Introduction
Aortic stenosis (AS) is the most common type of
heart valve disease with an increasing prevalence; in 11,911 people
with different age, sex and ethnic characteristics heart valve
disease accounted for 5.2% of the elderly (>75 years) population
(1). The narrowing of the exit of
the left ventricle (LV) of the heart results in chronic LV pressure
overloading (2). This sustained
myocardial stress leads to pathological LV remodeling,
characterized by concentric hypertrophy and interstitial and
perivascular fibrosis (3), which
increases the risk of heart failure and of mortality (25%)
(4). The average overall survival
rate in symptomatic patients with AS without aortic valve
replacement is 2–3 years (5,6).
Surgery to repair or replace the valve releases the biomechanical
stress and improves LV function, and is recommended as the only
lifesaving therapy for symptomatic patients (6). However, structural abnormalities in
severe cases are only partially reversed following valve
replacement (7). There is currently
no preventive therapy for LV structural damage secondary to AS
(2). Understanding the mechanisms
involved in pressure overload-induced cardiac remodeling may enable
development of novel drugs to delay the progression of structural
abnormality before surgery and promote recovery following valve
replacement.
Inflammation is a well-known contributing factor of
atherosclerotic cardiovascular disease (8). Chemokines and their receptors regulate
immune cell recruitment and activation and serve an important role
in atherosclerosis (9). Evidence
suggests that ‘atherosclerosis-like’ pathogenesis is involved in
the initiation of AS (10).
Chemokines and their receptors have also been implicated in the
pathophysiology of LV remodeling and cardiac dysfunction caused by
pressure overload (11–13). In cardiac-specific transgenic mice,
C-C chemokine receptor (CCR)9 knockout attenuates, whereas CCR9
overexpression enhances, pressure overload-induced cardiac
hypertrophy (12). Serum chemokine
(C-C motif) ligand (CCL) 21 levels are elevated in patients with AS
and in mice exposed to LV pressure overload. Moreover, knockout of
the CCL21 receptor CCR7 prevents pressure overload-induced LV wall
thickening and functional impairment (13). CCR5 is involved in a number of
autoimmune and inflammatory diseases, including rheumatoid
arthritis and juvenile idiopathic arthritis, and potent CCR5
antagonists (e.g., Maraviroc) have been developed as potential
therapeutics (14). CCR5
polymorphism has been linked to the degree of calcification of
stenotic aortic valves (15).
However, the role of CCR5 in cardiac remodeling and dysfunction
under pressure overload is unclear.
Transverse aortic constriction (TAC) in mice is a
common animal model used to study pressure overload-induced cardiac
hypertrophy and dysfunction (16).
In the present study, expression levels of CCR5 and its ligands
were assessed in mice subjected to TAC. The effects of CCR5
inhibition on TAC-induced cardiac hypertrophy and dysfunction, as
well as the molecular mechanisms involved, were also
investigated.
Materials and methods
TAC
C57BL/6 mice (female; age 8–10 weeks; weight 18–25
g) were purchased from Shanghai Laboratory Animal Center (Shanghai,
China). The breeding conditions were: 21±2°C, 30–70% humidity, 12-h
light/dark cycle and free access to food and water. TAC surgery was
performed as described previously (17). The mice were deeply anesthetized by
intraperitoneal injection of 10% chloral hydrate (300 mg/kg); mice
showed no signs of peritonitis following injection of chloral
hydrate. Anesthesia was indicated by decreased limb tension and
slow corneal reflex. Briefly, aortic occlusion was achieved by
tying a suture around the transverse aorta over a juxtaposed
27-gauge blunted needle placed between the innominate and left
carotid arteries. The needle was then removed, resulting in a
severely narrowed aortic lumen. Hearts were collected for analysis
at specific time-points during a study period of 28 days.
Sham-operated mice without TAC served as controls. Following TAC
surgery, mice received once-daily intraperitoneal injection of 0.5
µg/g goat anti-mouse CCR5 antibody (1:500; cat. no. ab65850;
Abcam), Rantes (CCR5 agonist 1:500; cat. no. ab189841; Abcam) or
isotype IgG control antibody (1:1,000; cat. no. ab190475; Abcam).
All animal studies were approved by the Medical Ethics Committee of
Baotou Central Hospital (Baotou, China) and complied with the
Principles of Laboratory Animal Care (National Society for Medical
Research). All experiments were performed in accordance with the
National Institutes of Health (NIH) guidelines (https://www.nih.gov/). Every effort was made to
minimize suffering and the number of animals. A total of 177 mice
were used in this experiment; experimental grouping and
distribution of mice are presented in Table I. A total of two mice were
anesthetized and died due to heart puncture and heavy bleeding due
to surgical errors during the modeling process, which was within
the acceptable limits of the ethical approval obtained for the
present study. Health of the mice was monitored according to the
time-points used in the experiment (immediately following and at 3,
7, 14, 21 and 28 days post-surgery). The humane endpoints were
complete loss of appetite for 24 h or inability to eat and drink on
their own, or, in the absence of anesthesia or sedation, depression
with hypothermia (<37°C). Euthanasia was performed via cervical
dislocation after sampling mice. Complete heartbeat cessation and
pupil dilation were considered to indicate death.
 | Table I.Distribution of experimental mice
(n=177). |
Table I.
Distribution of experimental mice
(n=177).
| Total | Groups | Type of experiment
performed | Figure |
|---|
| 45 | Sham (n=5) | RT-qPCR (CCL3,
CCL4, CCL5); WB (CCR5) | 1 |
|
| 3 days after TAC
(n=8) |
|
|
|
| 7 days after TAC
(n=8) |
|
|
|
| 14 days after TAC
(n=8) |
|
|
|
| 21 days after TAC
(n=8) |
|
|
|
| 28 days after TAC
(n=8) |
|
|
| 25 | Sham (n=5) | RT-qPCR
(atrial/brain natriuretic peptide) | 2A and B |
|
| TAC-Control
(n=5) |
|
|
|
| TAC-IgG (n=5) |
|
|
|
| TAC-CCR5 agonist
(n=5) |
|
|
|
| TAC-CCR5 Ab
(n=5) |
|
|
| 20 | Sham (n=5) | IHC (CCR5);
hematoxylin-eosin staining | 2C and D |
|
| TAC-Control
(n=8) |
|
|
|
| TAC-IgG (n=8) |
|
|
|
| TAC-CCR5Ab
(n=8) |
|
|
| 20 | Sham (n=5) | Echocardiographic
and hemodynamic measurements; | 2E-J; 3 |
|
|
| IHC (α-SMA);
Masson's staining; |
|
|
|
| RT-qPCR (collagen
I, MMP2, MMP9) |
|
|
| TAC-Control
(n=8) |
|
|
|
| TAC-IgG (n=8) |
|
|
|
| TAC-CCR5Ab
(n=8) |
|
|
| 20 | Sham (n=5) | WB (p-ERK, ERK,
p-p38, p38) | 4A |
|
| TAC-Control
(n=8) |
|
|
|
| TAC-IgG (n=8) |
|
|
|
| TAC-CCR5Ab
(n=8) |
|
|
| 18 | Normal control
(n=3) | WB (p-ERK, ERK,
p-p38, p38); immunofluorescence | 4B and C |
|
| Ang II (n=3) |
|
|
|
| Ang II + IgG
(n=3) |
|
|
|
| Ang II + CCR5 Ab
(n=3) |
|
|
|
| Ang II + U0126
(n=3) |
|
|
|
| Ang II +
SB203580 |
|
|
|
| (n=3) |
|
|
| 2 | Modeling failure
(n=2) | – | – |
| Total | n=177 | – | – |
Echocardiographic and hemodynamic
measurements
Mouse echocardiography was performed on a Vevo2100
imaging system (VisualSonics Inc.) with a 22–55 MHz probe. All mice
were anaesthetized with ketamine (50 mg/kg) by intraperitoneal
injection. Left ventricular end-systolic and -diastolic left
dimensions (LVESD and LVEDD, respectively) were measured according
the American Society of Echocardiography guidelines (18). LV end-diastolic and -systolic
volumes (EDV and ESV, respectively) were calculated using the
Simpson method of disks (19).
Ejection fraction (EF) was determined using the formula EF (%) =
(EDV-ESV)/EDVx100. LVEDD, LVESD and the maximal rates of rise and
fall in LV pressure (+ and -dP/dt, respectively) were recorded as
previously described (20). LV
fractional shortening (FS) was calculated using the formula FS (%)
= (LVEDD-LVESD)/LVEDDx100. All measurements were performed by an
investigator blinded to treatment conditions.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was isolated from mouse myocardial tissue
using TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.).
First-strand complementary DNA was synthesized using the
SuperScript III kit (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. CCL3, CCL4, CCL5,
collagen I, matrix metalloproteinase (MMP)2 and 9, atrial and brain
natriuretic peptide (ANP and BNP, respectively) and GAPDH mRNA
expression levels were determined using SYBR-Green Real-Time PCR
reagents (Invitrogen; Thermo Fisher Scientific, Inc.) on a MyiQ
Single-Color Real-Time PCR Detection System (Bio-Rad Laboratories,
Inc.). Primer sequences are listed in Table II. PCR conditions were 95°C for 10
min followed by 40 amplification cycles of 95°C for 10 sec and 72°C
for 30 sec. Data were normalized to GADPH. The relative mRNA
expression levels were calculated using the 2−ΔΔCq
method (21) and are presented as
fold-change compared with the control/sham group.
| Table II.Reverse transcription-quantitative
PCR primer sequences. |
Table II.
Reverse transcription-quantitative
PCR primer sequences.
| Primer | Forward sequence
(5′→3′) | Reverse sequence
(5′→3′) |
|---|
| CCL3 |
TACAAGCAGCAGCGAGTACC |
GAGCAAAGGCTGCTGGTTTC |
| CCL4 |
ACCAATGGGCTCTGACCCTC |
CTTGGAGCAAAGACTGCTGGT |
| CCL5 |
CCTCACCATATGGCTCGGAC |
TCTTCTCTGGGTTGGCACAC |
| Collagen I |
GGGTTCAGCATGAGGGGATT |
GGTGGAAGAGGTCAATGGGG |
| MMP2 |
CCCCATGAAGCCTTGTTTACC |
GAAGGGGAAGACACATGGGG |
| MMP9 |
AAACCCTGTGTGTTCCCGTT |
CGTCGCTGGTACAGGAAGAG |
| GAPDH |
CCATCTTCCAGGAGCGAGAC |
GCCCTTCCACAATGCCAAAG |
Western blot analysis
Mouse myocardial tissue or H9c2 cells were
homogenized in RIPA buffer (Beyotime Institute of Biotechnology).
Protein concentrations were determined using a bicinchoninic acid
(BCA) protein assay kit (Beyotime Institute of Biotechnology).
Samples (5 µl) were separated via 10% SDS-PAGE and transferred to
nitrocellulose membranes (Pall Life Sciences). The membranes were
blocked with 5% non-fat milk at room temperature for 2 h, followed
by incubation with primary antibodies against CCR5 (1:500; cat. no.
ab65850), phosphorylated (p)-ERK1 (T202) + ERK2 (T185) (1:1,000;
ab201015), ERK1/2 (1:1,000; cat. no. ab184699), p-P38 (Y182)
(1:500; cat. no. ab47363), P38 (1:1,000; cat. no. ab170099) and
β-actin (1:2,000; cat. no. ab8226; all from Abcam) overnight at
4°C. According to manufacturer's instructions, following incubation
(for 2 h at room temperature) with horseradish
peroxidase-conjugated secondary antibody (horseradish
peroxidase-conjugated anti-rabbit immunoglobulin G; cat. no. 7074;
1:2,000; Cell Signaling Technology, Inc.), protein bands were
visualized using enhanced chemiluminescence reagents (Thermo Fisher
Scientific, Inc.) and quantitated by densitometric analysis using a
Science Imaging system (version 4.1; Bio-Rad Laboratories, Inc.).
Data were normalized to β-actin.
Histology and
immunohistochemistry
The left ventricles were fixed (24 h at room
temperature) in 4% paraformaldehyde, embedded in paraffin, and
sliced into 4-µm sections. Tissue sections were deparaffinized in
xylene, rehydrated in a descending graded alcohol series and
incubated in Bouin's solution at 37°C for 2 h. The Bouin's solution
consisted of 75 ml saturated picric acid, 25 ml 10% formalin
solution (v/v) and 5 ml acetic acid. Tissue sections were then
stained using the Masson's Trichrome Staining kit (cat. no. G1340;
Beijing Solarbio Science & Technology Co., Ltd.) according to
manufacturer's protocols to assess interstitial fibrosis and
collagen deposition. The collagen volume fraction (CVF; percentage
of collagen-positive area) was estimated from five randomly
selected visual fields of the myocardial interstitium using ImageJ
imaging software (version 1.60; NIH). Collagen around blood vessels
was not included in the analysis. The expression levels and
distribution of CCR5 and α-smooth muscle actin (α-SMA) in
myocardial tissues were examined by immunostaining using the
streptavidin-biotin amplification method (22). In brief, following endogenous
peroxidase inactivation (3% hydrogen peroxide; Beyotime Institute
of Biotechnology) and microwave-based antigen retrieval (citric
acid buffer, pH 6.0, 98°C for a total of 30 min), sections were
probed overnight at 4°C with anti-CCR5 (1:500; cat. no. ab65850;
Abcam) or anti-α-SMA antibody (1:200; cat. no. ab124964; Abcam).
The staining intensity was scored as follows: 0, no staining or
similar to background; 1, light staining or slightly higher than
background; 2, moderate staining or significantly higher than
background; 3, strong staining; and 4 very strong staining. The
sections were then washed thoroughly in PBS three times for 5 min
each, stained in hematoxylin for 20 sec at room temperature,
dehydrated in an ascending absolute alcohol gradient, washed with
xylene and mounted in synthetic resin and ImageJ software was used
to assess cross-sectional areas of ventricular cardiomyocytes and
inflammatory cell infiltration. All scoring was performed
separately by two of the authors (WD and YL).
Cell culture and surface area
determination
H9c2 cells were obtained from the American Type
Culture Collection and cultured in DMEM (cat. no. C11885500BT;
Invitrogen; Thermo Fisher Scientific, Inc.) supplemented with 10%
FBS (cat. no. TBD31HB; Tian Jin Hao Yang Biological Manufacture
Co., Ltd.) and penicillin-streptomycin (100 IU/ml) in a humidified
incubator at 5% CO2 and 37°C. The cells were
pre-incubated (37°C for 30 min) with anti-CCR5 antibody (0.4 µg/ml;
cat. no. ab65850; Abcam), isotype IgG control antibody (0.4 µg/ml;
cat. no. ab190475; Abcam), according to supplier instructions, MEK
inhibitor U0126 (1 µM; Sigma-Aldrich; Merck KGaA) or P38 inhibitor
SB203580 (1 µM; Sigma-Aldrich; Merck KGaA) for 1 h prior to
stimulation with angiotensin II (Ang II; 1 µM; Sigma-Aldrich; Merck
KGaA) for 24 h at 37°C. Cell surface area was determined using the
F-actin staining method as previously described (23). In brief, according to supplier
instructions, cells were washed with PBS twice and fixed in 4%
paraformaldehyde for 10 min at 37°C followed by incubation with
0.1% Triton X-100 for 20 min at 37°C. After blocking with 5% BSA
(9048-46-8; Sigma-Aldrich; Merck KGaA) for 30 min at 37°C, the
cells were incubated with Actin-Tracker Green (Beyotime Institute
of Biotechnology) for 45 min at room temperature and subjected to
examination by confocal laser microscopy (magnification, ×100;
Zeiss LSM 510 META; Zeiss AG, Oberkochen). Nuclei were
counterstained with DAPI (90%; Sigma-Aldrich; Merck KGaA) at room
temperature for 10 min. Images were processed using Image-Pro Plus
6.0 (Media Cybernetics, Inc.).
Statistical analysis
All data are presented as the mean ± SEM. Each
experiment was repeated three times. Data were analyzed with SPSS
(version 22.0; IBM Corp.) or GraphPad Prism (version 5.0; GraphPad
Software, Inc.). One-way analysis of variance followed by Tukey's
post hoc test was applied to determine the statistical significance
of differences among groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
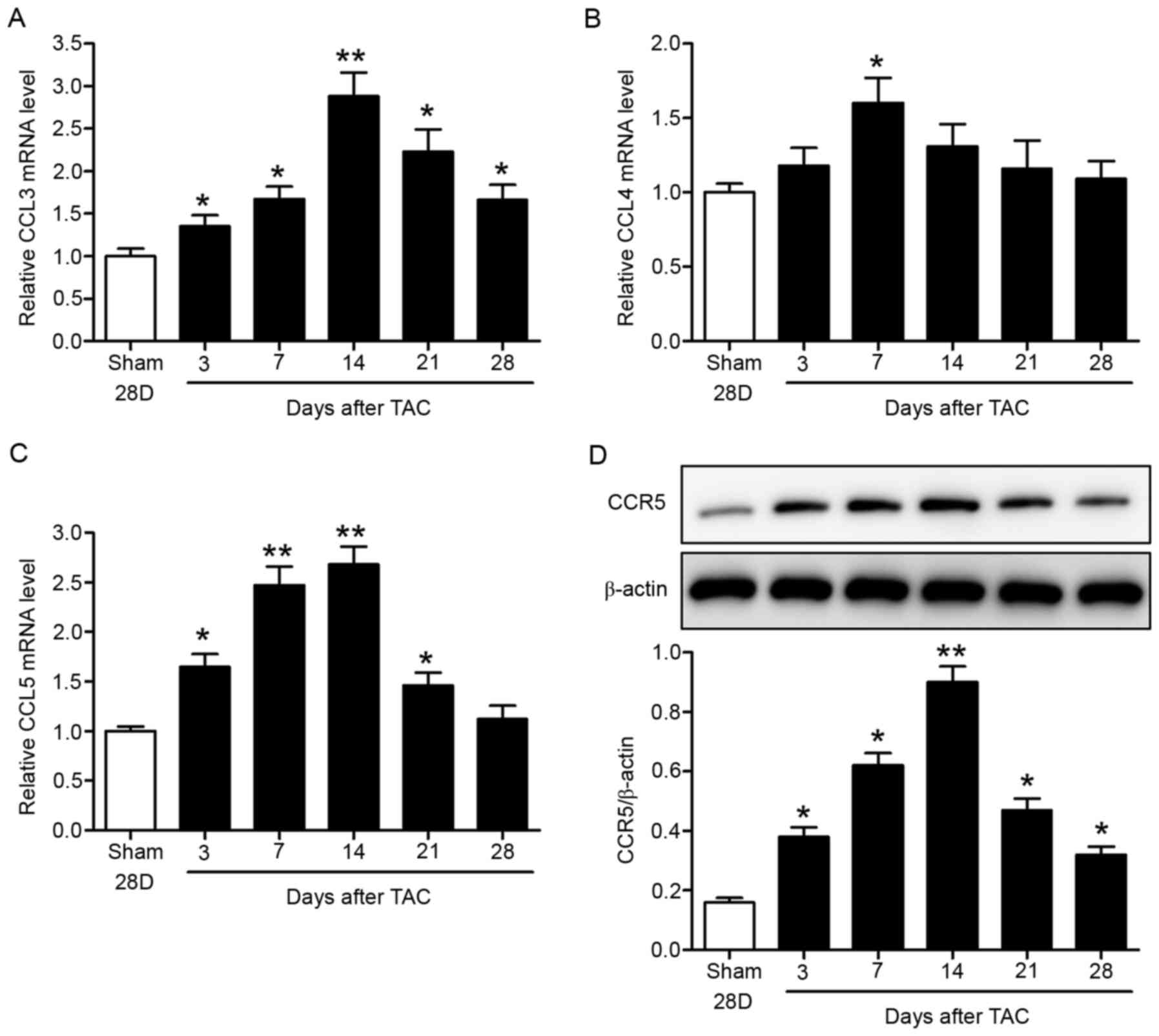
LV pressure overload upregulates CCR5
and its ligands in mice
Expression levels of CCR5 and its ligands (CCL3,
CCL4 and CCL5) were determined in the TAC mouse model. C57BL/6 mice
were subjected to TAC or sham surgery. The hearts were collected at
specific time-points up to 28 days after surgery for analysis.
RT-qPCR data showed time-dependent increases in myocardial CCL3,
CCL4 and CCL5 mRNA expression levels, which peaked at 1–2 weeks
after surgery (Fig. 1A-C). The
chemokine levels then declined with time. In addition, myocardial
CCR5 protein levels increased with time and peaked 2 weeks after
surgery, as indicated by western blotting (Fig. 1D). CCR5 levels subsequently
decreased (Fig. 1D).
CCR5 inhibition ameliorates pressure
overload-induced myocardial hypertrophy and cardiac
dysfunction
The effects of CCR5 inhibition on pressure
overload-induced myocardial hypertrophy and cardiac dysfunction
were investigated in TAC mice. ANP and BNP are two known biomarkers
of cardiac hypertrophy (24).
Inhibiting myocardial CCR5 decreased myocardial hypertrophy,
whereas overexpression of myocardial CCR5 increased myocardial
hypertrophy in the TAC heart (Fig. 2A
and B). At 28 days post-surgery, immunohistochemical staining
for CCR5 revealed increased myocardial CCR5 immunoreactivity in TAC
hearts (Fig. 2C). The increase in
myocardial CCR5 expression levels in response to TAC was
accompanied by visible leukocyte infiltration and cardiomyocyte
hypertrophy, as indicated by H&E staining (Fig. 2D). These histopathological changes
in the pressure-overloaded hearts were associated with altered
chamber dimension and impaired LV function. At 14 days
post-surgery, the TAC hearts displayed decreased FS, EF and + and
-dP/dt, along with increased LVEDD and LVESD (Fig. 2E-J). Compared with IgG control,
anti-CCR5 antibody-treated TAC hearts exhibited decreased CCR5
immunoreactivity, leukocyte infiltration and cardiomyocyte
hypertrophy and less severe cardiac dysfunction (Fig. 2A-J).
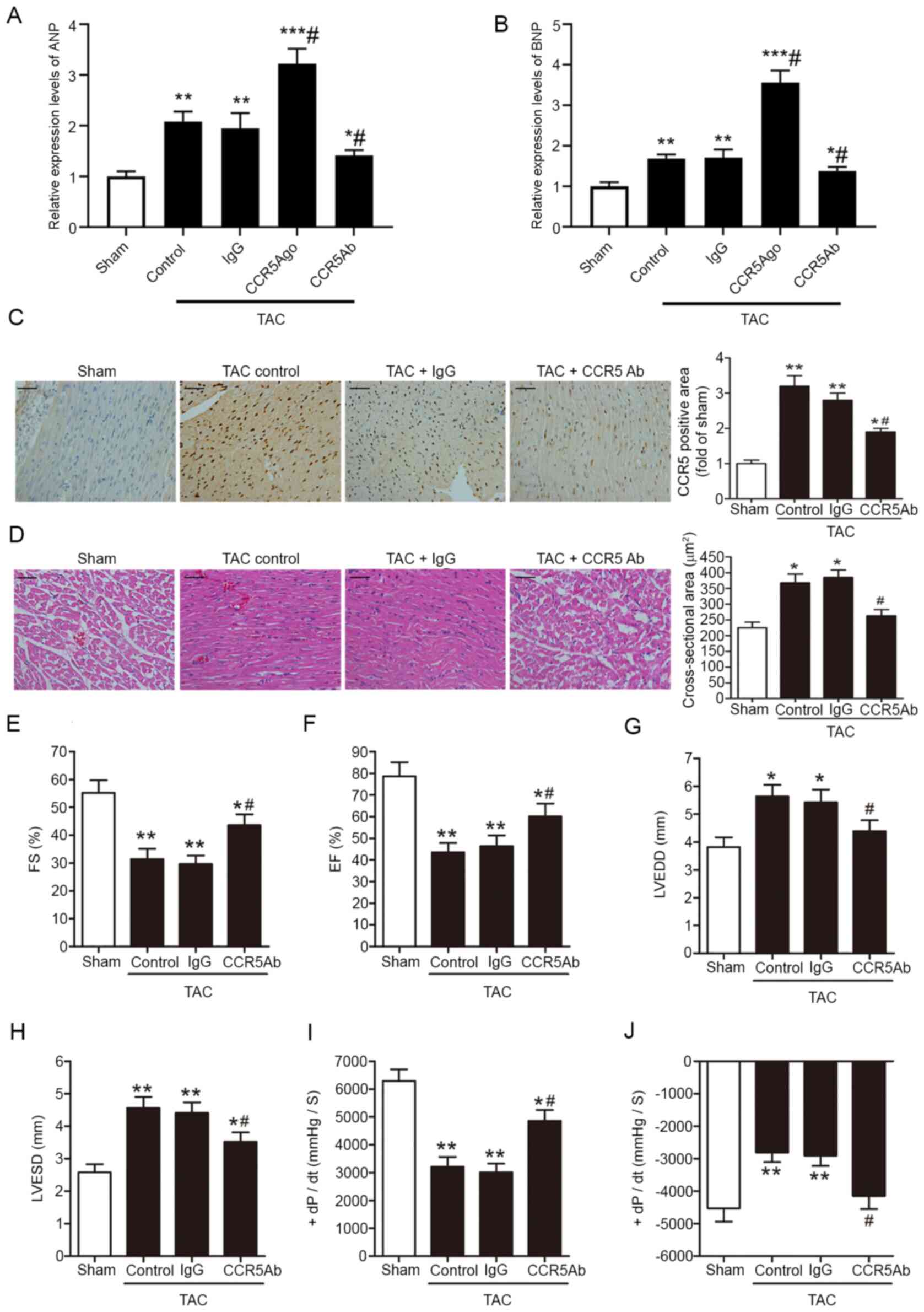 | Figure 2.CCR5 inhibition ameliorates pressure
overload-induced myocardial hypertrophy and cardiac dysfunction.
TAC or sham mice were treated with anti-CCR5 or IgG control
antibody. Relative (A) ANP and (B) BNP mRNA levels were assessed by
reverse transcription-quantitative PCR. (C) Myocardial CCR5
expression levels were assessed at 28 days after surgery by
immunohistochemical staining. Scale bar, 50 µm. (D) Myocardial
hypertrophy (indicated in cross-sectional areas of ventricular
cardiomyocytes) and inflammatory cell infiltration 28 days after
surgery were assessed by hematoxylin and eosin staining. Scale bar,
50 µm. Cardiac parameters were assessed 14 days after surgery by
echocardiographic and hemodynamic measurements. (E) FS, (F) EF, (G)
LVEDD, (H) LVESD, (I) +dP/dt and (J) -dP/dt are shown. TAC,
n=8/group; Sham, n=5. *P<0.05, **P<0.01 and ***P<0.001 vs.
Sham; #P<0.05 vs. TAC/IgG. C-C chemokine receptor 5;
TAC, transverse aortic constriction; ANP, atrial natriuretic
peptide; BNP, brain natriuretic peptide; FS, fractional shortening;
EF, ejection fraction; LVEDD, left ventricular end-diastolic
dimension; LVESD, left ventricular end-systolic dimension; +dP/dt,
maximal rate of rise in left ventricle pressure; -dP/dt, maximal
rate of fall in left ventricle pressure; Ago, agonist; Ab,
antibody. |
CCR5 inhibition decreases pressure
overload-induced myocardial fibrosis
Myocardial fibrosis mediated by
trans-differentiation of fibroblasts into myofibroblasts is a
pathological response to cardiac insult such as pressure overload
(25). Immunohistochemical staining
revealed higher myocardial expression levels of the myofibroblast
marker α-SMA in the TAC heart, compared with sham (Fig. 3A). Collagen I, MMP2 and MMP9 are
known contributing factors of cardiac fibrosis and remodeling in
heart disease (26). Masson's
trichrome staining of LV sections showed greater collagen
deposition in TAC hearts compared with sham (Fig. 3B). In addition, RT-qPCR assay
revealed higher myocardial mRNA expression levels of collagen I,
MMP2 and MMP9 in the TAC heart compared with sham hearts (Fig. 3C-E). Compared with IgG control, the
anti-CCR5 antibody-treated TAC hearts exhibited significantly
decreased myocardial α-SMA immunoreactivity and collagen
deposition, as well as lower mRNA expression levels of collagen I,
MMP2 and MMP9 (Fig. 3A-E). These
results supported the role of CCR5 as a contributing factor in
pressure overload-induced cardiac fibrosis.
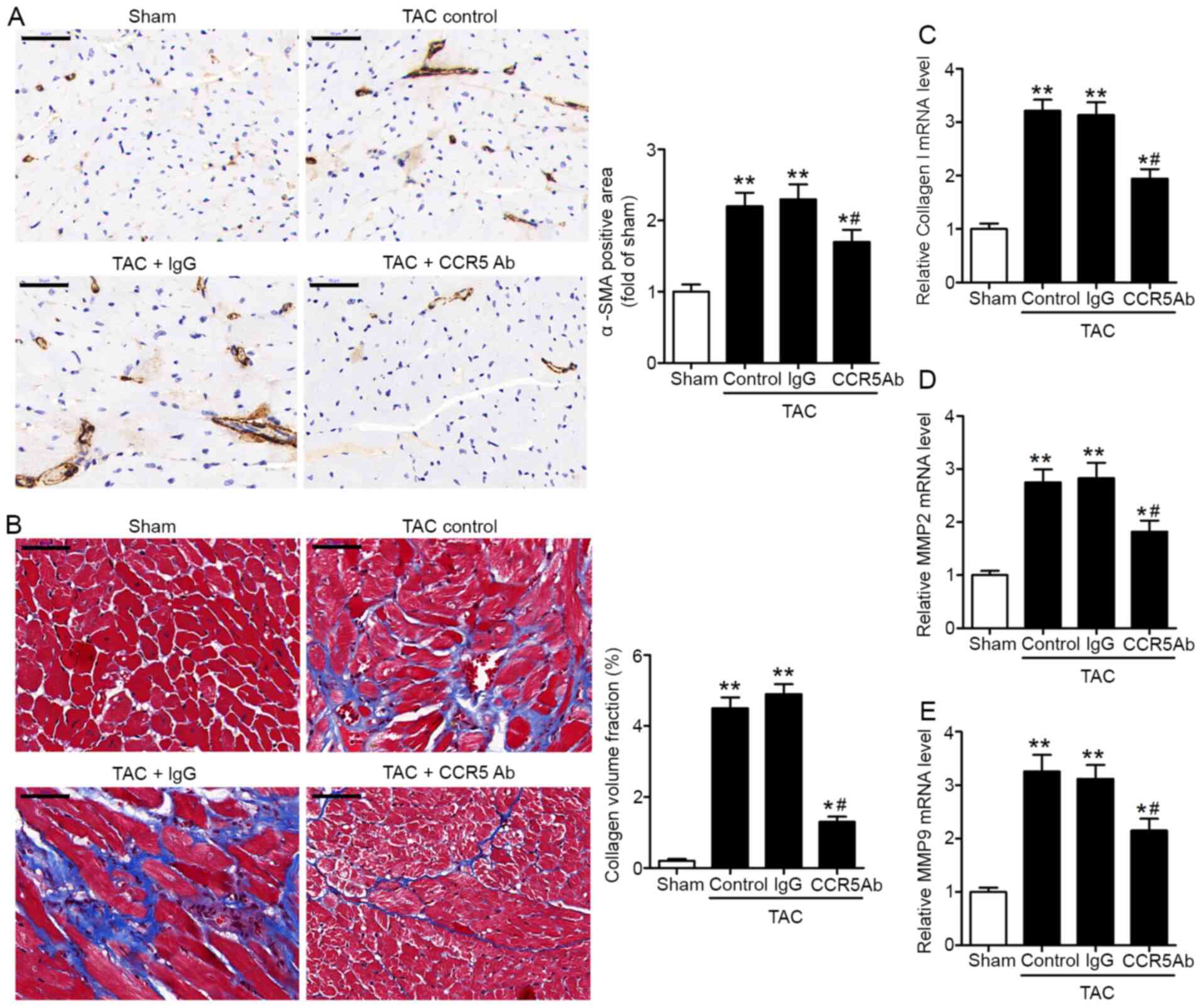 | Figure 3.CCR5 inhibition decreases pressure
overload-induced myocardial fibrosis. TAC or sham mice were treated
with anti-CCR5 or IgG control antibody. Myocardial tissues were
collected 28 days after surgery for analysis. (A) Detection of
myocardial α-SMA by immunohistochemical staining. Scale bar, 50 µm.
(B) Detection of left ventricle collagen deposition by Masson's
trichrome staining. Scale bar, 50 µm. Collagen volume fraction was
defined as the percentage of collagen-positive area. The mRNA
expression levels of (C) collagen I, (D) MMP2 and (E) MMP9 in
myocardial tissue were detected by reverse
transcription-quantitative PCR. TAC, n=8/group; Sham, n=5.
*P<0.05, **P<0.01 vs. Sham; #P<0.05 vs. TAC +
IgG. CCR5, C-C chemokine receptor 5; TAC, transverse aortic
constriction; α-SMA, α-smooth muscle actin; MMP, matrix
metalloproteinase; Ab, antibody. |
CCR5 inhibition ameliorates pressure
overload-induced cardiac abnormality via MAPK signaling
ERK1/2 and P38 MAPKs are considered to be central
regulators of cardiac hypertrophy (27). Studies have shown that MAPK
signaling induces myocardial diastolic dysfunction (28) and that activated p38 MAPK induces
CCR5 to cause damage to the cell morphology and increases the
percentage of cell death in rat cardiomyocytes (29). Therefore, studying the CCR5-MAPK
pathway is necessary to improve understanding of cardiovascular
physiology. In order to investigate the signaling mechanisms
involved in the cardioprotective effects of CCR5 inhibition,
myocardial ERK1/2 and P38 phosphorylation were assessed by western
blot analysis. The data revealed significantly higher myocardial
protein expression levels of p-ERK1/2 and p-P38 in the TAC heart
compared with sham hearts 28 days after surgery (Fig. 4A). Administration of anti-CCR5 (but
not IgG control antibody) inhibited ERK1/2 and P38 phosphorylation
induced by TAC (Fig. 4A). In order
to elucidate the molecular mechanisms involved, the effect of CCR5
inhibition on Ang II-induced hypertrophy of cultured H9c2
cardiomyocytes was investigated. Ang II significantly increased
p-ERK1/2 and p-P38 levels in H9c2 cells and induced cell
hypertrophy, as indicated by F-actin staining (Fig. 4B and C). Blockade of ERK1/2 by the
MEK inhibitor U0126 or P38 by the P38 inhibitor SB203580
effectively attenuated cell hypertrophy induced by Ang II (Fig. 4B and C). Moreover, pre-treatment
with anti-CCR5, but not IgG control antibody, inhibited ERK1/2 and
P38 phosphorylation as well as cell hypertrophy induced by Ang II
(Fig. 4B and C).
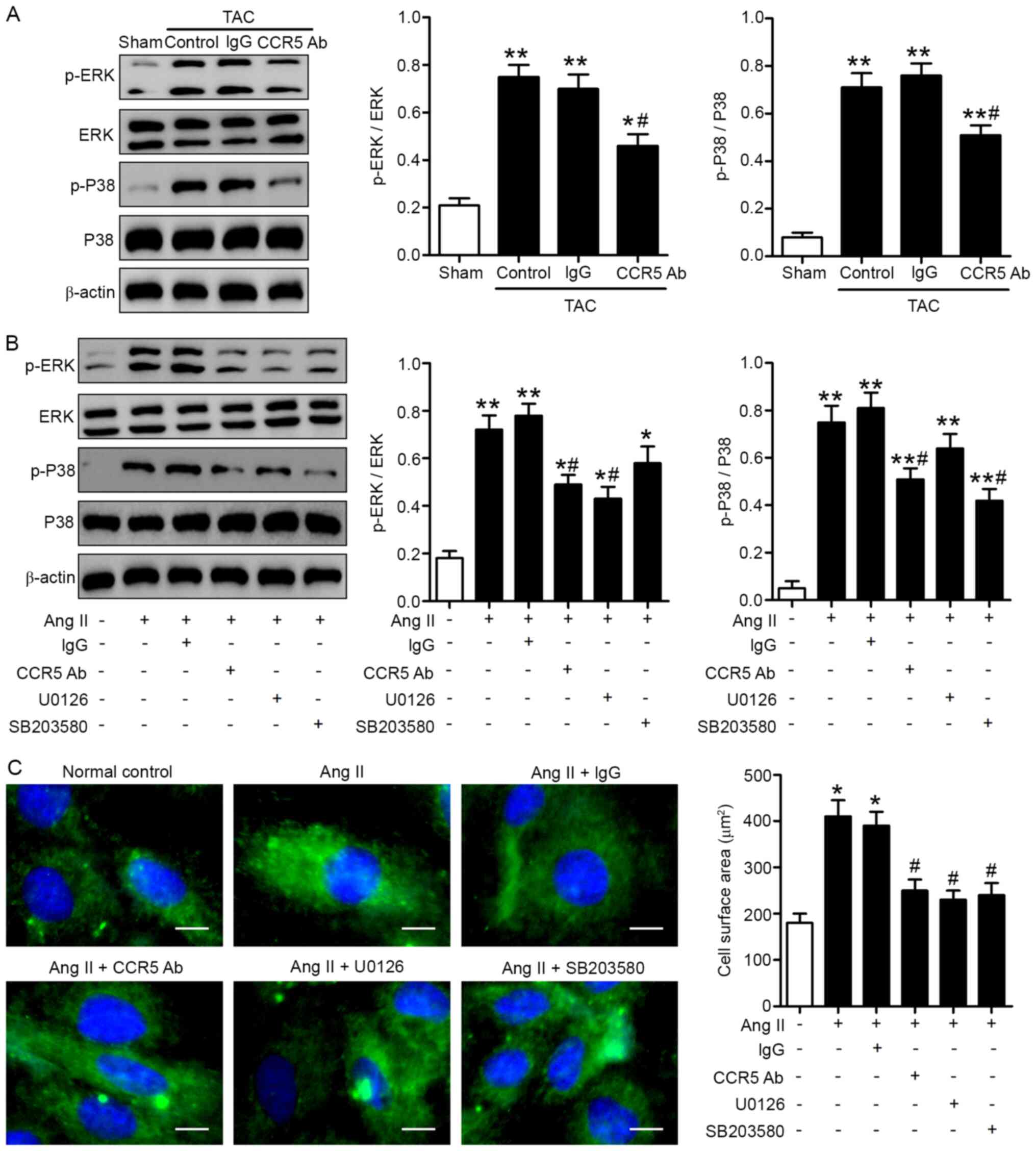 | Figure 4.ERK and P38 signaling pathways are
involved in the cardioprotective effects of CCR5 inhibition. (A)
TAC or sham mice were treated with anti-CCR5 or IgG control
antibody. Myocardial tissues were collected 28 days after surgery.
Protein expression levels of p-ERK, ERK, p-P38 and P38 were
determined by western blot analysis. TAC, n=8/group; Sham, n=5.
*P<0.05, **P<0.01 vs. Sham; #P<0.05 vs. TAC +
IgG. H9c2 cardiomyocytes were stimulated with Ang II for 24 h in
the presence or absence of 1 h pretreatment with anti-CCR5
antibody, U0126 or SB203580. (B) p-ERK, ERK, p-P38 and P38 levels
and (C) cell surface areas were determined by western blot analysis
and F-actin immunofluorescence, respectively. Scale bar, 20 µm.
n=3. *P<0.05, **P<0.01 vs. normal control;
#P<0.05 vs. Ang II alone. CCR5, C-C chemokine
receptor 5; TAC, transverse aortic constriction; p-,
phosphorylated; Ang II, angiotensin II, Ab, antibody. |
Discussion
CCR5 is predominantly expressed on natural killer
and dendritic cells, T lymphocytes and macrophages (14). CCR5 serves a key role in human
immunodeficiency virus (HIV)-1 entry into target cells, and
CCR5-targeted pharmaceutical and gene therapies have been developed
for clinical use against HIV (30).
Research has shown that CCR5 and its ligands serve a role in
autoimmune and inflammatory disease via regulation of immune cell
activation and migration (14). For
example, CCR5+ inflammatory cells and increased CCR5
ligands (CCL3, CCL4 and CCL5) are found within multiple sclerosis
lesions and may contribute to recruitment of inflammatory cells to
the inflamed tissue and their activation (31,32).
In patients with rheumatoid arthritis, CCR5+ mononuclear
cells and increased expression levels of CCR5 ligands CCL3, CCL4
and CCL5 are found in the inflamed synovium (33,34),
and a non-functional CCR5 variant (CCR5 d32) is considered to
exhibit a protective effect (35).
Moreover, CCL5 expression levels are increased in inflammatory
bowel disease (36), and CCR5
inhibition by genetic ablation or by pharmacological inhibition
ameliorates both acute and chronic colitis in mice (37). CCR5 has also been implicated in
development of multiple types of cancer, such as breast, prostate
and colon cancer, as well as melanoma and Hodgkin lymphoma
(38). The pro-tumorigenic function
of CCR5 is primarily mediated by it ligand CCL5, which promotes
proliferation of CCR5-positive tumor cells, recruits
immunosuppressive T-regulatory cells and favors metastasis
(39). Hence, the CCL5/CCR5 axis
has become a potential target for development of novel anticancer
therapy (40,41).
Arthrosclerosis is increasingly recognized to be a
chronic inflammatory disease that involves leukocytes, such as
monocytes/macrophages and T cells (42). Studies have indicated a role of CCR5
in the initiation and progression of arthrosclerosis and associated
cardiovascular disease (43,44).
Specifically, the CCR5 polymorphism CCR5 d32 has been linked to
decreased risk of cardiovascular disease (45), and antagonism of either CCR5 or CCL5
decreases atherosclerosis in animal models (43). Additionally, CCR5 is implicated in
Ang II-induced hypertension and vascular dysfunction (46). Based on these previous findings, it
was speculated that CCR5 may be involved in the pathogenesis of AS,
a heart disease characterized by pressure overload-induced LV
remodeling and dysfunction.
The present study investigated the role of CCR5 in
the pathophysiology of pressure overload-induced cardiac remodeling
and dysfunction in a TAC mouse model. Higher myocardial CCL3, CCL4,
CCL5 and CCR5 expression levels were detected in the TAC heart
compared with sham mice. Echocardiographic and hemodynamic
measurements revealed cardiac abnormality in TAC mice,
characterized by decreased FS, EF and maximal rates of rise and
fall in LV pressure, along with LV hypertrophy. Histological
examination of TAC hearts showed evidence of inflammatory cell
infiltration as well as myocardial hypertrophy and fibrosis.
TAC-induced LV remodeling and dysfunction were effectively
ameliorated by administration of anti-CCR5 antibody, supporting
CCR5 as a key factor in pressure overload-induced cardiac
abnormality. Thus, pharmacological blockade of CCR5 signaling may
delay the progression of cardiac remodeling in patients with AS
before valve replacement surgery.
Mechanistically, increased ERK1/2 and P38
phosphorylation was detected in TAC mice but not in sham mice in
the present study, consistent with previous findings that ERK1/2
and P38 MAPKs are key regulators of cardiac hypertrophy (47,48).
In the present study CCR5 inhibition attenuated myocardial ERK1/2
and P38 phosphorylation induced by TAC. By contrast, there are
reports that the ERK pathway promotes compensatory hypertrophy,
enhances contractile function, decreases fibrosis, and protects the
heart (49,50). Studies have shown that in
drug-induced liver injury, CCR5 activates M1 polarization and
prevents M2 polarization via the MAPK pathway (51,52).
Another study demonstrated that CCR5 silencing inhibits the
inflammatory response in rheumatoid arthritis rats by inhibiting
the MAPK pathway, and also inhibits viability and promotes
synoviocyte apoptosis (53). Ang II
has been shown to promote cardiac tissue growth and contribute to
pressure overload-induced cardiac hypertrophy (54,55).
In the present study, in cultured H9c2 cardiomyocytes, CCR5
inhibition decreased Ang II-induced ERK1/2 and P38 phosphorylation
and cell hypertrophy. Taken together, the findings of the present
study suggested that the ERK1/2 and P38 MAPK pathways were involved
in the cardioprotective effects of CCR5 inhibition.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National
Natural Science Foundation of China (grant nos. 81760077 and
81860447), Natural Science Foundation of Inner Mongolia Autonomous
Region, China (grant nos. 2018MS08053), The National Key Research
and Development Program of China (grant no. 2016YFC1102502),
Science and Technology Project Fund of Baotou (grant nos.
2018C2007-2-7 and 2018C2007-2-8) and Science Program Fund of Inner
Mongolia Health Commission (grant no. 201703181).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XW, WL, RZ and JH designed the entire experimental
study, XW, QY and JH conducted TAC modeling, WL and RZ conducted
echocardiographic and hemodynamic measurements, FL and LY conducted
PCR and western blotting, WD and YL performed histology and
immunohistochemistry, LX and LY performed cell culture and surface
area determination, QY and WD performed data analysis, XW, RZ and
JH wrote the manuscript and RZ and JH made important revisions to
the manuscript and approved the published version. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the institutional review
committee of the Baotou Central Hospital in accordance with the
ethical standards formulated in the Declaration of Helsinki.
Standard animal care and laboratory guidelines were followed
according to the IACUC protocol.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nkomo VT, Gardin JM, Skelton TN,
Gottdiener JS, Scott CG and Enriquez-Sarano M: Burden of valvular
heart diseases: A population-based study. Lancet. 368:1005–1011.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rassi AN, Pibarot P and Elmariah S: Left
ventricular remodelling in aortic stenosis. Can J Cardiol.
30:1004–1011. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Badiani S, van Zalen J, Treibel TA,
Bhattacharyya S, Moon JC and Lloyd G: Aortic stenosis, a left
ventricular disease: Insights from advanced imaging. Curr Cardiol
Rep. 18:802016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Carabello BA: Aortic stenosis: From
pressure overload to heart failure. Heart Fail Clin. 2:435–442.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pellikka PA, Sarano ME, Nishimura RA,
Malouf JF, Bailey KR, Scott CG, Barnes ME and Tajik AJ: Outcome of
622 adults with asymptomatic, hemodynamically significant aortic
stenosis during prolonged follow-up. Circulation. 111:3290–3295.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grimard BH and Larson JM: Aortic stenosis:
Diagnosis and treatment. Am Fam Physician. 78:717–724.
2008.PubMed/NCBI
|
|
7
|
Yarbrough WM, Mukherjee R, Ikonomidis JS,
Zile MR and Spinale FG: Myocardial remodeling with aortic stenosis
and after aortic valve replacement: Mechanisms and future
prognostic implications. J Thorac Cardiovasc Surg. 143:656–664.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gregersen I, Holm S, Dahl TB, Halvorsen B
and Aukrust P: A focus on inflammation as a major risk factor for
atherosclerotic cardiovascular diseases. Expert Rev Cardiovasc
Ther. 14:391–403. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Koenen RR and Weber C: Manipulating the
chemokine system: Therapeutic perspectives for atherosclerosis.
Curr Opin Investig Drugs. 11:265–272. 2010.PubMed/NCBI
|
|
10
|
Carità P, Coppola G, Novo G, Caccamo G,
Guglielmo M, Balasus F, Novo S, Castrovinci S, Moscarelli M,
Fattouch K, et al: Aortic stenosis: Insights on pathogenesis and
clinical implications. J Geriatr Cardiol. 13:489–498.
2016.PubMed/NCBI
|
|
11
|
Shioi T, Matsumori A, Kihara Y, Inoko M,
Ono K, Iwanaga Y, Yamada T, Iwasaki A, Matsushima K and Sasayama S:
Increased expression of interleukin-1 beta and monocyte chemotactic
and activating factor/monocyte chemoattractant protein-1 in the
hypertrophied and failing heart with pressure overload. Circ Res.
81:664–671. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yin X, Gao Y, Shi HS, Song L, Wang JC,
Shao J, Geng XH, Xue G, Li JL and Hou YN: Overexpression of SIRT6
in the hippocampal CA1 impairs the formation of long-term
contextual fear memory. Sci Rep. 6:189822016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Finsen AV, Ueland T, Sjaastad I, Ranheim
T, Ahmed MS, Dahl CP, Askevold ET, Aakhus S, Husberg C, Fiane AE,
et al: The homeostatic chemokine CCL21 predicts mortality in aortic
stenosis patients and modulates left ventricular remodeling. PLoS
One. 9:e1121722014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Turner J, Steinmetz O, Stahl R and Panzer
U: Targeting of Th1-associated chemokine receptors CXCR3 and CCR5
as therapeutic strategy for inflammatory diseases. Mini Rev Med
Chem. 7:1089–1096. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ortlepp JR, Schmitz F, Mevissen V, Weiss
S, Huster J, Dronskowski R, Langebartels G, Autschbach R, Zerres K,
Weber C, et al: The amount of calcium-deficient hexagonal
hydroxyapatite in aortic valves is influenced by gender and
associated with genetic polymorphisms in patients with severe
calcific aortic stenosis. Eur Heart J. 25:514–522. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rockman HA, Ross RS, Harris AN, Knowlton
KU, Steinhelper ME, Field LJ, Ross J Jr and Chien KR: Segregation
of atrial-specific and inducible expression of an atrial
natriuretic factor transgene in an in vivo murine model of cardiac
hypertrophy. Proc Natl Acad Sci USA. 88:8277–8281. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Balasubramanian S, Pleasant DL,
Kasiganesan H, Quinones L, Zhang Y, Sundararaj KP, Roche S,
O'Connor R, Bradshaw AD and Kuppuswamy D: Dasatinib attenuates
pressure overload induced cardiac fibrosis in a murine transverse
aortic constriction model. PLoS One. 10:e01402732015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Henry WL, DeMaria A, Gramiak R, King DL,
Kisslo JA, Popp RL, Sahn DJ, Schiller NB, Tajik A, Teichholz LE, et
al: Report of the American Society of Echocardiography Committee on
nomenclature and standards in two-dimensional echocardiography.
Circulation. 62:212–217. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ünlüer EE, Karagöz A, Bayata S and Akoğlu
H: An alternative approach to the bedside assessment of left
ventricular systolic function in the emergency department:
Displacement of the aortic root. Acad Emerg Med. 20:367–373. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Toldo S, Bogaard HJ, Van Tassell BW,
Mezzaroma E, Seropian IM, Robati R, Salloum FN, Voelkel NF and
Abbate A: Right ventricular dysfunction following acute myocardial
infarction in the absence of pulmonary hypertension in the mouse.
PLoS One. 6:e181022011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vachon CM, Sasano H, Ghosh K, Brandt KR,
Watson DA, Reynolds C, Lingle WL, Goss PE, Li R, Aiyar SE, et al:
Aromatase immunoreactivity is increased in mammographically dense
regions of the breast. Breast Cancer Res Treat. 125:243–252. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang K, Long B, Zhou J and Li PF: miR-9
and NFATc3 regulate myocardin in cardiac hypertrophy. J Biol Chem.
285:11903–11912. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Lan Y, Wang Y, Nie M, Lu Y and Zhao
E: Telmisartan suppresses cardiac hypertrophy by inhibiting
cardiomyocyte apoptosis via the NFAT/ANP/BNP signaling pathway. Mol
Med Rep. 15:2574–2582. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chin CWL, Everett RJ, Kwiecinski J, Vesey
AT, Yeung E, Esson G, Jenkins W, Koo M, Mirsadraee S, White AC, et
al: Myocardial fibrosis and cardiac decompensation in aortic
stenosis. JACC Cardiovasc Imaging. 10:1320–1333. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fan D, Takawale A, Lee J and Kassiri Z:
Cardiac fibroblasts, fibrosis and extracellular matrix remodeling
in heart disease. Fibrogenesis Tissue Repair. 5:152012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Berzingi C, Chen F and Finkel MS: p38 MAP
kinase inhibitor prevents diastolic dysfunction in rats following
HIV gp120 injection in vivo. Cardiovasc Toxicol. 9:142–150. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gao R, Fang Q, Zhang X, Xu Q, Ye H, Guo W,
He J, Chen Y, Wang R, Wu Z, et al: R5 HIV-1 gp120 activates p38
MAPK to induce rat cardiomyocyte injury by the CCR5 coreceptor.
Pathobiology. 86:274–284. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Haworth KG, Peterson CW and Kiem HP:
CCR5-edited gene therapies for HIV cure: Closing the door to viral
entry. Cytotherapy. 19:1325–1338. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Balashov KE, Rottman JB, Weiner HL and
Hancock WW: CCR5(+) and CXCR3(+) T cells are increased in multiple
sclerosis and their ligands MIP-1alpha and IP-10 are expressed in
demyelinating brain lesions. Proc Natl Acad Sci USA. 96:6873–6878.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Boven LA, Montagne L, Nottet HS and De
Groot CJ: Macrophage inflammatory protein-1alpha (MIP-1alpha),
MIP-1beta, and RANTES mRNA semiquantification and protein
expression in active demyelinating multiple sclerosis (MS) lesions.
Clin Exp Immunol. 122:257–263. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mack M, Brühl H, Gruber R, Jaeger C, Cihak
J, Eiter V, Plachý J, Stangassinger M, Uhlig K, Schattenkirchner M,
et al: Predominance of mononuclear cells expressing the chemokine
receptor CCR5 in synovial effusions of patients with different
forms of arthritis. Arthritis Rheum. 42:981–988. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Patel DD, Zachariah JP and Whichard LP:
CXCR3 and CCR5 ligands in rheumatoid arthritis synovium. Clin
Immunol. 98:39–45. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pokorny V, McQueen F, Yeoman S, Merriman
M, Merriman A, Harrison A, Highton J and McLean L: Evidence for
negative association of the chemokine receptor CCR5 d32
polymorphism with rheumatoid arthritis. Ann Rheum Dis. 64:487–490.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCormack G, Moriarty D, O'Donoghue DP,
McCormick PA, Sheahan K and Baird AW: Tissue cytokine and chemokine
expression in inflammatory bowel disease. Inflamm Res. 50:491–495.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mencarelli A, Cipriani S, Francisci D,
Santucci L, Baldelli F, Distrutti E and Fiorucci S: Highly specific
blockade of CCR5 inhibits leukocyte trafficking and reduces mucosal
inflammation in murine colitis. Sci Rep. 6:308022016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Aldinucci D and Colombatti A: The
inflammatory chemokine CCL5 and cancer progression. Mediators
Inflamm. 2014:2923762014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
de Oliveira CE, Oda JM, Losi Guembarovski
R, de Oliveira KB, Ariza CB, Neto JS, Banin Hirata BK and Watanabe
MA: CC chemokine receptor 5: The interface of host immunity and
cancer. Dis Markers. 2014:1269542014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Velasco-Velázquez M, Xolalpa W and Pestell
RG: The potential to target CCL5/CCR5 in breast cancer. Expert Opin
Ther Targets. 18:1265–1275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bronte V and Bria E: Interfering with
CCL5/CCR5 at the tumor-stroma interface. Cancer Cell. 29:437–439.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Weber C, Zernecke A and Libby P: The
multifaceted contributions of leukocyte subsets to atherosclerosis:
Lessons from mouse models. Nat Rev Immunol. 8:802–815. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jones KL, Maguire JJ and Davenport AP:
Chemokine receptor CCR5: From AIDS to atherosclerosis. Br J
Pharmacol. 162:1453–1469. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
de Jager SC, Kraaijeveld AO, Grauss RW, de
Jager W, Liem SS, van der Hoeven BL, Prakken BJ, Putter H, van
Berkel TJ, Atsma DE, et al: CCL3 (MIP-1 alpha) levels are elevated
during acute coronary syndromes and show strong prognostic power
for future ischemic events. J Mol Cell Cardiol. 45:446–452. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Afzal AR, Kiechl S, Daryani YP,
Weerasinghe A, Zhang Y, Reindl M, Mayr A, Weger S, Xu Q and Willeit
J: Common CCR5-del32 frameshift mutation associated with serum
levels of inflammatory markers and cardiovascular disease risk in
the Bruneck population. Stroke. 39:1972–1978. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mettimano M, Specchia ML, Ianni A, Arzani
D, Ricciardi G, Savi L and Romano-Spica V: CCR5 and CCR2 gene
polymorphisms in hypertensive patients. Br J Biomed Sci. 60:19–21.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rose BA, Force T and Wang Y:
Mitogen-activated protein kinase signaling in the heart: Angels
versus demons in a heart-breaking tale. Physiol Rev. 90:1507–1546.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Marber MS, Rose B and Wang Y: The p38
mitogen-activated protein kinase pathway - a potential target for
intervention in infarction, hypertrophy, and heart failure. J Mol
Cell Cardiol. 51:485–490. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mutlak M, Schlesinger-Laufer M, Haas T,
Shofti R, Ballan N, Lewis YE, Zuler M, Zohar Y, Caspi LH and Kehat
I: Extracellular signal-regulated kinase (ERK) activation preserves
cardiac function in pressure overload induced hypertrophy. Int J
Cardiol. 270:204–213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Schiattarella GG: Extracellular
signal-regulated kinase (ERK) in left ventricular pathological
hypertrophy: Not a new kid on the block anymore. Int J Cardiol.
271:260–261. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li M, Sun X, Zhao J, Xia L, Li J, Xu M,
Wang B, Guo H, Yu C, Gao Y, et al: CCL5 deficiency promotes liver
repair by improving inflammation resolution and liver regeneration
through M2 macrophage polarization. Cell Mol Immunol. 17:753–764.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Suarez-Carmona M, Chaorentong P, Kather
JN, Rothenheber R, Ahmed A, Berthel A, Heinzelmann A, Moraleda R,
Valous NA, Kosaloglu Z, et al: CCR5 status and metastatic
progression in colorectal cancer. OncoImmunology. 8:e16261932019.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lan YY, Wang YQ and Liu Y: CCR5 silencing
reduces inflammatory response, inhibits viability, and promotes
apoptosis of synovial cells in rat models of rheumatoid arthritis
through the MAPK signaling pathway. J Cell Physiol.
234:18748–18762. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhai P, Yamamoto M, Galeotti J, Liu J,
Masurekar M, Thaisz J, Irie K, Holle E, Yu X, Kupershmidt S, et al:
Cardiac-specific overexpression of AT1 receptor mutant lacking G
alpha q/G alpha i coupling causes hypertrophy and bradycardia in
transgenic mice. J Clin Invest. 115:3045–3056. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu CJ, Tang LL, Liang C, Chen X, Song SY,
Ding XQ, Zhang KY, Song BL, Zhao D, Zhu XY, et al:
Angiotensin-converting enzyme 3 (ACE3) protects against pressure
overload-induced cardiac hypertrophy. J Am Heart Assoc. 5:52016.
View Article : Google Scholar : PubMed/NCBI
|