Introduction
Epithelial ovarian cancer (EOC) is one of the most
common and frequently fatal gynecological malignancies worldwide.
Annually, ~230,000 women are diagnosed with EOC, resulting in
150,000 deaths (1). Since the
1980s, debulking surgery followed by chemotherapy has been the
primary treatment approach for EOC, and paclitaxel (PTX) is the
recommended first-line drug of choice. Although PTX treatment is
initially effective, patients with advanced or relapsing tumors
frequently develop chemoresistance, which further contributes to
disease progression (2); however,
the mechanisms underlying PTX resistance are unclear. Notably, PTX
has been shown to induce autophagy in various cancer cell types,
resulting in chemoresistance and a decreased rate of tumor cell
death (3,4). Therefore, the discovery of novel
therapeutic targets is required to overcome the autophagic effects
of PTX in tumor cells.
Autophagy is an evolutionarily conserved
intracellular self-defense mechanism that maintains homeostasis
during a variety of stress situations, such as starvation,
chemotherapy, aging-related disease and cancer (5). When autophagy is initiated, organelles
and proteins are sequestered into autophagosomes that subsequently
combine with lysosomes to form autolysosomes, in which cytoplasmic
materials are degraded (6).
Accumulating data have indicated that autophagy may be involved in
the development of chemotherapeutic resistance and tumor
progression in various types of cancer (7,8).
In humans, the WW domain-containing oxidoreductase
(WWOX) gene is located at the ch16q23.2 or ch16q23.3–24.1
chromosomal sites, which span the common fragile site FRA16D. Due
to the fragility of this genetic locus, WWOX expression is
frequently lost or decreased, together with the loss of
heterozygosity associated with ovarian, esophageal, breast and lung
cancer (9–13). In response to stressful stimuli,
WWOX is phosphorylated at Tyr33, which enables the formation of a
complex containing p53 and JNK1 (14,15).
Tyr33 phosphorylation and nuclear localization results in
WWOX-mediated apoptosis (16). Tsai
et al (17) revealed that
WWOX may suppress autophagy and enhance methotrexate-induced death
of tongue squamous cancer cells. However, studies focusing on the
association between WWOX and autophagy are limited, and further
analysis is required to confirm whether WWOX exerts an inhibitory
effect on autophagy.
mTOR is a serine/threonine kinase, which has been
reported to regulate cellular metabolism and promote proliferation
in response to environmental stimuli (18). Elevated levels of mTOR
phosphorylation have been predicted to be associated with
diminished autophagy (18).
Increasing evidence has also suggested that the relationship
between mTOR signaling and autophagy is inhibitory, and that this
association may act as a potential treatment target in cancer
research (19,20). However, further investigation is
required to confirm these findings, such as the potential mechanism
of mTOR in regulating autophagy, and whether combining mTOR with
WWOX may affect its downstream factors and ultimately inhibit the
occurrence of autophagy.
The present study aimed to demonstrate the role of
WWOX in apoptosis and autophagy during PTX treatment and to
investigate the underlying mechanisms of WWOX-regulated autophagy
and PTX resistance in ovarian carcinoma.
Materials and methods
Cell culture and treatment
The human EOC cell lines A2780 and SKOV3 were
purchased from the American Type Culture Collection, and A2780/T
cells were obtained from the Medical College of Shandong University
(Jinan, China). A2780 and A2780/T cells were maintained in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.) and SKOV3
cells were cultured in McCoy's 5A medium (Gibco; Thermo Fisher
Scientific, Inc.). A2780/T is a PTX-resistant cell line; to
maintain its drug-resistant properties, PTX (800 ng/ml) was added
to the culture medium. PTX was dissolved in DMSO and then diluted
with medium to the specified concentration. The SKOV3 cell line
originated from the ascites of an elderly woman with moderately
differentiated ovarian adenocarcinoma, which has certain tolerance
to TNF and several cytotoxic drugs. A2780 cells were derived from
the ovarian tumor tissues of an untreated patient (21). PTX resistance of A2780/T cells is
induced by artificial PTX treatment of A2780 cells. All of the
cells were cultured at 37°C in an atmosphere containing 5%
CO2 without any antibiotics, and the media were
supplemented with 10% fetal bovine serum (Moregate; Bovogen
Biologicals Pty Ltd.). PTX and chloroquine (CQ) were obtained from
Beijing Solarbio Science & Technology Co., Ltd. and
Sigma-Aldrich (Merck KGaA), respectively. PTX and CQ were dissolved
in cell medium to the specified concentration prior to use.
Cell viability detection
EOC cells were seeded (8,000 cells/well) in 96-well
plates; after 24 h adhering to the plate, the cells were exposed to
the indicated concentrations of PTX for 48 h in the incubator at
37°C. The treatment concentrations of PTX for A2780 and SKOV3 cells
were 0.0, 3.0, 6.0, 12.5, 25.0, 50.0, 100.0 and 200.0 ng/ml. The
concentrations of PTX for A2780/T cells were 0, 100 200, 400, 800,
1,600, 2,400 and 3,200 ng/ml. Each drug concentration was repeated
in five wells. Subsequently, 10 µl Cell Counting Kit-8 (Eno Gene
Biotech. Co., Ltd.) solution was mixed into each well and incubated
for 1 h, according to the manufacturer's protocol. A microplate
reader (Thermo Fisher Scientific, Inc.) was then used to measure
the optical density values at a wavelength of 450 nm. Each time
point was tested in triplicate.
Western blotting
Total protein was extracted from cells using RIPA
lysis buffer (Beyotime Institute of Biotechnology) and protein
concentrations were quantified using a bicinchoninic acid protein
kit (Pierce; Thermo Fisher Scientific, Inc.). Total proteins (30
µg) were separated by SDS-PAGE, on 15% gels for LC3 and caspase-3
detection, and 10% gels for the detection of other proteins.
Proteins were transferred to PVDF membranes (EMD Millipore), which
were blocked with 5% non-fat dry milk (FUJIFILM Wako Pure Chemical
Corporation) or 5% bovine serum albumin (Beijing Solarbio Science
& Technology Co., Ltd.) on a shaker at room temperature for 1
h, and washed in TBS-Tween (0.1% Tween-20) (Beijing Solarbio
Science & Technology Co., Ltd.) for 5 sec. Then, the blots were
incubated with primary antibodies at 4°C overnight. The following
antibodies were used for western blot analysis:
Anti-WWOX-N-terminal (cat. no. ab189410; 1:1,000; Abcam),
anti-phosphorylated (p)-Y33-WWOX (cat. no. ab193624; 1:1,000;
Abcam), anti-caspase-3 (cat. no. 9662; 1:1,000; Cell Signaling
Technology, Inc.), anti-poly ADP-ribose polymerase (PARP; cat. no.
9542; 1:1,000; Cell Signaling Technology, Inc.), mTOR sampler kit
(cat. no. 9862; 1:1,000; Cell Signaling Technology, Inc.),
anti-LC3B (cat. no. 2775; 1:1,000; Cell Signaling Technology,
Inc.), autophagy antibody sampler kit (cat. no. 4445; 1:1,000; Cell
Signaling), anti-p62/SQSTM1 (cat. no. 18420-1-AP; 1:1,000;
Proteintech Group, Inc.), anti-GAPDH (cat. no. 2605479; 1:2,000;
EMD Millipore). Subsequently, membranes were washed 3 times with
TBS-Tween (0.1% Tween-20) for 10 min each and incubated with a
HRP-labeled secondary anti-rabbit antibody (1:1,000; cat. no.
ZF-0311; OriGene Technologies, Inc.) at room temperature for 1 h.
Antibody binding was visualized using ECL Plus Detection reagent
(cat. no. 34080; Thermo Fisher Scientific, Inc.) and ECL film
(Carestream Health, Inc.). Images were captured using Image Lab
v5.2.1 (Bio-Rad Laboratories, Inc.).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
PBS was used to wash EOC cells twice at 4°C, and
then added 1 ml TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) was added to each well of the 6-well plate
according to the manufacturer's protocol. Total RNA was obtained
from cells. RT was performed using the PrimeScript RT Master Mix
(Perfect Real-Time) (Takara Bio, Inc.) according to the
manufacturer's instructions. qPCR was conducted using SYBR Green
Master Mix (Beijing Solarbio Science & Technology Co., Ltd.)
and primers designed by Wanleibio Co., Ltd. The primer sequences
were as follows: WWOX, forward, 5′-GTAGAATACGCAGAACTACCAG-3′ and
reverse, 5′-GAACTGAGACTGTGATTGACAGAC-3′; and β-actin, forward,
5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and reverse,
5′-CTGTCACCTTCACCGTTCCAGTTT-3′. The reaction conditions were 95°C
for 5 min, followed by 35 cycles at 95°C for 30 sec, 63°C for 30
sec and 72°C for 30 sec, and a final extension step at 72°C for 5
min. All qPCR analyses were performed in triplicate. The
amplification and melting curves of the end products were obtained
to confirm PCR specificity. Relative gene expression data were
analyzed using RT-qPCR and the 2−ΔΔCq method (22). Relative expression levels of genes
were obtained through sequential normalization of the values
against β-actin.
Flow cytometric analysis of
apoptosis
EOC cells were treated with the indicated doses of
PTX for 48 h in the incubator at 37°C, then harvested and
resuspended in binding buffer. A2780 cells were treated with 150
ng/ml PTX; A2780/T cells were treated with 1,600 ng/ml PTX. The
cells were washed twice and adjusted to a concentration of
1×106 cells/ml with cold D-Hanks buffer. Annexin
V-Pacific Blue and propidium iodide (BioLegend, Inc.) were then
added to the cell suspension and incubated for 15 min at room
temperature in the dark, according to the manufacturer's
instructions. Finally, 400 µl binding buffer from the kit was added
to each sample without washing, and apoptosis was analyzed using
flow cytometry (BD FACSCalibur; BD Biosciences) and the data were
analyzed with FlowJo software (version 7.6.1; FlowJo LLC). Each
experiment was performed in triplicate.
Transfection
The eukaryotic expression vector pcDNA3.1 (cat. no.
V790-20; Invitrogen; Thermo Fisher Scientific, Inc.) carrying the
WWOX gene sequence (4 µg) was transfected into A2780 and A2780/T
cells (50% confluence) at room temperature and the empty pcDNA3.1
vector was used as a mock control. The WWOX mRNA fragment was
synthesized by Sangon Biotech Co., Ltd. A plasmid construct
containing a small interfering RNA (siRNA) targeting WWOX was
generated using the mammalian expression vector pRNA-H1.1/Neo
(GenScript), which was transfected into A2780 cells (50-60%
confluence) with a final siRNA concentration of 100 nM at 37°C for
6 h. The WWOX-siRNA sequence was as follows:
5′-GCTGGGTTTACTACGCCAA-3′. The vector containing a scrambled
sequence (GenScript) was used as a negative control (sense,
5′-TAATACGACTCACTATAGGG-3′ and antisense,
5′-TAGAAGGCACAGTCGAGG-3′). The EOC cells were transfected using
Lipofectamine® 2000 transfection kit (Invitrogen; Thermo
Fisher Scientific, Inc.), according to the manufacturer's
instructions. Following 24 h incubation at 37°C, the cells were
collected for protein extraction for analysis of WWOX expression
levels by western blotting as aforementioned. Monomeric red
fluorescent protein (mRFP)-GFEP-LC3 adenovirus was purchased from
Hanbio Biotechnology Co., Ltd. and was transfected into A2780 (MOI,
1,000) and A2780/T (MOI, 3,500) cells in 24-well dishes with 50–70%
of confluence for 2 h at 37°C according to the manufacturer's
protocol. The adenovirus was used to mark LC3 protein and trace
autophagosome formation and degradation. After 24 h, the efficiency
of adenovirus infection was confirmed by fluorescence microscopy
(magnification, ×200).
Confocal microscopy
A2780-mRFP-GFEP-LC3 and A2780/T-mRFP-GFEP-LC3 cells
were seeded into 24-well cell culture plates at 4×104
cells/well. After treatment with PTX (150 ng/ml for A2780, 1,600
ng/ml for A2780/T) for 48 h at 37°C, the cells were washed with
cold PBS and then fixed with 4% paraformaldehyde in PBS for 30 min
at room temperature. Fixed cells were washed with PBS twice and
examined under a laser scanning fluorescence confocal microscope
(Leica TCS SP5; Leica Microsystems, Inc.). A total of eight images
were randomly selected to determine the average number of
mRPF-GFP-LC3 puncta per cell.
Transmission electron microscopy
(TEM)
A2780 and A2780/T cells were washed 3 times with PBS
at 4°C. The adherent cells were removed and centrifuged at 1,000 ×
g at room temperature for 10 min and fixed with 2.5% glutaraldehyde
solution (Sigma-Aldrich; Merck KGaA) at 4°C for 2 h, dehydrated in
a graded series of ethanol and finally embedded in resin (Ted
Pella, Inc.). Ultrathin sections (70–80 nm) were prepared with an
ultramicrotome (Reichert, Inc.). The thin sections were sliced and
stained with 2% uranyl acetate at room temperature for 3 min, then
detected using the Tecnai 10 transmission electron microscope
(Philips Healthcare).
Statistical analysis
Data are presented as the mean ± standard error of
at least three experimental repeats. Comparisons with the untreated
control were conducted using unpaired Student's t-test for analysis
of RT-qPCR data and one-way ANOVA followed by Dunnett's multiple
comparisons test for analysis of cell viability data (as shown in
Fig. 1). The differences in cell
viability between groups were evaluated by one-way ANOVA followed
by Tukey's multiple comparisons test (as shown in Fig. S1). All statistical analyses were
performed using GraphPad Prism version 8.0.2 (263) software
(GraphPad Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
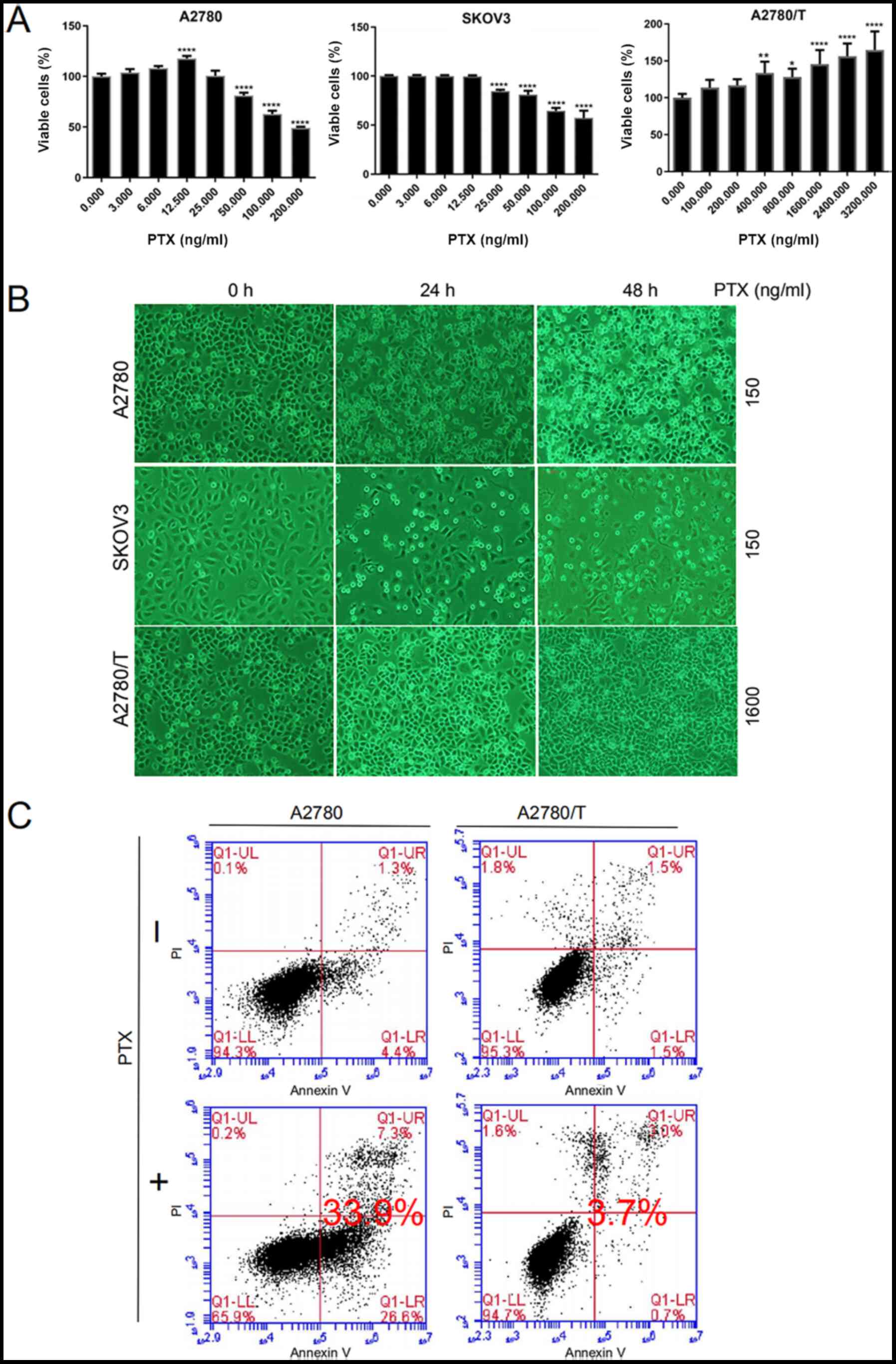 | Figure 1.PTX mediates apoptotic death of EOC
cells. (A) A2780, SKOV3 and A2780/T cells were seeded in 96-well
plates. After 24 h, cells were treated with the indicated doses of
PTX for 48 h. Cell viability was determined by Cell Counting Kit-8
assay. The percentages of viable cells were calculated vs.
untreated control cells (PTX, 0 ng/ml). Representative results of
three independent experiments are shown. Data are presented as the
mean ± SD. Comparisons with the untreated control group were
performed by one-way ANOVA followed by Dunnett's multiple
comparison test. *P<0.05, **P<0.01 and ****P<0.0001 vs.
untreated control group. (B) A2780 and SKOV3 cells were treated
with 150 ng/ml PTX for 24 and 48 h. A2780/T cells were treated with
1,600 ng/ml PTX for the indicated times. Cell morphology was
observed under a light microscope (magnification, ×400). Dead cells
were visualized as round suspended whitish cells. (C) Annexin V and
PI staining of A2780 and A2780/T cells treated with PTX for 48 h.
Lower left quadrants, viable cells; lower right quadrants, necrotic
cells; upper left quadrants, early apoptotic cells; upper right
quadrants, nonviable late apoptotic cells. The apoptotic cell rates
in A2780 and A2780/T cells were 33.9 and 3.7%, respectively. Data
are representative of three independent experiments. PTX,
paclitaxel; PI, propidium iodide. |
Results
PTX inhibits cellular proliferation
and induces cell death in A2780 and SKOV3 cells, but not A2780/T
cells
The susceptibility of the A2780, A2780/T and SKOV3
cell lines was assessed following PTX treatment for 48 h. At 48 h,
the half maximal inhibitory concentration (IC50) of PTX
was 167 and 371 ng/ml in A2780 and SKOV3 cells, respectively
(Fig. 1A). Ultimately, a
concentration of 150 ng/ml PTX was selected for further
experimentation, as it was close to the 48-h IC50 of
A2780 cells, and is readily available. Due to their characteristic
resistance to PTX, A2780/T cells were treated with a higher
concentration of 1,600 ng/ml PTX, such that the observed trends
were more apparent and easily interpreted. As shown in Fig. 1A and B, PTX inhibited cellular
viability and induced cell death in a dose- and time-dependent
manner in A2780 and SKOV3 cells, but not in A2780/T cells. Further
quantification of Fig. 1B is shown
in Fig. S1. In the present study,
A2780/T cells proliferated well and minimal apoptosis was observed,
even at the highest PTX dosage of 3,200 ng/ml. Conversely, the
proliferation of A2780/T cells was accelerated with increasing drug
concentrations. In addition, flow cytometric analysis revealed that
PTX significantly induced cell death in A2780 cells; however, no
apparent apoptosis was observed in A2780/T cells (Fig. 1C). These data clearly indicated that
PTX treatment may significantly induce cell death in A2780 and
SKOV3 cells, but not A2780/T cells.
Anticancer drug-induced WWOX
activation reduces chemoresistance and triggers cancer cell
death
There is substantial evidence to suggest that WWOX
may serve a significant role in apoptosis (23,24).
In the present study, the protein expression levels of WWOX were
examined in three EOC cell lines and the highest protein expression
levels of WWOX were observed in A2780/T cells; these cells
exhibited the lowest degree of PTX sensitivity. A moderate level of
WWOX expression was detected in A2780 cells and the lowest level
was observed in SKOV3 cells (Fig.
2A). These observations are in contrast to those presented in
previous studies, which reported the deficiency or loss of WWOX
expression in more aggressive, refractory and advanced tumor cells
(11,25,26).
In A2780 cells, PTX upregulated the protein expression levels of
WWOX in a dose- and time-dependent manner (Fig. 2C and D). In addition, the expression
levels of p-WWOX (Tyr33) were increased following PTX treatment,
which was accompanied by an accumulation of the cleaved forms of
caspase-3 and PARP (Fig. 2D). By
contrast, PTX did not increase the expression levels of WWOX or its
phosphorylation in A2780/T cells (Fig.
2C and D), and the activated forms of caspase-3 and PARP were
not observed at any of the post-treatment time points (Fig. 2D). The mRNA expression levels of
WWOX in PTX-treated A2780 and A2780/T cells were determined using
RT-qPCR analysis (Fig. 2E). PTX
increased the mRNA expression levels of WWOX in A2780 cells, but
downregulated its expression in A2780/T cells. Compared with in
A2780 cells, a higher endogenous level of WWOX mRNA was observed in
A2780/T cells. Thus, it was hypothesized that failure to induce
WWOX upregulation and activation may result in chemoresistance.
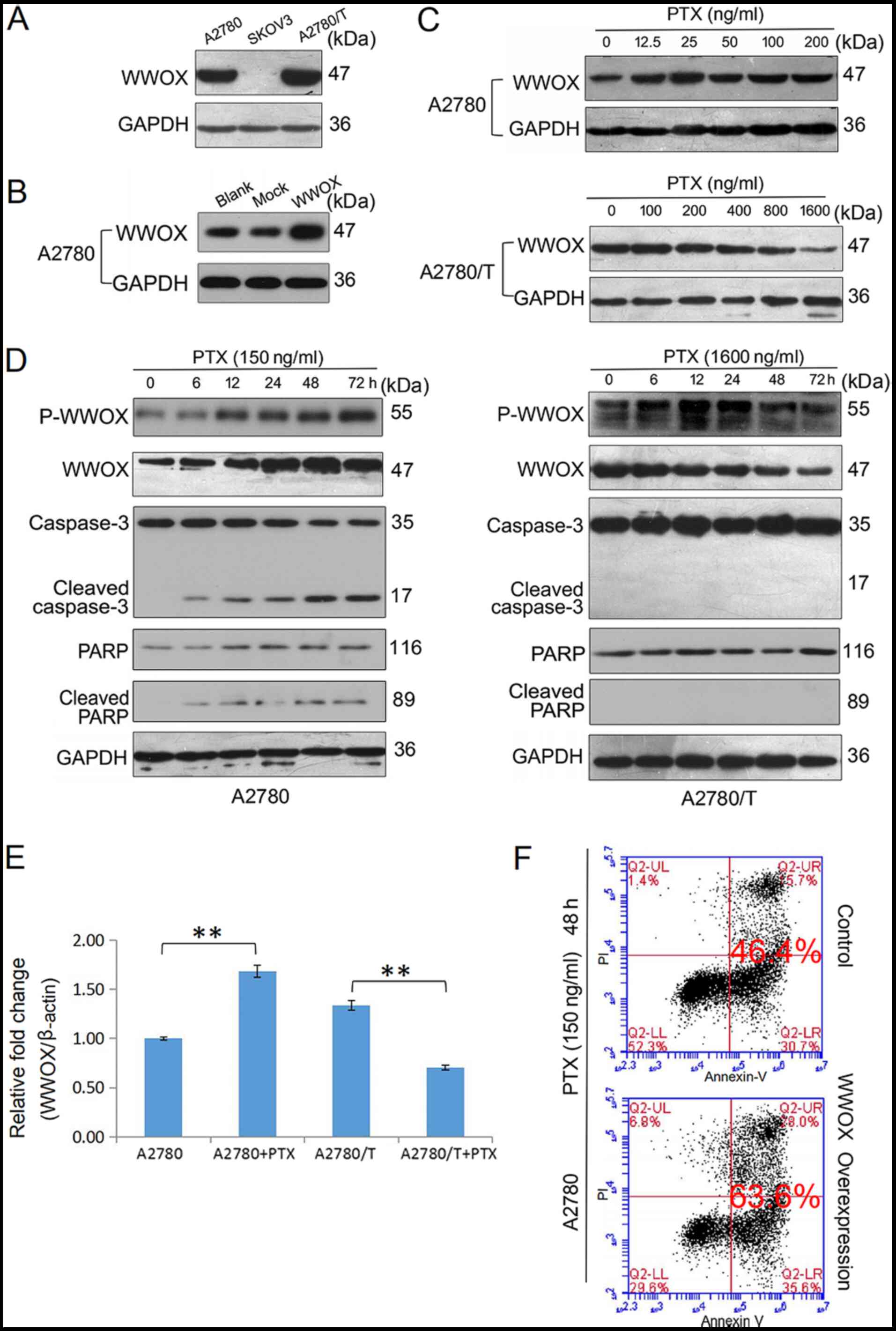 | Figure 2.PTX-induced apoptotic cell death is
associated with endogenous WWOX expression and activation of WWOX.
(A) Western blot analysis of the protein expression levels of
endogenous WWOX in A2780, SKOV3 and A2780/T cells. (B) A2780 cells
were transfected with a control vector and a vector encoding human
WWOX. After incubating at 37°C for 24 h, total lysates of A2780
(blank control) and transfected cells were examined for expression
of WWOX by western blotting using an anti-WWOX antibody. GAPDH was
used as a loading control. (C) A2780 and A2780/T cells were treated
with the indicated doses of PTX for 48 h, the protein expression
levels of WWOX were detected using western blotting. GAPDH was used
as the internal control. (D) A2780 and A2780/T cells were treated
with 150 and 1,600 ng/ml PTX, respectively. The expression levels
of p-WWOX, WWOX, cleaved-caspase-3 and cleaved-PARP were determined
by western blotting. GAPDH was used as the loading control. (E)
A2780 and A2780/T cells were treated with 150 and 1,600 ng/ml PTX
for 48 h, respectively. Quantification of WWOX mRNA expression in
PTX-treated epithelial ovarian cancer cells was performed by
reverse transcription-quantitative PCR analysis, and data were
analyzed using the 2−ΔΔCq method. Date are presented as
the mean ± SD. Comparisons with the untreated control were made by
unpaired Student's t-test. **P<0.01. (F) A2780 cells were
transfected with a vector encoding WWOX and cultured at 37°C for 24
h. Cell apoptosis was detected using Annexin V and PI staining in
A2780 and transfected cells via flow cytometry. Control and WWOX
overexpression cells were treated with PTX for 48 h The apoptotic
rate is indicated. Data are representative of three independent
experiments. PTX, paclitaxel; WW domain-containing oxidoreductase;
p-WWOX, phosphorylated-WWOX; PARP, poly (ADP-ribose)
polymerase. |
WWOX was subsequently overexpressed in EOC cells
using plasmid transfection (Fig.
2B). Flow cytometric analysis indicated that a higher level of
WWOX expression enhanced PTX-induced apoptosis of A2780 cells
(Fig. 2F). Due to ambiguously high
levels of intrinsic WWOX mRNA and protein expression, and an
insensitivity to PTX-induced cell death, A2780/T cells were not
used for apoptosis-associated transfection experiments. In
conclusion, these findings suggested that WWOX may have an
important role in the induction of drug-sensitive cancer cell
apoptosis.
PTX increases autophagic flux in human
EOC cells
To further investigate the autophagic alterations in
EOC cells, the expression levels of LC3, Beclin-1,
autophagy-related protein (Atg)12-5 complex and P62 were
determined. Pro-LC3 is proteolytically transformed to LC3-I, which
is converted to LC3-II by ubiquitin proteases; the overall level of
LC3-II indicates the extent of autophagy (27). Beclin-1 is an autophagy-specific
protein that regulates autophagosome formation (28), and P62, as an autophagy-specific
substrate, is incorporated into autophagosomes and consequently
degraded during autophagy (29). In
A2780 cells, PTX enhanced the expression levels of LC3, Beclin-1
and Atg12-5 in a time-dependent manner (Fig. 3A). Similar changes were observed in
chemoresistant A2780/T cells (Fig.
3A). The expression levels of P62 were decreased in both cell
lines (Fig. 3A). The endogenous
levels of LC3 and Beclin-1 were higher, and the change in P62
expression occurred at an earlier stage in A2780/T cells, which
implies active and persistent autophagic flux. In A2780/T cells,
P62 expression was abruptly decreased after 12-h exposure to PTX.
By contrast, the protein expression levels of P62 in A2780 cells
were unaltered until 48 h, and was markedly decreased after 72 h
(Fig. 3A). Thus, it was
hypothesized that increased autophagic flux may facilitate the
refractory features of A2780/T cells. Taken together, these data
demonstrated that PTX could enhance the expression of multiple
central autophagy-related proteins in PTX-sensitive and -resistant
human EOC cells.
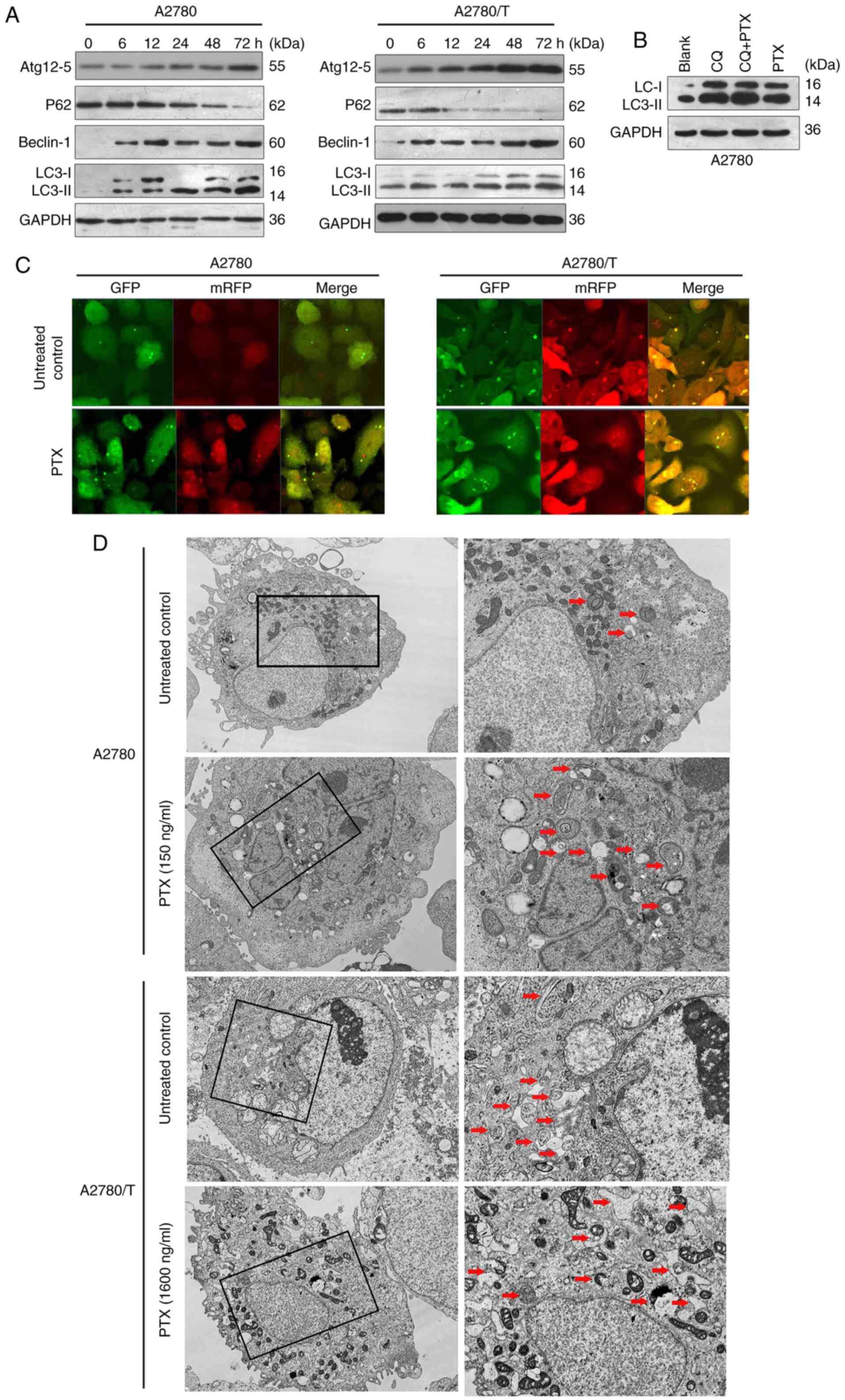 | Figure 3.PTX enhances autophagy in epithelial
ovarian cancer cells. (A) Total cell lysates were prepared from
A2780 and A2780/T cells following treatment with 150 and 1,600
ng/ml PTX for indicated durations, respectively. The expression
levels of Atg12-5, p62, Beclin-1 and LC3 were examined using
western blotting. GAPDH was used as an internal control. (B) A2780
cells were treated with or without 150 ng/ml PTX in the presence or
absence of CQ (25 µM). After culturing for 24 h, cytosolic protein
extracts were analyzed for LC3 protein expression. (C) A2780 and
A2780/T cells were transiently transfected with GFP-mRFP-LC3
adenovirus overnight and were analyzed. After 48-h exposure to the
indicated dosage of PTX (A2780, 150 ng/ml; A2780/T, 1,600 ng/ml),
representative images of GFP- and mRFP-positive LC3 puncta were
captured with a confocal fluorescence microscope. Cells without PTX
treatment were assigned as the control. Scale bar, 20 µm. (D) A2780
and A2780/T cells were treated with PTX for 48 h. Autophagosome and
autolysosome vesicles were visualized by transmission electron
microscopy. Left, magnification, ×10,000; scale bar, 1 µm. Right,
magnification, ×30,000; scale bar, 2 µm. Cells without PTX
treatment were assigned as the control. The typical images of
autophagosomes and autolysosomes at high magnification are
indicated with red arrows. PTX, paclitaxel; CQ, chloroquine;
ATg12-5, autophagy-related protein 12-5 complex. |
To monitor autophagic flux, the turnover of LC3-II
protein was assessed by western blotting. Increased LC3-II levels
can be interpreted as either enhanced autophagosome synthesis or
reduced autophagosome turnover. In order to monitor autophagic flux
induced by PTX, A2780 cells were co-treated with CQ (an autophagy
inhibitor) and PTX to assess the changes in LC3-II expression, and
thus, autophagic flux. LC3-II was upregulated following exposure to
PTX or CQ, and was further enhanced following simultaneous
treatment with both drugs, suggesting that autophagic flux was
amplified by PTX, not the slow lysosomal turnover in A2780 cells
(Fig. 3B).
To monitor autophagosome progression, fluorescence
microscopy was used to detect GFP- and mRFP-tagged LC3-II
(GFP-mRFP-LC3). Compared with those detected in untreated cells,
the levels of GFP and mRFP puncta were markedly increased in A2780
cells following treatment with 150 ng/ml PTX for 48 h, indicating
induced autophagic flux (Fig. 3C).
By contrast, an increased number of larger sized puncta were
observed in untreated A2780/T cells, which implies active basal
autophagy. The number of fluorescent puncta was slightly elevated
in A2780/T cells following stimulation with 1,600 ng/ml PTX for 48
h (Fig. 3C). These observations
were separately validated by TEM. An increasing number of
phagocytic vacuoles was also observed in the A2780 cell cytoplasm
following PTX treatment (Fig. 3D).
In A2780/T cells, autophagosome formation was apparent in untreated
cells, and was still active following exposure to PTX (Fig. 3D). It was therefore inferred that
PTX treatment may augment autophagy in EOC cells, including
drug-sensitive variants.
PTX mediates the mTOR signaling
pathway in EOC cells
mTOR, GβL (mLST8) and raptor comprise the mTOR
complex 1 (mTORC1), which is a cellular sensor for nutritional
conditions that has been confirmed to negatively regulate autophagy
(30). Therefore, mTOR/p70S6K
signaling [mTORC1 and its downstream substrates, p-p70S6K and
p-eukaryotic translation initiation factor 4E-binding protein 1
(4E-BP-1)] has been researched extensively in tumor-related studies
(17,31). In the present study, the expression
levels of mTOR and p-mTOR were detected, along with the downstream
factors p-p70S6K kinase (Thr389 and Ser371) and p-4E-BP-1
(Thr37/46). The alterations in these target proteins during mTOR
signaling were observed to be similar in PTX-sensitive and
-resistant EOC cells (Fig. 4A).
Furthermore, PTX suppressed the expression of p-mTOR and increased
the expression levels of p-4E-BP-1 (Thr37/46). p-p70S6K kinase (at
Thr389 and Ser371) was barely detected (Fig. 4A). Furthermore, to identify the
potential effects of WWOX on autophagy in EOC cells, siRNA
knockdown of WWOX was conducted in A2780 cells (Fig. 4B), and ectopic overexpression of
WWOX was performed in A2780 and PTX-resistant A2780/T cells
(Fig. 2B and 4B). The expression levels of p-mTOR in
A2780 cells were decreased following siRNA-mediated WWOX-knockdown
(Fig. 4C). Moreover, ectopic
overexpression of WWOX increased the expression levels of p-mTOR
(Fig. 4D). Thus, it was
hypothesized that WWOX may interact with mTOR to mediate mTOR
signaling, thus influencing its downstream targets.
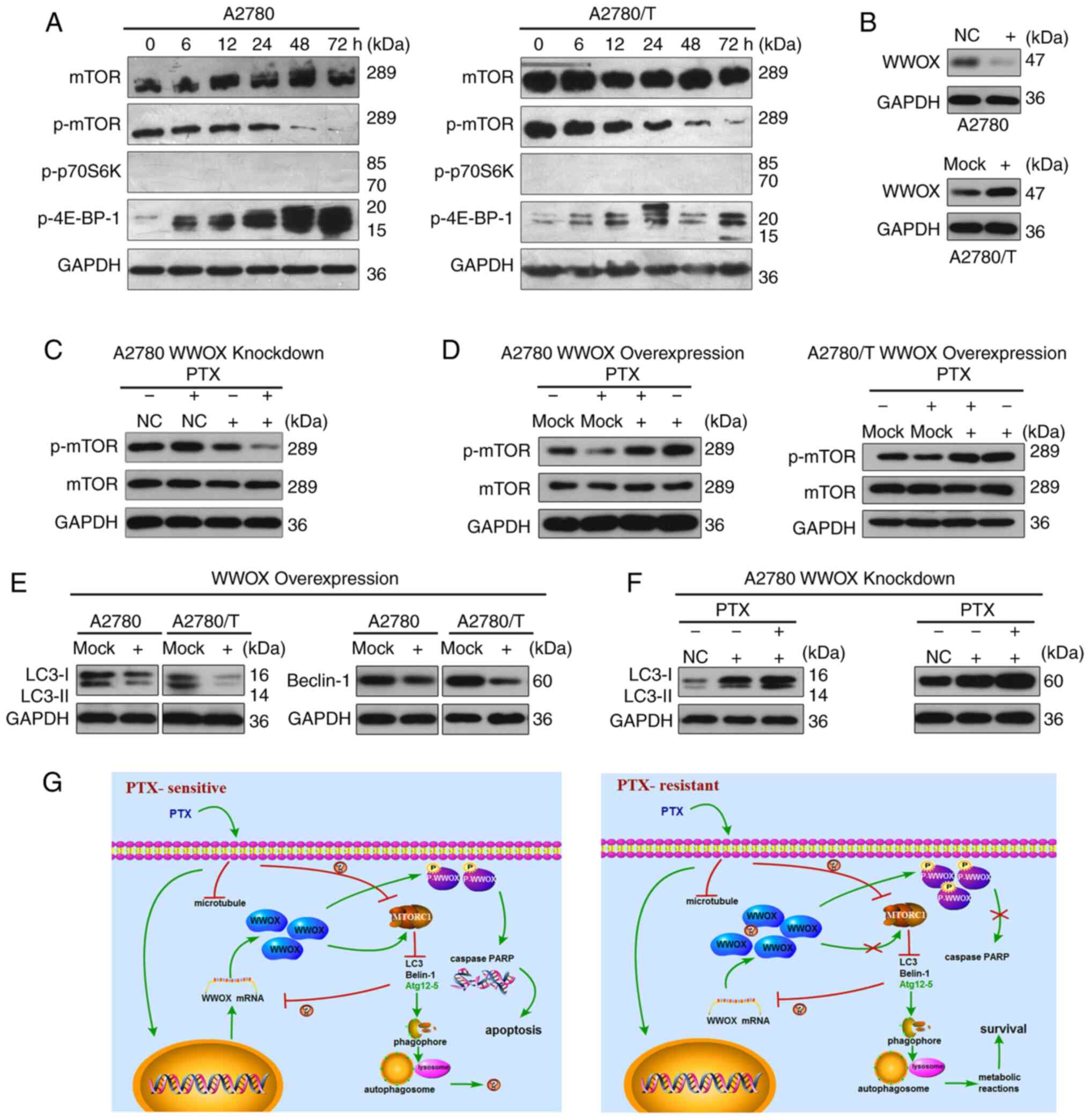 | Figure 4.WWOX mediates the inhibitory effects
of PTX on mTOR signaling and suppresses autophagy in epithelial
ovarian cancer cells. (A) PTX reduced the expression levels of
p-mTOR and increased the expression levels of p-4E-BP-1 in A2780
and A2780/T cells. Total protein extracts were prepared and
analyzed using western blotting. GAPDH was used as the loading
control. (B) A2780 cells were transfected with a siRNA construct
targeting WWOX or a control scrambled control. A2780/T cells were
transfected with a vector encoding WWOX or a control vector. (C)
A2780 cells were transfected with WWOX siRNA or a control scrambled
sequence; after 24 h, cells were treated with or without 150 ng/ml
PTX. After culturing for 48 h, total cell lysates were detected for
phosphorylation of mTOR by western blotting. GAPDH was used as the
loading control. (D) A2780 and A2780/T cells were transfected with
a vector encoding WWOX or a control vector and treated with 150 or
1,600 ng/ml PTX after 24 h, respectively, for 48 h. Total cell
lysates were detected for phosphorylation of mTOR by western
blotting. GAPDH was used as the loading control. (E) A2780 and
A2780/T cells were transfected with a vector encoding WWOX or a
control vector. After incubating at 37°C for 24 h, cells were
cultured for another 24 h, and total cell lysates were examined for
LC3 and Beclin-1 protein using western blotting. GAPDH was used as
a loading control. (F) A2780 cells were infected with WWOX siRNA or
a control scrambled sequence, 24 h later cells were treated with or
without 150 ng/ml PTX. After culturing for 48 h, total cell lysates
were detected for protein LC3 and Beclin-1. (G) Diagrams explaining
the potential effects of WWOX on PTX. Left, PTX induced the
upregulated level of WWOX, the phosphorylation of WWOX and
increased autophagy in PTX-sensitive cells, thus resulting in
apoptosis. Right, in PTX-resistant cells, PTX failed to increase
the expression of WWOX and the phosphorylation of WWOX, but
enhanced autophagy, resulting in cancer cell survival. PTX,
paclitaxel; WW domain-containing oxidoreductase; p-,
phosphorylated; siRNA, small interfering RNA; NC, negative
control. |
WWOX inhibits autophagy in EOC
cells
To ascertain the association between WWOX and
autophagy, EOC cells were transiently transfected with WWOX cDNA,
and the expression levels of Beclin-1 and LC3 were assessed by
western blotting. Since high levels of WWOX did not promote
apoptosis and activate mTOR, it was unclear whether WWOX was
inactivated in A2780/T cells, thus WWOX-knockdown was conducted in
A2780 cells by siRNA transfection. Upregulating WWOX expression
inhibited autophagy, as evidenced by the notable reduction in
Beclin-1 and LC3 in both cell lines (Fig. 4E). Conversely, the expression levels
of Beclin-1 and LC3 were increased in A2780 cells following
WWOX-knockdown, and further upregulated to a moderate degree
following PTX administration (Fig.
4F). These findings clearly demonstrated that WWOX may suppress
autophagy in human EOC cells.
Discussion
The investigation of pivotal molecules involved in
drug sensitivity is an essential approach to treating cancer and
targeting drug resistance; however, the related key molecules and
associated mechanisms in EOC remain to be elucidated. Previous
studies on WWOX have revealed some of the causes of tumorigenesis
and highlighted potential methods to reverse drug resistance
(16,32). WWOX is a proapoptotic protein that
is believed to be tumor suppressive (33,34).
In response to various stimuli, such as chemotherapeutic drug
treatment and TNF, the synthesis of cytosolic WWOX has been
reported to be increased and apoptosis has been shown to be induced
(24). WWOX may also act as a tumor
suppressor in numerous human cancer cell lines, following
regulation of cancer cell biological behaviors (35). Notably, the aberrant expression of
WWOX (either altered or lost) has been reported to be apparent in
numerous tumor types, and may influence loss of heterozygosity
(36), gene deletion, loss of
protein expression (13) and
epigenetic mechanisms (10).
Ectopic restoration of the WWOX gene has been shown to inhibit the
growth of cervical (37),
pancreatic (38) and breast cancer
(39). In addition, therapeutic
WWOX activation reversed temozolomide resistance and induced cancer
cell death in glioblastoma (34).
The results of the present study demonstrated that PTX increased
WWOX expression at both the mRNA and protein levels, which was
accompanied by WWOX phosphorylation and resulted in A2780 cell
apoptosis. A previous study demonstrated that p-WWOX, which is
generated in the cytoplasm, is primarily located in the
mitochondria and translocates to the nucleus following stress
stimulation, which is a requirement for WWOX-mediated apoptosis
(16) p-WWOX interacts with p53,
TNFR1-associated DEATH domain protein and Fas-associating death
domain protein to activate caspase-8, which in turn activates
caspase-9, caspase-3 and downstream PARP, ultimately resulting in
cell death (16). By contrast,
reduced levels of WWOX and unaltered levels of p-WWOX were detected
in PTX-resistant (A2780/T) cells treated with PTX. It was
hypothesized that autophagy was responsible for the accelerated
A2780/T cell proliferation observed with increasing doses of PTX.
Furthermore, due to its ability to support cellular metabolism via
diverse nutrient sources, autophagy has been implied to promote the
growth of certain tumor types (40). In the present study, an increased
rate of apoptosis was observed in A2780 cells transfected with WWOX
cDNA, which further verified the role of WWOX in apoptosis. These
results suggested that the activation of WWOX by anticancer drugs
may be essential for overcoming chemoresistance and promoting
cancer cell death (6,24).
Notably, the results of the present study revealed
that WWOX was substantially expressed in drug-resistant cells
(A2780/T), moderately expressed in A2780 cells and largely
unexpressed in SKOV3 cells; the mechanism underlying differences in
WWOX expression levels in the cells is unclear. The present study
was unable to explain this phenomenon (Fig. 4G), and the mechanisms associated
with the high levels of WWOX and p-WWOX in resistant cells remain
undetermined. However, this expression appears to be redundant, as
high levels of WWOX did not effectively promote mTORC1 to inhibit
autophagy in PTX-resistant cells. In addition, activated WWOX
(p-WWOX) failed to elicit tumor cell death. This phenomenon cannot
be explained from existing literature or the results of the present
study, and the only conceivable, though somewhat implausible
explanation, is the difference in cellular morphology. Taking cell
line features into consideration, SKOV3 cells are morphologically
more similar to interstitial cells, which may indicate a role for
WWOX in epithelial-mesenchymal transition. Increased expression of
WWOX may promote the retention of A2780/T cells in a normal ovarian
epithelial phenotype. It has been observed that WWOX-p53Δosx1
double knockout mice develop poorly differentiated osteosarcomas
(14). However, differences between
the expression levels of WWOX between A2780 and A2780/T cells
cannot currently be explained. In resistant A2780/T cells, it was
hypothesized that certain unidentified factors may cause p-WWOX to
lose its apoptosis-inducing ability, and when exposed to PTX,
nuclear WWOX synthesis is inhibited (Fig. 4G).
The present study did not observe apoptosis in
A2780/T cells when treated with 3,200 ng/ml PTX and apoptosis
induction experiments with higher concentrations of PTX were not
performed. DMSO was used to prepare stock solutions of PTX. Due to
its cytotoxicity, the dosage of DMSO should be as low as possible;
therefore, the concentration of PTX could not be further increased.
In addition, it was hypothesized that resistance of A2780/T cells
may be enhanced due to prolonged PTX exposure.
As a potent microtubule-targeting agent, PTX is the
recommended first-line chemotherapeutic drug against EOC. However,
the complex mechanisms underlying PTX resistance have not yet been
clarified. Cytoprotective autophagy serves an important role in
chemoresistance in various types of cancer cells (41). In the present study, a higher level
of intrinsic autophagy was detected in A2780/T cells compared with
in A2780 cells. Thus the acquired resistance to PTX may be
attributed to the robust autophagy of A2780/T cells. However, PTX
induced autophagy in both susceptible and resistant EOC cells. A
previous study suggested that thioredoxin domain containing
17-induced autophagy may result in PTX resistance in ovarian cancer
via Beclin-1 (42). Therefore, it
was hypothesized that PTX-mediated autophagy may protect EOC cells
against induced apoptosis and thus promote PTX resistance.
Autophagic modulation has become increasingly
important for effectively treating tumors. In numerous types of
cancer, inhibiting autophagy has been shown to attenuate cellular
viability and increase apoptosis (43,44).
The results of the present study demonstrated that WWOX suppressed
autophagy by downregulating the expression of LC3-II and Beclin-1.
Furthermore, WWOX activated mTOR/p70S6K signaling and prevented the
inhibitory effects of PTX. Hence, WWOX was speculated to negatively
regulate autophagy by activating the mTOR signaling cascade and
potentially modulating mTORC1. In view of the regulatory role of
the phosphorylated proteins, western blotting was only performed
for the downstream factors of mTOR (p-mTOR), p-P70S6K and
p-4E-BP-1; total protein expression levels were not detected. A
previous study confirmed the interaction between WWOX and mTOR
(17); however, little is currently
known of the exact mechanisms involved. Furthermore, the potential
effects of WWOX on late-stage autophagy (i.e., the coalescence of
autophagic vacuoles and liposomes) remain to be elucidated.
To the best of our knowledge, the present study is
the first to indicate that in PTX-sensitive EOC cells, PTX may
concurrently induce suppression of mTOR signaling and upregulation
of WWOX. WWOX was demonstrated to activate mTORC1, and thus inhibit
the autophagic process. From this point of view, PTX was determined
to modulate autophagy by inhibiting mTORC1 in a WWOX-independent
manner (Fig. 4G). Certain
PTX-induced cytoplasmic factors may competitively inhibit the
binding of WWOX to mTORC1. Furthermore, as PTX decreased the
expression of WWOX in A2780/T cells, active autophagy may also
negatively regulate WWOX (Fig.
4G).
As the mechanisms underlying the high levels of WWOX
expression in drug-resistant cells are currently unknown, the
existence of factors that antagonize its function cannot currently
be determined. Therefore, WWOX-knockdown and the related
experimental studies were not conducted in A2780/T cells in the
present study. As such, the significance of WWOX in drug-resistant
EOC cells requires further investigation in follow-up studies.
In conclusion, WWOX may be critical for the
PTX-induced apoptosis of EOC cells, and could suppress autophagy by
downregulating essential autophagic effectors via mTOR signaling
(Fig. 4G). Thus, to a certain
extent, robust basal autophagy may promote PTX-resistance in
A2780/T cells (Fig. 4G).
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to acknowledge the support
and valuable guidance of Dr Jiangang Gao (Laboratory of Department
of Life Sciences, Shandong University Central Campus).
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SZ and YZ conceived and designed the study. YZ, WW,
WP, WH, YZ, YY and JG performed the experiments. YZ and SZ analyzed
the data. SZ and YZ organized and wrote the manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mozzetti S, Ferlini C, Concolino P,
Filippetti F, Raspaglio G, Prislei S, Gallo D, Martinelli E,
Ranelletti FO, Ferrandina G, et al: Class III beta-tubulin
overexpression is a prominent mechanism of paclitaxel resistance in
ovarian cancer patients. Clin Cancer Res. 11:298–305.
2005.PubMed/NCBI
|
|
3
|
Veldhoen RA, Banman SL, Hemmerling DR,
Odsen R, Simmen T, Simmonds AJ, Underhill DA and Goping IS: The
chemotherapeutic agent paclitaxel inhibits autophagy through two
distinct mechanisms that regulate apoptosis. Oncogene. 32:736–746.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee Y, Na J, Lee MS, Cha EY, Sul JY, Park
JB and Lee JS: Combination of pristimerin and paclitaxel additively
induces autophagy in human breast cancer cells via ERK1/2
regulation. Mol Med Rep. 18:4281–4288. 2018.PubMed/NCBI
|
|
5
|
Zhan L, Zhang Y, Wang W, Song E, Fan Y, Li
J and Wei B: Autophagy as an emerging therapy target for ovarian
carcinoma. Oncotarget. 7:83476–83487. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hale AN, Ledbetter DJ, Gawriluk TR and
Rucker EB III: Autophagy: Regulation and role in development.
Autophagy. 9:951–972. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang C, Yang Y, Sun L, Wang J, Jiang Z, Li
Y, Liu D, Sun H and Pan Z: Baicalin reverses radioresistance in
nasopharyngeal carcinoma by downregulating autophagy. Cancer Cell
Int. 20:352020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tyutyunyk-Massey L and Gewirtz DA: Roles
of autophagy in breast cancer treatment: Target, bystander or
benefactor. Semin Cancer Biol. 66:155–162. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao K, Yin J and Dong J: Deregulated WWOX
is involved in a negative feedback loop with microRNA-214-3p in
osteosarcoma. Int J Mol Med. 38:1850–1856. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yan H, Yu N and Tong J: Effects of
5-Aza-2′-deoxycytidine on the methylation state and function of the
WWOX gene in the HO-8910 ovarian cancer cell line. Oncol Lett.
6:845–849. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Guo W, Wang G, Dong Y, Guo Y, Kuang G and
Dong Z: Decreased expression of WWOX in the development of
esophageal squamous cell carcinoma. Mol Carcinog. 52:265–274. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li J, Liu J, Ren Y and Liu P: Roles of the
WWOX in pathogenesis and endocrine therapy of breast cancer. Exp
Biol Med (Maywood). 240:324–328. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baykara O, Demirkaya A, Kaynak K, Tanju S,
Toker A and Buyru N: WWOX gene may contribute to progression of
non-small-cell lung cancer (NSCLC). Tumour Biol. 31:315–320. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Del MS, Husanie H, Iancu O, Abu-Odeh M,
Evangelou K, Lovat F, Volinia S, Gordon J, Amir G, Stein J, et al:
WWOX and p53 dysregulation synergize to drive the development of
osteosarcoma. Cancer Res. 76:6107–6117. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Aderca I, Moser CD, Veerasamy M, Bani-Hani
AH, Bonilla-Guerrero R, Ahmed K, Shire A, Cazanave SC, Montoya DP,
Mettler TA, et al: The JNK inhibitor SP600129 enhances apoptosis of
HCC cells induced by the tumor suppressor WWOX. J Hepatol.
49:373–383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang NS, Doherty J, Ensign A, Lewis J,
Heath J, Schultz L, Chen ST and Oppermann U: Molecular mechanisms
underlying WOX1 activation during apoptotic and stress responses.
Biochem Pharmacol. 66:1347–1354. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsai CW, Lai FJ, Sheu HM, Lin YS, Chang
TH, Jan MS, Chen SM, Hsu PC, Huang TT, Huang TC, et al: WWOX
suppresses autophagy for inducing apoptosis in methotrexate-treated
human squamous cell carcinoma. Cell Death Dis. 4:e7922013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kong W, Mao J, Yang Y, Yuan J, Chen J, Luo
Y, Lai T and Zuo L: Mechanisms of mTOR and autophagy in human
endothelial cell infected with dengue Virus-2. Viral Immunol.
33:61–70. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jia J, Abudu YP, Claude-Taupin A, Gu Y,
Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, et al:
Galectins control MTOR and AMPK in response to lysosomal damage to
induce autophagy. Autophagy. 15:169–171. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Munson MJ and Ganley IG: MTOR, PIK3C3, and
autophagy: Signaling the beginning from the end. Autophagy.
11:2375–2376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
simplehttps://www.atcc.org/products/all/HTB-77.aspx#characteristics
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using Real-Time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu BS, Tan JW, Zhu GH, Wang DF, Zhou X and
Sun ZQ: WWOX induces apoptosis and inhibits proliferation of human
hepatoma cell line SMMC-7721. World J Gastroenterol. 18:3020–3026.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lo JY, Chou YT, Lai FJ and Hsu LJ:
Regulation of cell signaling and apoptosis by tumor suppressor
WWOX. Exp Biol Med (Maywood). 240:383–391. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ekizoglu S, Bulut P, Karaman E, Kilic E
and Buyru N: Epigenetic and genetic alterations affect the WWOX
gene in head and neck squamous cell carcinoma. PLoS One.
10:e01153532015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Li P, Yang Z and Mo WN: Expression
of fragile histidine triad (FHIT) and WW-domain oxidoreductase gene
(WWOX) in nasopharyngeal carcinoma. Asian Pac J Cancer Prev.
14:165–171. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barth S, Glick D and Macleod KF:
Autophagy: Assays and artifacts. J Pathol. 221:117–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schmitz KJ, Ademi C, Bertram S, Schmid KW
and Baba HA: Prognostic relevance of autophagy-related markers LC3,
p62/sequestosome 1, Beclin-1 and ULK1 in colorectal cancer patients
with respect to KRAS mutational status. World J Surg Oncol.
14:1892016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li RF, Chen G, Ren JG, Zhang W, Wu ZX, Liu
B, Zhao Y and Zhao YF: The adaptor protein p62 is involved in
RANKL-induced autophagy and osteoclastogenesis. J Histochem
Cytochem. 62:879–888. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Noureldein MH and Eid AA: Gut microbiota
and mTOR signaling: Insight on a new pathophysiological
interaction. Microb Pathog. 118:98–104. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yan H, Tong J, Lin X, Han Q and Huang H:
Effect of the WWOX gene on the regulation of the cell cycle and
apoptosis in human ovarian cancer stem cells. Mol Med Rep.
12:1783–1788. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin JT, Li HY, Chang NS, Lin CH, Chen YC
and Lu PJ: WWOX suppresses prostate cancer cell progression through
cyclin D1-mediated cell cycle arrest in the G1 phase. Cell Cycle.
14:408–416. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chiang MF, Chou PY, Wang WJ, Sze CI and
Chang NS: Tumor suppressor WWOX and p53 alterations and drug
resistance in glioblastomas. Front Oncol. 3:432013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Płuciennik E, Nowakowska M, Gałdyszyńska
M, Popęda M and Bednarek AK: The influence of the WWOX gene on the
regulation of biological processes during endometrial
carcinogenesis. Int J Mol Med. 37:807–815. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baryła I, Styczeń-Binkowska E and Bednarek
AK: Alteration of WWOX in human cancer: A clinical view. Exp Biol
Med (Maywood). 240:305–314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hung PS, Chuang FJ, Chen CY, Chou CH, Tu
HF and Lo SS: miR-187* enhances SiHa cervical cancer cell
oncogenicity via suppression of WWOX. Anticancer Res. 40:1427–1436.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nakayama S, Semba S, Maeda N, Aqeilan RI,
Huebner K and Yokozaki H: Role of the WWOX gene, encompassing
fragile region FRA16D, in suppression of pancreatic carcinoma
cells. Cancer Sci. 99:1370–1376. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Khawaled S, Suh SS, Abdeen SK, Monin J,
Distefano R, Nigita G, Croce CM and Aqeilan RI: WWOX inhibits
metastasis of triple-negative breast cancer cells via modulation of
miRNAs. Cancer Res. 79:1784–1798. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kimmelman AC and White E: Autophagy and
tumor metabolism. Cell Metab. 25:1037–1043. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
O'Donovan TR, O'Sullivan GC and McKenna
SL: Induction of autophagy by drug-resistant esophageal cancer
cells promotes their survival and recovery following treatment with
chemotherapeutics. Autophagy. 7:509–524. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang SF, Wang XY, Fu ZQ, Peng QH, Zhang
JY, Ye F, Fu YF, Zhou CY, Lu WG, Cheng XD and Xie X: TXNDC17
promotes paclitaxel resistance via inducing autophagy in ovarian
cancer. Autophagy. 11:225–238. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Li LQ, Xie WJ, Pan D, Chen H and Zhang L:
Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity
of gastric cancer cells. Tumour Biol. 37:653–659. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Polishchuk EV, Merolla A, Lichtmannegger
J, Romano A, Indrieri A, Ilyechova EY, Concilli M, De Cegli R,
Crispino R, Mariniello M, et al: Activation of autophagy, observed
in liver tissues from patients with Wilson disease and from
ATP7B-deficient animals, protects hepatocytes from copper-induced
apoptosis. Gastroenterology. 156:1173–1189. 2019. View Article : Google Scholar : PubMed/NCBI
|














