Introduction
Microglia cells are resident macrophages of the
central nervous system (CNS) that are essential components of the
innate immune response to external pathogen infections, cell debris
and CNS injuries (1,2). In response to stimuli, microglia
express extensive pattern recognition receptors belonging to the
Toll-like receptor (TLR) family and rapidly release various
pro-inflammatory and neurotoxic mediators, including reactive
oxygen species (ROS), prostaglandin E2
(PGE2), nitric oxide (NO) and cytokines, including
interleukin (IL)-6, IL-12, tumor necrosis factor-α (TNF-α) and
IL-1β. Excessive stimulation leads to neuronal death, resulting in
neurodegenerative diseases (3),
including Alzheimer's disease (AD), Parkinson's disease (PD),
cerebral ischemia, multiple sclerosis and stroke (4–6).
Lipopolysaccharides (LPSs) have been demonstrated to
induce the activation of microglial cell pathways (7). Activation of Toll-like receptor 4
(TLR4) is preceded by the binding of LPS, which regulates
microglial activation of various signal transduction pathways
involved in inflammatory reactions (8). Stimulated TLR4 delivers signals via
two major downstream pathways: i) the Toll/IL-1 receptor
domain-containing adapter-mediated induction of the interferon-β
(TRIF)-dependent pathway (9); and
ii) the TLR4-mediated myeloid differentiation factor 88
(MyD88)-dependent pathway. MyD88 regulates the signaling pathway
for the majority of TLRs that mediate the activation of nuclear
factor-κB (NF-κB) and mitogen-activated protein kinases (MAPKs)
(10).
NF-κB serves a critical role in the regulation of
inflammatory and immune reactions. When inactive, NF-κB is resident
as a cytoplasmic p50/p65 dimer complexed with the NF-κB inhibitor
κB (IκB), forming the NF-κB-IκB complex. By contrast, activation of
NF-κB by LPS leads to proteasomal degradation and phosphorylation
of IκB, allowing the p50/p65 dimer to translocate to the nucleus.
The translocated p50/p65 dimer binds to κB, a DNA-binding site,
leading to transcription of various genes, including those encoding
ROS, PGE2, NO, IL-6, IL-12, TNF-α and IL-1β (11).
Among the intracellular signaling pathways
associated with pro-inflammatory mediator production, MAPK cascades
appear to perform vital functions (12,13).
Cellular processes, including stress response, differentiation,
apoptosis and immune defense, are associated with MAPKs. MAPK has
three major subfamilies that are associated with modulation of the
inflammatory process: extracellular signal-regulated kinase (ERK),
c-Jun N-terminal kinase (JNK) and p38. Previous studies have
reported natural products with anti-neuroinflammatory effects that
function via inhibiting the NF-κB and MAPK signaling pathways
(14,15).
In Tibet, India and China, Nardostachys
jatamansi DC (Valerianaceae) is a vital traditional medicinal
plant used to treat mental disorders, hypertension, convulsions and
hyperlipidemia (16). In a recent
study, N. jatamansi extracts were reported to decrease beta
amyloid-activated toxicity in SH-SY5Y cells (17). Its aqueous extracts appeared to
protect against cardiac hypertrophy in a 2K1C-induced rat model
(18). Furthermore, various
sesquiterpenoids isolated from N. jatamansi have been
demonstrated to mediate the serotonin transport system (19). N. jatamansi contains various
potentially bioactive chemical components, including
monoterpenoids, sesquiterpenoids, triterpenoids and lignans.
Recently, sesquiterpenoids from N. jatamansi were
demonstrated to inhibit the increase in the levels of
pro-inflammatory cytokines, including IL-1β, IL-12 and TNF-α, as
well as of pro-inflammatory mediators, including NO and
PGE2, in LPS-stimulated BV2 microglial cells (20,21).
Materials and methods
Instruments and isolation of
nardostachin. Nuclear magnetic resonance (NMR) spectra were
obtained in CDCl3 using a JEOL JNM ECP-400 spectrometer
Chemical shifts were referenced relative to the
residual solvent peak (δH/δC = 7.26/77.2). Electrospray ionization
mass spectrometry (ESI-MS) data were obtained using an ESI
quadrupole time-of-flight MS/MS system (AB SCIEX Pte. Ltd.).
High-performance liquid chromatography (HPLC) separation was
performed using a YOUNGLIN-YL9100 system (Young Lin Instrument Co.,
Ltd.) and a Phenomenex Synergi 4 µm Polar-RP 80A, AXIA packed
column (21.2×150 mm, 5 µm particle size, 2 mg of the sample
injection, 25°C) with a flow rate of 5 ml/min. All solvents used
for HPLC were of analytical grade.
N. jatamansi hexane fraction (GSH-H) was
subjected to Si column chromatography (75×330 mm) and eluted with
CHCl3-MeOH (70:1-1:1), which yielded seven sub-fractions
(GSH-H1-7). Sub-fraction GSH-H3 was subjected to Si column
chromatography (70×300 mm) and eluted with hexane-EtOAc (20:1-1:1),
which yielded six sub-fractions (GSH-H3-1-6). Sub-fraction GSH-H3-5
was subjected to Si column chromatography (70×280 mm) and eluted
with hexane-EtOAc (10:1-1:1), which yielded 12 sub-fractions
(GSH-H3-5-1-12). Sub-fraction GSH-H3-5-9 (370.7 mg) was subjected
to Si column chromatography (180×30 mm) and eluted with
MeOH-CHCl3 (30:1-1:1), which yielded nine sub-fractions
(GSH-H3-5-9-1-9). Sub-fraction GSH-H3-5-9-2 (22.0 mg) was further
purified using semi-preparative reversed-phase HPLC (20×150 mm),
eluting in a gradient of 35–100% ACN in H2O for over 50
min, which yielded GSH-H3-5-9-2-2 (4.0 mg, tR = 30.9 min).
Nardostachin: Pale yellow powder; 1H-NMR
(400 MHz, CDCl3,): δ 6.30 (1H, brs, H-3), 5.94 (1H, d,
J = 4.2, H-1), 4.57 (1H, d, J = 12.2, H-11), 4.37
(1H, d, J = 12.2, H-11), 4.17 (1H, m, H-7), 2.92 (1H, q,
H-5), 1.12 (3H, d, J = 6.8, H-10), 0.97 (3H, d, J =
6.59, H-4′), 0.97 (3H, d, J = 6.59, H-5′), 0.97 (3H, d,
J = 6.59, H-4″), 0.97 (3H, d, J = 6.59, H-5″);
13C-NMR (100 MHz, CDCl3) δ: 172.1 (C-1″),
172.1 (C-1′), 139.6 (C-3), 114.3 (C-4), 91.6 (C-1),74.7 (C-7), 63.8
(C-11), 44.9 (C-9), 43.6 (C-2′), 43.6 (C-2″), 40.6 (C-8), 39.8
(C-6), 32.1 (C-5), 25.9 (C-3′), 25.8 (C-3″), 22.5 (C-4′), 22.5
(C-5′), 22.5 (C-4″), 22.5(C-5″), 12.8 (C-10); HRESIMS m/z
391.2092 [M + Na+] (calculated for
C20H32O6Na 391.2097).
Chemicals and reagents
Tissue culture reagents, including RPMI-1640 medium
and fetal bovine serum (FBS), were purchased from Gibco; Thermo
Fisher Scientific, Inc. All other chemicals were obtained from
Sigma-Aldrich; Merck KGaA. Primary antibodies against IкB-α (cat.
no. 4812, 1:1,000, rabbit), p-IкB-α (cat. no. 2859, 1:1,000,
rabbit), p65 (cat. no. 8242, 1:1,000, rabbit), TLR4 (cat. no.
14358, 1:1,000, rabbit), MyD88 (cat. no. 4283, 1:1,000, rabbit),
p-ERK (cat. no. 9101, 1:1,000, rabbit), ERK (cat. no. 9102,
1:1,000, rabbit), p-JNK (cat. no. 9251, 1:1,000, rabbit), JNK (cat.
no. 9252, 1:1,000, rabbit), p-p38 (cat. no. 9211, 1:1,000, rabbit)
and p38 (cat. no. 9212, 1:1,000, rabbit) were obtained from Cell
Signaling Technology, Inc. Antibody against inducible nitric oxide
synthase (iNOS; cat. no. 160862, 1:1,000, rabbit) was obtained from
Cayman Chemical and against cyclooxygenase-2 (COX-2; cat. no.
ab15191, 1:1,000, rabbit) from Abcam. Antibodies against p50 (cat.
no. sc-7178; 1:1,000, rabbit), β-actin (cat. no. sc-47778; 1:1,000,
rabbit) and PCNA (cat. no. sc-56, 1:1,000, mouse) were purchased
from Santa Cruz Biotechnology. Anti-mouse, anti-goat, and
anti-rabbit secondary antibodies were purchased from Merck
Millipore ELISA kits for PGE2 were purchased from
R&D Systems, Inc (cat. no. KGE004B).
Cell culture and viability assay
BV2 microglial cells were maintained at a density of
5×105 cells/ml in RPMI-1640 medium, supplemented with
10% heat-inactivated FBS, penicillin G (100 U/ml), streptomycin
(100 mg/ml) and L-glutamine (2 mM), and were incubated at 37°C in a
humidified atmosphere containing 5% CO2. Primary
microglial cells were cultured from the cerebral cortices or
substantia nigra of 1-day-old Sprague-Dawley rats weighing 6–8 g
which were purchased from the Orient Co., Ltd. Rats were housed in
standard cages in a climate-controlled room with an ambient
temperature of 23±2°C only for 3 h and then were sacrificed. A
total of 15 rats were used in the isolation of primary microglial
cells. Brain tissues were triturated into single cells in
Dulbecco's modified Eagle's medium containing 10% FBS and cultured
in 75-cm2 T-flasks for 2 weeks in a humified incubator
at 37°C. The microglial cells were detached from the flasks by mild
shaking and applied to a strainer to remove astrocytes and cell
clumps. Cells were plated onto 6-well plates (5×105
cells/ml) with B27® supplement (Thermo Fisher
Scientific, Inc.) in the absence of insulin for 2–3 days. Cells
were washed to remove unattached cells prior to use in subsequent
experiments.
Cell viability was evaluated by determining
mitochondrial reductase function using an assay based on reducing
MTT into formazan crystals. The formation of formazan is
proportional to the number of functional mitochondria in living
cells. Cells were maintained at 1×105 cells/ml in each
well of the 96-well plate to determine cell viability. Following
incubation for 6 h, the cells were treated for 24 h with the
indicated concentrations of 1.0. 4.0 µM nardostachin with or
without LPS (1 µg/ml). Next, 50 µl MTT (2.5 mg/ml) was added to
each well in fresh medium at a final concentration of 0.5 mg/ml,
and the mixture was further incubated for 3–4 h at 37°C, following
which the liquid was removed from the wells. Next, the formazan
formed was dissolved in 150 µl dimethyl sulfoxide, and the optical
density was measured at 590 nm wavelength. The optical density of
the formazan formed in control (untreated) cells was considered to
indicate 100% viability.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated from the cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocols, and quantified
spectrophotometrically (at 260 nm wavelength). Total RNA (1 µg) was
reverse transcribed using the High Capacity RNA-to-cDNA kit for 65
min at 100°C (Applied Thermo Fisher Scientific, Inc.). The cDNA was
then amplified using the SYBR Premix Ex Taq kit (Takara Bio, Inc.)
on a StepOnePlus Real-Time PCR system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). In brief, each 20 ml reaction volume
contained 10 ml SYBR Green PCR Master Mix, 0.8 mM each primer and
diethylpyrocarbonate-treated water. Primer sequences were designed
using PrimerQuest (Integrated DNA Technologies, Inc.) and were as
follows: TNF-α forward, 5′-CCAGACCCTCACACTCACAA-3′ and reverse,
5′-ACAAGGTACAACCCATCGGC-3′; IL-1β forward,
5′-AATTGGTCATAGCCCGCACT-3′ and reverse, 5′-AAGCAATGTGCTGGTGCTTC-3′;
IL-6 forward, 5′-ACTTCCAAGTCGGAGGCTT-3′ and reverse,
5′-TGCAAGTGCATCATCGTTGT-3′; and IL-12 forward,
5′-AGTGACATGTGGAATGGCGT-3′ and reverse, 5′-CAGTTCAATGGGCAGGGTCT-3′.
The optimal conditions for PCR amplification of the cDNA were
established according to the manufacturer's protocols. The data
were analyzed using StepOne software (Applied Biosystems; Thermo
Fisher Scientific, Inc.), and the cycle numbers at the linear
amplification threshold (Ct) for the endogenous control gene
(GAPDH) and target gene were recorded. Relative gene expression
(target gene expression normalized to endogenous control gene
expression) was calculated using the comparative Ct method
(2−ΔΔCt) (22). The
analysis was conducted independently three times.
DNA-binding activity of NF-κB
The DNA-binding activity of NF-κB in nuclear
extracts was measured using the TransAM kit (cat. no. 40096; Active
Motif, Inc.), according to the manufacturer's protocols. BV2 and
rat primary microglial cells were pre-treated for 3 h with the
indicated concentrations of nardostachin and stimulated for 30 min
with LPS (1 µg/ml) at 37°C, and the nuclear fractions were
extracted. The nuclear extracts were added to the
oligonucleotide-coated plate and reacted with primary and secondary
antibodies each for 1 h at room temperature provided in the kit. A
developing buffer was then added to each well, and this buffer
reacted with the horseradish peroxidase-conjugated secondary
antibodies provided in the kit, yielding a blue color. Next, the
stop solution was added, and the optical density was measured at
450 nm wavelength. The assay was conducted independently three
times.
Preparation of cytosolic and nuclear
fractions
The cytosolic and nuclear fractions were extracted
using the Caiman Nuclear Extraction kit from Cayman Chemical (cat.
no. 10009277) and each fraction was lysed according to the
manufacturer's protocols.
Nitrite (NO production)
determination
The nitrite concentration, an indicator of NO
production, was measured using the Griess reaction of the culture
medium. The detailed procedure for this assay is described in our
previous study (23).
PGE2 assay
The level of PGE2 present in each sample
was determined using a commercially available kit (cat. no.
KGE004B) from R&D Systems, Inc. Three independent assays were
performed according to the manufacturer's protocols.
Western blot analysis
Western blot analysis was performed as previously
described (23). In brief, BV2 and
primary microglial cells were harvested by centrifugation at 200 ×
g for 3 min at 4°C, washed with phosphate-buffered saline, and
lysed using radioimmunoprecipitation assay (RIPA) buffer (Thermo
Fisher Scientific, Inc.). The protease and phosphatase inhibitor
cocktail (Thermo Fisher Scientific, Inc.) was mixed with RIPA
buffer at a 3:100 ratio to evaluate phosphorylation.
Protein concentrations were measured using Bradford
protein assay (Bio-Rad Laboratories, Inc.) and normalized to ensure
equal amounts of protein were loaded. Subsequently, 30 µg protein
from each sample was resolved using 7.5 and 12% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis. Proteins were
electrophoretically transferred onto a Hybond enhanced
chemiluminescence (ECL) nitrocellulose membrane. The membrane was
blocked with 5% skimmed milk at 4°C for 1 h and sequentially
incubated with the primary antibodies diluted at 1:1,000 at 4°C for
1.5 h, and horseradish peroxidase-conjugated secondary antibodies
diluted at a rate of 1:1,000 at 4°C for 1 h, followed by ECL
detection. The intensity of the protein signals was quantified
using ImageJ software (ver. 1.47; National Institutes of Health).
The data represent the means of three independent experiments.
NF-κB Localization and
Immunofluorescence
BV2 and primary rat microglial cells were cultured
on Lab-Tek II chamber slides and treated as described in the figure
legends. The cells were treated with 4.0 µM nardostachin for 1 h,
fixed in formalin, permeabilized with cold acetone, and then probed
with a primary antibody against NF-κB and a fluorescein
Isothiocyanate (FITC)-labeled secondary antibody (Alexa Fluor 488;
Invitrogen; Thermo Fisher Scientific, Inc.). To visualize the
nuclei, the cells were treated with DAPI (1 µg/ml) for 30 min,
washed with PBS for 5 min, and treated with 50 µl VectaShield
(Vector Laboratories, Inc.). The stained cells were visualized and
images captured by using a confocal laser microscope (Olympus
Corporation).
Statistical analysis
Data are expressed as the mean ± standard deviation
of at least three independent experiments. To compare three or more
groups, one-way analysis of variance, followed by Tukey's multiple
comparison tests, was used. Statistical analysis was performed
using GraphPad Prism software, version 3.03 (GraphPad Software,
Inc.) (24).
Results
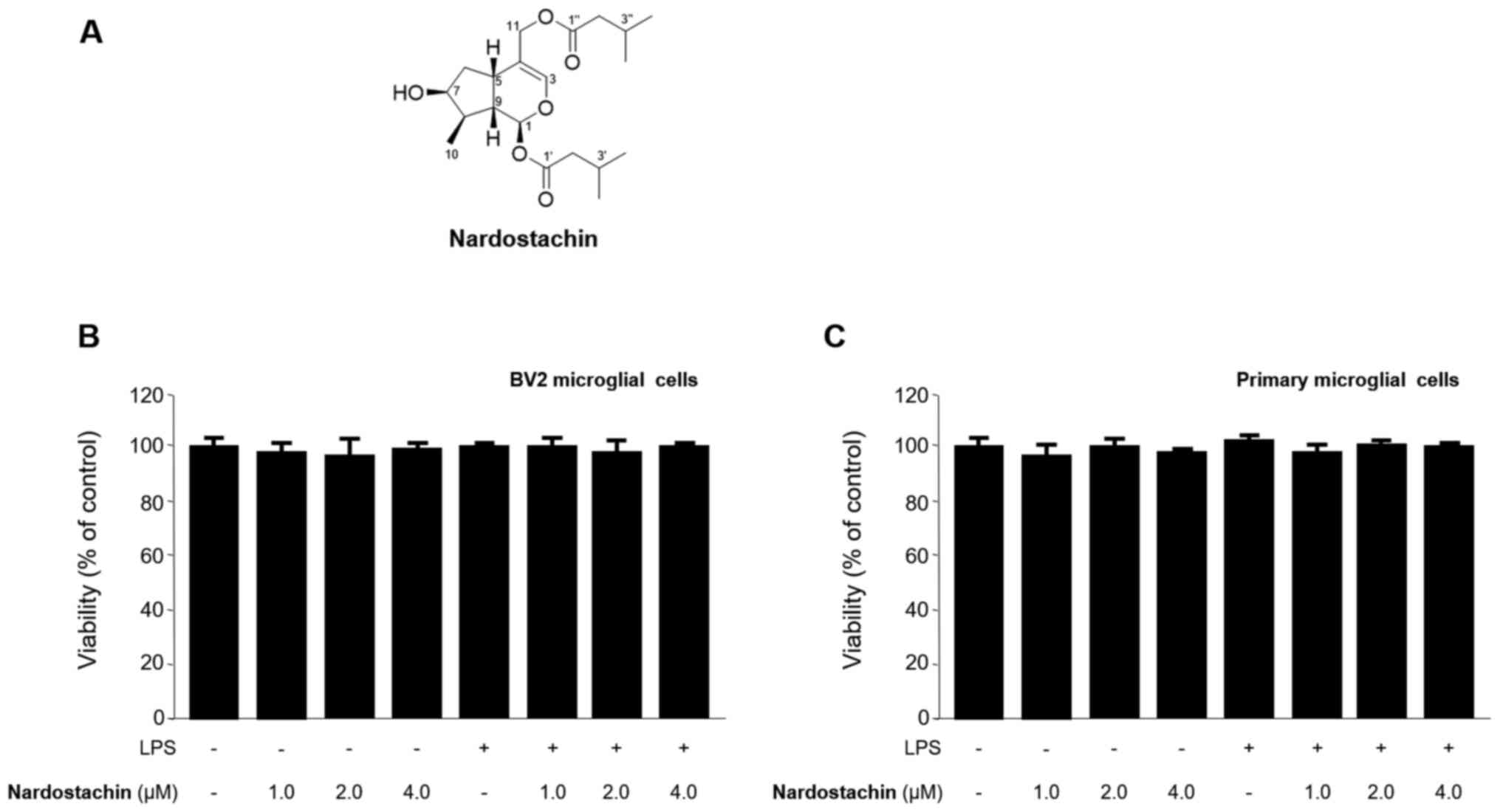
Isolation and structure determination
of nardostachin and its effect on BV2 and rat primary microglial
cell viability
Nardostachin was isolated from the hexane-soluble
fraction of the methanol extracts of N. jatamansi using
C18 flash column chromatography and semi-preparative
HPLC (Fig. 1A). The structure of
nardostachin was elucidated by analysis of MS and NMR data and a
comparison of its spectral data with those reported in the
literature (25).
A conventional MTT assay was used to analyze BV2 and
rat primary microglial cells treated with different concentrations
of nardostachin (1.0–4.0 µM) for 24 h in the absence of LPS to
determine the cytotoxicity of nardostachin. As shown in Fig. 1B and C, cell viability was not
affected by nardostachin compared with that in the control group.
Based on the MTT assay results, the concentration range of
nardostachin from 1.0 to 4.0 µM was selected for subsequent
experiments.
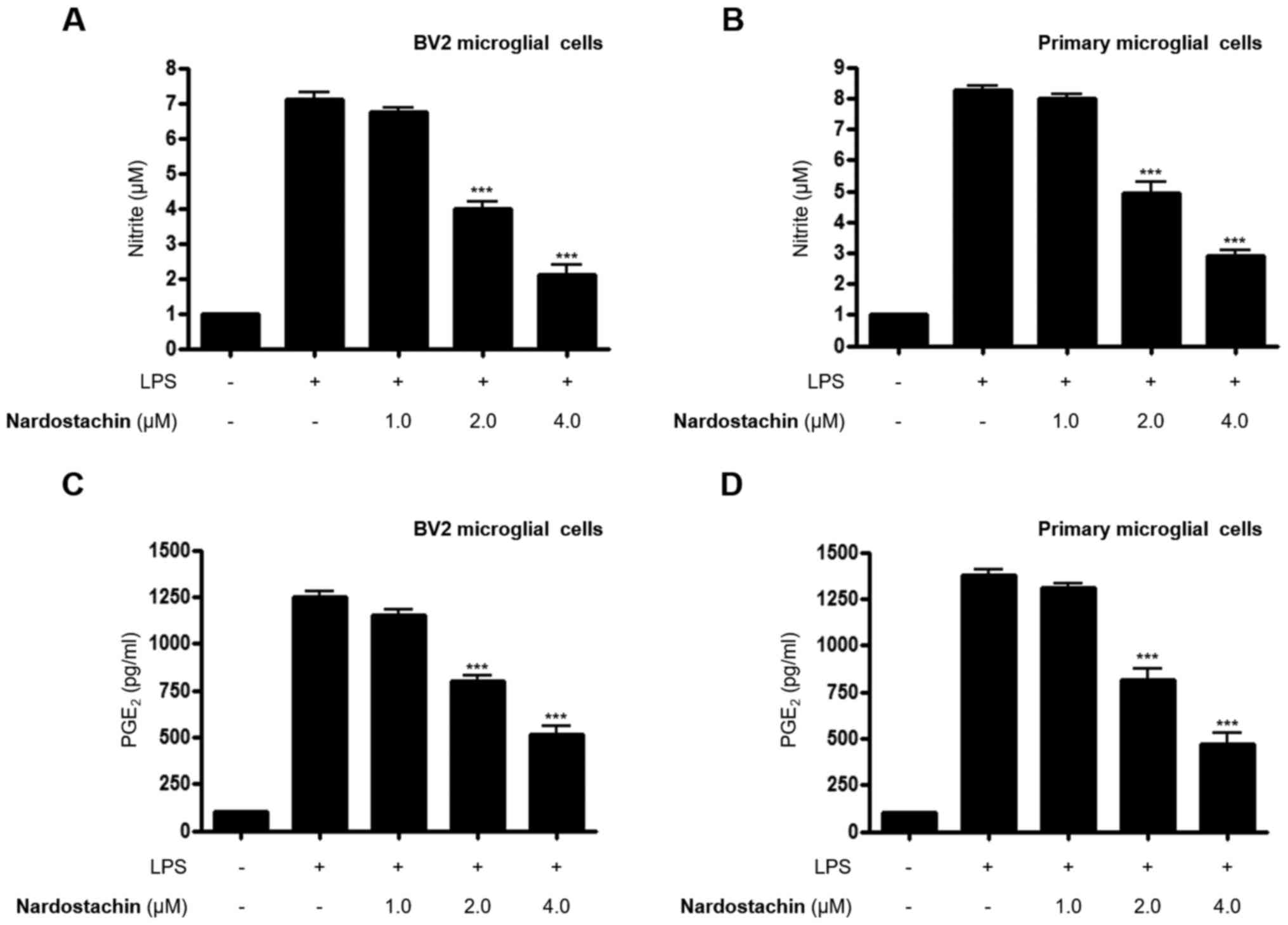
Effect of nardostachin on NO and
PGE2 production in LPS-stimulated BV2 and rat primary
microglial cells
The effects of nardostachin on LPS-induced secretion
of NO and PGE2 was subsequently analyzed using the
Griess method and ELISA, respectively, at non-cytotoxic
concentrations. As shown in Fig.
2A-D, nardostachin significantly downregulated the excessive
secretion of NO and PGE2 in LPS-stimulated BV2 and rat
primary microglial cells at concentrations ranging from 1.0 to 4.0
µM.
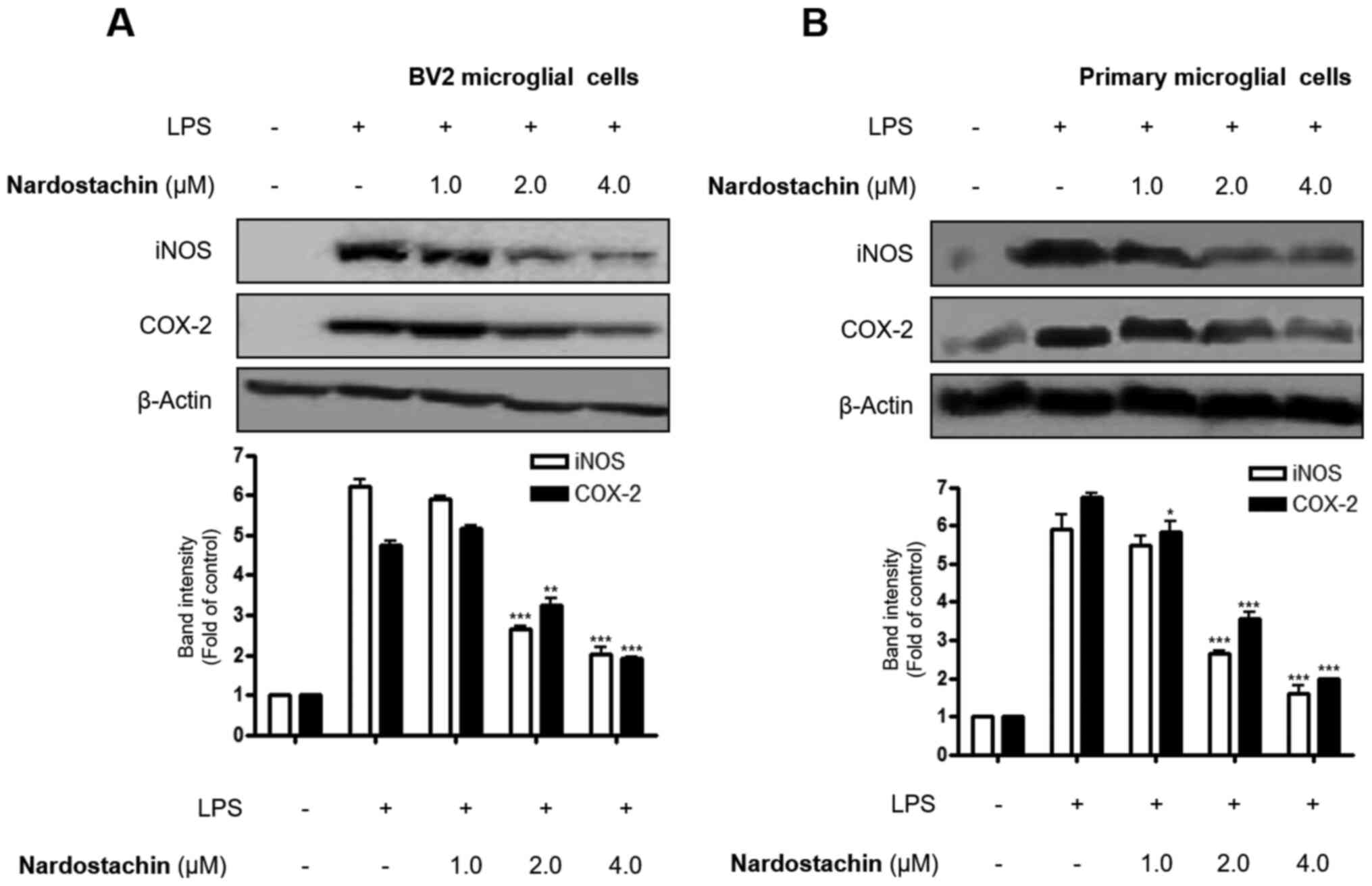
Effects of nardostachin on iNOS
expression and COX-2 activation in LPS-stimulated BV2 cells and rat
primary microglial cells
Since nardostachin was demonstrated to suppress the
overproduction of NO and PGE2 in LPS-induced BV2 and rat
primary microglial cells, its effects on iNOS and COX-2 expression
in activated BV2 and rat primary microglial cells were
investigated. As shown in Fig. 3,
LPS (1 µg/ml) induced a 6–7-fold increase in iNOS and COX-2
expression after 24 h of treatment. However, nardostachin treatment
(1.0–4.0 μM) attenuated LPS-mediated iNOS and COX-2 expression in a
dose-dependent manner.
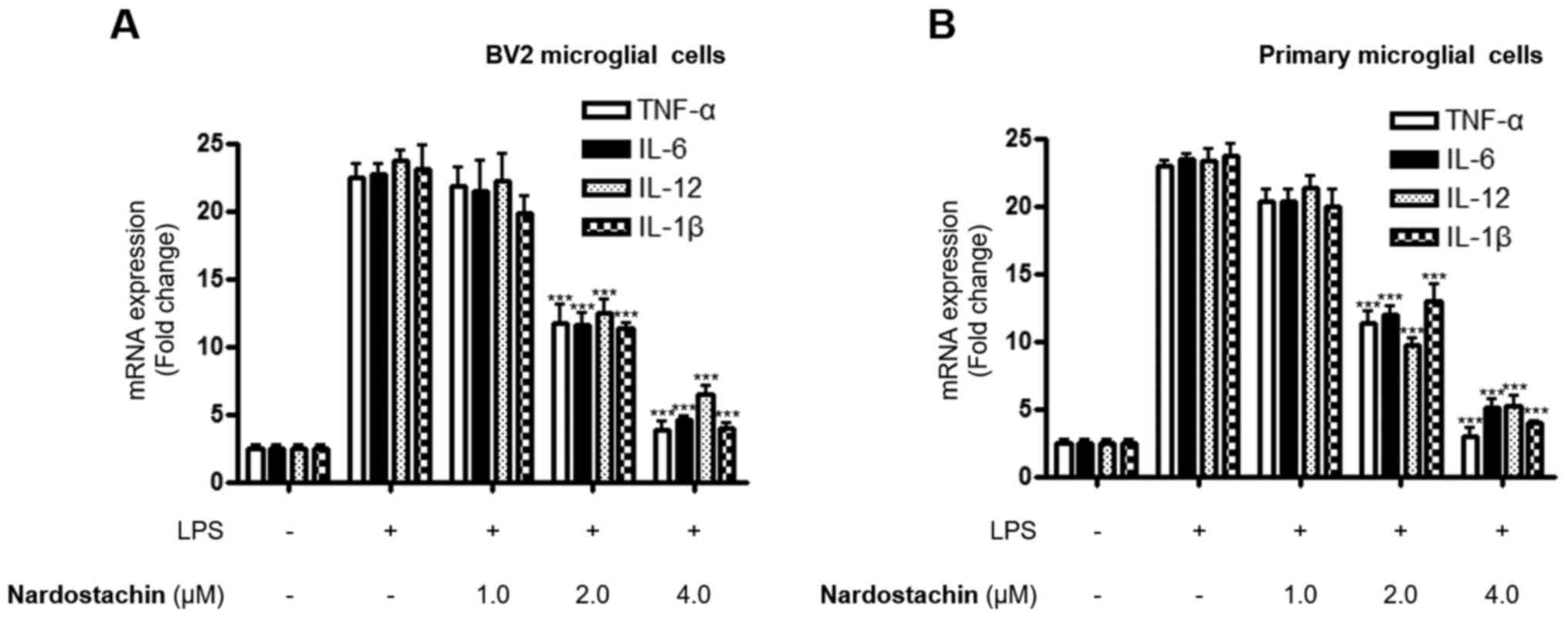
Effects of nardostachin on
pro-inflammatory cytokines in LPS-stimulated BV2 and rat primary
microglial cells
Next, the effects of nardostachin on LPS-induced BV2
and rat primary microglial cells were investigated. The mRNA levels
of pro-inflammatory cytokines, including TNF-α, IL-6, IL-12 and
IL-1β were assayed at non-cytotoxic concentrations. The production
of TNF-α, IL-6, IL-12 and IL-1β was increased in the cells treated
with LPS alone, whereas the co-treatment of nardostachin with LPS
downregulated the overproduction of TNF-α, IL-6, IL-12 and IL-1β
mRNA in a dose-dependent manner (Fig.
4).
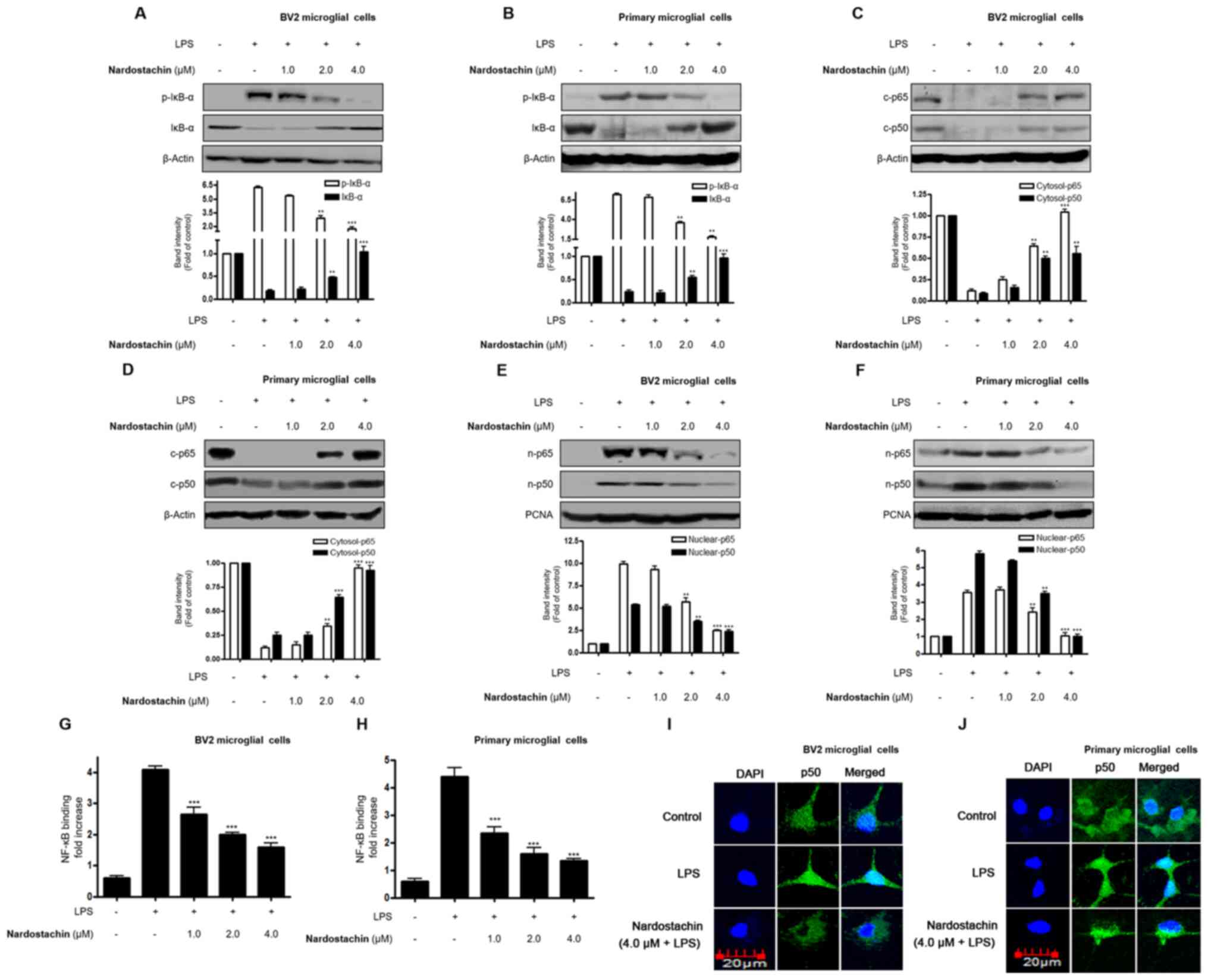
Effect of nardostachin on NF-κB
activation in LPS-stimulated BV2 and rat primary microglial
cells
Since stimulation of the NF-κB pathway is a critical
step in the inflammatory reaction, whether nardostachin inhibits
LPS-induced NF-κB activation in LPS-stimulated BV2 and rat primary
microglial cells was investigated by examining its effects on NF-κB
translocation. As shown in Fig. 5A and
B, LPS-stimulated phosphorylation of IκB-α was significantly
suppressed by nardostachin. Furthermore, IκB-α was degraded after
the cells were stimulated with LPS for 1 h. However, IκBα
degradation was markedly inhibited by the pretreatment of the cells
with nardostachin. It was also demonstrated to block LPS-stimulated
nuclear translocation of the NF-κB p50/p65 dimer. As shown in
Fig. 5C-F, the protein expression
of cytosolic p65/p50 was decreased, and protein levels of nuclear
p65/p50 were increased following stimulation with LPS for 1 h.
However, treatment with nardostachin suppressed the LPS-enhanced
protein levels of nuclear p65/p50 in a dose-dependent manner. The
results of the NF-κB binding assay also indicated that treatment
with nardostachin markedly inhibited the DNA-binding activity of
NF-κB in the nucleus (Fig. 5G and
H). Immunofluorescence analysis indicated the significant
translocation of NF-κB/p50 into the nucleus upon LPS stimulation.
However, nardostachin pretreatment blocked nuclear translocation in
the LPS-induced BV2 and rat primary microglial cells (Fig. 5I and J).
Effects of nardostachin on MAPK
phosphorylation in LPS-stimulated BV2 cells and rat primary
microglial cells
NF-κB activation is alternatively regulated by the
intracellular signaling proteins, MAPKs, in LPS-induced BV2 and rat
primary microglial cells (26).
Therefore, it was investigated whether nardostachin regulates the
phosphorylation of these signaling proteins, which may be activated
by LPS in BV2 and rat primary microglial cells. As shown in
Fig. 6, LPS-induced phosphorylation
of JNK was significantly suppressed by pretreatment of the cells
with nardostachin for 3 h. This effect was dose-dependent,
suggesting an additional characteristic of nardostachin in
regulating LPS-induced inflammation. However, ERK and p38
phosphorylation were unaffected upon exposure to nardostachin
(Fig. 6).
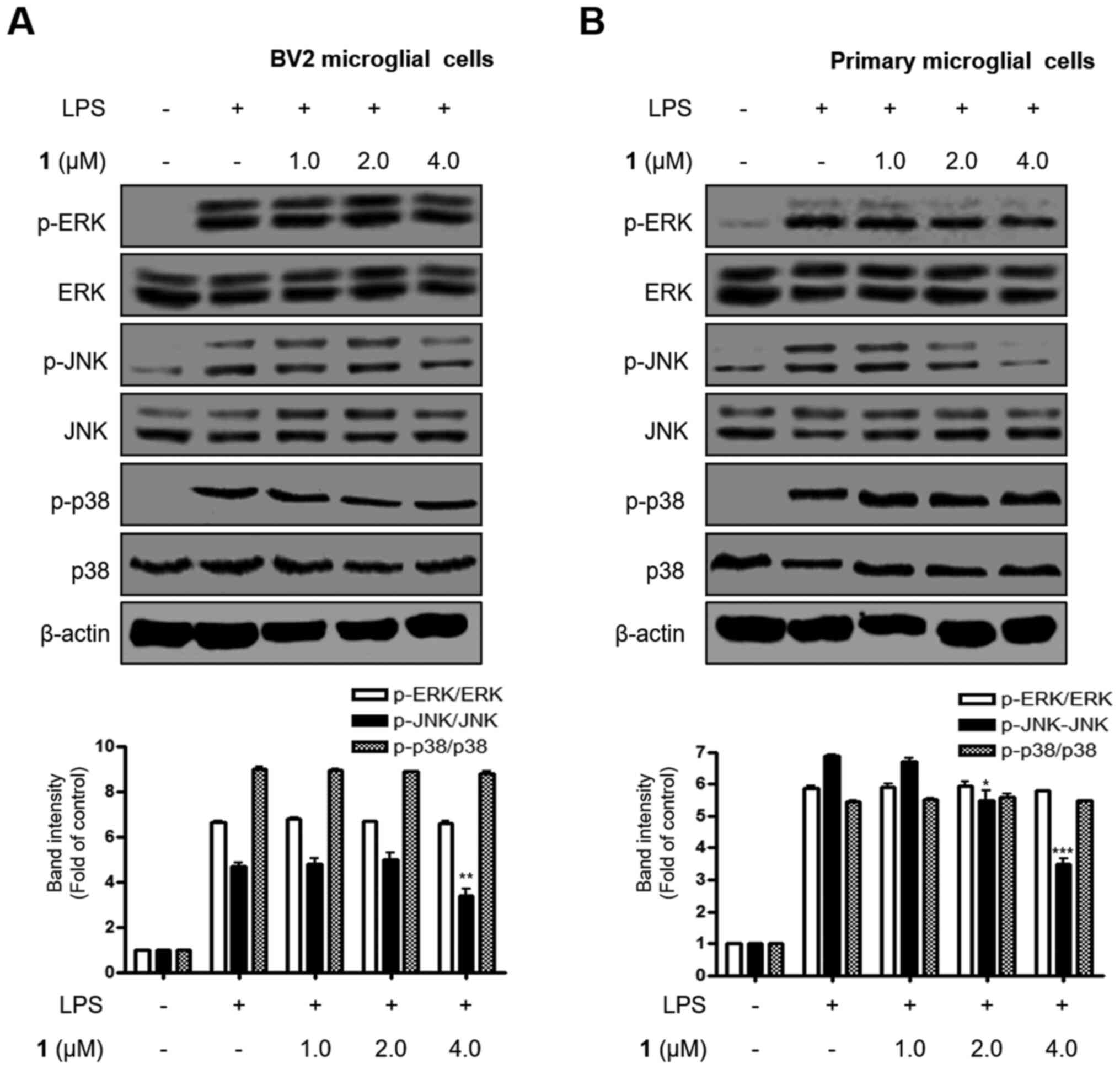 | Figure 6.Effects of nardostachin on ERK, JNK
and p38 MAPK phosphorylation and protein expression. (A) BV2 cells
were pre-treated for 3 h with the indicated concentration of
nardostachin, and for 1 h with LPS (1 µg/ml), and (B) primary
microglial cells were treated under the same conditions. The levels
of p-ERK, p-JNK and p-p38 MAPK were determined by western blot
analysis. Representative blots from three independent experiments
with similar results and densitometric evaluations are shown. Band
intensity was quantified by densitometry and normalized to the
total form of each MAPK, the values presented at the bottom of each
band. Data represent the mean values of three experiments ±
standard deviation. *P<0.05, **P<0.01, ***P<0.001 vs. the
group treated with LPS. LPS, lipopolysaccharide; p-ERK,
phosphorylated-ERK; p-JNK, phosphorylated-JNK; p-p38 MAPK,
phosphorylated-p38 MAPK. |
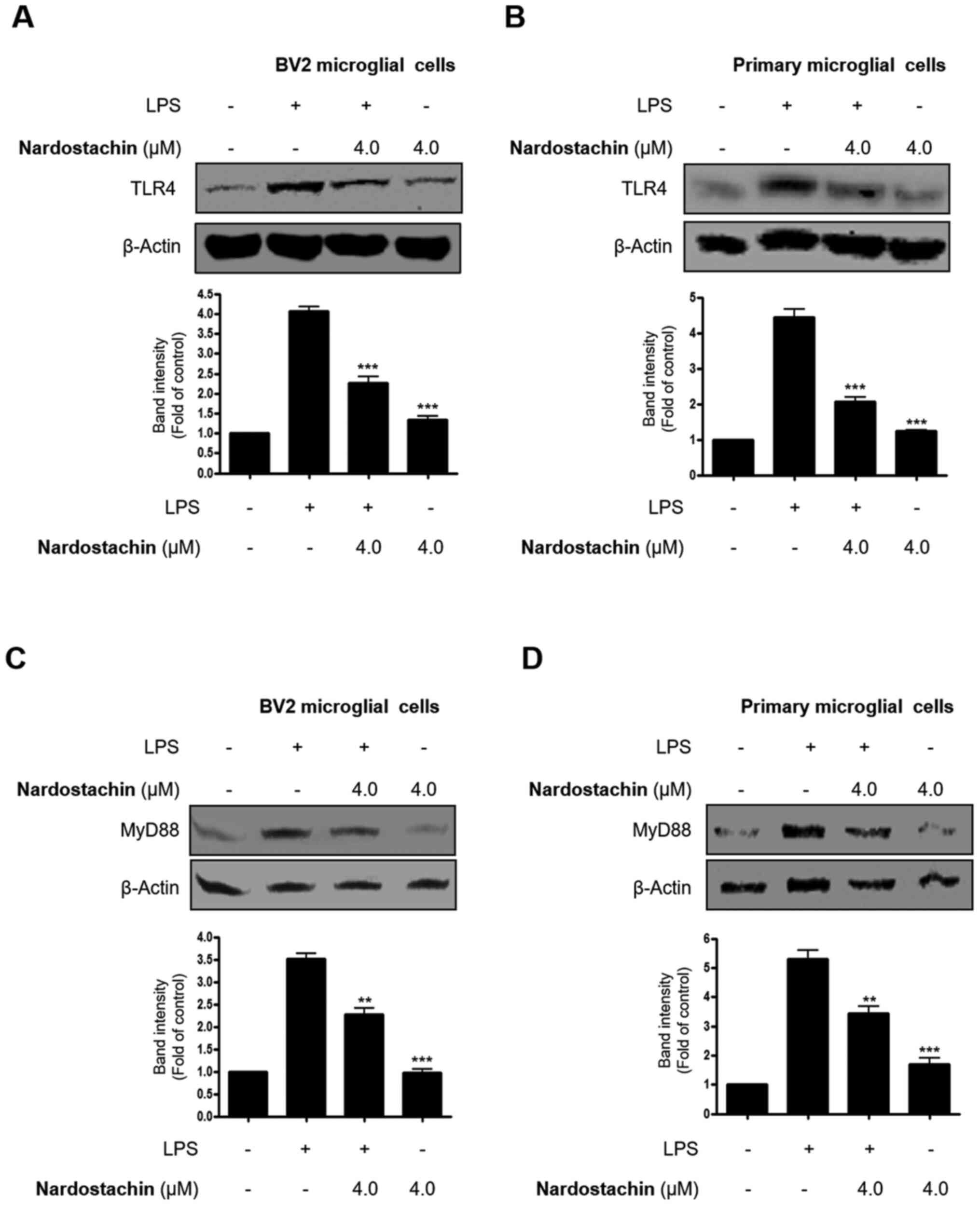
Effect of nardostachin on TLR4
expression and its interaction with MyD88 in LPS-induced BV2 and
rat primary microglial cells
TLR4 is involved in the regulation of LPS-stimulated
inflammatory mediators through stimulation of the NF-κB and MAPK
pathways. Therefore, the effect of nardostachin on protein
expression of TLR4 was investigated to gain further insight into
the mechanism underlying its anti-neuroinflammatory activity. As
shown in Fig. 7, TLR4 and MyD88
protein expression was increased in BV2 and rat primary microglial
cells upon LPS treatment, whereas pretreatment with nardostachin
for 3 h attenuated the LPS-induced TLR4 and MyD88 protein
expression in a dose-dependent manner.
Discussion
The activation of microglial cells through
TLR-mediated signaling pathways leads to the production of several
types of inflammatory mediators, and thus to the development of
various neurological disorders, including AD, PD, Huntington's
disease, multiple sclerosis and amyotrophic lateral sclerosis
(27). When the brain is
excessively stimulated, microglia release extensive pattern
recognition receptors belonging to the TLR family and their
MyD88-dependent pathway is activated. MyD88 mediates representative
inflammatory pathways like those of NF-κB and MAPK, which leads to
the production of various pro-inflammatory cytokines and mediators,
including NO and PGE2, ROS and cytokines, including
IL-12, IL-6, TNF-α and IL-1β (28).
The present study demonstrated that nardostachin suppressed the
TLR4-MyD88-NF-κB and MAPK pathways in LPS-induced BV2 and rat
primary microglial cells. iNOS and COX-2 produced inflammatory
mediators, including NO and PGE2 under inflammatory
conditions, indicative of inflammation. Nardostachin suppressed the
inflammatory reaction via repression of pro-inflammatory mediators,
including iNOS-derived NO and COX-2-derived PGE2 in
LPS-induced BV2 and rat primary microglial cells (Fig. 2). LPS stimulated iNOS and COX-2
overproduction in microglial cells, and this overproduction was
inhibited by pretreatment with nardostachin (Fig. 3). Furthermore, nardostachin
decreased the mRNA overproduction of pro-inflammatory cytokines,
including IL-12, IL-6, TNF-α and IL-1β (Fig. 4), which are involved in severe
chronic inflammatory disease (29).
NF-κB is triggered by LPS, which activates
transcription molecules that regulate the expression of various
inflammatory genes. NF-κB heterodimers consist of p65 and p50 that
generally exist in combination with IκB-α in the cytoplasm. In
response to stimuli, NF-κB is modulated by various proteins,
including TRIF, TRAF6, CD14 and TAK1, which have vital roles in
phosphorylation and degradation of IκB and translocation to
DNA-binding sites (30).
Furthermore, translocated NF-κB binds to the κB site, causing the
transcription and translation of various inflammatory genes.
Following treatment with nardostachin, LPS-induced IκB-α
degradation and NF-κB translocation were decreased in
LPS-stimulated BV2 and rat primary microglial cells (Fig. 5A-F). However, nardostachin repressed
the NF-κB DNA-binding activity (Fig. 5G
and H) and blocked the localization of p50 into the nucleus in
LPS-induced BV2 and rat primary microglial cells (Fig. 5G and H).
MAPK intracellular signaling pathways are involved
in modulating inflammatory mediators (31). Therefore, whether the
anti-inflammatory effects of nardostachin in LPS-challenged
microglial cells affected the expression of MAPKs was investigated.
The results demonstrated that nardostachin has potent inhibitory
effects on JNK MAPK phosphorylation in LPS-stimulated BV2 and rat
primary microglial cells (Fig.
6).
TLR family receptors and MyD88 mediate NF-κB and
MAPK and regulate the inflammatory reaction in microglial cells.
The present study demonstrated that nardostachin suppressed TLR4
and MyD88 protein expression in LPS-stimulated BV2 and rat primary
microglial cells (Fig. 7).
There are certain limitations to the present study.
The only in vitro test performed in the present study was
unable to sufficiently elucidate the mechanism of nardostachin.
Additionally, experiments using inhibitors associated with the
mechanisms identified in the present study were not conducted. For
further research, it is necessary to conduct additional experiments
using an in vivo model and use appropriate inhibitors to
explain the detailed mechanisms through which nardostachin exhibits
anti-neuroinflammatory effects. In conclusion, the present study
demonstrated that nardostachin isolated from N. jatamansi
showed anti-neuroinflammatory effects through TLR4/MyD88-mediated
regulation of the NF-κB and JNK MAPK signaling pathways in
LPS-stimulated BV2 and rat-derived primary microglial cells, and
these findings suggested that nardostachin has potential for the
design and development of a therapeutic agent against
neurodegenerative diseases.
Acknowledgements
Not applicable.
Funding
The present study was supported by a Korea Research
Foundation of Korea Grant funded by the Korean Government (grant
no. NRF-2017R1A5A2015805).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DCK performed the experiments associated with the
biological evaluation of the compound and wrote the manuscript. JSP
and CSY contributed toward the isolation of the compound. YCK
organized this study, contributed toward the biological evaluation
of the compound, and wrote the manuscript. HO organized this work,
contributed toward the compound's isolation and structure
determination, and wrote the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All experiments were performed according to
protocols approved by the Animal Care Committee of Wonkwang
University and were approved by the Institution Animal Care and Use
Committee (IACUC) Certification of the Wonkwang University, Korea
(approval no. WKU18-04)
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ransohoff RM and Perry VH: Microglial
physiology: Unique stimuli, specialized responses. Annu Rev
Immunol. 27:119–145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Perry VH and Holmes C: Microglial priming
in neurodegenerative disease. Nat Rev Neurol. 10:217–224. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lehnardt S: Innate immunity and
neuroinflammation in the CNS: The role of microglia in Toll-like
receptor-mediated neuronal injury. Glia. 58:253–263.
2010.PubMed/NCBI
|
|
4
|
Lukiw WJ: Bacteroides fragilis
lipopolysaccharide and inflammatory signaling in Alzheimer's
disease. Front Microbiol. 7:15442016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Long-Smith CM, Sullivan AM and Nolan YM:
The influence of microglia on the pathogenesis of Parkinson's
disease. Prog Neurobiol. 89:277–287. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perry VH, Nicoll JA and Holmes C:
Microglia in neurodegenerative disease. Nat Rev Neurol. 6:193–201.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fan K, Li D, Zhang Y, Han C, Liang J, Hou
C, Xiao H, Ikenaka K and Ma J: The induction of neuronal death by
up-regulated microglial cathepsin H in LPS-induced
neuroinflammation. J Neuroinflammation. 12:542015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Medzhitov R: Toll-like receptors and
innate immunity. Nat Rev Immunol. 1:135–145. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Noman AS, Koide N, Khuda II, Dagvadorj J,
Tumurkhuu G, Naiki Y, Komatsu T, Yoshida T and Yokochi T:
Thalidomide inhibits lipopolysaccharide-induced nitric oxide
production and prevents lipopolysaccharide-mediated lethality in
mice. FEMS Immunol Med Microbiol. 56:204–211. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park EJ, Cheenpracha S, Chang LC,
Kondratyuk TP and Pezzuto JM: Inhibition of
lipopolysaccharide-induced cyclooxygenase-2 and inducible nitric
oxide synthase expression by
4-[(2′-O-acetyl-α-L-rhamnosyloxy)benzyl]isothiocyanate from
Moringa oleifera. Nutr Cancer. 63:971–982. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kaltschmidt B and Kaltschmidt C: NF-kappaB
in the nervous system. Cold Spring Harb Perspect Biol.
1:a0012712009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mehan S, Meena H, Sharma D and Sankhla R:
JNK: A stress-activated protein kinase therapeutic strategies and
involvement in Alzheimer's and various neurodegenerative
abnormalities. J Mol Neurosci. 43:376–390. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Corrêa SA and Eales KL: The role of p38
MAPK and its substrates in neuronal plasticity and
neurodegenerative disease. J Signal Transduct. 2012:6490792012.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ha SK, Moon E, Ju MS, Kim DH, Ryu JH, Oh
MS and Kim SY: 6-Shogaol, a ginger product, modulates
neuroinflammation: A new approach to neuroprotection.
Neuropharmacology. 63:211–223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Park SY, Jin ML, Kim YH, Kim Y and Lee SJ:
Anti-inflammatory effects of aromatic-turmerone through blocking of
NF-κB, JNK, and p38 MAPK signaling pathways in amyloid β-stimulated
microglia. Int Immunopharmacol. 14:13–20. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song MY, Bae UJ, Lee BH, Kwon KB, Seo EA,
Park SJ, Kim MS, Song HJ, Kwon KS, Park JW, et al: Nardostachys
jatamansi extract protects against cytokine-induced beta-cell
damage and streptozotocin-induced diabetes. World J Gastroenterol.
16:3249–3257. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu QF, Jeon Y, Sung YW, Lee JH, Jeong H,
Kim YM, Yun HS, Chin YW, Jeon S, Cho KS, et al: Nardostachys
jatamansi ethanol extract ameliorates Aβ42 cytotoxicity. Biol
Pharm Bull. 41:470–477. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aisa R, Yu Z, Zhang X, Maimaitiyiming D,
Huang L, Hasim A, Jiang T and Duan M: The effects of aqueous
extract from Nardostachys chinensis batalin on blood pressure and
cardiac hypertrophy in two-kidney one-clip hypertensive rats. Evid
Based Complement Alternat Med. 2017:40319502017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen YP, Ying SS, Zheng HH, Liu YT, Wang
ZP, Zhang H, Deng X, Wu YJ, Gao XM, Li TX, et al: Novel serotonin
transporter regulators: Natural aristolane- and nardosinane- types
of sesquiterpenoids from Nardostachys chinensis Batal. Sci Rep.
7:151142017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yoon CS, Kim KW, Lee SC, Kim YC and Oh H:
Anti-neuroinflammatory effects of sesquiterpenoids isolated from
Nardostachys jatamansi. Bioorg Med Chem Lett. 28:140–144.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hwang JS, Lee SA, Hong SS, Han XH, Lee C,
Lee D, Lee CK, Hong JT, Kim Y, Lee MK, et al: Inhibitory
constituents of Nardostachys chinensis on nitric oxide production
in RAW 264.7 macrophages. Bioorg Med Chem Lett. 22:706–708. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rao X, Huang X, Zhou Z and Lin X: An
improvement of the 2ˆ(-delta delta CT) method for quantitative
real-time polymerase chain reaction data analysis. Biostat
Bioinforma Biomath. 3:71–85. 2013.PubMed/NCBI
|
|
23
|
Yoon CS, Kim DC, Lee DS, Kim KS, Ko W,
Sohn JH, Yim JH, Kim YC and Oh H: Anti-neuroinflammatory effect of
aurantiamide acetate from the marine fungus Aspergillus sp.
SF-5921: Inhibition of NF-κB and MAPK pathways in
lipopolysaccharide-induced mouse BV2 microglial cells. Int
Immunopharmacol. 23:568–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim DC, Lee HS, Ko W, Lee DS, Sohn JH, Yim
JH, Kim YC and Oh H: Anti-inflammatory effect of methylpenicinoline
from a marine isolate of Penicillium sp. (SF-5995):
Inhibition of NF-κB and MAPK pathways in lipopolysaccharide-induced
RAW264.7 macrophages and BV2 microglia. Molecules. 19:18073–18089.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
An RB, Min BS, Na MK, Chang HW, Son KH,
Kim HP, Lee HK, Bae K and Kang SS: Iridoid esters from Patrinia
saniculaefolia. Chem Pharm Bull (Tokyo). 51:583–585. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y and Dong C: Regulatory mechanisms
of mitogen-activated kinase signaling. Cell Mol Life Sci.
64:2771–2789. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nguyen MD, Julien JP and Rivest S: Innate
immunity: The missing link in neuroprotection and
neurodegeneration? Nat Rev Neurosci. 3:216–227. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lappas M, Permezel M, Georgiou HM and Rice
GE: Nuclear factor kappa B regulation of proinflammatory cytokines
in human gestational tissues in vitro. Biol Reprod. 67:668–673.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Agostinho P, Cunha RA and Oliveira C:
Neuroinflammation, oxidative stress and the pathogenesis of
Alzheimer's disease. Curr Pharm Des. 16:2766–2778. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang G and Ghosh S: Toll-like
receptor-mediated NF-kappaB activation: A phylogenetically
conserved paradigm in innate immunity. J Clin Invest. 107:13–19.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bennett AM and Tonks NK: Regulation of
distinct stages of skeletal muscle differentiation by
mitogen-activated protein kinases. Science. 278:1288–1291. 1997.
View Article : Google Scholar : PubMed/NCBI
|