-HCl, RIPA buffer was
obtained from Beijing Dingguo Changsheng Biotechnology Co., Ltd. A
BCA Protein Quantitative Analysis kit was purchased from
Fudebio-tech. BSA, penicillin, streptomycin, NaCl, SDS, formamide,
and SSC were purchased from Sigma-Aldrich, Merck KGaA.
For immunofluorescence analysis, antibodies against
cardiac troponin T (cTnT; cat. no. ab8295; 1:100), ki67 (cat. no.
ab15580; 1:100) and histone H3 phosphorylated at serine 10 (pH3;
cat. no. ab47297; 1:100) were obtained from Abcam, and goat
anti-mouse IgG/Alexa Fluor 488 (cat. no. bs-0296G-A488; 1:100) and
goat anti-rabbit IgG/Alexa Fluor 555 (cat. no. bs-0295G-A555;
1:100) antibodies were purchased from Beijing Biosynthesis
Biotechnology Co., Ltd.
For western blotting, antibodies against CDK6 (cat.
no. 3136; 1:2,000) were purchased from Cell Signaling Technology,
Inc., and β-actin (cat. no. bs-0061R; 1:2,000) antibodies were
purchased from Beijing Biosynthesis Biotechnology Co., Ltd. Donkey
anti-mouse IgG H&L (cat. no. ab6820; 1:10,000) and donkey
anti-rabbit IgG H&L (cat. no. ab16284; 1:10,000) antibodies
were purchased from Abcam.
Laboratory animals
A total of 30 pregnant, 500 postnatal day (P) 1 (1–2
g), 30 P4 (3–4 g), 80 P7 (5–6 g), 20 P28 (18–20 g) and 20 P56 male
(20–25 g) C57BL/6J mice were purchased from the Experimental Animal
Center of Guizhou Medical University. Animals were housed in a
temperature-controlled (23±1°C), humidity-controlled (40–60%)
environment with a 12-h light/dark cycle and food and water
available ad libitum. All animal experiments were approved
by the Guizhou University Subcommittee of Experimental Animal
Ethics (Guizhou, China). The current study followed the guidelines
of the Guide for the Care and Use of Laboratory Animals published
by the US National Institutes of Health (22).
Tissue collection
Ventricular heart tissues were isolated from
embryonic day 16.5 (E16.5), P1, P7, P28 and P56 C57BL/6J mice. Mice
were deeply anesthetized with 2% inhaled isoflurane and euthanized
by cervical dislocation. Then, hearts were dissected out and washed
with 0.9% NaCl. The hearts were further processed for total RNA
isolation and reverse transcription-quantitative PCR (RT-qPCR).
Ventricular CM and cardiac fibroblast
(CF) isolation
CMs were isolated on P1, P4 or P7 from C57BL/6J
mice, as previously described (10,11).
Briefly, the mice were anesthetized using 2% isoflurane inhalation.
The ventricles were separated from the atria, cut into pieces and
digested with 0.25% trypsin at 4°C overnight. Then, digestion was
performed two to three times using 10 mg collagenase type II and 50
mg BSA in 10 ml PBS at 37°C for 15 min under constant stirring.
Following digestion, the supernatant was collected with DMEM/F12
1:1 medium supplemented with 10% FBS. The collected supernatant was
centrifuged (500 × g) at room temperature for 5 min to harvest the
cells, which were resuspended in DMEM/F12 1:1 medium supplemented
with 10% FBS, 100 U/ml penicillin and 100 mg/ml streptomycin. The
collected cells, containing CMs and CFs were seeded onto 100-mm
plastic dishes for 2 h at 37°C in a humidified atmosphere of 5%
CO2. CMs and CFs were cultured via differential
adhesion, as described previously (23,24).
The CFs adhered to the uncoated cell-culture dish. The supernatant,
composed mostly of CMs, was then collected and pelleted. Following
this, the CMs were resuspended in DMEM/F12 containing 10% FBS,
counted using a BA210 Digital light microscope (magnification,
×100), and plated at the appropriate density (70–80%
confluency).
RNA isolation and RT-qPCR
Total RNA was isolated from cultured P1, P4, or P7
CMs or E16.5, P1, P7, P28 or P56 ventricular heart tissue using an
E.Z.N.A. Total RNA kit II, according to the manufacturer's
protocol. Nuclear and cytoplasmic RNAs were isolated using an
RNeasy Midi kit according to the manufacturer's protocol. RNAs were
treated with DNase I (1 µl for 1 µg RNA) for 15 min at room
temperature to exclude DNA contamination.
For the quantification of mRNA expression,
PrimeScript™ RT Master mix was used, according to the
manufacturer's protocol, to synthesize cDNA at 37°C for 15 min and
85°C for 5 sec. RT-qPCR was then performed with a SYBRs Premix Ex
Taq™ kit on a Lightcycler 480 (Roche Diagnostics). A total of 20 µl
reaction mixture was incubated at 95°C for 30 sec for initial
denaturation followed by 95°C for 5 sec and 60°C for 30 sec for 40
cycles. To normalize gene expression, β-actin was used as the
reference gene. For the quantification of miRNA expression, miRNA
First Strand cDNA Synthesis and miRNAs Quantitation PCR kits were
used according to the manufacturers' protocol. U6 was used as a
reference gene to normalize miRNA expression, according to previous
studies (25,26).
mRNA and miRNA relative expression levels were
analyzed using the 2−ΔΔCq method (27). The difference between the Cq values
of the target gene and internal reference gene in each group were
used as the ΔCq of each group, and the average value of ΔCq of each
group was subtracted from that of the control group to obtain the
ΔΔCq of each group; next, 2−ΔΔCq was used to calculate
the relative expression level of each group. To determine the
endogenous efficiency, cDNA samples were diluted to four
concentration gradients (cDNA dilution: 0.001, 0.01, 0.1, 1),
amplified with primers specific to β-actin and CDK6 and the average
Cq values of β-actin and CDK6, as well as the ΔΔCq values, were
calculated. Finally, ΔΔCq values were mapped using log values of
the cDNA concentration gradient. If the absolute slope of the
obtained straight line is close to zero, the amplification
efficiency of β-actin and CDK6 primers is equal. All primers were
designed by Sangon Biotech Co., Ltd. The primer sequences are
provided in Table I.
 | Table I.List of the sequences of the primers
used in the current study. |
Table I.
List of the sequences of the primers
used in the current study.
| Primer | Forward
(5′→3′) | Reverse
(5′→3′) |
|---|
| miR-449a-5p reverse
transcription primer |
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACCAGC |
| miR-449a-5p |
CGCGCGTGGCAGTGTATTGTTA |
ATCCAGTGCAGGGTCCGAGG |
| U6 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
| CDK6 |
TAGCTGTCTCCACCACCCAC |
GGCCATCTGTCGTTAGCCAG |
| RUNX1 |
AACCAGGTAGCGAGATTCAACGAC |
CAACTTGTGGCGGATTTGTAAAGA |
| c-kit |
CGGGCTAGCCAGAGACATCA |
TCTCTGGTGCCATCCACTTCA |
| DAB2 |
TAGTCCAACAGAAAGCAAAG |
GAGGTGACTCCATTTGTTAAG |
| β-actin |
TGCTGTCCCTGTATGCCTCTG |
TTGATGTCACGCACGATTTCC |
Fluorescence in situ hybridization
(FISH)
Isolated CMs were cultured on coverslips, fixed in
4% PFA for 30 min and washed with PBS at room temperature.
Following this, cells were permeabilized in 0.2% Triton X-100 in
PBS for 10 min at room temperature, washed with PBS three times,
hybridized with a hybridization solution (containing 20 mM
Tris-HCl, 0.9 M NaCl, 0.01% SDS, and 40% formamide) and incubated
with 10 µM labeled miR-449a-5p probe at 37°C for 12 h. The
miR-449a-5p probe was synthesized by Sangon Biotech Co., Ltd. The
cells were washed with 2X SSC (5 min), 1X SSC (5 min) and 0.5X SSC
(5 min) at room temperature, and incubated with DAPI for 2 h at
37°C. Image acquisition was performed using an LSM 880 confocal
microscope (magnification, ×1,000; Zeiss GmbH).
Transfection of plasmids, miRNA mimics
and inhibitors
The plasmid pcDNA-CDK6 and pcDNA-NC was synthesized
by Vigene. miR-449a-5p mimics, mimic-NCs (cat. no.
miR1N0000001-1-5), miR-449a-5p inhibitors, and inhibitor-NCs (cat.
no. miR2N0000001-1-5) were synthesized by Guangzhou Ribobio Co.,
Ltd. The sequences of miR-449a-5p mimics were
5′-UGGCAGUGUAUUGUUAGCUGGU-3′. The sequences of miR-449a-5p
inhibitors were 5′-ACCAGCUAACAAUACACUGCCA-3′. After isolated P1 CMs
were cultured for 48 h at 37°C, 5 µl Lipofectamine® 2000
and 50 nM plasmids, mimics or inhibitors were added to Opti-MEM.
The mixture was added to the cells following incubation at room
temperature for 20 min. This medium was replaced after 6 h at 37°C
with the same volume of DMEM/F12 medium. RNA or protein was
isolated and immunofluorescence analysis was conducted following 48
h.
Immunofluorescence analysis
For cultured CMs in 24-well plates, the culture
medium was washed with PBS, fixed with 4% PFA for 30 min at room
temperature, permeabilized with 0.2% Triton X-100 in PBS for 10 min
at room temperature, and blocked with PBS containing 1% BSA at room
temperature for 30 min. Cells were then incubated with primary
antibodies, including cTnT, ki67 or pH3, for 2 h at room
temperature, followed by washing with PBS and incubation with goat
anti-mouse IgG/Alexa Fluor 488 or goat anti-rabbit IgG/Alexa Fluor
555 secondary antibodies for 1 h at room temperature. Subsequently,
cells were washed with PBS and incubated with Hoechst 33342 at room
temperature for 30 min. Apoptotic cell death was determined via
TUNEL staining (Roche Diagnostics) at room temperature for 1 h,
according to the manufacturer's protocol. Image acquisition was
performed using an LSM 880 confocal microscope (magnification,
×200). To quantify CM proliferation, ki67 and pH3 marked cell
proliferation, while cTnT labeled CM areas. Hoechst 33342 was used
for nuclear counterstaining. Quantitative data were obtained by
measuring co-localization of Hoechst 33342 staining with ki67 or
pH3 in cTnT areas using ImageJ software (version no. 1.6.0;
National Institutes of Health). In total, four fields of each
section were randomly selected to examine for quantification.
Luciferase reporter assay
Luciferase reporter assays were performed as
previously described (28).
Wild-type (WT) CDK6 3′-untranslated region (UTR; CDK6-WT) and CDK6
3′-UTR mutant (MUT) derivatives devoid of the miR-449a-5p binding
site (CDK6-MU1 and CDK6-MU2) were cloned into the psiCHECKTM-2
vector (SaichengBio Co. Ltd.). After isolated CMs were cultured for
48 h, 5 µl Lipofectamine® 2000 and 50 nM
mimic-miR-449a-5p, CDK6-WT or CDK6-MU plasmids were added to
Opti-MEM. The mixture was added to the cells following incubation
at room temperature for 20 min. The medium was replaced after
incubation for 6 h at 37°C with the same volume of DMEM/F12 medium.
Cells were harvested at 48 h post-transfection and luciferase
activity was determined using a luciferase reporter system (Promega
Corporation), according to the manufacturer's protocol. The firefly
luciferase activity was normalized to Renilla luciferase
activity
Western blotting
Isolated CMs were lysed in ice-cold RIPA with
protease inhibitors and protein concentrations were detected using
a BCA Protein Quantitative Analysis kit. A total of 30 µg of
protein samples were electrophoresed using 10% SDS-PAGE and
transferred to PVDF membranes (EMD Millipore). The PVDF membranes
were incubated in blocking buffer [5% BSA in TBS-0.1%
Tween-20-(TBST) buffer] for 2 h at room temperature. Following
blocking, the PVDF membranes were incubated with primary antibodies
(CDK6, 36 kDa or β-actin, 42 kDa) in 5% BSA at 4°C overnight. After
washing with TBST, the PVDF membranes were incubated with donkey
anti-mouse IgG H&L or donkey anti-rabbit IgG H&L antibodies
for 1 h at room temperature. Images were acquired using the
chemiluminescence imager GeneGnome XRQ (Syngene). ImageJ software
(version no. 1.6.0; National Institutes of Health) was used to
calculate the relative density. The intensity of each protein was
normalized to β-actin.
miRNA target prediction
Putative miRNA targets were identified using the
miRNA target prediction tools miRmap (version no. 1.1) (29) and miRanda (version no. 3.3)
(30).
Statistical analysis
All experiments were repeated at least three times.
Data are presented as the mean ± SEM. Statistical analyses were
performed using SPSS software (version no. 20.0; IBM Corp.). For
comparison between two groups, an unpaired two-tailed Student's
t-test was used. For the comparison of ≥3 groups, one-way ANOVA
followed by the Least Significant Difference or Tukey's post hoc
test was used. For correlation analysis, Spearman's test was used.
P<0.05 was considered to indicate a statistically significant
difference.
Results
miR-449a-5p is upregulated in aged
mouse CMs
Firstly, the expression levels of miR-449a-5p in
embryonic day 16.5 (E16.5), P1, P7, P28 and P56 were detected
ventricular heart tissues. RT-qPCR results demonstrated that
miR-449a-5p expression was significantly increased during cardiac
development (Fig. 1A).
Additionally, miR-449a-5p expression progressively increased in
isolated P1, P4 and P7 CMs (Fig.
1B). These results suggested that miR-449a-5p expression
gradually increased with age. miR-449a-5p was highly expressed in
CMs compared with CFs (Fig. 1C).
RT-qPCR results identified that miR-449a-5p was mainly expressed in
the cytoplasm of CMs (Fig. 1D).
This was further demonstrated via FISH assays results, which
indicated that miR-449a-5p was primarily located in the cytoplasm
of CMs (Fig. 1E).
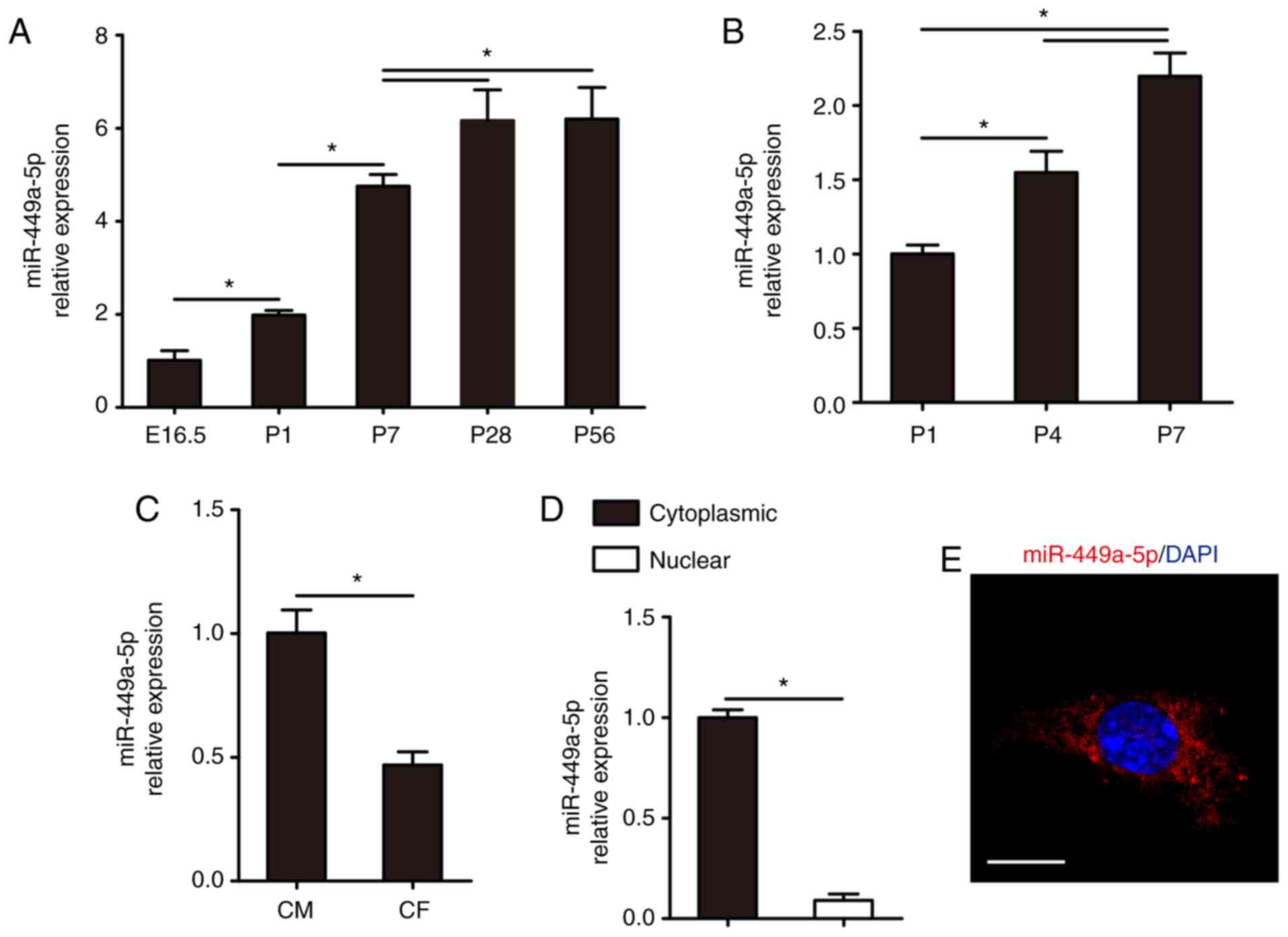 | Figure 1.Expression of miR-449a-5p in mouse
hearts and CMs. RT-qPCR results for miR-499a-5p expression in the
(A) hearts from E16.5, P1, P7, P28 and P56 mice (n=5) and in (B)
CMs isolated from P1, P4 and P7 mice (n=3). (C) RT-qPCR results of
miR-449a-5p expression in isolated P1 CMs and CFs (n=3). (D)
Cytoplasmic and nuclear fractions for the abundance of miR-449a-5p
expression in isolated P1 CMs (n=3). (E) RNA FISH assay results for
miR-449a-5p detection in isolated P1 CMs (n=3). Scale bar, 20 µm.
Statistical significance was calculated using one-way ANOVA
followed by Tukey's test in (A), Least Significant Difference post
hoc test in (B) and two-tailed unpaired Student's t-test in (C and
D). Error bars represent mean ± SEM. *P<0.05 vs. indicated
groups. miR, microRNA; CMs, cardiomyocytes; RT-qPCR, reverse
transcription-quantitative PCR; E, embryonic; P, postnatal; CFs,
cardiac fibroblasts. |
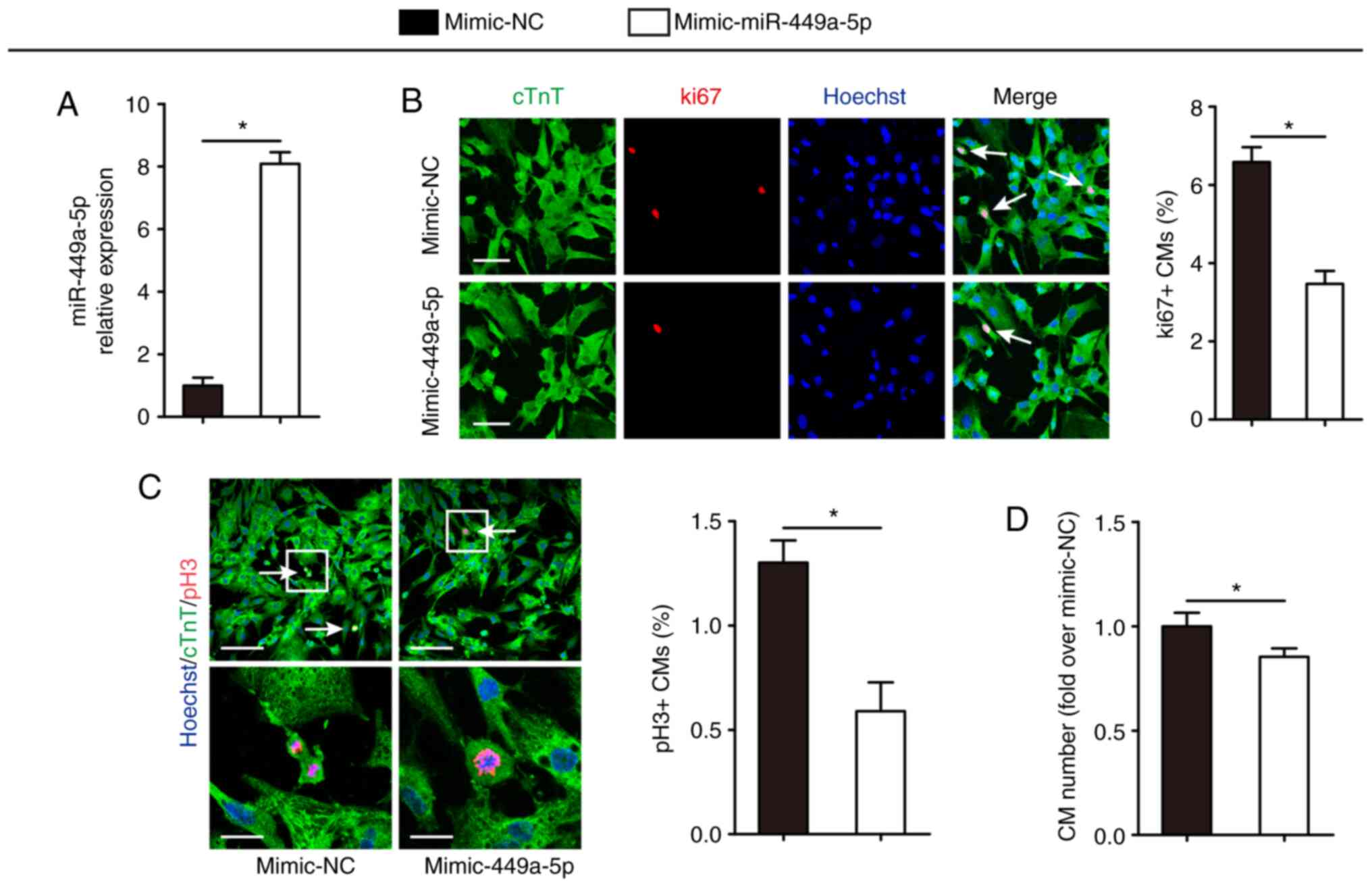
Overexpression of miR-449a-5p inhibits
CM proliferation
To investigate the potential role of miR-449a-5p in
CM proliferation, isolated P1 CMs were transfected with mimic-NCs
or mimic-miR-449a-5p. RT-qPCR results demonstrated that
mimic-miR-449a-5p significantly overexpressed miR-449a-5p in
isolated P1 CMs compared with the mimic-NC group (Fig. 2A). Following this, CM proliferation
was examined using the cell cycle activity marker ki67 and the
mitosis marker pH3. Compared with the mimic-NC, miR-449a-5p
overexpression significantly decreased the percentage of
ki67-positive CMs (6.58±0.38% vs. 3.47±0.33%, respectively;
Fig. 2B) and the percentage of
pH3-positive CMs (1.30±0.18% vs. 0.59±0.24%, respectively; Fig. 2C). Furthermore, miR-449a-5p
overexpression decreased the number of P1 CMs to 0.85±0.04-fold
(Fig. 2D).
Whether miR-449a-5p had an effect on CM
dedifferentiation was then examined. Previous studies have reported
that the stem/progenitor markers Runt related transcription factor
1 (RUNX1), mast/stem cell growth factor receptor kit (c-kit), and
differentially-expressed protein 2 (DAB2) identify dedifferentiated
CMs (31,32). RT-qPCR results indicated that
miR-449a-5p mimic significantly inhibited RUNX1, c-kit and DAB2
expression levels in isolated P1 CMs compared with the mimic-NC
group (Fig. S1A), suggesting that
miR-449a-5p inhibited CM dedifferentiation. TUNEL staining results
identified that miR-449a-5p overexpression significantly increased
isolated P1 CM apoptosis compared with the mimic-NC group (Fig. S1B). These findings demonstrated
that miR-449a-5p may be an endogenous regulator of neonatal CM
proliferation.
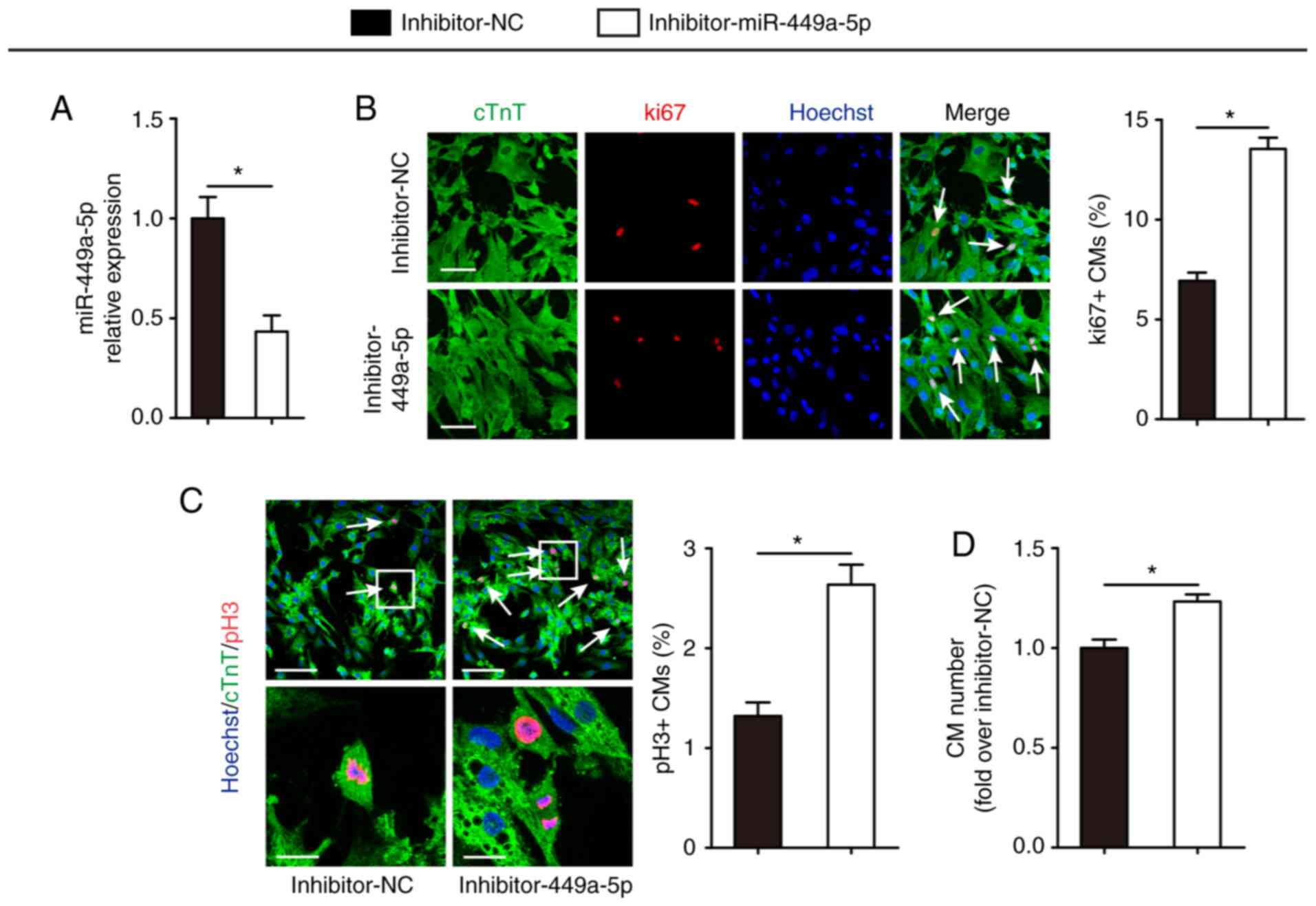
Knockdown of miR-449a-5p stimulates CM
proliferation
Having established that miR-449a-5p overexpression
inhibited CM proliferation, it was then evaluated whether knockdown
of miR-449a-5p had an effect on CM proliferation. Transfection of
isolated P1 CMs with inhibitor-miR-449a-5p led to a significant
decrease in miR-449a-5p expression compared with the inhibitor-NC
group (Fig. 3A). The effect of
miR-449a-5p knockdown on CM proliferation was examined using ki67
and pH3 immunofluorescence staining. Compared with the inhibitor-NC
group, silencing of miR-449a-5p significantly increased the number
of ki67-positive (6.93±0.41% vs. 13.55±0.56%, respectively;
Fig. 3B) and pH3-positive CMs
(1.32±0.14% vs. 2.64±0.20%; Fig.
3C). Compared with the inhibitor-NC group, miR-449a-5p
knockdown demonstrated a 1.23±0.04-fold increase in the numbers of
CMs (Fig. 3D). These data indicated
that miR-449a-5p regulated neonatal CM proliferation.
CDK6 is a direct target gene of
miR-449a-5p
Since miRNAs regulate biological function mainly by
binding to the 3′-UTR of the target miRNAs (33), miRmap (29) and miRanda (30) were used to perform a bioinformatics
search. CDK6, an important cell cycle regulator that induces cell
cycle progression and cell proliferation (34,35),
was identified as a potential target of miR-449a-5p (Fig. 4A). Using a reporter construct with
the putative miR-449a-5p binding site of CDK6 3′-UTR downstream of
the luciferase gene, the results demonstrated that miR-449a-5p
overexpression significantly decrease the luciferase activity of
the reporter gene of CDK6 from 100.00±12.22 to 55.63±6.77%, whereas
this reduction was abrogated when the seed sequences of the 3′-UTR
binding site were mutated (Fig.
4B). These results suggested that CDK6 was a target for
miR-449a-5p.
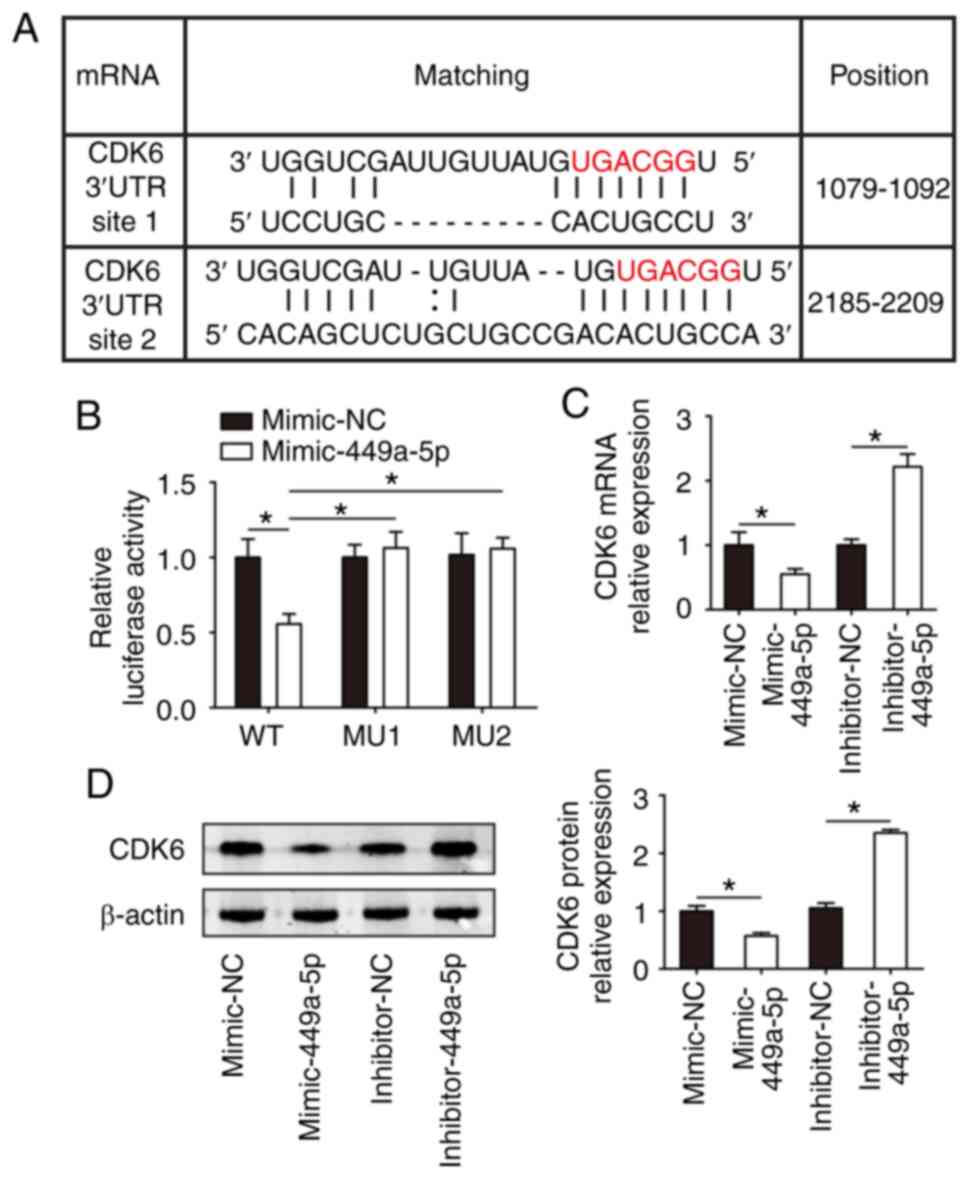 | Figure 4.CDK6 is a target gene of miR-449a-5p.
(A) Binding sites for miR-449a-5p on the 3′-UTR of CDK6 as
predicted using miRmap and miRanda. (B) Luciferase assays of
isolated P1 CMs transfected with luciferase-CDK6-WT,
luciferase-CDK6-MU site 1 and luciferase-CDK6-MU site 2 (n=3). (C)
Reverse transcription-quantitative PCR assays detected CDK6 mRNA
expression in isolated P1 CMs transfected with mimic-NC,
mimic-miR-449a-5p, inhibitor-NC or inhibitor-miR-449a-5p (n=3). (D)
Western blotting results and semi-quantitative analyses of CDK6
protein expression in isolated P1 CMs transfected with mimic-NC,
mimic-miR-449a-5p, inhibitor-NC or inhibitor-miR-449a-5p (n=3).
Statistical significance was calculated using one-way ANOVA
followed by Tukey's test in (B) or Least Significant Difference
post hoc test in (C and D) Error bars represent mean ± SEM.
*P<0.05 vs. indicated groups. miR, microRNA; UTR, untranslated
region; P, postnatal; CMs, cardiomyocytes; WT, wild-type; MU,
mutant; NC, negative control. |
Subsequently, the endogenous efficiency of the
RT-qPCR samples was tested according to a previous study that
analyzed relative gene expression data using the 2−ΔΔCq
method (27). The results revealed
that the efficiency of primer amplification of CDK6 and β-actin was
equal in the RT-qPCR experiments (Fig.
S2). Therefore, β-actin as was used as the internal
reference.
Then, whether miR-449a-5p had an effect on CDK6 in
CMs was evaluated. CDK6 mRNA and protein expression was
significantly decreased in isolated CMs transfected with
mimic-miR-449a-5p compared with the NC group (Fig. 4C and D); however, the expression
levels were significantly increased in the inhibitor-miR-449a-5p
group compared with the inhibitor-NC group (Fig. 4C and D). These results indicated
that miR-449a-5p bound directly to CDK6 mRNA and inhibited CDK6
expression.
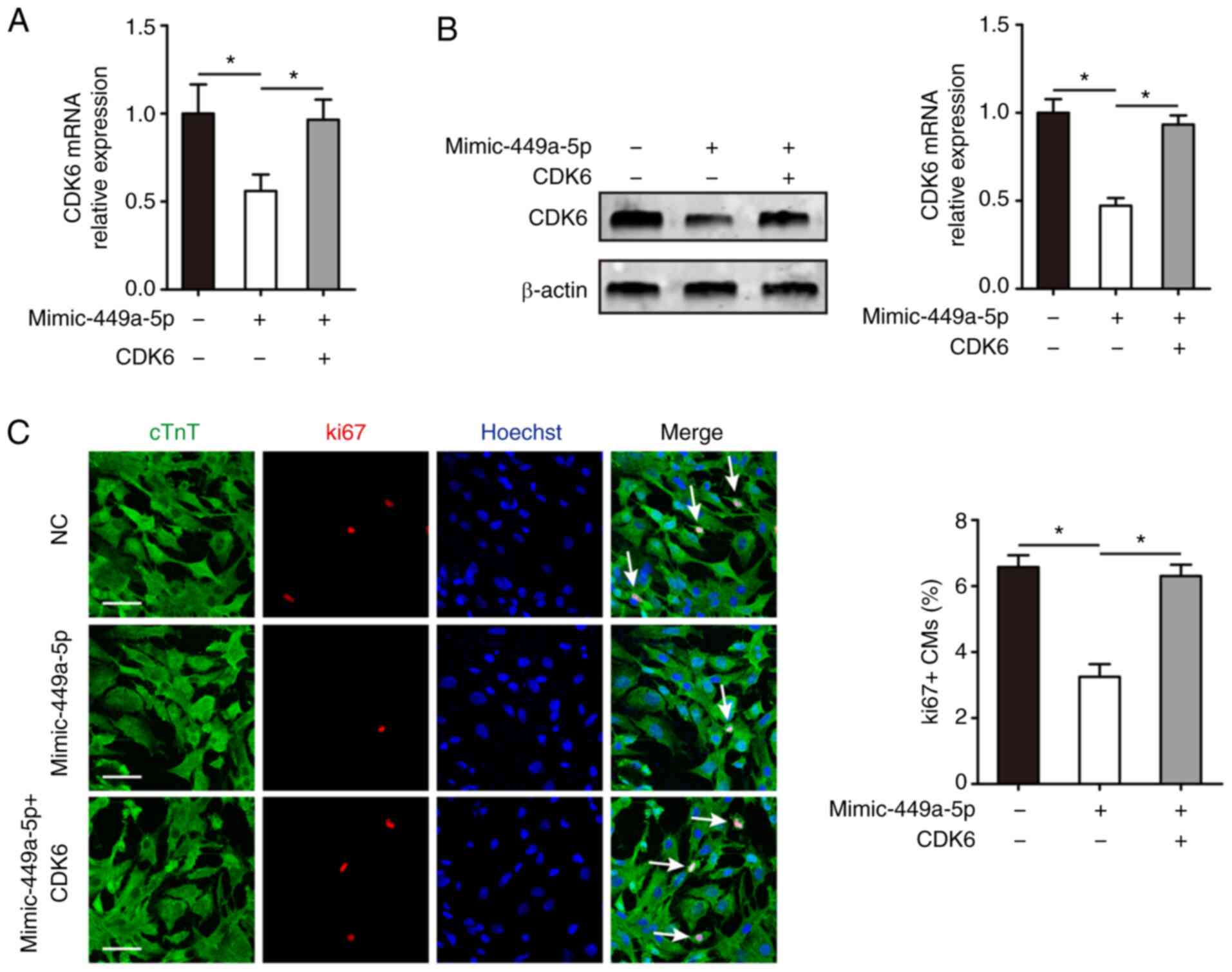
CDK6 overexpression induces CM
proliferation
The effects of CDK6 on CM proliferation were
evaluated. RT-qPCR confirmed that CDK6 mRNA expression
significantly decreased during mouse heart development (Fig. 5A and B). Following this, the
correlation between miR-449a-5p and CDK6 was determined by
examining their expression levels in 15 ventricular heart samples
from mice of different ages (E16.5, P1, P7, P28 or P56). A very
strong negative correlation was found between miR-449a-5p and CDK6
mRNA expression levels (r=−0.957) in the samples (Fig. 5C). Furthermore, it was detected
whether CDK6 induced CM proliferation. RT-qPCR results demonstrated
that CDK6 mRNA expression was upregulated in isolated CMs
transfected with pcDNA-CDK6 compared with pcDNA-NC (Fig. 5D). Western blotting confirmed that
CDK6 protein expression was upregulated in the pcDNA-CDK6 group
compared with the pcDNA-NC group (Fig.
5E). CM proliferation was assessed via ki67 immunofluorescence
staining, which revealed that CDK6 promoted CM proliferation in the
pcDNA-CDK6-NC vs. the pcDNA-CDK6 group (6.35±0.51% vs. 12.52±0.60%,
respectively; Fig. 5F).
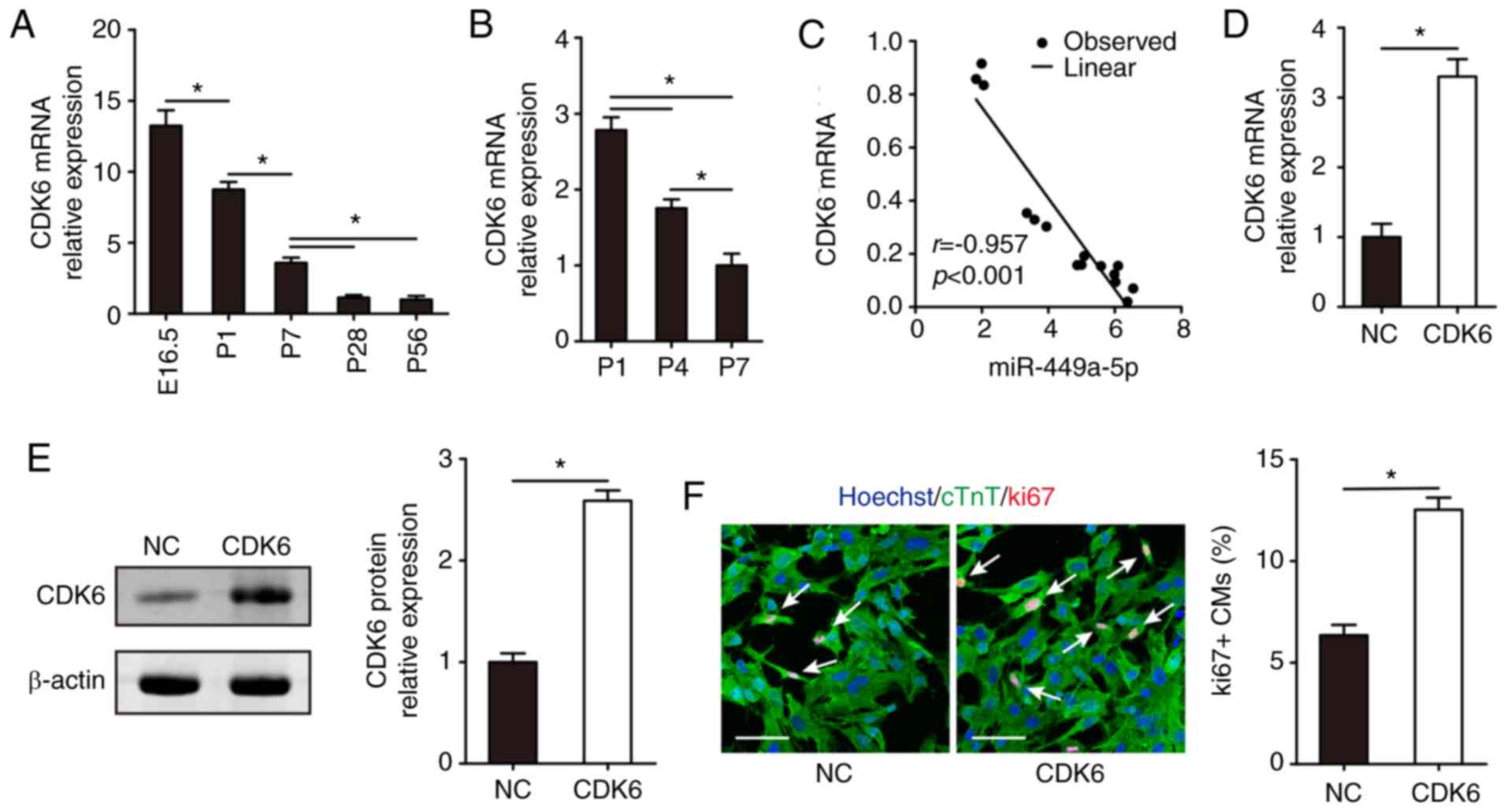 | Figure 5.CDK6 overexpression promotes CM
proliferation. (A) Reverse transcription-quantitative PCR results
of (A) CDK6 mRNA expression in the hearts of E16.5, P1, P7, P28 and
P56 mice (n=5) and (B) CDK6 mRNA expression in CMs isolated from
P1, P4 and P7 mice (n=3). (C) Correlation between miR-449a-5p and
CDK6 mRNA expression levels in 15 ventricular heart samples from
different aged mice. (D) CDK6 mRNA expression in isolated P1 CMs
transfected with NC or pcDNA-CDK6 (n=3). (E) Western blotting
results and semi-quantitative analyses of CDK6 protein expression
in isolated P1 CMs transfected with NC or pcDNA-CDK6 (n=3). (F)
Ki67 immunofluorescence staining in isolated P1 CMs transfected
with NC or pcDNA-CDK6 and quantification of ki67-positive CMs
(n=3). Ki67-positive CMs were indicated by arrows. Scale bar, 50
µm. Statistical significance was calculated using one-way ANOVA
followed by Tukey's test in (A), Least Significant Difference post
hoc test in (B), Spearman's test in (C) and two-tailed unpaired
Student's t-test in (D-F). Error bars represent mean ± SEM.
*P<0.05 vs. indicated groups. CM, cardiomyocyte; E, embryonic;
P, postnatal; miR, microRNA; NC, negative control; cTnT, cardiac
troponin T. |
miR-449a-5p inhibits CM proliferation
by targeting CDK6
Whether CDK6 was involved in the functional role of
miR-449a-5p in CM proliferation was also investigated.
Mimic-miR-449a-5p and pcDNA-CDK6 were co-transfected into isolated
P1 CMs. RT-qPCR and western blotting results demonstrated that CDK6
mRNA and protein expression was significantly decreased in CMs
transfected with mimic-miR-449a-5p alone compared with the control
group, but significantly increased in the mimic-miR-449a-5p +
pcDNA-CDK6 group compared with the mimic-miR-449a-5p group
(Fig. 6A and B). The ki67
immunofluorescence staining assay identified that co-transfection
with the mimic-miR-449a-5p and pcDNA-CDK6 abrogated the decline in
ki67-positive CMs induced by mimic-miR-449a-5p (6.30±0.34% vs.
3.25±0.38%, respectively; Fig. 6C).
Collectively, the results indicated that CDK6 mediated the
regulatory role of miR-449a-5p in CM proliferation.
Discussion
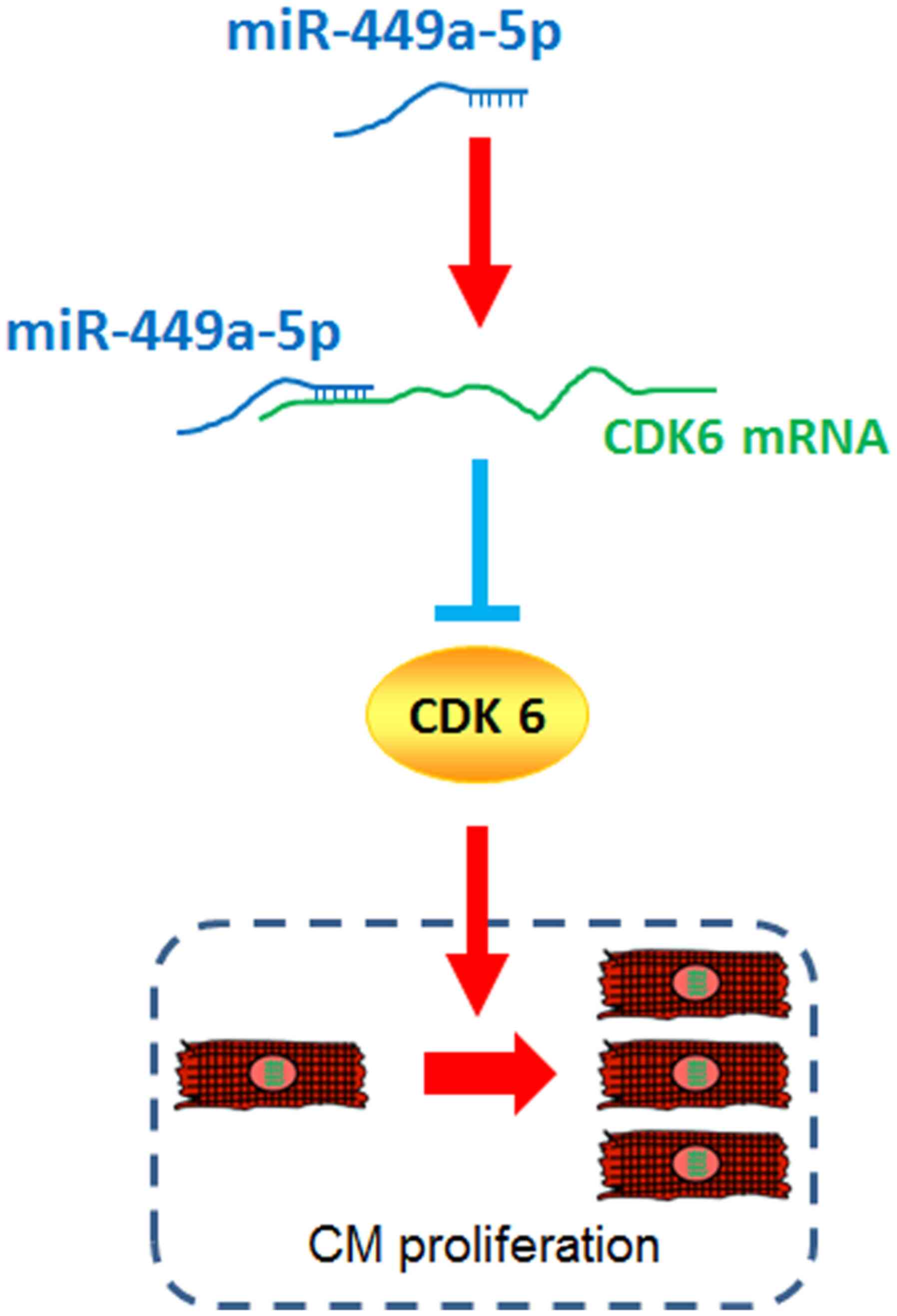
The present study demonstrated that miR-449a-5p was
a negative regulator of CM proliferation. Additionally, miR-449a-5p
directly bound the CDK6 mRNA 3′-UTR and inhibited CDK6 expression.
Thus, it was suggested that CDK6 mediated miR-449a-5p-inhibited CM
proliferation (Fig. 7). These
findings indicated that miR-449a-5p may be a valuable therapeutic
target for activating cardiac regeneration in the treatment cardiac
diseases.
Increasing numbers of miRNAs, including miR-17–92
(12), miR-128 (36) and miR-590 (11), have been observed to regulate CM
proliferation. miR-449a-5p has been reported to be associated with
CM hypoxia/reoxygenation injury (18). miR-449a-5p and miR-34a contain
distinct seed regions, which suggests that miR-449a-5p and miR-34a
may have similar functions (19,37).
miR-34a has been proven to regulate CM proliferation and cardiac
regeneration in post-myocardial infarction (10). Therefore, it was hypothesized that
miR-449a-5p may regulate CM proliferation. miR-449a-5p has been
identified to negatively regulate CM proliferation from previous
high-throughput functional screening data (11). In the present study, by quantifying
miR-449a-5p expression levels in CMs in mice at different ages,
miR-449a-5p was found to be increased during CM proliferation,
accompanied by the gradual decline in the proliferative ability of
CMs. There was low miR-449a-5p expression in the embryonic heart,
increased expression during the neonatal period and very high
expression at the adult stages.
To further investigate whether miR-449a-5p regulated
CM proliferation, CM proliferation in isolated mouse CMs was
evaluated using labeling proliferation-related markers. CMs were
immunostained with previously used markers, ki67 and pH3, which are
conventional markers of CM proliferation and mitosis, respectively
(11,31). The present results suggested that
miR-449a-5p overexpression led to a significant decrease in the
percentage of ki67- and pH3-positive CMs, as well as the numbers of
CMs. However, knockdown of miR-449a-5p caused an increase in ki67-
and pH3-positive CMs. The present study also investigated whether
miR-449a-5p caused physiological myocardial growth and development
or myocardial damage. Changes in stem/progenitor markers RUNX1,
c-kit and DAB2 expression levels and in CM apoptosis were detected
using TUNEL staining. The results demonstrated that miR-449a-5p
overexpression inhibited CM dedifferentiation markers and induced
CM apoptosis. Consistent with the present results, previous studies
have reported that miR-449a-5p inhibition protected CMs against
apoptosis (18,38). These results support the current
hypothesis that miR-449a-5p inhibits CM proliferation.
The present study found that miR-449a-5p regulated
CM proliferation by targeting CDK6. CDK6 is a cyclin D activated
kinase that phosphorylates the Rb in the G1 phase and
associates with E2F to regulate G1 to S transition in
the cell cycle (21). Consistent
with previous studies (39,40), the current results demonstrated that
the expression of CDK6 was high in the embryonic heart, decreased
during the neonatal period and was very low at the adult stages.
Thus, the expression pattern of CDK6 was opposite to that of
miR-449a-5p. A previous study reported that miR-449a-5p directly
targets CDK6 to suppress cell proliferation in neuroblastoma
(16). The current study also
observed that CDK6 was a target gene of miR-449a-5p in CM
proliferation. Additionally, the present results identified that
CDK6 overexpression promoted CM proliferation, and the
overexpression of CDK6 rescued the inhibitory effects caused by
miR-449a-5p in CM proliferation. Collectively, these results
suggested that miR-449a-5p inhibited CM proliferation via decreased
CDK6 expression.
The loss of CMs and their deficient regeneration
capability are major contributors to the pathogenesis of numerous
cardiac diseases, such as heart failure (41,42).
Promoting CM proliferation is hypothesized to be one of the most
effective regenerative therapeutic strategies to prevent
exacerbation of heart failure (3,4).
Previous studies have revealed that the inhibition of negative
miRNA regulators of proliferation, including the miR-15 family
(43) or, conversely, the
enhancement of proliferative miRNAs, such as miR-199a and −590
(11), lead to increased CM
proliferation and improved cardiac function post-myocardial injury.
Combined with the current results, these research studies highlight
the mechanistic and therapeutic significance of miRNA regulation in
CM proliferation. Therefore, treating the injured heart directly
with miRNAs with regenerative abilities or targeting miRNA with
small molecular drugs may improve cardiac function in patients with
cardiac injury (44,45).
The present study had several limitations. Firstly,
although the effects of miR-449a-5p on CM proliferation were
verified in vitro, additional comprehensive experiments
in vivo may aid to further detect the role of miR-449a-5p.
Secondly, whether miR-449a-5p regulates cardiac regeneration in
myocardial infarction and heart failure should be clarified in
future studies.
In conclusion, the present study identified that
miR-449a-5p regulated CM proliferation by inhibiting CDK6. These
results indicated that miR-449a-5p may act as a novel effective
gene target for treatment approaches for myocardial injury.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The current work was supported by grants from the
Science and Technology Plan of Guizhou Province [Guizhou Science
and Technology Cooperation Foundation; grant no. (2019)1260] and
the National Natural Science Foundation of China (grant no.
81960083).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
BL and XS conceived the current study and designed
experiments. BL, FY, JH and XH performed the experiments. ZW, SD
and MT analyzed the data. BL and XS wrote the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The animal study was approved by the Guizhou
University Subcommittee of Experimental Animal Ethics.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Organization WH, . The top 10 causes of
death. simplehttps://www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death2018.
|
|
2
|
Li M and Izpisua Belmonte JC: Mending a
faltering heart. Circ Res. 118:344–351. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yuan X and Braun T: Multimodal regulation
of cardiac myocyte proliferation. Circ Res. 121:293–309. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Leach JP and Martin JF: Cardiomyocyte
proliferation for therapeutic regeneration. Curr Cardiol Rep.
20:632018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li B, Hu Y, Li X, Jin G, Chen X, Chen G,
Chen Y, Huang S, Liao W, Liao Y, et al: Sirt1 antisense long
noncoding RNA promotes cardiomyocyte proliferation by enhancing the
stability of Sirt1. J Am Heart Assoc. 7:e0097002018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wojciechowska A, Braniewska A and
Kozar-Kaminska K: MicroRNA in cardiovascular biology and disease.
Adv Clin Exp Med. 26:865–874. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bernardo BC, Ooi JY, Lin RC and McMullen
JR: miRNA therapeutics: A new class of drugs with potential
therapeutic applications in the heart. Future Med Chem.
7:1771–1792. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Colpaert RMW and Calore M: MicroRNAs in
cardiac diseases. Cells. 8:7372019. View Article : Google Scholar
|
|
9
|
Tian Y, Liu Y, Wang T, Zhou N, Kong J,
Chen L, Snitow M, Morley M, Li D, Petrenko N, et al: A
microRNA-Hippo pathway that promotes cardiomyocyte proliferation
and cardiac regeneration in mice. Sci Transl Med. 7:279ra2382015.
View Article : Google Scholar
|
|
10
|
Yang Y, Cheng HW, Qiu Y, Dupee D, Noonan
M, Lin YD, Fisch S, Unno K, Sereti KI and Liao R: MicroRNA-34a
plays a key role in cardiac repair and regeneration following
myocardial infarction. Circ Res. 117:450–459. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eulalio A, Mano M, Dal Ferro M, Zentilin
L, Sinagra G, Zacchigna S and Giacca M: Functional screening
identifies miRNAs inducing cardiac regeneration. Nature.
492:376–381. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen J, Huang ZP, Seok HY, Ding J, Kataoka
M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, et al: mir-17-92 cluster
is required for and sufficient to induce cardiomyocyte
proliferation in postnatal and adult hearts. Circ Res.
112:1557–1566. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liang D, Li J, Wu Y, Zhen L, Li C, Qi M,
Wang L, Deng F, Huang J, Lv F, et al: miRNA-204 drives
cardiomyocyte proliferation via targeting Jarid2. Int J Cardiol.
201:38–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bou Kheir T, Futoma-Kazmierczak E,
Jacobsen A, Krogh A, Bardram L, Hother C, Grønbaek K, Federspiel B,
Lund AH and Friis-Hansen L: miR-449 inhibits cell proliferation and
is down-regulated in gastric cancer. Mol Cancer. 10:292011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sandbothe M, Buurman R, Reich N, Greiwe L,
Vajen B, Gürlevik E, Schäffer V, Eilers M, Kühnel F, Vaquero A, et
al: The microRNA-449 family inhibits TGF-β-mediated liver cancer
cell migration by targeting SOX4. J Hepatol. 66:1012–1021. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao Z, Ma X, Sung D, Li M, Kosti A, Lin
G, Chen Y, Pertsemlidis A, Hsiao TH and Du L: microRNA-449a
functions as a tumor suppressor in neuroblastoma through inducing
cell differentiation and cell cycle arrest. RNA Biol. 12:538–554.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yong-Ming H, Ai-Jun J, Xiao-Yue X,
Jian-Wei L, Chen Y and Ye C: miR-449a: A potential therapeutic
agent for cancer. Anticancer Drugs. 28:1067–1078. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng J, Wu Q, Lv R, Huang L, Xu B, Wang
X, Chen A and He F: MicroRNA-449a inhibition protects H9C2 cells
against Hypoxia/Reoxygenation-induced injury by targeting the
Notch-1 signaling pathway. Cell Physiol Biochem. 46:2587–2600.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song R, Walentek P, Sponer N, Klimke A,
Lee JS, Dixon G, Harland R, Wan Y, Lishko P, Lize M, et al:
miR-34/449 miRNAs are required for motile ciliogenesis by
repressing cp110. Nature. 510:115–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen H, Lin YW, Mao YQ, Wu J, Liu YF,
Zheng XY and Xie LP: MicroRNA-449a acts as a tumor suppressor in
human bladder cancer through the regulation of pocket proteins.
Cancer Lett. 320:40–47. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ezhevsky SA, Nagahara H, Vocero-Akbani AM,
Gius DR, Wei MC and Dowdy SF: Hypo-phosphorylation of the
retinoblastoma protein (pRb) by cyclin D:Cdk4/6 complexes results
in active pRb. Proc Nati Acad Sci USA. 94:10699–10704. 1997.
View Article : Google Scholar
|
|
22
|
National Research Council Committee for
the Update of the Guide for the C and Use of Laboratory A, . The
National Academies Collection: Reports funded by National
Institutes of Health. Guide for the Care and Use of Laboratory
Animals. National Academies Press (US). Copyright © 2011, National
Academy of Sciences, Washington (DC). 2011, simplehttps://www.ncbi.nlm.nih.gov/books/NBK4119/Feb
02–2020PubMed/NCBI
|
|
23
|
Ehler E, Moore-Morris T and Lange S:
Isolation and culture of neonatal mouse cardiomyocytes. J Vis Exp.
79:501542013.
|
|
24
|
Mbogo GW, Nedeva C and Puthalakath H:
Isolation of cardiomyocytes and cardiofibroblasts for ex vivo
analysis. Methods Mol Biol. 1419:117–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li X, He X, Wang H, Li M, Huang S, Chen G,
Jing Y, Wang S, Chen Y, Liao W, et al: Loss of AZIN2 splice variant
facilitates endogenous cardiac regeneration. Cardiovasc Res.
114:1642–1655. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu X, Xiao J, Zhu H, Wei X, Platt C,
Damilano F, Xiao C, Bezzerides V, Bostrom P, Che L, et al: miR-222
is necessary for exercise-induced cardiac growth and protects
against pathological cardiac remodeling. Cell Metab. 21:584–595.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang J, Gong Y, Cai J, Liu Q and Zhang Z:
Lnc-3215 suppression leads to calcium overload in selenium
deficiency-induced chicken heart lesion via the
lnc-3215-miR-1594-TNN2 pathway. Mol Ther Nucleic Acids. 18:1–15.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vejnar CE and Zdobnov EM: MiRmap:
Comprehensive prediction of microRNA target repression strength.
Nucleic Acids Res. 40:11673–11683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Enright AJ, John B, Gaul U, Tuschl T,
Sander C and Marks DS: MicroRNA targets in Drosophila. Genome Biol.
5:R12003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
D'Uva G, Aharonov A, Lauriola M, Kain D,
Yahalom-Ronen Y, Carvalho S, Weisinger K, Bassat E, Rajchman D,
Yifa O, et al: ERBB2 triggers mammalian heart regeneration by
promoting cardiomyocyte dedifferentiation and proliferation. Nat
Cell Biol. 17:627–638. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kubin T, Pöling J, Kostin S, Gajawada P,
Hein S, Rees W, Wietelmann A, Tanaka M, Lörchner H, Schimanski S,
et al: Oncostatin M is a major mediator of cardiomyocyte
dedifferentiation and remodeling. Cell Stem Cell. 9:420–432. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sherr CJ, Beach D and Shapiro GI:
Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov.
6:353–367. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tigan AS, Bellutti F, Kollmann K, Tebb G
and Sexl V: CDK6-a review of the past and a glimpse into the
future: From cell-cycle control to transcriptional regulation.
Oncogene. 35:3083–3091. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang W, Feng Y, Liang J, Yu H, Wang C,
Wang B, Wang M, Jiang L, Meng W, Cai W, et al: Loss of microRNA-128
promotes cardiomyocyte proliferation and heart regeneration. Nat
Commun. 9:7002018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mercey O, Popa A, Cavard A, Paquet A,
Chevalier B, Pons N, Magnone V, Zangari J, Brest P and Zaragosi
LE,s: Characterizing isomiR variants within the microRNA-34/449
family. FEBS Lett. 591:693–705. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang X, Dong H, Liu Y, Han J, Tang S and
Si J: Tetramethylpyrazine partially relieves hypoxia-caused damage
of cardiomyocytes H9c2 by downregulation of miR-449a. J Cell
Physiol. Feb 15–2019.(Epub ahead of print). doi:
10.1002/jcp.28151.
|
|
39
|
Kim WH, Joo CU, Ku JH, Ryu CH, Koh KN, Koh
GY and Ko JK: Cell cycle regulators during human atrial
development. Korean J Intern Med. 13:77–82. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ponnusamy M, Li PF and Wang K:
Understanding cardiomyocyte proliferation: An insight into cell
cycle activity. Cell Mol Life Sci. 74:1019–1034. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gill C, Mestril R and Samali A: Losing
heart: The role of apoptosis in heart disease-a novel therapeutic
target? FASEB J. 16:135–146. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Petrovic D: Cytopathological basis of
heart failure-cardiomyocyte apoptosis, interstitial fibrosis and
inflammatory cell response. Folia Biol (Praha). 50:58–62.
2004.PubMed/NCBI
|
|
43
|
Porrello ER, Mahmoud AI, Simpson E,
Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA,
Olson EN and Sadek HA: Regulation of neonatal and adult mammalian
heart regeneration by the miR-15 family. Proc Natl Acad Sci USA.
110:187–192. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lock MC and Tellam RL: The role of miRNA
regulation in fetal cardiomyocytes, cardiac maturation and the risk
of heart disease in adults. The J Physiol. 596:5625–5640. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Giacca M and Zacchigna S: Harnessing the
microRNA pathway for cardiac regeneration. J Mol Cell Cardiol.
89:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|