Introduction
Acute lung injury (ALI) is a serious disease with
diffuse alveolar injury, which has a high morbidity and mortality
rate in patients in intensive care (1). Uncontrolled acute inflammatory
response and excessive secretion of proinflammatory cytokines are
considered to be two of the primary pathological factors leading to
ALI (2). Previous studies have
confirmed that macrophages, neutrophils, lymphocytes, lung
epithelial fibroblasts and platelets are associated with the
occurrence and development of ALI (3,4). In
addition, the inflammatory cytokines produced by these cells form a
complex signaling network, which is activated by various external
stimuli and can regulate all stages of the inflammatory response in
ALI (5). Therefore, preventing the
release of inflammatory cytokines is of great significance for
treating ALI. As a component of Gram-negative bacterial cell walls,
lipopolysaccharide (LPS) is one of the most potent activators for
regulating the gene expression of inflammatory cytokines, and is
commonly utilized to induce ALI in animals models and cell lines
(6,7).
Salusin-β is a 20-amino acid peptide that is
translated from an alternatively spliced mRNA of prosalusin
(TOR2A), which encodes proteins of the torsion dystonia family
(8). The first 18 amino acids of
human salusin-β have high homology with the N-terminal sequence of
rat salusin (9). TOR2A is widely
expressed in the small intestine, stomach and lung, and salusin-β
can be detected in macrophages of the hematopoietic and immune
systems (9,10).
Over the past decade, salusin-β has been extensively
reported to be associated with inflammatory-related diseases
(11,12). For example, Xu et al
(13) found that salusin-β was
predominantly expressed in pulmonary macrophages and contributed to
vascular inflammation associated with pulmonary arterial
hypertension in rats. Salusin-β was also demonstrated to lead to
inflammation in diabetic cardiomyopathy (DCM), and its knockdown
could attenuate cardiac dysfunction, oxidative stress and
inflammation in DCM (14). Li et
al (15) suggested the
potential beneficial effects of salusin-β blockade in essential
hypertension via downregulation of inflammatory molecules and
oxidative stress. However, whether salusin-β could inhibit the
inflammatory response in LPS-induced alveolar macrophages remains
unknown.
Various molecular pathways participate in the
occurrence or development of ALI. Among them, the high mobility
group box-1 (HMGB1) protein and HMGB1-mediated NF-κB activation
play important roles in LPS-induced ALI (16,17).
Heme oxygenase-1 (HO-1) is a stress-response proteins, and can be
induced by stimulants, such as proinflammatory cytokines, heat
shock and oxidants (18). The
activation of HO-1 has been demonstrated to be required for
antioxidant and anti-inflammatory actions in LPS-induced ALI
(19,20).
The rat alveolar macrophage cell line, NR8383 is a
homogenous and expandable source of alveolar macrophage-like cells,
which has been shown to express functional characteristics of
alveolar macrophages, including properties of phagocytes,
production of proinflammatory cytokines and oxidative stress
(21). In the present study, LPS
was used to induce ALI in rats and alveolar macrophage inflammation
in NR8383 cells to investigate whether salusin-β was involved in
LPS-induced lung inflammation, as well as to uncover the potential
underlying mechanisms.
Materials and methods
Animals and protocols
A total of 15 specific pathogen-free male
Sprague-Dawley rats (age, 7–8 weeks; weight, 240–280 g) were
purchased from Guangdong Medical Laboratory Animal Center and
housed at room temperature under a controlled 12/12 h light/dark
cycle. All rats received food and water ad libitum. All
procedures were performed in accordance with the Care and Use Guide
of Laboratory Animals of the National Institutes of Health and with
the approval of the ethics committee of Fujian Medical University
Union Hospital (approval no. IACUC-20181212-09; Fuzhou, China).
Rats were randomly assigned to two groups (n=5 rats/group): Control
and LPS. LPS was intratracheally administered to rats as described
previously (22). After
anesthetization by intraperitoneal (i.p.) injection of 3% sodium
pentobarbital, the LPS-treated rats received 50 mg/kg LPS
(Sigma-Aldrich; Merck KGaA) by intratracheal instillation, while
control animals were instilled intratracheally with 200 µl normal
saline instead of LPS. At 24 h after LPS instillation, rats were
anesthetized for subsequent experiments, and then subjected to
cervical dislocation of the spine immediately.
Collection of broncho alveolar lavage
fluid (BALF)
BALF (n=5 rats/group) was collected at 24 h post-LPS
treatment. Before lavage, rats were anesthetized by i.p. injection
of sodium pentobarbital (3%, 10 ml/kg). After exposing the chest
cavity and intubating the trachea, the lungs were washed with 2 ml
normal saline three times, and the flushing fluid was collected.
The collected solution was centrifuged at 1,500 × g for 10 min at
4°C. The supernatant was collected and stored at −80°C for further
analysis.
Histological staining
At 24 h after LPS instillation, rats were
anesthetized by injection of sodium pentobarbital (i.p., 3%, 10
ml/kg). The left lung tissues were isolated and fixed with 4%
paraformaldehyde at 4°C for 24 h, embedded in paraffin, and then
cut into 5-µm thick sections. After staining with hematoxylin (5
min) and eosin (20 sec) at room temperature, the lung tissue
sections were observed under an optical microscope for pathological
examination (magnification, ×400).
Cell culture and treatment
The rat alveolar macrophage cell line NR8383
(American Type Culture Collection) was maintained in F12 medium
(Thermo Fisher Scientific, Inc.) supplemented with 15% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.), 100 µg/ml
streptomycin, 100 U/ml penicillin and 2 mmol L-glutamine (Beyotime
Institute of Biotechnology) at 37°C in a humid atmosphere of 5%
CO2. The medium was discarded and replaced by fresh
medium every 3 days until the NR8383 cells reached 60% confluence.
Before passaging, adherent cells were harvested, and then
centrifuged (100 × g, 4°C, 5 min) and transferred to new
microplates.
For induction of ALI in vitro, the cells were
stimulated with or without 1 µg/ml LPS for various times (6, 12, 24
and 48 h) at 37°C. Short hairpin (sh)RNA targeting salusin-β and
HO-1 together with shRNA control were designed and synthesized by
Shanghai GenePharma Co., Ltd. Then, 20 µg pcDNA 3.1 plasmids
(Thermo Fisher Scientific, Inc.) and 20 nM shRNAs were transfected
into cells at 70–80% confluence using Lipofectamine®
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions, as described previously (23). At 48 h post-transfection, cells were
selected for subsequent experiments.
ELISA
The concentrations of TNF-α (cat. no. ab236712),
IL-1β (cat. no. ab255730), IL-6 (cat. no. ab234570) and monocyte
chemotactic protein 1 (MCP-1; cat. no. ab219045) in the BALF or
cell culture medium were determined using specific ELISA kits
(Abcam), according to the manufacturer's instructions.
Western blotting
NR8383 cells were lysed, total protein was extracted
using RIPA lysis buffer (Beyotime Institute of Biotechnology) and
the total protein concentration was determined with a Bradford
assay (Bio-Rad Laboratories, Inc.). Equal quantities of protein (20
µg) in each sample were separated via 10% SDS-PAGE, and
subsequently transferred to a PVDF membrane. The membrane was
blocked with 5% non-fat milk at room temperature for 2 h and
incubated with primary antibodies against salusin-β (1:200; cat.
no. B-010-68; Phoenix Pharmaceuticals, Inc.), CD68 (1:1,000; cat.
no. ab125212; Abcam), HO-1 (1:2,000; cat. no. ab189491; Abcam),
HMGB1 (1:800; cat. no. ab18256; Abcam), IκBα (1:500; cat. no.
ab76429; Abcam), p-IκBα (1:10,000; cat. no. ab133462; Abcam), p65
(1:1,000; cat. no. ab16502; Abcam), p-p65 (1:1,000; cat. no.
ab76302; Abcam) and GAPDH (1:5,000; cat. no. ab8245; Abcam)
overnight at 4°C. Horseradish peroxidase-conjugated goat
anti-rabbit IgG (1:5,000; cat. no. ab6721; Abcam) and goat
anti-mouse IgG secondary antibodies (1:5,000; cat. no. ab6789;
Abcam) were used for detection (room temperature, 2 h). The protein
bands were visualized with an Enhanced Chemiluminescence Detection
kit (Thermo Fisher Scientific, Inc.). Protein expression levels
were semi-quantified using Image-Pro Plus software version 6.0
(Roper Technologies, Inc.).
Reverse transcription-quantitative PCR
(RT-qPCR)
RT-qPCR was used to analyze the expression of genes.
Total RNA was isolated from cells using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.). The PrimeScript RT
Master Mix kit (Takara Biotechnology, Co., Ltd.) was utilized to
synthesize cDNA according to the manufacturer's instructions.
Subsequently, qPCR was performed with SYBR-Green PCR Master Mix
(Roche Diagnostics) on an ABI Quantitative PCR 7500 system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The primers used were
as follows: Salusin-β forward, 5′-TCACTTCTCTCCTATCATCCACTCC-3′ and
reverse, 5′-GGCAGCTTGTCCATCTCATCG-3′; HO-1 forward,
5′-GTCCCAGGATTTGTCCGAGG-3′ and reverse,
5′-GGAGGCCATCACCAGCTTAAA-3′; and GAPDH forward,
5′-GTGGAGTCTACTGGCGTCTT-3′ and reverse, 5′-TGCTGACAATCTTGAGGGA-3′.
PCR reaction conditions were as follows: 95°C for 2 min, followed
by 40 cycles of 95°C for 20 sec and 65°C for 40 sec. Expression
levels of target genes were normalized to endogenous control GAPDH
using the 2−ΔΔCq method (24).
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 6.0 (GraphPad Software, Inc.). All experiments were repeated
at least three times and data are expressed as the mean ± standard
deviation. One-way ANOVA followed by Tukey's post hoc test was used
for multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
Salusin-β is upregulated in lung
tissues of LPS-induced rats and in the LPS-treated rat alveolar
macrophage cell line NR8383
First, to determine whether Salusin-β plays a role
in LPS-induced ALI, rats were treated with LPS via intratracheal
instillation, which is a previously published method to induce ALI
in animals (25,26). The lung tissues and BALF of rats
with ALI were collected at 24 h after treatment with or without
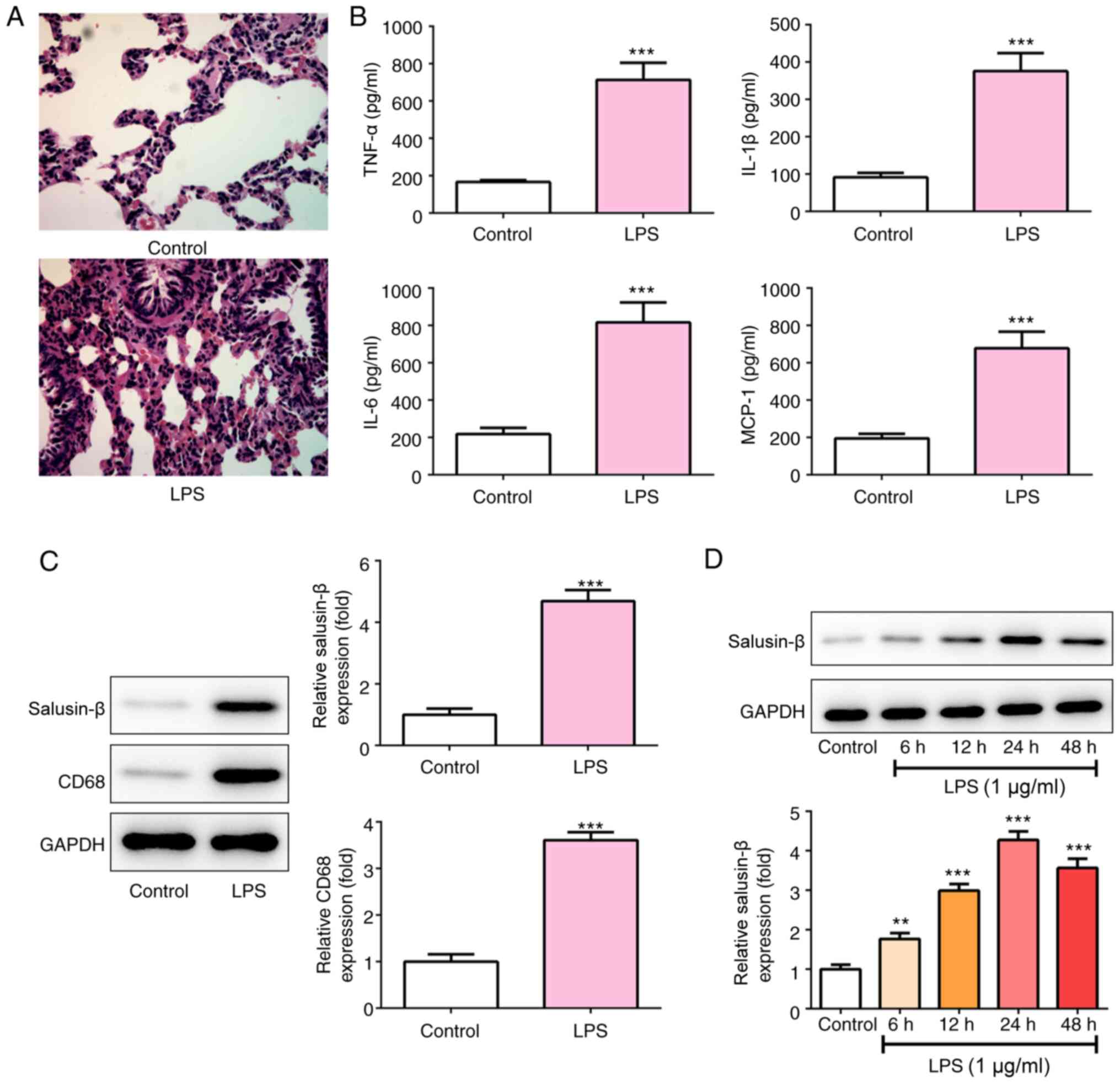
LPS. As shown in Fig. 1A, in the
control group, the lung tissue structure was complete, the alveolar
cavity was clear, the alveolar wall was not congested, and there
was no inflammatory cell infiltration in the lung interstitium. By
contrast, in the LPS group, the alveolar wall was diffusely
thickened, and part of the alveolar wall was destroyed with obvious
inflammatory cell infiltration, alveolar hemorrhage and structural
damage. At the same time, the concentration of inflammatory
cytokines, including TNF-α, IL-1β, IL-6 and MCP-1 in the BALF of
the LPS group was significantly increased to nearly 4-fold of that
of the control group (Fig. 1B).
These results confirmed the induction of ALI in rats. CD68 is a
marker of macrophages, and the results shown in Fig. 1C revealed that the lung tissues of
rats in the LPS group expressed significantly higher levels of
salusin-β and CD68. Additionally, NR8383 cells were exposed to 1
µg/ml LPS for 6, 12, 24 or 48 h, and the expression of salusin-β
was measured. LPS also significantly promoted salusin-β expression
in NR8383 cells (Fig. 1D).
Considering that the expression of salusin-β reached the highest
level (4.275±0.2177-fold of control) at 24 h post-LPS treatment,
cells were exposed to LPS for 24 h in the subsequent
experiments.
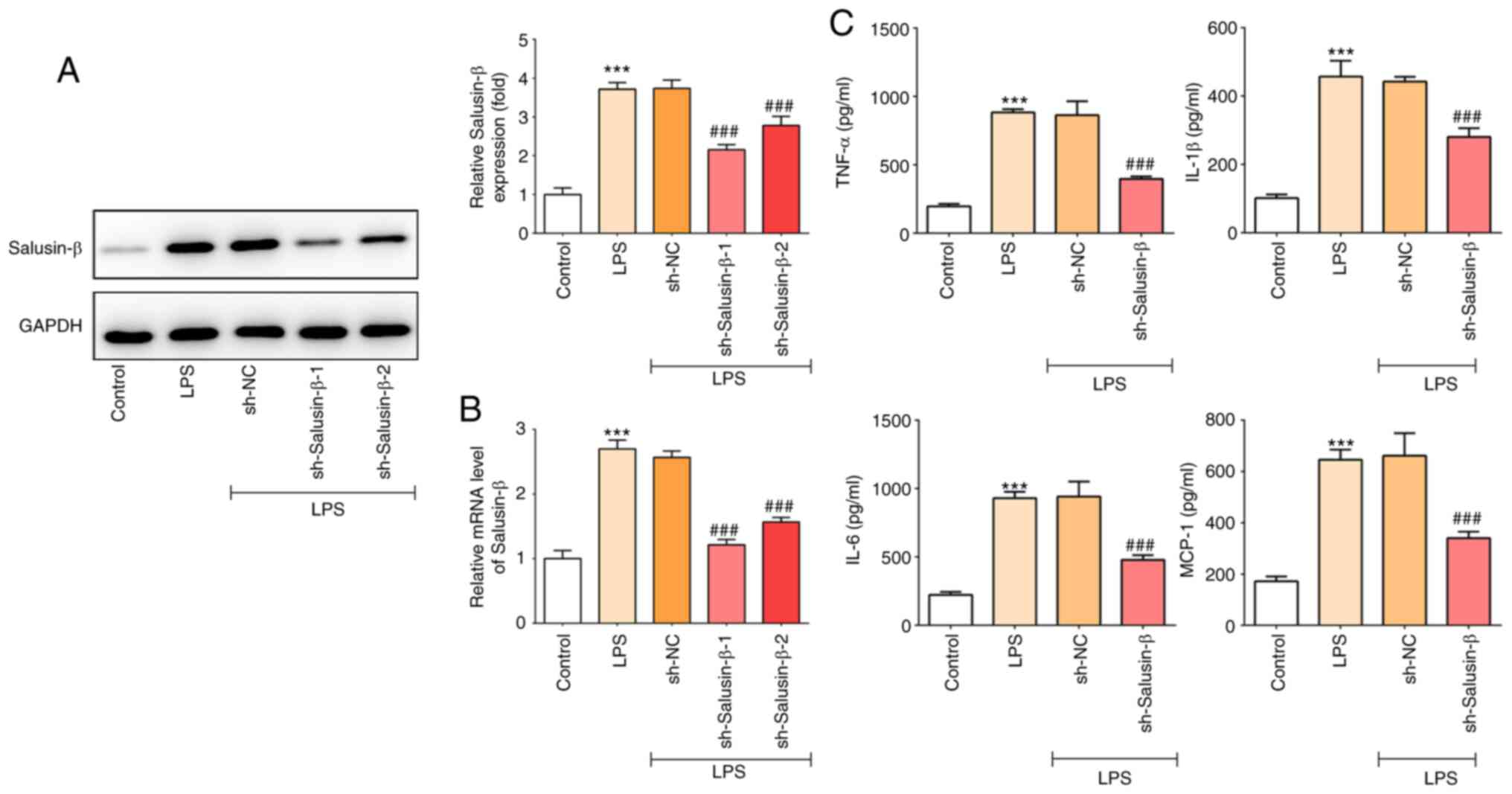
Knockdown of salusin-β inhibits the
LPS-induced release of inflammatory cytokines
Next, to further explore the role of salusin-β in
LPS-induced ALI, salusin-β was knocked down using shRNA, as
shRNA-salusin-β-1 showed the highest knock down effect, it was
selected for subsequent experiments (Fig. 2A and B). As shown in Fig. 2C, knockdown of salusin-β
significantly inhibited the concentration of inflammatory
cytokines, including TNF-α, IL-1β, IL-6 and MCP-1, compared with
the increase caused by LPS treatment, indicating the inhibitory
effect of salusin-β knockdown on LPS-induced inflammation in
alveolar macrophage cells.
Knockdown of salusin-β prevents
LPS-induced activation of NF-κB and inhibition of HO-1
The present study then aimed to investigate the
potential underlying mechanism involved in the action of salusin-β.
The protein expression of HMGB1, phosphorylated (p)-IκB α, p-p65
and HO-1 in the lung tissues of rats was determined. The results
from Fig. 3A show that, compared
with that of control rats, the expression levels of HMGB1, p-IκBα
and p-p65 were significantly upregulated, whereas that of HO-1 was
downregulated (0.393±0.0662-fold of control), in rats that were
subjected to LPS treatment. Consistently, LPS treatment also
increased HMGB1, p-IκBα and p-p65 expression, but reduced HO-1
expression, in NR8383 cells (Fig.
3B). However, the knockdown of salusin-β partially recovered
the LPS-induced expression changes of these proteins (Fig. 3B), suggesting that the knockdown of
salusin-β could prevent the LPS-induced activation of NF-κB and
inhibition of HO-1.
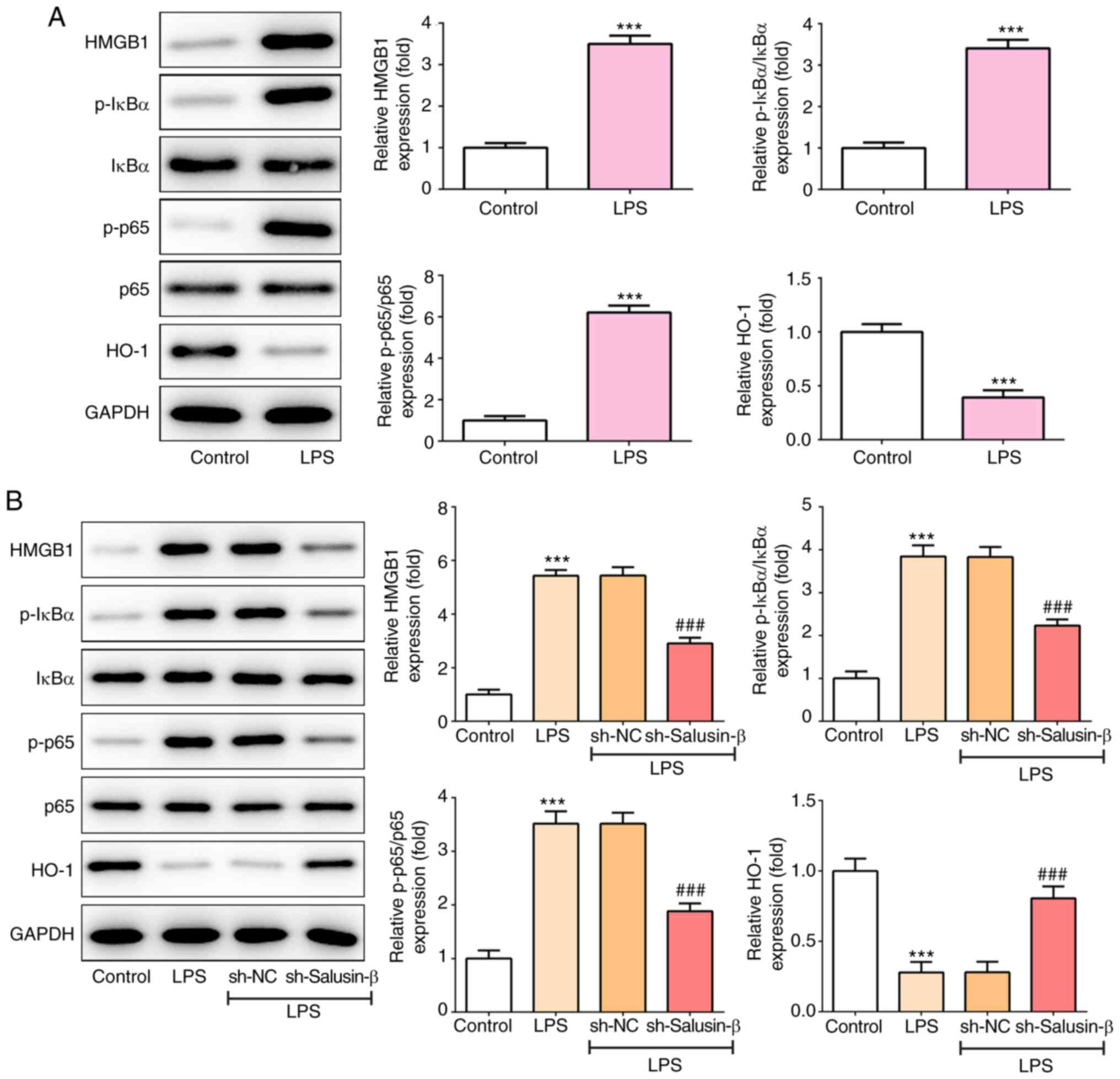 | Figure 3.Effect of salusin-β knockdown on
LPS-induced activation of NF-κB and inhibition of HO-1. (A) The
protein expression of HMGB1, p-IκBα, p-p65 and HO-1 in lung tissues
of rats in the LPS or control group (n=5). (B) The protein
expression of HMGB1, p-IκBα, p-p65 and HO-1 in LPS-induced NR8383
cells with or without salusin-β knockdown (n=3). ***P<0.001 vs.
control; ###P<0.001 vs. sh-NC. NC, negative control;
p-, phosphorylated; HO-1, heme oxygenase-1; HMGB1, high mobility
group box-1; LPS, lipopolysaccharide; shRNA, short hairpin RNA. |
Knockdown of HO-1 weakens the
inhibitory effect of shRNA-salusin-β on LPS-induced inflammation
and NF-κB activation
Finally, to further confirm the aforementioned
findings, the expression of HO-1 was silenced, and shRNA-HO-1-1 was
utilized to knock down the expression of HO-1 in subsequent
experiments, which was based on its higher efficacy (Fig. 4A and B). As shown in Fig. 4C, compared with cells that had been
transfected with shRNA-salusin-β, cells that were subjected to
co-treatment with shRNA-HO-1 produced a relatively higher
(P<0.05) concentration of inflammatory cytokines, including
TNF-α, IL-1β, IL-6 and MCP-1, under LPS stimulation. In addition,
shRNA-HO-1 also blocked the effect of shRNA-salusin-β on HMGB1,
p-IκB, p-p65 and HO-1 expression (P<0.01; Fig. 4D). These results indicated that the
inhibitory effect of shRNA-salusin-β on LPS-induced inflammation
and NF-κB activation was dependent on the activation of HO-1.
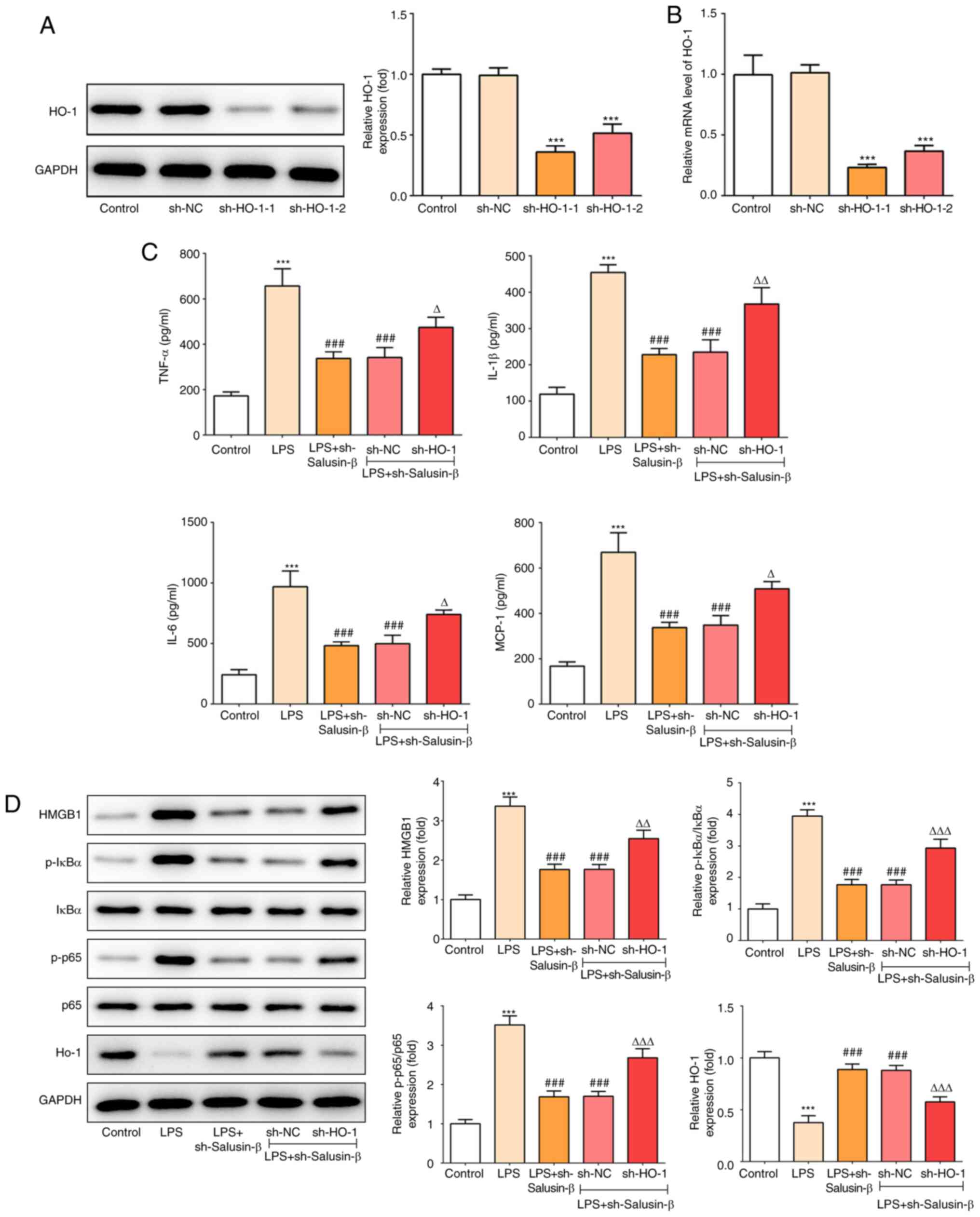 | Figure 4.Effect of HO-1 knockdown on
LPS-induced inflammation and NF-κB activation in the presence of
shRNA-salusin-β. (A) Protein expression of HO-1 in NR8383 cells
transfected with shRNA-HO-1-1/2 or sh-NC (n=3). (B) mRNA expression
of HO-1 in NR8383 cells transfected with shRNA-HO-1-1/2 or sh-NC
(n=3). (C) Concentrations of TNF-α, IL-1β, IL-6 and MCP-1 in the
culture medium of LPS-induced NR8383 cells with or without HO-1
knockdown in the presence of shRNA-salusin-β (n=3). (D) Protein
expression of HMGB1, p-IκBα, p-p65 and HO-1 in LPS-induced NR8383
cells with or without HO-1 knockdown in the presence of
shRNA-salusin-β (n=3). ***P<0.001 vs. control;
###P<0.001 vs. LPS; ΔP<0.05,
ΔΔP<0.01, ΔΔΔP<0.001 vs. LPS +
sh-salusin-β + sh-NC. HO-1, heme oxygenase-1; p-, phosphorylated;
NC, negative control; LPS, lipopolysaccharide; shRNA, short hairpin
RNA; MCP-1, monocyte chemotactic protein 1. |
Discussion
ALI is a common clinical critical illness, which is
usually induced by infection, trauma and shock. In the early stage
of ALI, diffuse alveolar injury and damage to the barrier function
of alveolar epithelial cells can activate various intracellular
signaling pathways involved in inflammatory responses, thus causing
a cascade of inflammatory factors and ultimately leading to
uncontrolled inflammation (27).
The present study showed that LPS caused obvious pathological
manifestations, including intra-alveolar hemorrhage, inter-alveolar
septum thickening, inflammatory cell infiltration in lung tissues
of rats, and increased production of inflammatory cytokines,
including TNF-α, IL-1β, IL-6 and MCP-1, in BALF. Therefore,
controlling the primary disease and preventing the inflammatory
response is an effective strategy and method for preventing and
treating ALI.
Salusin-β has been extensively reported to be
upregulated in inflammatory tissues and to play a proinflammatory
effect in various diseases (13,14).
Consistent with previous studies (13,14,28),
the present results revealed that salusin-β expression was
increased in LPS-induced lung tissues compared with that of normal
lung tissues of rats (nearly 5 times as much as the control group).
Moreover, LPS treatment could enhance salusin-β expression in a
time-dependent manner in a rat alveolar macrophage cell line. Of
note, the expression of salusin-β was highest at 24 h post-LPS
treatment, which could be because the stimulation of LPS for 48 h
caused damage to cells, leading to cell death, therefore the
relative expression of salusin-β at 48 h post-treatment was lower
than 24 h stimulation. These results indicated that salusin-β may
also have a proinflammatory effect on LPS-induced lung injury.
Therefore, in the present study, the expression of salusin-β was
knocked down to observe the alterations in LPS-induced alveolar
macrophage inflammation. The results demonstrated that knockdown of
salusin-β significantly reduced the release of inflammatory
cytokines, including TNF-α, IL-1β, IL-6 and MCP-1, in NR8383 cells,
indicating that salusin-β silencing could exert an
anti-inflammatory effect on LPS-induced lung injury.
Numerous studies have demonstrated that HO-1 and its
products can exhibit antioxidant, anti-apoptotic and
immunomodulatory functions in various models of cell and tissue
injury (29,30). In addition, HO-1 has been
demonstrated to significantly block the expression of the
proinflammatory mediator HMGB1 and the pro-inflammatory NF-κB
signaling pathway induced by LPS in animal models and cell lines,
thus alleviating the pathogenesis of ALI (19,31).
In accordance with the aforementioned findings, the present study
also confirmed that, following LPS stimulation, HMGB1, p-IκBα and
p-p65 expression increased, and HO-1 expression decreased. However,
the present study found that the knockdown of salusin-β
successfully inhibited the activation of NF-κB, but upregulated the
expression of HO-1. These results revealed that the
anti-inflammatory effect of salusin-β knockdown on LPS-induced lung
cell injury may be dependent on inactivating NF-κB, while
activating HO-1. Subsequently, HO-1 was also knocked down in the
presence of salusin-β knockdown, and the results revealed that
knockdown of HO-1 significantly blocked the inhibitory effect of
salusin-β knockdown on the generation of inflammatory cytokines. At
the same time, the LPS-induced expression of HMGB1, p-IκBα and
p-p65, which was reduced by salusin-β knockdown, was restored by
knockdown of HO-1. These data revealed that the loss of HO-1 could
partially reverse the anti-inflammatory effect of salusin-β
knockdown on LPS-induced lung cells, thus further confirming the
findings that the anti-inflammatory effect of salusin-β knockdown
on LPS-induced lung cell injury was dependent on HO-1 activation.
However, the knockdown of HO-1 did not completely reverse the
effects of salusin-β knockdown, which indicates that other
downstream mediators or pathways are also involved, which need to
be investigated in future studies.
To the best of our knowledge, the present study
reported for the first time that salusin-β is involved in
LPS-induced lung injury, and its knockdown can exert
anti-inflammatory effects on LPS-induced lung injury, potentially
via NF-κB inhibition and HO-1 activation. These findings provided a
novel target and an improved understanding of the potential
underlying mechanism of pathogenesis and a molecular therapeutic
strategy for ALI.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
PZ and SC contributed to the conception and design
of the present study; SC and YH contributed to the acquisition of
data; SC and JZ contributed to the analysis and interpretation of
data; and SC and PZ drafted the article and revised it critically
for important intellectual content. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
This study was approved by the ethics committee of
Fujian Medical University Union Hospital (approval no.
IACUC-20181212-09).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Summers C, Singh NR, Worpole L, Simmonds
R, Babar J, Condliffe AM, Gunning KE, Johnston AJ and Chilvers ER:
Incidence and recognition of acute respiratory distress syndrome in
a UK intensive care unit. Thorax. 71:1050–1051. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gouda MM and Bhandary YP: Acute lung
injury: IL-17A-mediated inflammatory pathway and its regulation by
curcumin. Inflammation. 42:1160–1169. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ware LB: Pathophysiology of acute lung
injury and the acute respiratory distress syndrome. Semin Respir
Crit Care Med. 27:337–349. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Laskin DL, Malaviya R and Laskin JD: Role
of macrophages in acute lung injury and chronic fibrosis induced by
pulmonary toxicants. Toxicol Sci. 168:287–301. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Johnson ER and Matthay MA: Acute lung
injury: Epidemiology, pathogenesis, and treatment. J Aerosol Med
Pulm Drug Deliv. 23:243–252. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu P, Yan H, Qi J, Jia W, Zhang W, Yao D,
Ding C, Zhang Y, Chen M and Cai X: L6H9 attenuates LPS-induced
acute lung injury in rats through targeting MD2. Drug Dev Res.
81:85–92. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iwamura H, Inushima K, Takeuchi K,
Kakutani M and Wakitani K: Prophylactic effect of JTE-607 on
LPS-induced acute lung injury in rats with CINC-1 inhibition.
Inflamm Res. 51:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sato K, Watanabe R, Itoh F, Shichiri M and
Watanabe T: Salusins: Potential use as a biomarker for
atherosclerotic cardiovascular diseases. Int J Hypertens.
2013:9651402013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun H, Zhang F, Xu Y, Sun S, Wang H, Du Q,
Gu C, Black SM, Han Y and Tang H: Salusin-β promotes vascular
calcification via nicotinamide adenine dinucleotide
phosphate/reactive oxygen species-mediated klotho downregulation.
Antioxid Redox Signal. 31:1352–1370. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun S, Zhang F, Pan Y, Xu Y, Chen A, Wang
J, Tang H and Han Y: A TOR2A gene product: Salusin-β contributes to
attenuated vasodilatation of spontaneously hypertensive rats.
Cardiovasc Drugs Ther. 2020.(Epub ahead of print). View Article : Google Scholar
|
|
11
|
Zhou CH, Pan J, Huang H, Zhu Y, Zhang M,
Liu L and Wu Y: Salusin-β, but not salusin-α, promotes human
umbilical vein endothelial cell inflammation via the p38
MAPK/JNK-NF-κB pathway. PLoS One. 9:e1075552014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhou CH, Liu L, Liu L, Zhang MX, Guo H,
Pan J, Yin XX, Ma TF and Wu YQ: Salusin-β not salusin-α promotes
vascular inflammation in ApoE-deficient mice via the I-kBα/NF-κB
pathway. PLoS One. 9:e914682014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu T, Zhang Z, Liu T, Zhang W, Liu J, Wang
W and Wang J: Salusin-β contributes to vascular inflammation
associated with pulmonary arterial hypertension in rats. J Thorac
Cardiovasc Surg. 152:1177–1187. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao MX, Zhou B, Ling L, Xiong XQ, Zhang
F, Chen Q, Li YH, Kang YM and Zhu GQ: Salusin-β contributes to
oxidative stress and inflammation in diabetic cardiomyopathy. Cell
Death Dis. 8:e26902017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li HB, Qin DN, Cheng K, Su Q, Miao YW, Guo
J, Zhang M, Zhu GQ and Kang YM: Central blockade of salusin β
attenuates hypertension and hypothalamic inflammation in
spontaneously hypertensive rats. Sci Rep. 5:111622015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Meng L, Li L, Lu S, Li K, Su Z, Wang Y,
Fan X, Li X and Zhao G: The protective effect of dexmedetomidine on
LPS-induced acute lung injury through the HMGB1-mediated TLR4/NF-κB
and PI3K/Akt/mTOR pathways. Mol Immunol. 94:7–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lan KC, Chao SC, Wu HY, Chiang CL, Wang
CC, Liu SH and Weng TI: Salidroside ameliorates sepsis-induced
acute lung injury and mortality via downregulating NF-κB and HMGB1
pathways through the upregulation of SIRT1. Sci Rep. 7:120262017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vijayan V, Wagener F and Immenschuh S: The
macrophage heme-heme oxygenase-1 system and its role in
inflammation. Biochem Pharmacol. 153:159–167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park J, Chen Y, Zheng M, Ryu J, Cho GJ,
Surh YJ, Sato D, Hamada H, Ryter SW, Kim UH, et al: Pterostilbene
4′-β-glucoside attenuates LPS-induced acute lung injury via
induction of heme oxygenase-1. Oxid Med Cell Longev.
2018:27470182018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin H, Li X, Yuan B, Zhang B, Hu S, Gu H,
Jin X and Zhu J: Heme oxygenase-1 ameliorates LPS-induced acute
lung injury correlated with downregulation of interleukin-33. Int
Immunopharmacol. 11:2112–2117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang X, Feng J, Zhu P and Zhao Z:
Ketamine inhibits calcium elevation and hydroxyl radical and nitric
oxide production in lipopolysaccharide-stimulated NR8383 alveolar
macrophages. Inflammation. 36:1094–1100. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ko IG, Hwang JJ, Chang BS, Kim SH, Jin JJ,
Hwang L, Kim CJ and Choi CW: Polydeoxyribonucleotide ameliorates
lipopolysaccharide-induced acute lung injury via modulation of the
MAPK/NF-κB signaling pathway in rats. Int Immunopharmacol.
83:1064442020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liang Y, Luo J, Yang N, Wang S, Ye M and
Pan G: Activation of the IL-1β/KLF2/HSPH1 pathway promotes STAT3
phosphorylation in alveolar macrophages during LPS-induced acute
lung injury. Biosci Rep. 40:BSR201935722020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fisher AB, Dodia C, Chatterjee S and
Feinstein SI: A peptide inhibitor of NADPH oxidase (NOX2)
activation markedly decreases mouse lung injury and mortality
following administration of lipopolysaccharide (LPS). Int J Mol
Sci. 20:23952019. View Article : Google Scholar
|
|
26
|
Liang Y, Yang N, Pan G, Jin B, Wang S and
Ji W: Elevated IL-33 promotes expression of MMP2 and MMP9 via
activating STAT3 in alveolar macrophages during LPS-induced acute
lung injury. Cell Mol Biol Lett. 23:522018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yan J, Li J, Zhang L, Sun Y, Jiang J,
Huang Y, Xu H, Jiang H and Hu R: Nrf2 protects against acute lung
injury and inflammation by modulating TLR4 and Akt signaling. Free
Radic Biol Med. 121:78–85. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Çakır M, Sabah-Özcan S and Saçmacı H:
Increased level of plasma salusin-α and salusin-β in patients with
multiple sclerosis. Mult Scler Relat Disord. 30:76–80. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Joe Y, Kim SK, Chen Y, Yang JW, Lee JH,
Cho GJ, Park JW and Chung HT: Tristetraprolin mediates
anti-inflammatory effects of carbon monoxide on
lipopolysaccharide-induced acute lung injury. Am J Pathol.
185:2867–2874. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sarady JK, Zuckerbraun BS, Bilban M,
Wagner O, Usheva A, Liu F, Ifedigbo E, Zamora R, Choi AMK and
Otterbein LE: Carbon monoxide protection against endotoxic shock
involves reciprocal effects on iNOS in the lung and liver. FASEB J.
18:854–856. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gong Q, Yin H, Fang M, Xiang Y, Yuan CL,
Zheng GY, Yang H, Xiong P, Chen G, Gong FL and Zheng F: Heme
oxygenase-1 upregulation significantly inhibits TNF-alpha and Hmgb1
releasing and attenuates lipopolysaccharide-induced acute lung
injury in mice. Int Immunopharmacol. 8:792–798. 2008. View Article : Google Scholar : PubMed/NCBI
|