Introduction
Diabetes mellitus, which is characterized by
hyperglycemia, affects >415 million people worldwide (1). The condition is a significant economic
burden and seriously affects the quality of life of diabetic
patients (2). Owing to
hyperglycemia, the dysfunction of the vascular endothelium may lead
to vascular lesions and cause further severe implications, such as
retinopathy (3), diabetic
nephropathy (4), cardiovascular
disease (5,6) and neuropathy (7). Previous studies have reported that
multiple factors, including mitochondrial dysfunction (8), oxidative stress (9) and the activity of various signaling
molecules, such as sirtuin 1 (10)
and p53 (11), were implicated in
the high glucose [HG (40 mM glucose)]-induced dysfunction of the
vascular endothelium. Accumulating evidence has suggested that p38
MAPK may be associated with diabetic complications; for example,
Song et al (12) discovered
that the p38 signaling pathway mediated renal injury in diabetic
nephropathy model mice. Furthermore, the activation of p38 MAPK was
identified in HK-2 cells stimulated with HG, while the inhibition
of p38 MAPK exerted a protective role over cell function (13). Notably, inflammation has also been
considered as a crucial player in the development of diabetic
mellitus, especially in vascular disorders (14–16).
In addition, accumulating studies have reported that p38 MAPK
contributed to the inflammatory process (17–19).
However, to the best of our knowledge, whether p38 MAPK contributes
to HG-induced injury and inflammation in human umbilical vein
endothelial cells (HUVECs) remains poorly understood. Thus, it is
of great significance to further determine the mechanism of
hyperglycemia-induced injury and inflammation in order to develop
effective therapeutic options for diabetes mellitus.
Necroptosis, which is also known as programmed
necrosis, has been demonstrated to serve a role in homeostasis,
inflammation and immunity (20–22).
Receptor-interacting protein 3 (RIP3) is the key determinant in
necroptosis pathway transduction (23,24).
Numerous previous studies have demonstrated that necroptosis was
involved in cytokine-induced cell death (25–27).
In addition, RIP3 knockout in vivo alleviated severe
inflammation induced by lipopolysaccharide (28). The findings of our previous study
also suggested that necroptosis was responsible for HG-induced
injury (29). Interestingly, the
p38 MAPK pathway has been identified to contribute to necroptosis
(30,31). For instance, Qin et al
(31) reported that the suppression
of the p38 MAPK signaling pathway contributed to the protective
effects of sulforaphane in neuronal necroptosis. In another in
vitro study, p38 MAPK was discovered to be activated in
TNF-α-induced necroptosis (32).
Accordingly, the relationship between p38 MAPK and the necroptosis
pathway in HG-induced injury of HUVECs should be further
investigated.
Hydrogen sulfide (H2S), the third gas
transmitter along with nitric oxide and carbon monoxide, has
attracted significant attention for its potential protective
endothelial effect (33). Previous
studies have revealed that H2S promoted
anti-inflammatory and vasculoprotective effects in various types of
disease model, such as ischemia/reperfusion injury and
atherosclerosis (34–36). Administration of sodium hydrosulfide
(NaHS), a H2S donor, was subsequently revealed to
prevent the inflammatory response, which was evidenced through the
decrease in the secretory levels of TNF-α, IL-6 and monocyte
chemoattractant protein-1, in hyperhomocysteinemia rats (37). Notably, recent research has reported
that the inhibition of p38 MAPK mediated by exogenous
H2S was conducive to ameliorating HG-induced damage in
myocardial cells (38). However,
whether p38 MAPK is associated with the protective effect of
H2S against HG-induced injury in HUVECs remains unclear.
Therefore, the present study exposed HUVECs to 40 mM HG to
establish an in vitro model of HG-induced injury and further
investigated the role of p38 MAPK and necroptosis in NaHS
protection in HUVECs.
Materials and methods
Bioinformatics analysis
A microarray dataset (GSE43950) containing five
healthy subjects and five patients with diabetes with microvascular
diseases was obtained from the Gene Expression Omnibus database
(http://www.ncbi.nlm.nih.gov/geo).
Principal component analysis (PCA) of the expression levels of
genes in 10 samples, including five healthy subjects and five
patients with diabetes with microvascular diseases, was conducted
using R package (version 3.6.3; http://cran.r-project.org/bin/windows/base/old/3.6.3/).
Gene set enrichment analysis (GSEA) of all genes was also performed
using R package. P<0.05 was considered as significance cut-off
levels.
Materials and reagents
NaHS, necrostatin-1 (Nec-1; RIP3 inhibitor),
SB203580 (p38 MAPK inhibitor), 2′,7′-dichlorofluorescein diacetate
(DCFH-DA) and rhodamine 123 (Rh123) were purchased from
Sigma-Aldrich; Merck KGaA. Glucose injection was purchased from
Hunan Kelun Pharmaceutical Co., Ltd. Small interfering RNA (siRNA)
targeting RIP3 and negative control (NC) siRNA were purchased from
Guangzhou RiboBio Co., Ltd. The anti-RIP3 primary antibody (cat.
no. ab56164) was purchased from Abcam. Anti-p38 (cat. no. 9212S)
and anti-phosphorylated (p)-p38 (cat. no. 4511S) were obtained from
Cell Signaling Technology, Inc. Anti-GAPDH primary antibody (cat.
no. 10494-1-AP) and HRP-conjugated secondary antibody (cat. no.
SA00001-2) were purchased from ProteinTech Group, Inc.
FITC-labelled anti-rabbit secondary antibody was purchased from
Invitrogen (cat. no. F-2765; Thermo Fisher Scientific, Inc.). Cell
Counting Kit-8 (CCK-8) reagent was obtained from Dojindo Molecular
Technologies, Inc. FBS and DMEM were obtained from Gibco; Thermo
Fisher Scientific, Inc. The BCA protein assay kit was obtained from
Kangchen BioTech Co., Ltd. IL-1β (cat. no. CSB-E08053h), IL-6 (cat.
no. CSB-E04638h), IL-8 (cat. no. CSB-E04641h) and TNF-α (cat. no.
CSB-E04740h) ELISA kits were purchased from Cusabio Technology LLC.
ECL solution was purchased from Nanjing KeyGen Biotech Co. Ltd.
RIPA lysis buffer was purchased from Beyotime Institute of
Biotechnology. HUVECs were supplied by Guangzhou Jiniou
Biotechnology Co., Ltd.
Cell culture and treatments
For each treatment, HUVECs were cultured in DMEM
supplemented with 10% FBS at 37°C in a humidified atmosphere and 5%
CO2. To determine the protective effects of
H2S on HG-induced injury, cells were pretreated with 400
µM NaHS (a well-known H2S donor) for 30 min prior to 40
mM HG for 24 h at 37°C. In order to determine the role of p38
MAPK/RIP3-mediated necroptosis pathways in HG-induced injury and
inflammation, HUVECs were either pretreated with 3 µM SB203580 (an
inhibitor of p38 MAPK) for 1 h prior to HG treatment or 100 µM
Nec-1 (an inhibitor of necroptosis) or transfection with RIP3-siRNA
for 24 h prior to HG treatment.
Cell transfection
HUVECs were cultured in 12-well-plates at a density
of 1×105 cells/ml and transfected at 70% confluence with
100 nM RIP3-siRNA (5′-CCAUGGCUUGUCUGGAUAA-3′) or 100 nM NC
(5′-CUAACUAUCUCGAACGCAA-3′) using Lipofectamine® 2000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Briefly, each
freeze-dried powder siRNA was dissolved in nuclease-free water to a
final concentration of 20 µM. Then, 5 µl siRNA and 5 µl
Lipofectamine reagent were added to 500 µl OptiMEM (Invitrogen;
Thermo Fisher Scientific, Inc.) to form a Lipofectamine 2000/siRNA
mixture. The mixture was maintained at room temperature for 30 min
to form complexes, and then the mixture was added to 12-well-plates
with 500 µl OptiMEM and incubated at 37°C in a 5% CO2
incubator. The medium was replaced after 24 h with DMEM with 10%
FBS, which contained neither siRNA nor the transfection
reagent.
Cell viability assay
HUVECs were cultured in 96-well plates at a density
of 1×104 cells/ml. Following incubation at 37°C for 24
h, cells received different treatments as described in ‘cell
culture and treatment’ section and then washed with PBS.
Subsequently, according to the manufacturer's protocol, 10 µl CCK-8
solution was added to each well and incubated at 37°C for 2 h. The
absorbance was measured at 450 nm with a microplate reader
(Multiskan MK3 Microplate reader; Thermo Fisher Scientific, Inc.).
The mean optical density (OD) of 3 wells in the indicated groups
was used to calculate the cell viability according to the following
formula: Percentage of cell viability (%)=(OD treatment
group/OD control group) ×100. The experiment was
repeated 5 times.
Measurement of intracellular reactive
oxygen species (ROS) generation
Intracellular ROS generation was measured by
determining the oxidation of DCFH-DA to fluorescent
dichlorofluorescein (DCF). Briefly, HUVECs at a density of
1×105 cells/ml were cultured at 37°C in DMEM with 10%
FBS on 6-well plates for 24 h. Following treatment as described
above, cells were collected for determination of ROS generation.
The cells were washed three times with PBS. Then, 10 µM DCFH-DA
solution in serum-free DMEM was added to the slides and incubated
at 37°C for 30 min. The cells were washed 5 times with PBS and DCF
fluorescence was measured over the entire field of vision using a
fluorescence microscope at ×40 magnification connected to an
imaging system (BX50-FLA; Olympus Corporation). The mean
fluorescence intensity (MFI) from five random fields, which is used
an index for ROS production, was analyzed using ImageJ (1.47i
software; National Institutes of Health). The experiment was
repeated 5 times.
Examination of mitochondrial membrane
potential (MMP)
The MMP was analyzed using a fluorescent dye named
Rh123. The depolarization of the MMP results in a loss of MMP and a
decrease in green fluorescence (39). Briefly, HUVECs at a density of
1×105 cells/ml were cultured at 37°C in DMEM with 10%
FBS on 6-well plates for 24 h. Following treatment as described
above, cells were collected for examination of MMP. The cells were
washed three times with PBS. The cells were subsequently incubated
with 1 µM Rh123 at 37°C for 30 min in an incubator and then washed
briefly with PBS 5 times. Fluorescence was then measured over the
entire field of vision using a fluorescence microscope at ×40
magnification connected to an imaging system (BX50-FLA). The MFI of
Rh123 from 5 randomly selected fields of view was analyzed using
ImageJ software, and the MFI indicated the levels of MMP. The lower
MFI, the higher loss of MMP. The experiment was repeated 5
times.
Western blotting
Following treatment, the HUVECs were harvested via
centrifugation for 5 min at 300 × g at 4°C and lysed with RIPA
lysis buffer for 30 min at 4°C. Total protein was quantified using
a BCA protein assay kit and 30 µg protein/lane was separated via
12% SDS-PAGE. The separated proteins were transferred onto a PVDF
membrane and blocked with 5% free-fat milk for 90 min at room
temperature. The membranes were then incubated with the following
primary antibodies at 4°C overnight with gentle agitation:
Anti-RIP3 (1:1,000), anti-p38 (1:1,000) and anti-p-p38 (1:1,000) or
anti-GAPDH (1:5,000). Following incubation with the primary
antibody, the membranes were washed with 0.1% TBS Tween-20 and then
incubated with the secondary antibody (1:5,000) for 60 min at room
temperature. The membranes were washed with 0.1% TBS Tween-20 and
protein bands were visualized using ECL and exposure to X-ray
films. To semi-quantify protein expression levels, the X-ray films
were scanned and analyzed with ImageJ software. The experiment was
repeated 3 times.
Immunofluorescence assay
HUVECs at a density of 1×105 cells/ml
were cultured at 37°C in DMEM with 10% FBS on glass coverslips for
24 h. Cells were fixed with 4% paraformaldehyde for 15 min at
temperature, permeabilized with PBS containing 0.2% Triton X-100
and blocked with 1% BSA (Seebio; http://www.seebio.cn) for 20 min at room temperature.
The slides were subsequently incubated with the anti-p-p38 primary
antibody (1:200) overnight at 4°C. Following the primary antibody
incubation, the glass coverslips were washed with 0.1% PBS-Tween 20
(PBST) and incubated with a FITC-labelled anti-rabbit secondary
antibody (1:1,000) for 1 h at 37°C, then washed by PBST and stained
with DAPI for 5 min at temperature. Stained cells were visualized
using a fluorescence microscope at ×40 magnification (Axio Imager
Z1).
Measurement of the secretory levels of
IL-1β, IL-6, IL-8 and TNF-a using ELISAs
HUVECs were cultured in 96-well growth-medium plates
at 37°C for 24 h. Then, after receiving different treatments as
described above, the secretory levels of IL-1β, IL-6, IL-8 and
TNF-a in the culture supernatant, which were acquired via
centrifuging at 500 × g for 5 min at 4°C, were analyzed using their
respective ELISA kits, according to the manufacturers' protocols.
The experiment was performed 5 times.
Statistical analysis
All data are presented as the mean ± SEM.
Statistical differences between groups were determined using a
one-way ANOVA followed by a LSD post-hoc test for 3 groups and by a
Tukey's post-hoc test for ≥3 groups using SPSS 20.0 software (IBM
Corp.). P<0.05 was considered to indicate a statistically
significant difference. Data in each experiment were obtained from
at least three independent experimental repeats.
Results
NaHS attenuates HG (40 mM
glucose)-induced upregulation of RIP3 and p-p38 expression levels
in HUVECs
As shown in Fig. 1A,
PCA of the GSE43950 dataset was performed. The results revealed an
obvious separation between healthy controls and patients with
diabetes with microvascular diseases. The results of the GSEA in
Fig. 1B identified that the
necroptosis pathway was enriched in samples with diabetes with
microvascular disease compared with healthy controls, implying the
significant role of the necroptosis pathway in diabetes with
vasculopathy. In addition, our previous study demonstrated that
necroptosis contributed to HG-induced injury (29). The present results revealed that
HUVECs treated with 40 mM HG for 24 h significantly upregulated the
expression levels of RIP3 and p-p38/p38 compared with the control
group, as detected using western blotting (Fig. 1C and D). According to our previous
study, 400 µM NaHS was determined as the effective therapeutic dose
for treatment of HUVECs without promoting cell cytotoxicity
(29). Thus, the present study also
used 400 µM NaHS for cell treatment. To determine the effect of
NaHS on the HG-induced necroptosis and the activity of p38, HUVECs
were pretreated with 400 µM NaHS for 30 min prior to exposure to
HG. The treatment of cells with 400 µM NaHS for 30 min before
exposure to HG significantly downregulated the upregulated
expression levels of RIP3 and p-p38/p38 induced by HG (Fig. 1C and D). However, the treatment with
400 µM NaHS alone did not affect the expression levels of RIP3 and
p-p38/p38 compared with the control group. In addition, an
immunofluorescence assay was performed with HUVECs. Similarly, the
results in Fig. 1E revealed that
p-p38 expression levels were upregulated by HG treatment, which
were subsequently markedly reduced in HUVECs pretreated with 400 µM
NaHS. These findings suggested that NaHS may inhibit HG-induced
necroptosis and the activity of p38 in HUVECs.
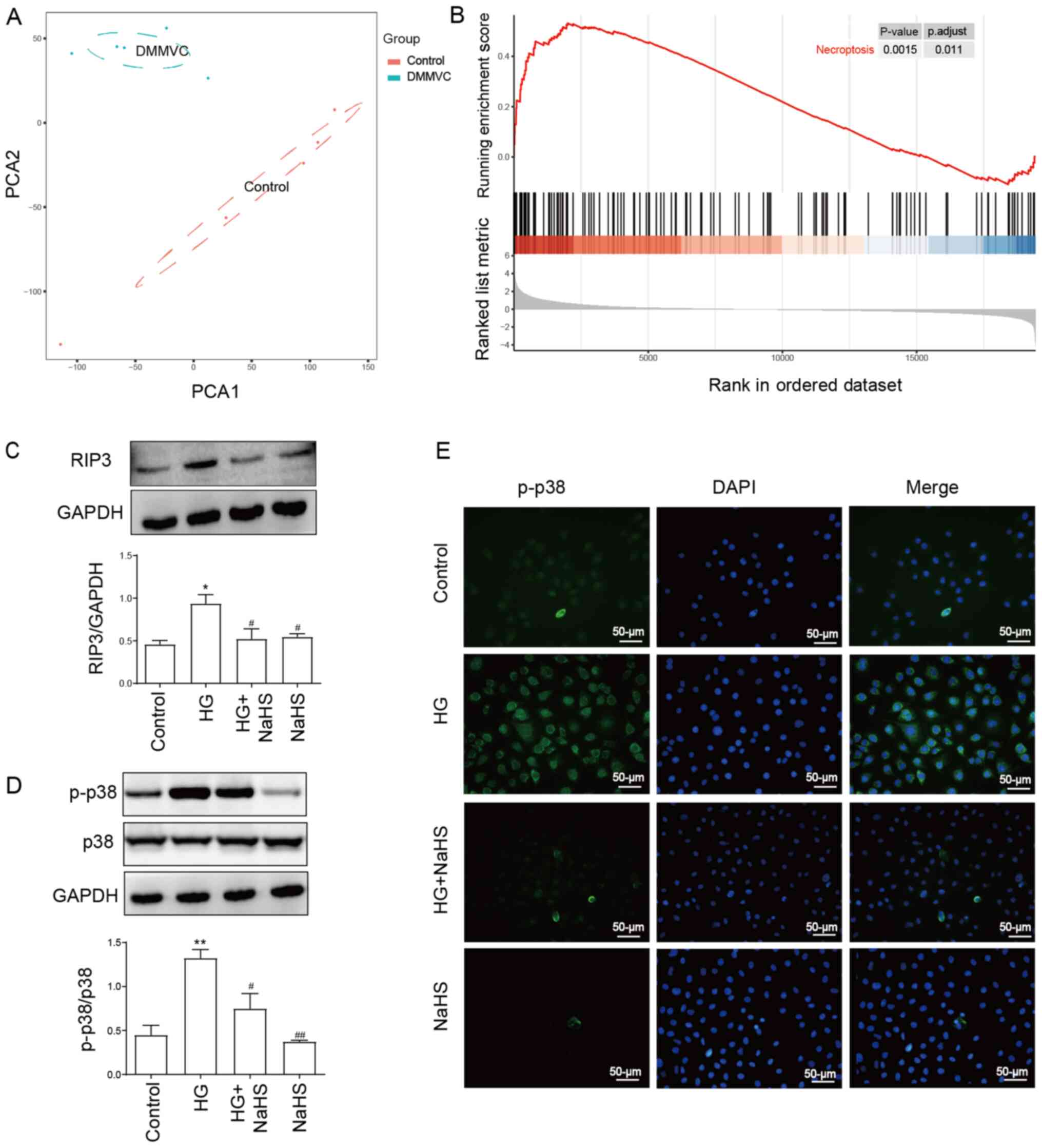 | Figure 1.Exogenous NaHS attenuates the
HG-induced upregulation of the expression levels of RIP3 and p-p38
in HUVECs. (A) PCA of the GSE43950 dataset obtained from the Gene
Expression Omnibus database. Samples including DMMVC and control
were separated into two cluster. (B) Gene Set Enrichment Analysis
for all genes in the GSE43950 dataset. Genes involved in the
necroptosis pathway were enriched in diabetes with microvascular
diseases. Expression levels of (C) RIP3 and (D) p-p38/p38 ratio
were analyzed and semi-quantified using western blotting. HUVECs
were pretreated with or without 400 µM NaHS for 30 min prior to
exposure to 40 mM HG. (E) Representative micrographs of
immunofluorescence staining of HUVECs with an anti-p-p38 antibody
(green) and the fluorescent nuclear stain DAPI (blue) following the
indicated treatments. Scale bar, 50 µm. Data are presented as the
mean ± SEM (n=3). *P<0.05, **P<0.01 vs. control group;
#P<0.05, ##P<0.01, vs. HG group. RIP3,
receptor-interacting protein 3; p-, phosphorylated; PCA, principal
component analysis; DMMVC, diabetes with microvascular disease;
HUVECs, human umbilical vein endothelial cells; NaHS, sodium
hydrosulfide; HG, high glucose. |
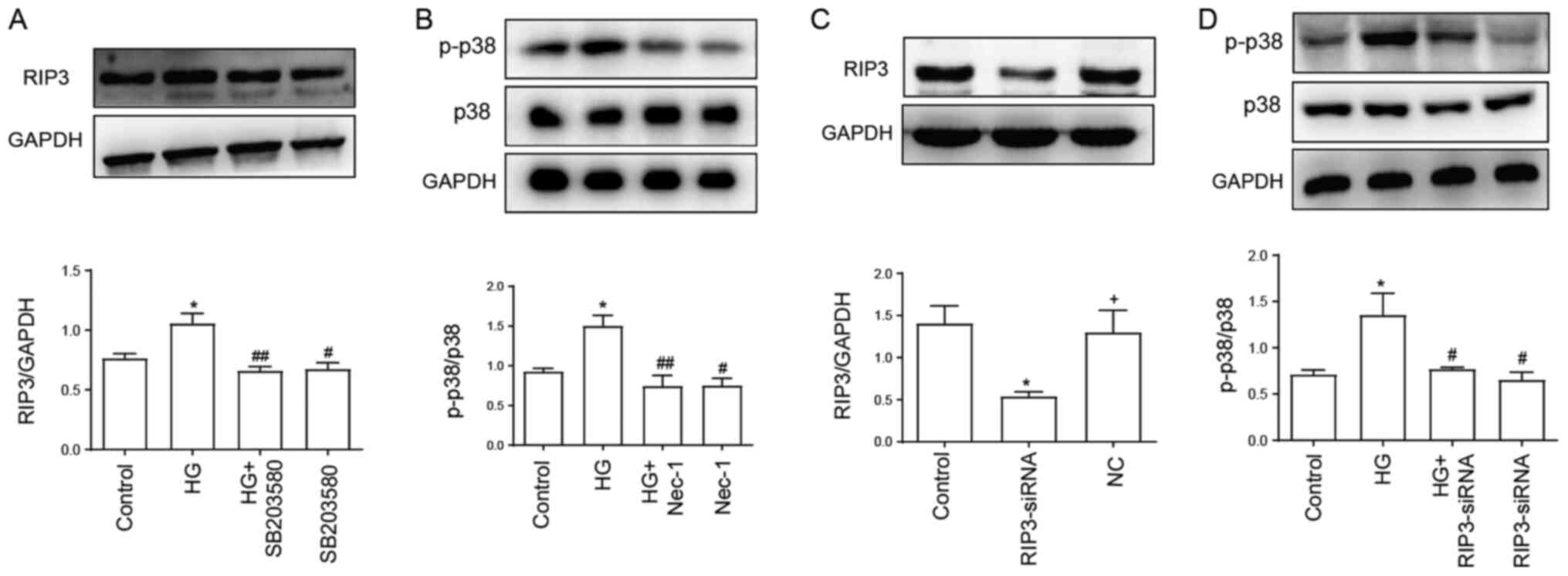
Identification of a positive feedback
loop between p38 MAPK and necroptosis pathways in HUVECs
As shown in Fig. 2A,
the expression levels of RIP3 were significantly upregulated
following the treatment with 40 mM HG compared with the control
group. However, the upregulated expression levels of RIP3 were
suppressed by the treatment with SB203580 (Fig. 2A), the inhibitor of p38 MAPK
pathway, which indicated that HG may mediate necroptosis through
activating the p38 MAPK signaling pathway. In addition,
administration of SB203580 alone had no effect on RIP3
expression.
In addition, the HG-induced upregulation of
p-p38/p38 expression levels were significantly downregulated
following Nec-1 treatment, while treatment with Nec-1 alone had no
effect on p-p38 and p38 expression levels (Fig. 2B). The transfection efficiency of
RIP3-siRNA was subsequently verified; the expression levels of RIP3
were downregulated by ~60% in the RIP3-siRNA group compared with
the control and negative control groups (Fig. 2C). Furthermore, the expression ratio
of p-p38/p38 was analyzed while inhibiting necroptosis via
RIP3-siRNA. The transfection of RIP3-siRNA promoted a significant
downregulation in the p-p38/p38 expression ratio compared with the
HG group (Fig. 2D). Thus, these
results suggested the existence of a positive feedback loop between
the necroptotic and p38 MAPK signaling pathways, which may serve an
important role in HG-induced injury.
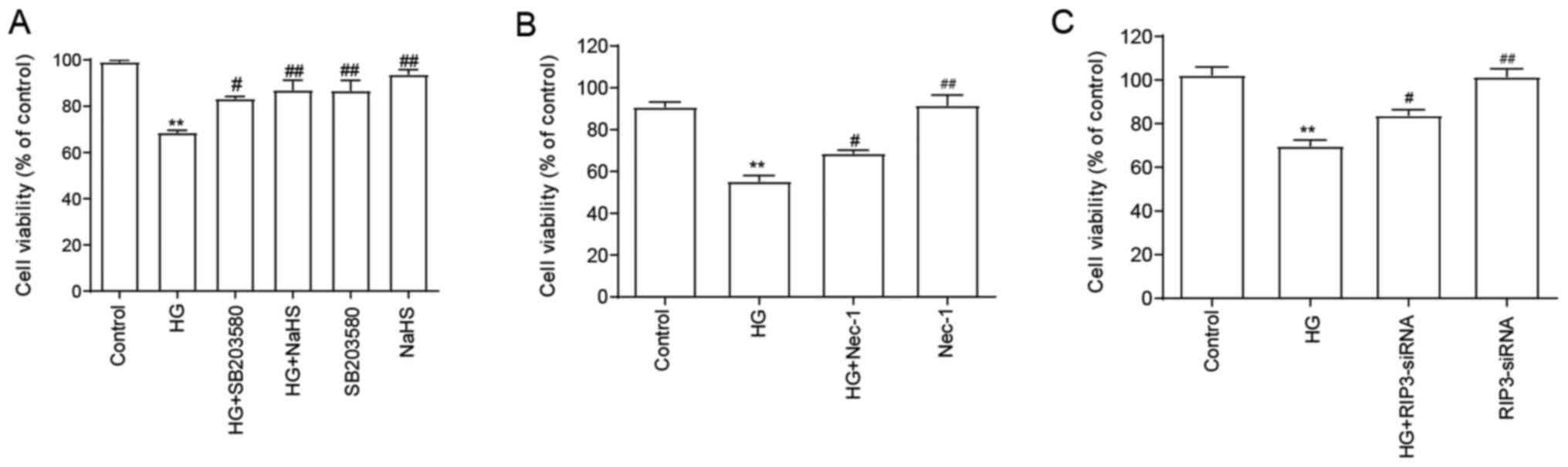
NaHS protects HUVECs against
HG-induced cytotoxicity by inhibiting necroptosis and p38 MAPK
signaling pathway activation
As shown in Fig. 3A,
compared with the HG group, the cell viability was significantly
increased following the pretreatment with NaHS, indicating that
NaHS may exert cytoprotective effects in HG-induced injury. Since
it was demonstrated that the expression levels of RIP3 and the
p-p38/p38 expression ratio were upregulated by HG, the role of RIP3
and p38 in the HG-induced cytotoxicity was subsequently
investigated. Thus, HUVECs were subsequently treated with SB203580
(Fig. 3A), Nec-1 (Fig. 3B) and RIP3-siRNA (Fig. 3C). The results suggested that all of
treatments/transfections in HUVECs treated with HG significantly
reversed the HG-induced cytotoxicity, suggesting that both
necroptosis and p38 MAPK may be involved in HG-induced
cytotoxicity. When administered alone, NaHS, SB203580, Nec-1 and
RIP3-siRNA did not affect the viability of HUVECs.
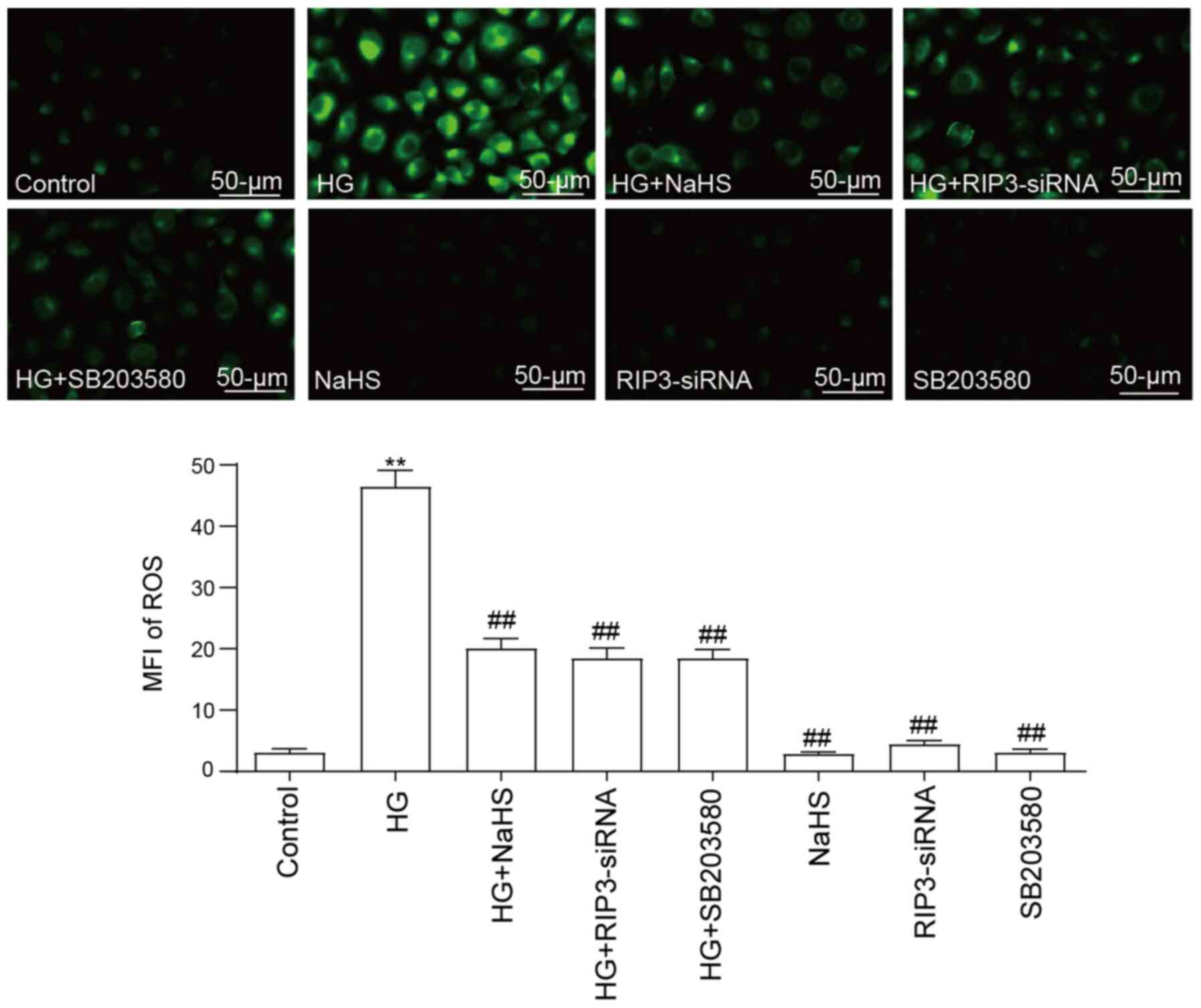
NaHS protects HUVECs against
HG-induced ROS generation by inhibiting necroptosis and p38 MAPK
activation
The treatment of HUVECs with 40 mM HG was discovered
to significantly increase ROS generation compared with the control
group (Fig. 4). Notably, the
pretreatment with 400 µM NaHS significantly decreased the
production of ROS induced by HG. Furthermore, both RIP3-siRNA
transfection and SB203580 treatment prior to HG treatment could
significantly attenuate the ROS generation compared with the HG
group. However, neither RIP3-siRNA, NaHS nor SB203580 treatment
alone influenced ROS generation compared with the control
group.
NaHS protects HUVECs against
HG-induced loss of MMP by inhibiting necroptosis and p38 MAPK
activation
As illustrated in Fig.
5, HG promoted a significant decrease in the MMP compared with
the control group, indicating that HG may induce mitochondrial
damage. However, the loss of MMP was significantly reversed by the
pretreatment with 400 µM NaHS. Similarly, following the
transfection with RIP3-siRNA or pretreatment with SB203580, the
HG-induced loss of MMP also could be inhibited, which suggested
that both necroptosis and p38 MAPK may contribute to HG-induced MMP
loss. In addition, administration of NaHS, RIP3-siRNA and SB203580
did not elicit a loss of MMP.
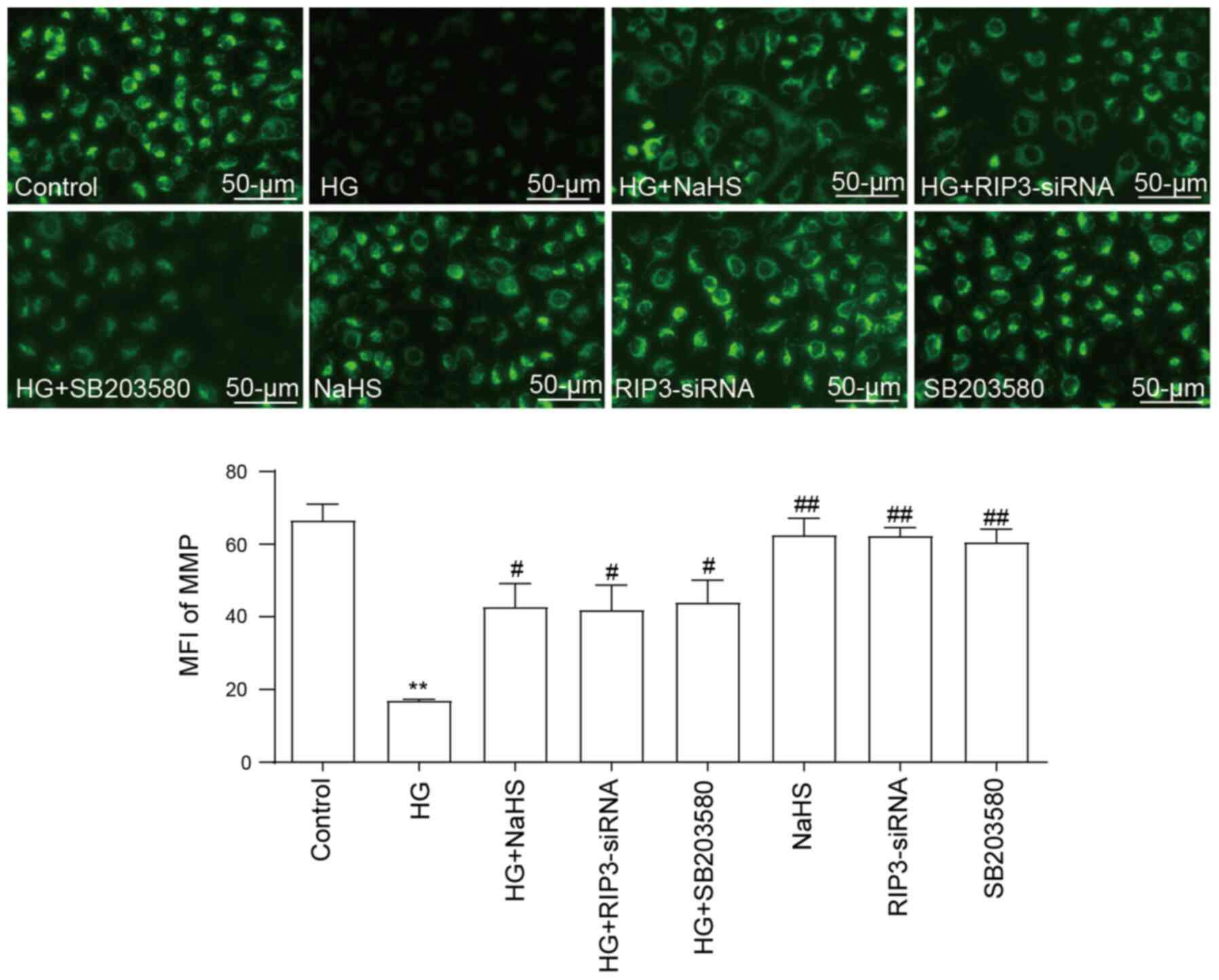 | Figure 5.Role of necroptosis and p38 MAPK
inhibition on the protective effects of NaHS against the HG-induced
loss of MMP in HUVECs. After the HUVECs were treated with the
indicated treatments, the MMP was analyzed using the fluorescent
dye, Rhodamine 123, followed by fluorescence microscopy. Scale bar,
50 µm. Data are presented as the mean ± SEM (n=5). **P<0.01 vs.
control group; #P<0.05, ##P<0.01 vs. HG
group. NaHS, sodium hydrosulfide; HG, high glucose; HUVECs, human
umbilical vein endothelial cells; RIP3, receptor-interacting
protein 3; siRNA, small interfering RNA; MMP, mitochondrial
membrane potential; MFI, mean fluorescence intensity. |
NaHS protects HUVECs against
HG-induced inflammation by inhibiting necroptosis and p38 MAPK
activation
Inflammatory cytokine levels were identified to be
increased in patients with type 2 diabetes mellitus compared with
normal glucose-tolerant subjects (40). Consistent with these findings, the
present results demonstrated that the secretory levels of IL-1β,
IL-6, IL-8 and TNF-α were significantly increased following the
exposure to HG compared with the control group (Fig. 6). However, the pretreatment with 400
µM NaHS prior to the exposure to HG significantly downregulated the
secretory levels of IL-1β, IL-6, IL-8 and TNF-α compared with the
HG group (Fig. 6A-D), suggesting
the anti-inflammatory effect of NaHS in HG-induced inflammation.
Similarly, the treatment with Nec-1 (Fig. 6E-H), the transfection with
RIP3-siRNA (Fig. 6I-L) and the
treatment with SB203580 (Fig. 6M-P)
also ameliorated the increase in HG-induced proinflammatory
cytokine secretion, further suggesting that necroptosis and the p38
MAPK signaling pathway may mediate the HG-induced secretion of
proinflammatory cytokines in HUVECs. When administered alone, NaHS,
Nec-1, RIP3-siRNA and SB203580 had no effect on the secretory
levels of the cytokines. Taken together, these results indicated
that NaHS may protect HUVECs against HG-induced inflammation by
inhibiting necroptosis and p38 MAPK activation.
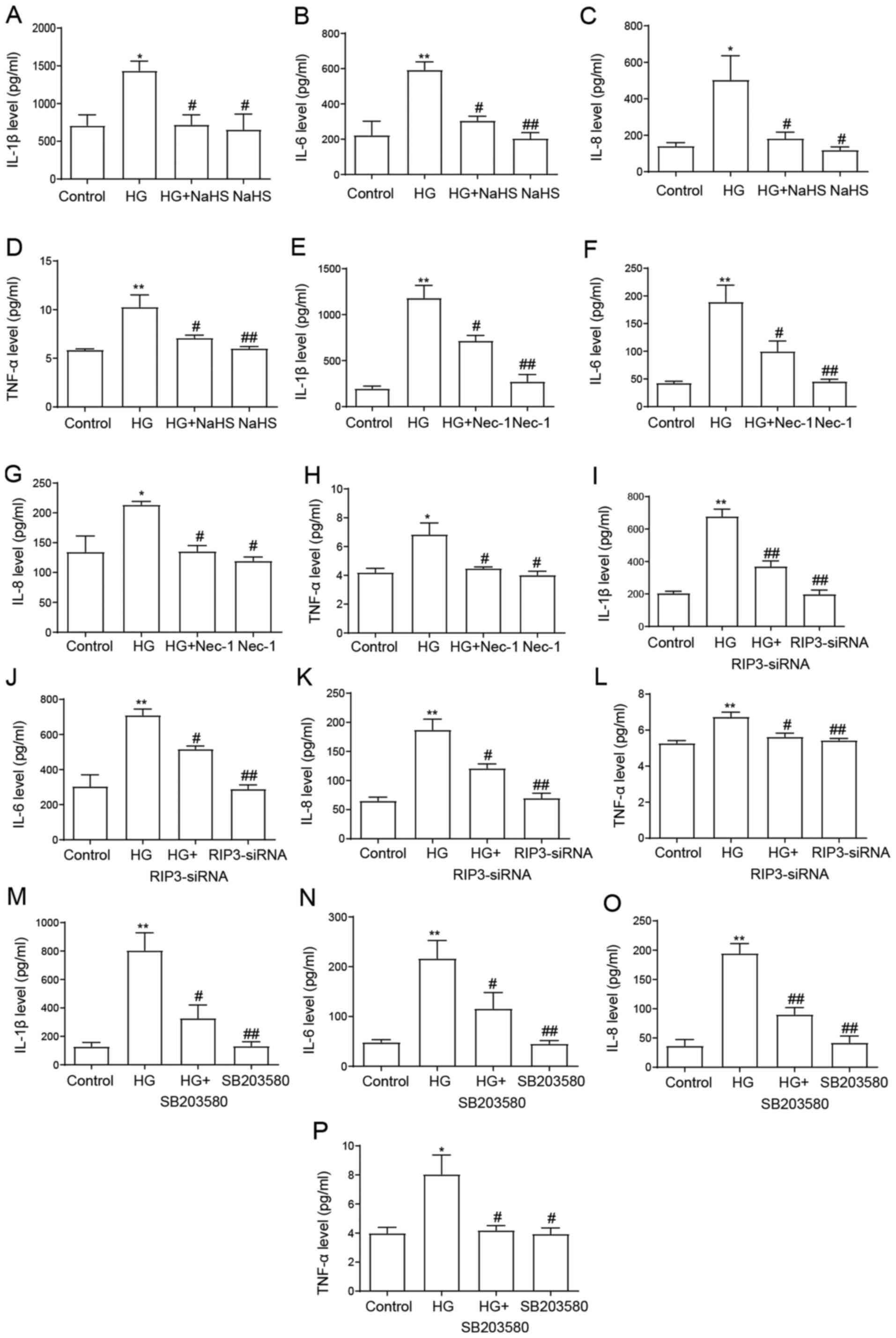 | Figure 6.Role of necroptosis and p38 MAPK
inhibition on the protective effects of NaHS against HG-induced
inflammation in HUVECs. HUVECs were treated with 40 mM HG for 24 h
with or without pretreatment with 400 µM NaHS for 30 min and ELISAs
were used to analyze the secretory levels of (A) IL-1β, (B) IL-6,
(C) IL-8 and (D) TNF-α. HUVECs were treated with 40 mM HG for 24 h
with or without pretreatment with 100 µM Nec-1 for 24 h and ELISAs
were used to analyze the secretory levels of (E) IL-1β, (F) IL-6,
(G) IL-8 and (H) TNF-α. HUVECs were treated with 40 mM HG for 24 h
with or without transfection with RIP3-siRNA for 24 h and ELISAs
were used to analyze the secretory levels of (I) IL-1β, (J) IL-6,
(K) IL-8 and (L) TNF-α. HUVECs were treated with 40 mM HG for 24 h
with or without pretreatment with 3 µM SB203580 for 1 h and ELISAs
were used to analyze the secretory levels of (M) IL-1β, (N) IL-6,
(O) IL-8 and (P) TNF-α. Data are presented as the mean ± SEM (n=5).
*P<0.05, **P<0.01 vs. control group; #P<0.05,
##P<0.01 vs. HG group. NaHS, sodium hydrosulfide; HG,
high glucose; HUVECs, human umbilical vein endothelial cells; RIP3,
receptor-interacting protein 3; siRNA, small interfering RNA;
Nec-1, necrostatin-1. |
Discussion
To investigate the potential mechanism of NaHS
cytoprotection, an in vitro HG-induced injury model was
established in HUVECs. The findings of the present study revealed
that 40 mM HG induced severe injury, including cytotoxicity, ROS
generation, MMP loss, as well as inducing inflammation in HUVECs.
In addition, the activation of p38 and necroptosis contributed to
HG-induced injury. The protective effect of NaHS in HUVECs was
found to be associated with necroptosis inhibition by repressing
the activation of p38 and a positive feedback loop was identified
between necroptosis and the p38 MAPK signaling pathway.
H2S has long been considered as a
poisonous gas, characterized by a sharp odor of rotten eggs
(41). Emerging evidence has
demonstrated that H2S exerted cardioprotective and
anti-atherosclerotic effects both in vivo and in
vitro (35,42,43).
It was previously reported that the production of H2S
was reduced in diabetes patients and diabetic model mice (44). In apolipoprotein E knockout mice,
H2S blocked the progression of atherosclerosis (45). Moreover, H2S improved
atherosclerotic lesions via suppressing ROS induction, indicating
the vasculoprotective effects of H2S. In our previous
study, NaHS was found to protect HUVECs against HG-induced injury
(29). Consistent with the previous
findings, the present results revealed that the pretreatment with
NaHS reduced cytotoxicity, ROS generation and MMP dissipation in
HUVECs. Previous studies have shown that H2S exerted
anti-inflammatory effects in doxorubicin-treated cardiac cells or
chemical hypoxia-treated HaCaT cells (46,47).
The current results identified that NaHS displayed strong
anti-inflammatory properties, as evidenced by the reduced secretory
levels of IL-1β, IL-6, IL-8 and TNF-α.
Necroptosis, also named programmed necrosis, was
identified as a new form of cell death (48). Previous evidence has indicated that
necroptosis was involved in the development of atherosclerosis
(49), retinopathy (50) and myocardial damage (51). GSEA analysis of the GSE43950 dataset
revealed a significant enrichment in the necroptosis pathway during
diabetes with microvascular disease, suggesting the important role
of the necroptosis pathway in diabetes with vasculopathy. Notably,
in our previous study, it was discovered that necroptosis
contributed to the protective effect of NaHS (29). The present study also demonstrated
that HG induced injuries, including cytotoxicity, ROS production
and mitochondrial injury, were subsequently ameliorated following
the transfection with RIP3-siRNA or pretreatment with NaHS.
Overall, these findings supported the hypothesis that NaHS may
ameliorate HG-induced damage, including cytotoxicity, ROS
accumulation, MMP loss and inflammation, at least in part, via
inhibiting necroptosis. However, the specific mechanism remains
unclear.
Cao et al (52) reported that the p38 MAPK pathway was
activated in HG-induced pancreatic cancer development. In H9c2
cells, which were exposed to 35 mM glucose, p38 MAPK was found to
mediate HG-induced damages, such as apoptosis, ROS overgeneration
and the loss of MMP (53). Hence,
the present study investigated the expression levels of p38 MAPK in
HUVECs treated with HG using western blotting and
immunofluorescence. Similar to the previous findings, the results
revealed a significant upregulation of p-p38 expression levels
following the treatment with 40 mM HG, indicating that the
activation of p38 may be induced by HG exposure. A previous study
demonstrated that the p38 MAPK pathway was responsible for the
protective effect of H2S in HG-induced injury in H9c2
cells (53). Therefore, the current
study further investigated whether the p38 MAPK pathway contributed
to the protective effect of NaHS in HUVECs. The results showed that
the upregulated p-p38 expression levels induced by HG were reduced
following the pretreatment with NaHS, implying the regulatory
effect of NaHS on p-p38 activation. Hence, these findings further
suggested that p38 MAPK signaling pathway inhibition may contribute
to NaHS protection in HG-induced injury. These results are
consistent with previous studies. For example, it was previously
reported that H2S exerted anti-inflammatory effects
repressing p38 phosphorylation (54). These results suggested that the
inhibition of p38 MAPK may be the main mechanism of H2S
protection. Another study demonstrated that p38 was implicated in
zVADfmk-mediated necroptosis (55).
Thus, the role of p38 in HG-induced necroptosis was subsequently
investigated. The upregulated expression levels of RIP3 induced by
HG were significantly reduced following the treatment with
SB203580, which indicated that p38 MAPK participated in HG-mediated
necroptosis. Taken together, these above results suggested that
NaHS may protect HUVECs against HG-induced necroptosis by
inhibiting the p38 MAPK pathway.
Further experiments were performed to clarify the
role of p38 in HG-induced injury in HUVECs. A previous study
reported that following the application of a p38 pathway inhibitor,
HG-induced oxidative stress was attenuated in cerebral endothelial
cells (18). Furthermore, another
study also demonstrated that the p38 inhibitor improved HG-induced
dysfunction of endothelial cells (56). Similar to these observations, the
present results illustrated that HG-induced injury in HUVECs was
significantly inhibited following the treatment with SB203580, as
evidenced by decreased cytotoxicity, MMP dissipation and ROS
generation, as well as the severity of inflammation. These results
demonstrated that the inhibition of the p38 MAPK signaling pathway
attenuated the injury induced by HG, including cytotoxicity, ROS
production, mitochondrial injury and inflammation damage in HUVECs,
indicating that the activation of p38 MAPK may be a potential
mechanism accounting for HG-induced injury.
In our previous study, it was revealed that NaHS
mediated a protective effect in HG-induced apoptosis and
necroptosis in HUVECs (29).
Consistent with this previous study, the present results suggested
that NaHS exerted significant cytoprotection in HUVECs, as
evidenced by an increase in cell survival, reduction in ROS
production and loss of MMP. In addition, RIP3-siRNA was used to
further determine the role of necroptosis pathway in HG-induced
injuries in the present study. Notably, both NaHS and RIP3-siRNA
effectively suppressed the inflammatory response induced by HG. Our
previous study reported the protective effect of NaHS (29), but the specific signaling pathways
via which it operates remained unknown. Therefore, the present
study was conducted to further investigate the specific mechanism.
The results demonstrated that the p38 MAPK signaling pathway was
implicated in protection of NaHS. Furthermore, in the present
study, a positive feedback loop was identified between necroptosis
and the p38 MAPK signaling pathway. The inhibitor of p38 MAPK,
SB203580, significantly inhibited the HG-induced expression levels
of RIP3. Combined with the aforementioned findings that both p38
MAPK and necroptosis contributed to cell dysfunction, the present
study indicated that necroptosis mediated HG-induced injury by
activating p38 MAPK. Interestingly, the HG-induced upregulated
expression levels of p-p38 were discovered to be diminished by
Nec-1 and RIP3-siRNA. This positive feedback may play a crucial
role in regulating HG-induced injury and inflammation. For example,
the positive feedback of necroptosis and p38 MAPK may trigger the
cascade amplification effect, resulting in the aggravation of the
inflammatory reaction and tissue injury. Therefore, the present
results provide two potential therapeutic targets for diabetic
vascular complications.
However, there are some limitations to the present
study. Firstly, only in vitro investigations were performed;
therefore, further in vivo experiments using model mice, for
example, are required to validate the results. Secondly, a
constitutively activated p38 activator, such as hesperetin, should
be used in further experiments to further strengthen the
conclusions.
In conclusion, the findings of the present study
elucidated the mechanism underlying the protective effect of NaHS
against HG-induced injury and inflammation. The results suggested
that NaHS H2S may protect HUVECs against HG-induced
injury by inhibiting necroptosis, which may be mediated through the
p38 MAPK signaling pathway. Furthermore, to the best of our
knowledge, this was the first study to identify the positive
feedback loop between necroptosis and the p38 MAPK signaling
pathway. These findings highlighted the vasculoprotective effects
of NaHS and may provide an improved understanding for developing
effective therapeutic strategies for diabetic vascular
complications.
Acknowledgements
Not applicable.
Funding
The present study was funded by a grant provided
from the National Natural Science Foundation of China (grant no.
81450062).
Availability of data and materials
The datasets analyzed during the current study are
available in the Gene Expression Omnibus repository (http://www.ncbi.nlm.nih.gov/geo).
Authors' contributions
All authors contributed to the study conception and
design. Experiments were performed by XL and YL. Data collection
and analysis were performed by WW. The manuscript was written,
drafted and designed by JL, who also performed the experiments.
Bioinformatics analysis was performed by ZH. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jeffery N and Harries LW: β-cell
differentiation status in type 2 diabetes. Diabetes Obes Metab.
18:1167–1175. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
da Rocha Fernandes J, Ogurtsova K,
Linnenkamp U, Guariguata L, Seuring T, Zhang P, Cavan D and
Makaroff LE: IDF diabetes atlas estimates of 2014 global health
expenditures on diabetes. Diabetes Res Clin Pract. 117:48–54. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sahajpal NS, Goel RK, Chaubey A, Aurora R
and Jain SK: Pathological perturbations in diabetic retinopathy:
Hyperglycemia, AGEs, oxidative stress and inflammatory pathways.
Curr Protein Pept Sci. 20:92–110. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu H, Wang X, Liu S and Li H, Yuan X,
Feng B, Bai H, Zhao B, Chu Y and Li H: Effects and mechanism of
miR-23b on glucose-mediated epithelial-to-mesenchymal transition in
diabetic nephropathy. Int J Biochem Cell Biol. 70:149–160. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Laakso M and Kuusisto J: Insulin
resistance and hyperglycaemia in cardiovascular disease
development. Nat Rev Endocrinol. 10:293–302. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Shelat H, Wu H, Zhu M, Xu J and Geng
YJ: Low circulating level of IGF-1 is a distinct indicator for the
development of cardiovascular disease caused by combined
hyperglycemia and dyslipidemia. Int J Cardiol. 171:272–273. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yerra VG, Areti A and Kumar A: Adenosine
monophosphate-activated protein kinase abates
hyperglycaemia-induced neuronal injury in experimental models of
diabetic neuropathy: Effects on mitochondrial biogenesis, autophagy
and neuroinflammation. Mol Neurobiol. 54:2301–2312. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Suzuki K, Olah G, Modis K, Coletta C, Kulp
G, Gerö D, Szoleczky P, Chang T, Zhou Z, Wu L, et al: Hydrogen
sulfide replacement therapy protects the vascular endothelium in
hyperglycemia by preserving mitochondrial function. Proc Natl Acad
Sci USA. 108:13829–13834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ceriello A, Novials A, Ortega E, Canivell
S, La Sala L, Pujadas G, Esposito K, Giugliano D and Genovese S:
Glucagon-like peptide 1 reduces endothelial dysfunction,
inflammation, and oxidative stress induced by both hyperglycemia
and hypoglycemia in type 1 diabetes. Diabetes Care. 36:2346–2350.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen H, Wan Y, Zhou S, Lu Y, Zhang Z,
Zhang R, Chen F, Hao D, Zhao X, Guo Z, et al: Endothelium-specific
SIRT1 overexpression inhibits hyperglycemia-induced upregulation of
vascular cell senescence. Sci China Life Sci. 55:467–473. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yokoyama M, Shimizu I, Nagasawa A, Yoshida
Y, Katsuumi G, Wakasugi T, Hayashi Y, Ikegami R, Suda M, Ota Y, et
al: p53 plays a crucial role in endothelial dysfunction associated
with hyperglycemia and ischemia. J Mol Cell Cardiol. 129:105–117.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song W, Wei L, Du Y, Wang Y and Jiang S:
Protective effect of ginsenoside metabolite compound K against
diabetic nephropathy by inhibiting NLRP3 inflammasome activation
and NF-κB/p38 signaling pathway in high-fat
diet/streptozotocin-induced diabetic mice. Int Immunopharmacol.
63:227–238. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen P, Yuan Y, Zhang T, Xu B, Gao Q and
Guan T: Pentosan polysulfate ameliorates apoptosis and inflammation
by suppressing activation of the p38 MAPK pathway in high
glucose-treated HK-2 cells. Int J Mol Med. 41:908–914.
2018.PubMed/NCBI
|
|
14
|
Chen Y, Wang JJ, Li J, Hosoya KI, Ratan R,
Townes T and Zhang SX: Activating transcription factor 4 mediates
hyperglycaemia-induced endothelial inflammation and retinal
vascular leakage through activation of STAT3 in a mouse model of
type 1 diabetes. Diabetologia. 55:2533–2545. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Perkins JM, Joy NG, Tate DB and Davis SN:
Acute effects of hyperinsulinemia and hyperglycemia on vascular
inflammatory biomarkers and endothelial function in overweight and
obese humans. Am J Physiol Endocrinol Metab. 309:E168–E176. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jung UJ and Choi MS: Obesity and its
metabolic complications: The role of adipokines and the
relationship between obesity, inflammation, insulin resistance,
dyslipidemia and nonalcoholic fatty liver disease. Int J Mol Sci.
15:6184–6223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li J, Bao L, Zha D, Zhang L, Gao P, Zhang
J and Wu X: Oridonin protects against the inflammatory response in
diabetic nephropathy by inhibiting the TLR4/p38-MAPK and TLR4/NF-κB
signaling pathways. Int Immunopharmacol. 55:9–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Arcambal A, Taïlé J, Rondeau P,
Viranaïcken W, Meilhac O and Gonthier MP: Hyperglycemia modulates
redox, inflammatory and vasoactive markers through specific
signaling pathways in cerebral endothelial cells: Insights on
insulin protective action. Free Radic Biol Med. 130:59–70. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shanmuganathan S and Angayarkanni N:
Chebulagic acid chebulinic acid and Gallic acid, the active
principles of Triphala, inhibit TNFα induced pro-angiogenic and
pro-inflammatory activities in retinal capillary endothelial cells
by inhibiting p38, ERK and NFkB phosphorylation. Vascul Pharmacol.
108:23–35. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dhuriya YK and Sharma D: Necroptosis: A
regulated inflammatory mode of cell death. J Neuroinflammation.
15:1992018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhe-Wei S, Li-Sha G and Yue-Chun L: The
role of necroptosis in cardiovascular disease. Front Pharmacol.
9:7212018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chan FKM, Luz NF and Moriwaki K:
Programmed necrosis in the cross talk of cell death and
inflammation. Annu Rev Immunol. 33:79–106. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FKM: Phosphorylation-driven assembly of
the RIP1-RIP3 complex regulates programmed necrosis and
virus-induced inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Newton K and Manning G: Necroptosis and
inflammation. Annu Rev Biochem. 85:743–763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Silke J, Rickard JA and Gerlic M: The
diverse role of RIP kinases in necroptosis and inflammation. Nat
Immunol. 16:689–697. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Negroni A, Colantoni E, Pierdomenico M,
Palone F, Costanzo M, Oliva S, Tiberti A, Cucchiara S and Stronati
L: RIP3 AND pMLKL promote necroptosis-induced inflammation and
alter membrane permeability in intestinal epithelial cells. Dig
Liver Dis. 49:1201–1210. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie
W, Feng Y, Socorburam T, Wu G, Xia Z and Wu Q: Receptor interacting
protein 3-mediated necroptosis promotes lipopolysaccharide-induced
inflammation and acute respiratory distress syndrome in mice. PLoS
One. 11:e01557232016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin J, Chen M, Liu D, Guo R, Lin K, Deng
H, Zhi X, Zhang W, Feng J and Wu W: Exogenous hydrogen sulfide
protects human umbilical vein endothelial cells against high
glucoseinduced injury by inhibiting the necroptosis pathway. Int J
Mol Med. 41:1477–1486. 2018.PubMed/NCBI
|
|
30
|
Feng T, Chen W, Zhang C, Xiang J, Ding HM,
Wu LL and Geng D: The p38/CYLD pathway is involved in necroptosis
induced by oxygen-glucose deprivation combined with ZVAD in primary
cortical neurons. Neurochem Res. 42:2294–2304. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Qin S, Yang C, Huang W, Du S, Mai H, Xiao
J and Lü T: Sulforaphane attenuates microglia-mediated neuronal
necroptosis through down-regulation of MAPK/NF-κB signaling
pathways in LPS-activated BV-2 microglia. Pharmacol Res.
133:218–235. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang D, Zhao M, Chen G, Cheng X, Han X,
Lin S, Zhang X and Yu X: The histone deacetylase inhibitor
vorinostat prevents TNFα-induced necroptosis by regulating multiple
signaling pathways. Apoptosis. 18:1348–1362. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Łowicka E and Bełtowski J: Hydrogen
sulfide (H2S)-the third gas of interest for pharmacologists.
Pharmacol Rep. 59:4–24. 2007.PubMed/NCBI
|
|
34
|
Gemici B, Elsheikh W, Feitosa KB, Costa
SK, Muscara MN and Wallace JL: H2S-releasing drugs:
Anti-inflammatory, cytoprotective and chemopreventative potential.
Nitric Oxide. 46:25–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Citi V, Piragine E, Testai L, Breschi MC,
Calderone V and Martelli A: The role of hydrogen sulfide and
H2S-donors in myocardial protection against ischemia/reperfusion
injury. Curr Med Chem. 25:4380–4401. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xu S, Liu Z and Liu P: Targeting hydrogen
sulfide as a promising therapeutic strategy for atherosclerosis.
Int J Cardiol. 172:313–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kumar M and Sandhir R: Hydrogen sulfide
suppresses homocysteine-induced glial activation and inflammatory
response. Nitric Oxide. 90:15–28. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Huang Z, Dong X, Zhuang X, Hu X, Wang L
and Liao X: Exogenous hydrogen sulfide protects against high
glucose-induced inflammation and cytotoxicity in H9c2 cardiac
cells. Mol Med Rep. 14:4911–4917. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang L, Jia YH, Zhao XS, Zhou FH, Pan YY,
Wan Q, Cui XB, Sun XG, Chen YY, Zhang Y and Cheng SB:
Trichosanatine alleviates oxidized low-density lipoprotein induced
endothelial cells injury via inhibiting the LOX-1/p38 MAPK pathway.
Am J Transl Res. 8:5455–5464. 2016.PubMed/NCBI
|
|
40
|
Daniele G, Guardado Mendoza R, Winnier D,
Fiorentino TV, Pengou Z, Cornell J, Andreozzi F, Jenkinson C,
Cersosimo E, Federici M, et al: The inflammatory status score
including IL-6, TNF-α, osteopontin, fractalkine, MCP-1 and
adiponectin underlies whole-body insulin resistance and
hyperglycemia in type 2 diabetes mellitus. Acta Diabetol.
51:123–131. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Smith RP and Gosselin RE: Hydrogen sulfide
poisoning. J Occup Med. 21:93–97. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chatzianastasiou A, Bibli SI, Andreadou I,
Efentakis P, Kaludercic N, Wood ME, Whiteman M, Di Lisa F, Daiber
A, Manolopoulos VG, et al: Cardioprotection by H2S donors: Nitric
oxide-dependent and -independent mechanisms. J Pharmacol Exp Ther.
358:431–440. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu X, Ma D, Zheng S, Zha K, Feng J, Cai
Y, Jiang F, Li J and Fan Z: The roles of nitric oxide and hydrogen
sulfide in the anti-atherosclerotic effect of atorvastatin. J
Cardiovasc Med (Hagerstown). 16:22–28. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bełtowski J, Wójcicka G and
Jamroz-Wiśniewska A: Hydrogen sulfide in the regulation of insulin
secretion and insulin sensitivity: Implications for the
pathogenesis and treatment of diabetes mellitus. Biochem Pharmacol.
149:60–76. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang Y, Zhao X, Jin H, Wei H, Li W, Bu D,
Tang X, Ren Y, Tang C and Du J: Role of hydrogen sulfide in the
development of atherosclerotic lesions in apolipoprotein E knockout
mice. Arterioscler Thromb Vasc Biol. 29:173–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Guo R, Wu K, Chen J, Mo L, Hua X, Zheng D,
Chen P, Chen G, Xu W and Feng J: Exogenous hydrogen sulfide
protects against doxorubicin-induced inflammation and cytotoxicity
by inhibiting p38MAPK/NFκB pathway in H9c2 cardiac cells. Cell
Physiol Biochem. 32:1668–1680. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang C, Yang Z, Zhang M, Dong Q, Wang X,
Lan A, Zeng F, Chen P, Wang C and Feng J: Hydrogen sulfide protects
against chemical hypoxia-induced cytotoxicity and inflammation in
HaCaT cells through inhibition of ROS/NF-κB/COX-2 pathway. PLoS
One. 6:e219712011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lin J, Li H, Yang M, Ren J, Huang Z, Han
F, Huang J, Ma J, Zhang D, Zhang Z, et al: A role of RIP3-mediated
macrophage necrosis in atherosclerosis development. Cell Rep.
3:200–210. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Murakami Y, Matsumoto H, Roh M, Suzuki J,
Hisatomi T, Ikeda Y, Miller JW and Vavvas DG: Receptor interacting
protein kinase mediates necrotic cone but not rod cell death in a
mouse model of inherited degeneration. Proc Natl Acad Sci USA.
109:14598–14603. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Luedde M, Lutz M, Carter N, Sosna J,
Jacoby C, Vucur M, Gautheron J, Roderburg C, Borg N, Reisinger F,
et al: RIP3, a kinase promoting necroptotic cell death, mediates
adverse remodelling after myocardial infarction. Cardiovasc Res.
103:206–216. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cao L, Chen X, Xiao X, Ma Q and Li W:
Resveratrol inhibits hyperglycemia-driven ROS-induced invasion and
migration of pancreatic cancer cells via suppression of the ERK and
p38 MAPK signaling pathways. Int J Oncol. 49:735–743. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Xu W, Wu W, Chen J, Guo R, Lin J, Liao X
and Feng J: Exogenous hydrogen sulfide protects H9c2 cardiac cells
against high glucose-induced injury by inhibiting the activities of
the p38 MAPK and ERK1/2 pathways. Int J Mol Med. 32:917–925. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Perry MM, Tildy B, Papi A, Casolari P,
Caramori G, Rempel KL, Halayko AJ, Adcock I and Chung KF: The
anti-proliferative and anti-inflammatory response of COPD airway
smooth muscle cells to hydrogen sulfide. Respir Res. 19:852018.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Koike A, Hanatani M and Fujimori K:
Pan-caspase inhibitors induce necroptosis via ROS-mediated
activation of mixed lineage kinase domain-like protein and p38 in
classically activated macrophages. Exp Cell Res. 380:171–179. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mazrouei S, Sharifpanah F, Caldwell RW,
Franz M, Shatanawi A, Muessig J, Fritzenwanger M, Schulze PC and
Jung C: Regulation of MAP kinase-mediated endothelial dysfunction
in hyperglycemia via arginase I and eNOS dysregulation. Biochim
Biophys Acta Mol Cell Res. 1866:1398–1411. 2019. View Article : Google Scholar : PubMed/NCBI
|