Introduction
Alzheimer's disease (AD) has become a global health
issue; >44 million people are currently living with AD and the
global health care cost in 2010 was ~US$818 billion (1). AD is a severe neurodegenerative
disease associated with progressive cognitive decline (2). The pathogenesis of AD is complex, and
the precise underlying mechanism has not yet been elucidated. The
amyloid hypothesis is widely accepted for the pathogenesis of AD
(3). As per this hypothesis,
accumulation of β-amyloid (Aβ) peptides in brain cells is the
primary pathological change in AD. Therefore, drugs such as
Semagacestat (4) and Tramiprosate
(5) targeting inhibition of Aβ
aggregation have been developed. However, no significant
therapeutic effects have been observed in phase III trials of these
drugs (6). Thus, identifying the
precise molecular mechanisms of AD progression is necessary.
Transmembrane immune signaling adaptor TYROBP
(TYROBP) is a type I transmembrane protein expressed in numerous
types of immune cell, such as osteoclasts, macrophages and
monocytes (7). The gene encoding
TYROBP is located on the long arm of chromosome 19 at position 13.1
and contains five exons (8). TYROBP
is a necessary component of activating signal transduction
(9). It is typically expressed on
microglial cells in the brain (10). Certain TYROBP-associated immune
receptors, such as triggering receptor expressed on myeloid cells
(TREM)2, signal regulatory protein β1 and complement receptor
(CR)3, have been identified (10).
The interaction between spleen tyrosine kinase and phosphorylated
TYROBP leads to intracellular calcium mobilization and modulates
immune cell function (11). In
addition, TYROBP suppresses the inflammatory response by
downregulating cytokine production and secretion (12). TYROBP-mediated signaling is
considered to participate in regulating the expression levels of
multiple genes, including TREM1 and TREM2, in the brain (13).
Modern second-generation sequencing technology can
reveal genetic changes between healthy individuals and patients
(14). This technique has been used
in AD to identify a number of relevant genes such as TREM1 and
TYROBP (15). However, owing to the
large number of altered genes, a single AD-promoting gene in AD has
not been identified. Bioinformatics analysis for identifying hub
genes may facilitate the identification of key diagnosis biomarkers
and molecular drug targets (16).
Before bioinformatics analysis, exclusion of irrelevant information
caused by individual differences (17) is necessary. In addition,
bioinformatics analysis results need to be verified
experimentally.
The present study integrated two AD profiles and
analyzed the co-expression of differentially expressed genes (DEGs)
in both profiles. Bioinformatics analysis was used to identify the
hub genes and relevant microRNAs (miRNAs or miRs). Finally, the
function of these identified miRNAs in AD was verified
experimentally.
Materials and methods
Preliminary analysis of GSE113141
(18) and GSE104249 (19)
A total of two AD profiles were acquired from the
Gene Expression Omnibus database (ncbi.nlm.nih.gov/gds/): GSE113141, containing
information from six APP/PS1 transgenic and six normal mice, and
GSE104249, containing information from three APP/PS1 transgenic and
three normal mice. R software (version R 3.2.3) (20) and R studio (version R 3.5.2)
(21) with the LIMMA package was
used for homogenizing the raw data and identifying co-expressed
DEGs.
Bioinformatics analysis
The Database for Annotation, Visualization and
Integrated Discovery online tool (david.ncifcrf.gov/summary.jsp) was used for Gene
Ontology (GO; http://david.ncifcrf.gov/) and Kyoto Encyclopedia of
Genes and Genomes (KEGG; http://david.ncifcrf.gov/) (22) pathway analysis of all co-expressed
DEGs. These genes were used as input for the Search Tool for the
Retrieval of Interacting Genes/Proteins (STRING) database
(string-db.org/cgi/input.pl) to
acquire a protein-protein interaction (PPI) network, and Cytoscape
(23) was used to visualize the
network. The cytoHubba plugin in Cytoscape (version 3.6.1) was used
to calculate the hub genes of AD. GeneMANIA (genemania.org/) was used to identify the interactions
between the co-expressed genes. TargetScan (targetscan.org/) was utilized to predict the relevant
miRNAs that bind the hub gene. All the predicted miRNAs were
integrated with GSE138382, a miRNA gene profile associated with AD
containing information from three APP/PS1 transgenic mice and three
normal mice.
Reverse transcription-quantitative
(RT-q)PCR validation
RNA (from cells, tissues and blood) was extracted
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). NanoDrop ND-1000 (Thermo Fisher Scientific,
Inc.) was used to detect the integrity and concentration of the RNA
samples. Total RNA was reverse-transcribed to cDNA using
PrimeScript RT reagent kit with gDNA Eraser (Takara Bio, Inc.)
according to the manufacturer's instructions. RT-qPCR was performed
using FastStart Universal SYBR Green Master (ROX) (Roche
Diagnostics) using an Applied Biosystems 7500 Fast Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.) to
confirm the relative expression levels of miR-628-5p and
TYROBP in mouse brain tissue. In addition, TYROBP and
miR-628-5p were detected in blood samples from patients with AD.
qPCR thermocycling conditions were 95°C for 5 min, followed by 40
cycles at 95°C for 10 sec, 55°C for 20 sec and 72°C for 20 sec with
an extension step of 72°C for 2 min. Results were normalized to
those of U6 or GAPDH and calculated using the 2−ΔΔCq
(24) method. The primers were as
follows: Human TYROBP forward, 5′-ACTGAGACCGAGTCGCCTTAT-3′ and
reverse, 5′-ATACGGCCTCTGTGTGTTGAG-3′; moue TYROBP forward,
5′-GAGTGACACTTTCCCAAGATGC-3′ and reverse,
5′-CCTTGACCTCGGGAGACCA-3′; human GAPDH forward,
5′-CGGACCAATACGACCAAATCCG-3′ and reverse,
5′-AGCCACATCGCTCAGACACC-3′; mouse GAPDH forward,
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse, 5′-GGGGTCGTTGATGGCAACA-3′;
human miR-628-5p forward, 5′-GATGCTGGATGCTGACATATTTAC-3′ and
reverse, 5′-TATGGTTGTTCTGCTCTCTGTCTC-3′; and U6 forward,
5′-GCGCGTCGTGAAGCGTTC-3′ and reverse, 5′-GTGCAGGGTCCGAGGT-3′.
Participants
The present study was performed in accordance with
the Declaration of Helsinki and was approved by the Ethics
Committee of Changchun University of Chinese Medicine (Changchun,
China; approval no. IRB:2019024421). Blood (4 ml) was obtained from
median cubital vein of patients with AD or healthy controls in
Department of Geriatrics or Medical Examination Center (Affiliated
Hospital to Changchun University of Chinese Medicine, Changchun,
China). The participants were enrolled between February 2019 and
July 2019. The inclusion and exclusion criteria of AD was based on
the criteria from the National Institute of Neurological and
Communicative Diseases and Stroke-Alzheimer's Disease and Related
Disorders Association (25).
Participants were divided into two groups, AD and healthy control,
based on the criteria from the National Institute of Neurological
and Communicative Diseases and Stroke-Alzheimer's Disease and
Related Disorders Association (25). Blood samples were collected from 20
participants, including 10 patients with AD and 10 healthy
controls. The AD group comprised six females and four males (age,
71–87 years; mean age, 77.6±4.6 years). The control group comprised
five females and five males (age, 73–82 years; mean age, 76.9±2.7
years). All patients agreed to the use of their samples in
scientific research and provided written informed consent.
Animals
Male 6-month-old APP/PS1 (n=3) and C57/6J (normal)
(n=3) mice (weight 24–28 g) were purchased from the Chinese Academy
of Medical Sciences (Shanghai, China). All mice were housed in 12 h
light/dark cycle conditions at a temperature of 22±1°C and a
humidity of 55±2% and given food and water ad libitum. All
mice were anesthetized with isoflurane (induction, 5%; maintenance,
1.5–2.5%). The hippocampus and cortex were dissected for subsequent
experiments, including RT-qPCR and western blotting. Then all the
mice were euthanized via cervical dislocation. Animal experiments
were performed in accordance with the National Institutes of Health
Guide for the Care and Use of Laboratory Animals (26) and approved by The Changchun
University of Chinese Medicine Animal Care and Use Committee
(Changchun, China; approval no. KT202009096).
Cell culture
U251 glioblastoma cells were purchased from
Institute of Biochemistry and Cell Biology and cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) with 10% FBS (cat. no.
0010; Gibco; Thermo Fisher Scientific, Inc.) at 37°C in a
humidified 5% CO2 incubator. The medium was replaced
every 3 days. Mimic and inhibitor were designed by Shanghai
GenePharma Co., Ltd. For transfection of miR-628-5p mimics (250
pmol; forward, 5′-AUGCUGACAUAUUUACUAGAGG-3′ and reverse,
5′-UCUAGUAAAUAUGUCAGCAUUU-3′) and inhibitor (250 pmol,
5′-CCUCUAGUAAAUAUGUCAGCAU-3′), cells were transfected with
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.). In the control group, cells were transfected
with Lipofectamine® 2000 alone. The time interval
between transfection and subsequent experimentation was 48 h. After
media was replaced with serum-free Opti-MEM (Gibco; Thermo Fisher
Scientific, Inc.), the transfection complexes were added for 6 h at
37°C in a humidified 5% CO2 incubator. Then the medium
was replaced with the standard culture medium (DMEM with 10% FBS)
and cells harvested following incubation for 48 h at 37°C.
Western blotting
The protein was extracted using RIPA buffer (Beijing
Solarbio Science & Technology Co., Ltd.) and quantified using a
BCA assay (Thermo Fisher Scientific, Inc.). Total cell protein
content (30 µg/lane) was separated by 10% SDS-PAGE (cat. no. 1200;
Beijing Solarbio Science & Technology Co., Ltd.) and
transferred to nitrocellulose membranes (0.45 µm; EMD Millipore).
Blots were blocked using 5% non-fat milk with 0.1% Tween-20
(Sigma-Aldrich; Merck KGaA) for 2 h at 37°C. Diluted antibodies
against TYROBP (cat. no. ab124834), Aβ (cat. no. ab224275)and Aβ
precursor protein (APP; cat. no. ab32136; all at 1:1,000 and all
from Abcam) were used to incubate the membranes overnight at 4°C,
followed by incubation with secondary antibody (β-actin; 1:5,000;
cat. no. bs-0061R, Bioss) at room temperature for 2.5 h. The blots
were then visualized with ECL using a Series Molecular Imaging
Biosystem (Azure Biosystems, Inc.). Protein expression levels were
semi-quantified using ImageJ software (version 4.62; National
Institutes of Health).
Dual luciferase reporter assay
293T cells were purchased from Institute of
Biochemistry and Cell Biology. A total of 2×104 293T
cells per well were seeded into 6-well plates for 24 h at 37°C
before transfection. The wild-type (WT) 3′-UTR sequence (16–23 bp)
of TYROBP that can bind to miR-628-5p or the mutant (MUT) 3′-UTR
sequence was amplified and then cloned into a pGL3 vector (Promega
Corporation). pGL6-TYROBP-WT, pGL6-TYROBP-MUT and miR-628-5p mimics
as well as mimic controls were co-transfected into 293T cells for
24 h at 37°C using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.). At 24 h post-transfection, the
absorbance of the luciferase was measured at 560 nm. A dual
luciferase reporter gene assay kit (Promega Corporation) was used
to measure luciferase activities according to the manufacturer's
protocol. Luciferase activity was normalized to Renilla
luciferase.
Statistical analysis
Continuous data are expressed as the mean ± standard
deviation. All experiments were performed in triplicate. One-way
ANOVA followed by post hoc Tukey's test was performed to assess
differences between multiple groups. All statistical analysis was
performed using SPSS 22.0 software (IBM Corp.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Integration of GSE113141 and
GSE104249
Raw data were normalized using R software and
integrated (Fig. 1). The
integration included information from nine APP/PS1 transgenic mice
and nine normal mice. The genes co-expressed in both GSE113141
and GSE104249 were selected. There were 22 different
co-expressing mRNAs in both AD profiles, including Lyz1, Clec7a,
Cybb, Thbs1, Pilra, Itgb2, Tyrobp, Rac2, Cst7, Lcp1, Klk6, Wfdc17,
Top2a, Lilrb4a, Lyz2, Ctse, Ptprc, Cd36, Igsf6, Mpeg1, Tnni2 and
Cd52. These 22 genes were subjected to bioinformatics analysis
following homogenization. These results demonstrated that these 22
genes might participate in the progress of AD.
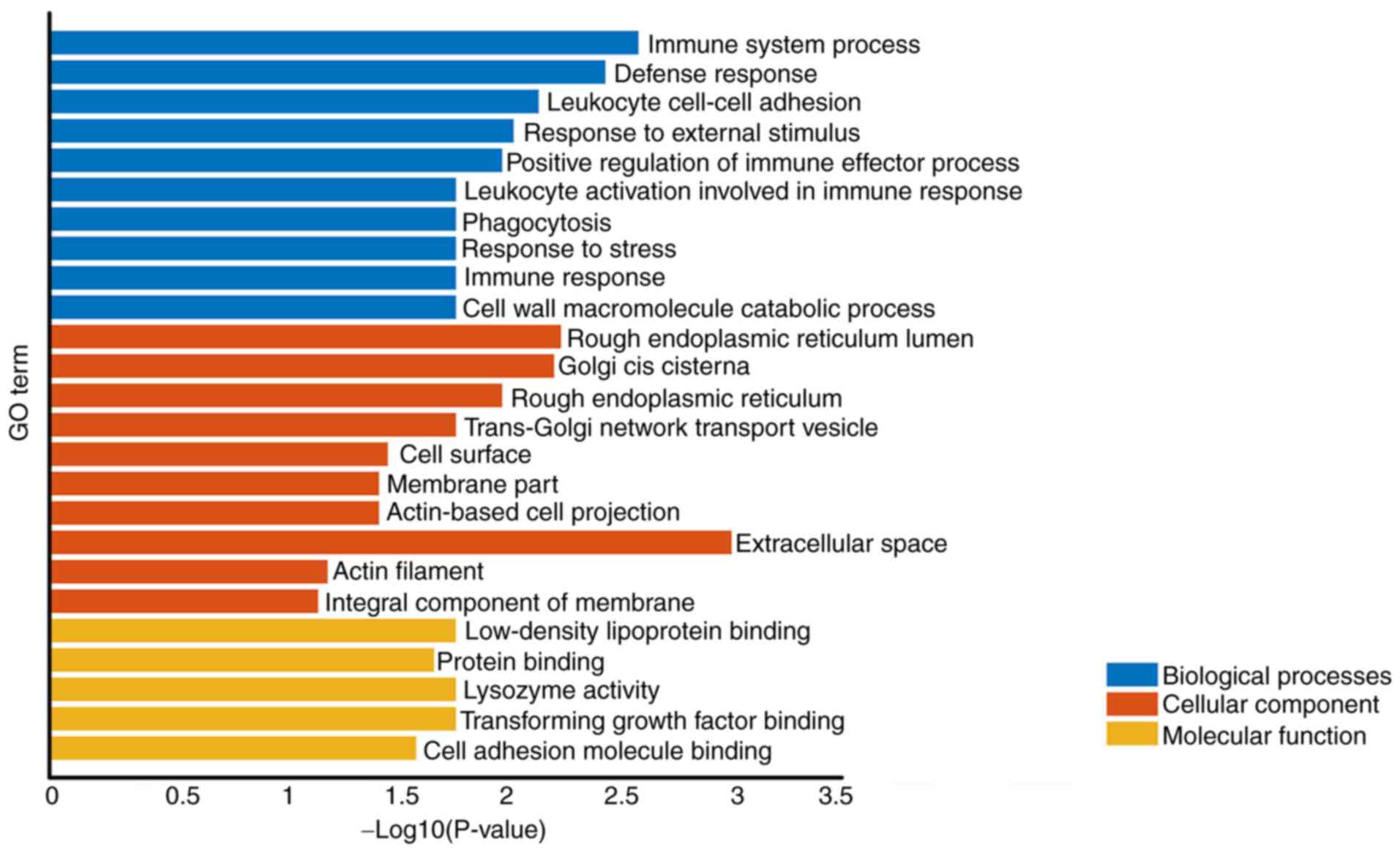
GO analysis
Co-expression of different mRNAs for biological
processes (‘immune system process’ and ‘defense response’) was
observed. The cellular component was associated with ‘Extracellular
space’, ‘Rough endoplasmic reticulum lumen’ and ‘Golgi cis
cisterna’. The molecular function primarily corresponded to
‘lysozyme activity’, ‘Low-density lipoprotein particle binding’ and
‘Transforming growth factor β binding’ (Table I; Fig.
2). These results demonstrated that immune system process,
rough endoplasmic reticulum lumen and lysozyme activity might
participate in the progress of AD.
 | Table I.Enrichment analysis. |
Table I.
Enrichment analysis.
| A, Biological
processes |
|---|
|
|---|
| Term | Description | Gene count | P-value |
|---|
| Immune system
process | GO:0002376 | 10 | 0.00250 |
| Defense
response | GO:0006952 | 8 | 0.00350 |
| Leukocyte cell-cell
adhesion | GO:0007159 | 3 | 0.00690 |
| Response to
external stimulus | GO:0009605 | 9 | 0.00890 |
| Positive regulation
of immune effector process | GO:0002699 | 4 | 0.01000 |
| Leukocyte
activation involved in immune response | GO:0002366 | 3 | 0.01600 |
| Phagocytosis | GO:0006909 | 3 | 0.01600 |
| Response to
stress | GO:0006950 | 10 | 0.01600 |
| Immune
response | GO:0006955 | 6 | 0.01600 |
| Cell wall
macromolecule catabolic process | GO:0016998 | 2 | 0.01600 |
|
| B, Cellular
component |
|
| Term |
Description | Gene
count | P-value |
|
| Extracellular
space | GO:0005615 | 7 | 0.00097 |
| Rough endoplasmic
reticulum lumen | GO:0048237 | 2 | 0.00550 |
| Golgi cis
cisterna | GO:0000137 | 2 | 0.00590 |
| Rough endoplasmic
reticulum | GO:0005791 | 3 | 0.01000 |
| Trans-Golgi network
transport vesicle | GO:0030140 | 2 | 0.01600 |
| Cell surface | GO:0009986 | 5 | 0.03200 |
| Membrane part | GO:0044425 | 13 | 0.03500 |
| Actin-based cell
projection | GO:0098858 | 3 | 0.03500 |
| Actin filament | GO:0005884 | 2 | 0.05900 |
| Integral component
of membrane | GO:0016021 | 10 | 0.06500 |
|
| C, Molecular
function |
|
| Term |
Description | Gene
count | P-value |
|
| Lysozyme
activity | GO:0003796 | 2 | 0.01600 |
| Low-density
lipoprotein particle binding | GO:0030169 | 2 | 0.01600 |
| Transforming growth
factor β binding | GO:0050431 | 2 | 0.01600 |
| Protein
binding | GO:0005515 | 14 | 0.02000 |
| Cell adhesion
molecule binding | GO:0050839 | 3 | 0.02400 |
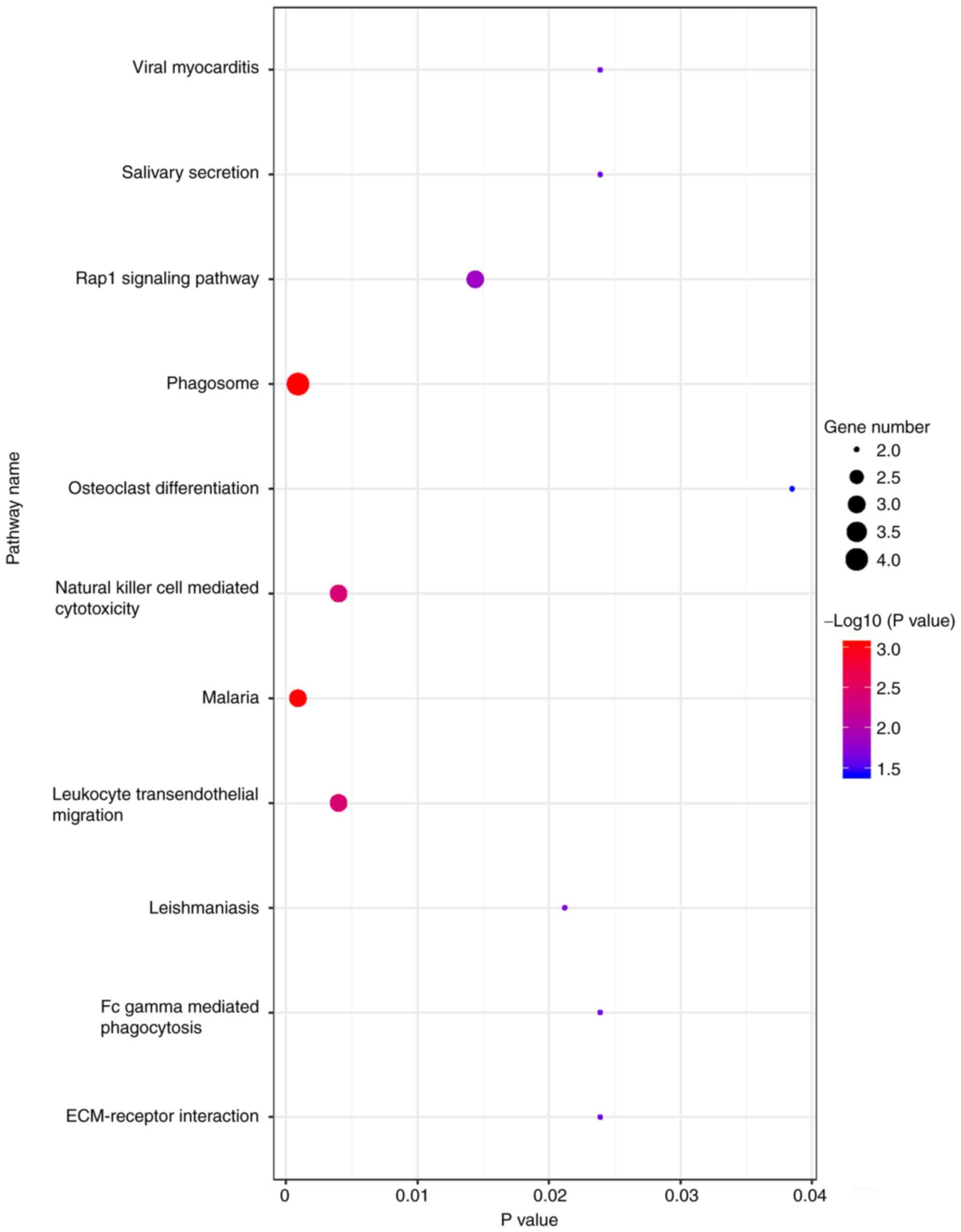
KEGG signaling pathway
All co-expressed DEG were enriched for ‘phagosome’
and ‘malaria’ (Fig. 3).
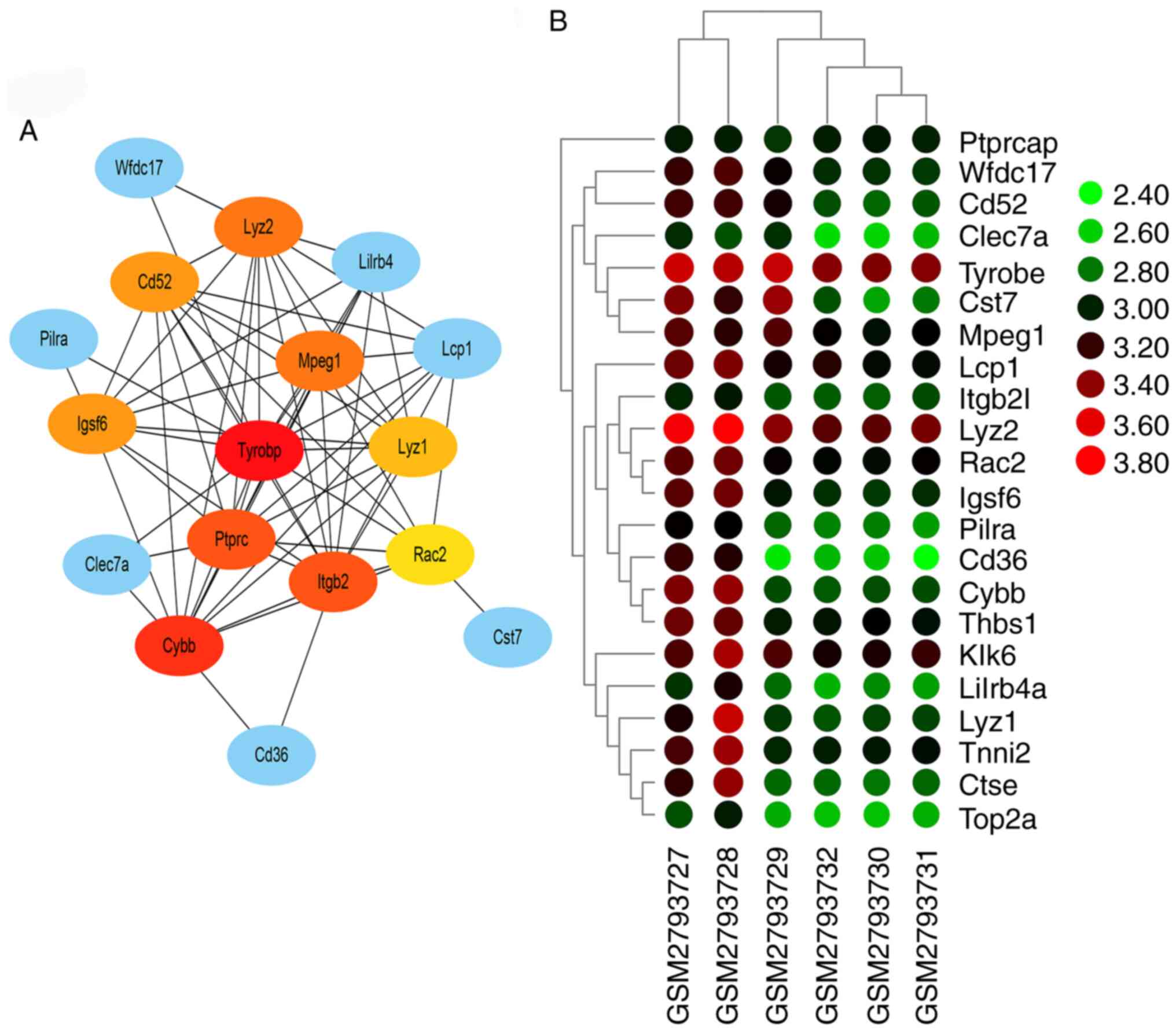
PPI network and hub genes
Interaction analysis using the STRING database
demonstrated 22 nodes with 69 edges. These nodes and edges were
utilized by Cytoscape for pictorial depiction of the network. The
five hub genes predicted by CytoHubba analysis were those encoding
TYROBP (score=14), cytochrome b-245 β chain (score=13), integrin
sub-unit β 2 (score=12), protein tyrosine phosphatase receptor type
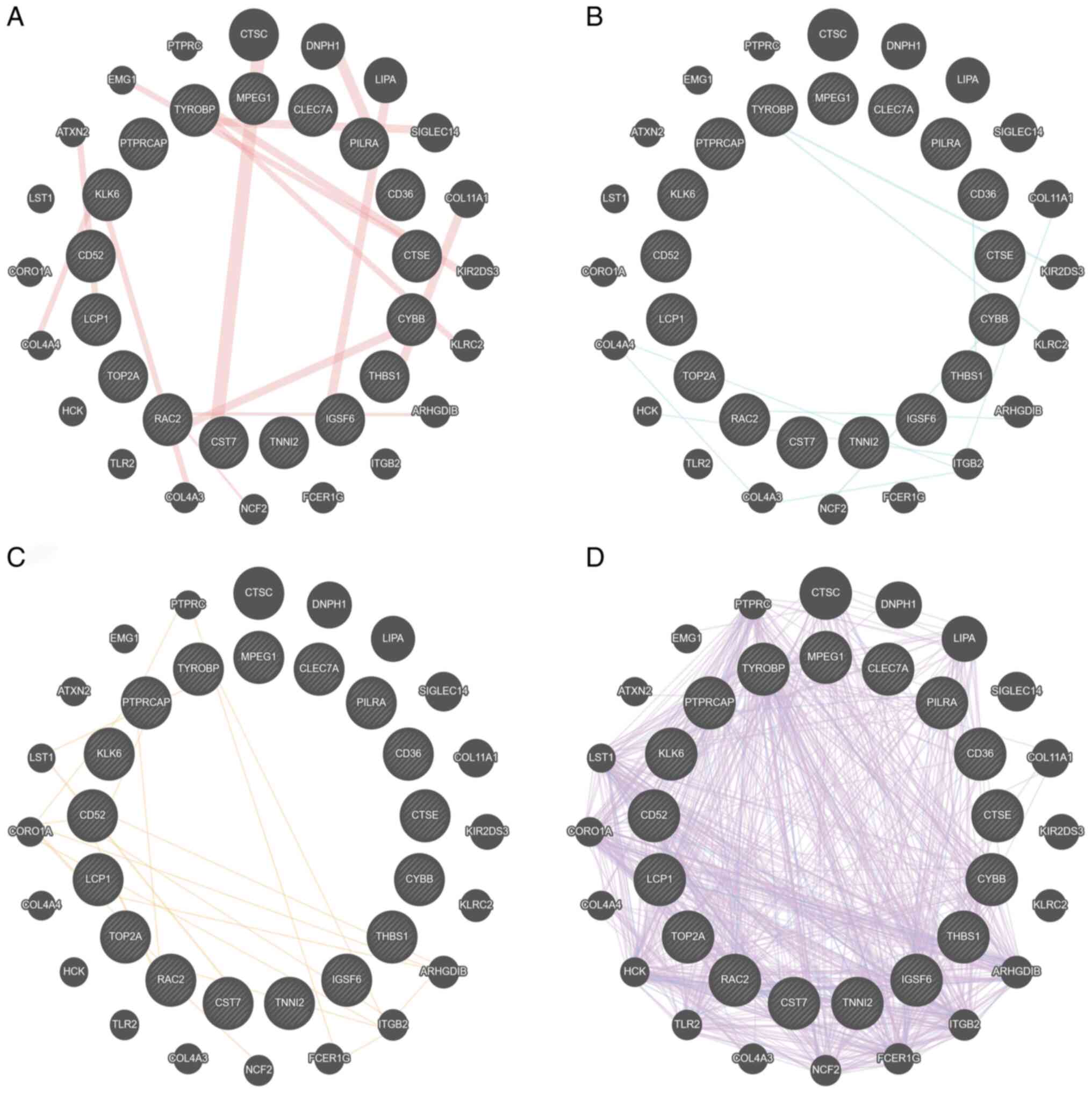
C (score=12) and lysozyme 2 (score 11) (Fig. 4). GeneMANIA database also
demonstrated that TYROBP served a central role in ‘physical
interactions’, ‘pathway’, ‘predicted’ and ‘co-expression’ networks
(Fig. 5A-D, respectively). These
results demonstrated that the function of TYROBP acted as a hub
gene in AD and required further investigation.
TargetScan prediction
TargetScan (version 7.2; www.targetscan.org/vert_72) was used to predict miRNAs
that target TYROBP (Table
SI). In total, there were 77 miRNAs having binding sites for
TYROBP. Subsequent integration of GSE138382 with the 77
predicted miRNAs demonstrated only one miRNA, miR-628-5p, that was
downregulated in GSE138382; this was also predicted to target
TYROBP. These results demonstrated miR-628-5p/TYROBP
may be used as an AD biomarker and molecular drug candidate.
High expression levels of TYROBP in
APP/PS1 mice
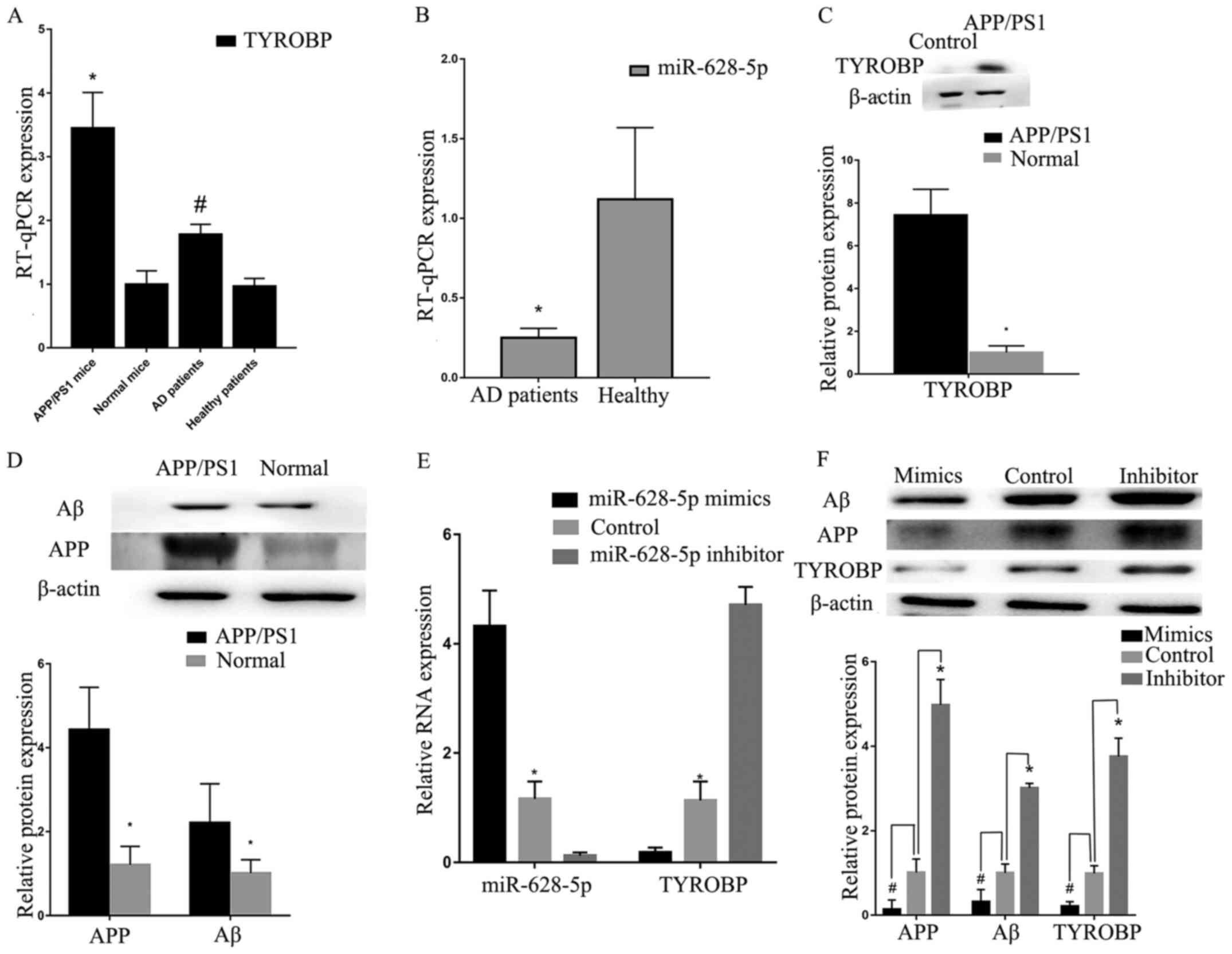
RT-qPCR verification was performed using samples
taken from mice (three APP/PS1 and three normal) and patients with
AD. Analysis using RT-qPCR indicated that TYROBP was
increased in APP/PS1 mice (Fig.
6A). Low miR-628-5p expression levels were detected in blood
from patients with AD (Fig. 6A and
B), which was consistent with GSE147232 (a miRNA microarray
profile in the plasma of patients with mild cognitive impairment
due to AD). Western blot analysis also confirmed that TYROBP, APP
and Aβ were more highly expressed in APP/PS1 mice compared with
normal mice (Fig. 6C and D). These
results demonstrated that high expression of TYROBP fulfilled
important roles in AD.
Increased miR-628-5p expression levels
inhibit TYROBP in vitro
The transfection efficiency of miR-628-5p was
confirmed by RT-qPCR. U251 cells transfected with miR-628-5p mimic
demonstrated increased expression levels of miR-628-5p (Fig. 6E), whereas those transfected with
miR-628-5p inhibitor exhibited upregulated TYROBP, compared
with the control. Western blot analysis demonstrated that decreased
expression of miR-628-5p resulted in the upregulation of TYROBP,
APP and Aβ compared with control group (Fig. 6F). These results demonstrated that
high expression of miR-628-5p could inhibit TYROBP in U251
cells.
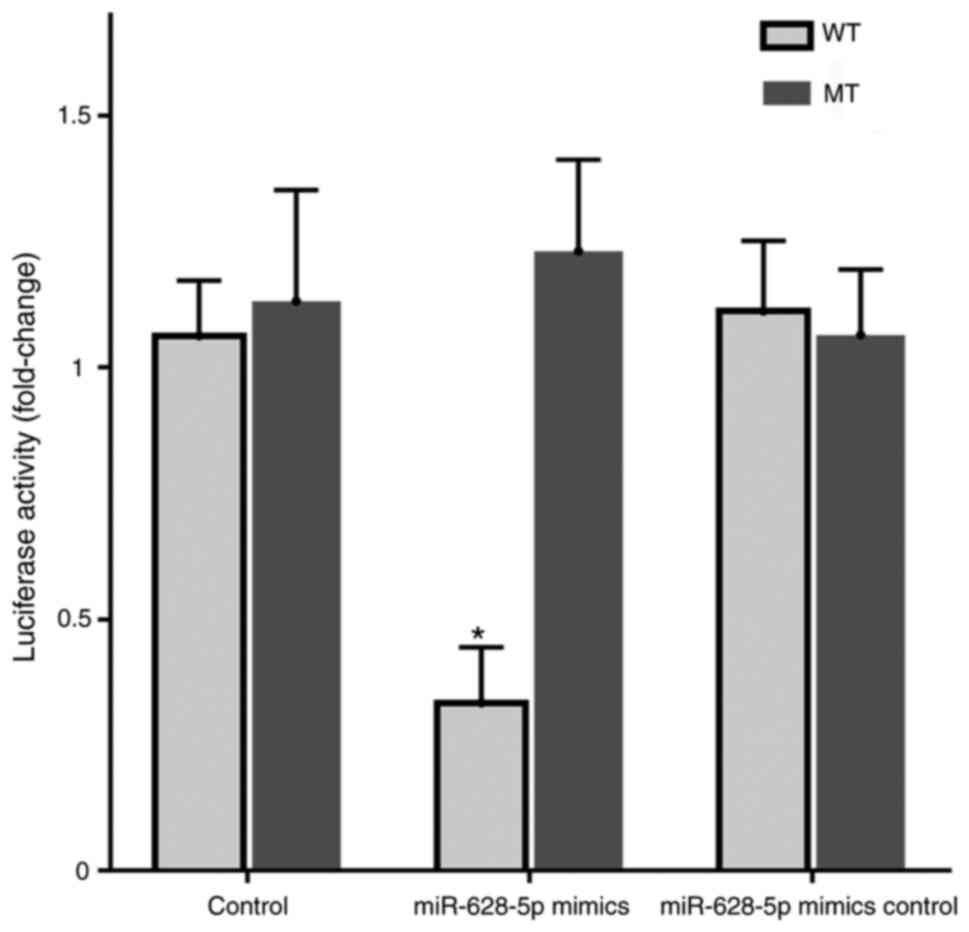
TYROBP targeting by miR-628-5p
Dual-luciferase reporter analysis demonstrated that
co-transfection with miR-628-5p mimics significantly decreased the
luciferase activity of the wild-type TYROBP reporter compared with
that in the normal or mimic inhibitor samples and the mutant
reporter (Fig. 7). These results
demonstrated that miR-628-5p had binding sites with TYROBP and
could inhibit translation of TYROBP in AD.
Discussion
AD has become one of the worldwide disorders
involving central nervous system neurodegeneration and cognitive
decline (27). However, its
detailed molecular pathogenesis remains unclear. Although a number
of hypotheses such as the cholinergic hypothesis (28), amyloid hypothesis (29) and tau hypothesis (30) have been proposed, the amyloid
hypothesis is the most widely accepted (29). A number of drugs, including
Semagacestat (4) and Tramiprosate
(5) have been developed to inhibit
Aβ; however, results have been unsatisfactory. For example,
Semagacestat failed phase III clinical trials for AD treatment
(4). Thus, further studies on the
molecular pathogenesis of AD and novel drug targets are
necessary.
Second-generation sequencing technology provides a
fast method to screen DEGs between normal and impaired tissue such
as brain, liver and kidney (14).
Differential expression levels of both coding and non-coding RNAs
may provide novel biomarkers for diagnosis and treatment targets
(31). However, the abundance of
DEGs obtained by this technique may mask core pathogenic genes. In
addition, ‘junk’ information caused by individual differences
(17) may be included among
sequencing results. Therefore, normalization of raw data and
integration analysis are necessary to provide reliable data for
bioinformatics analysis.
The present GO analysis indicated that biological
processes of AD were associated with immune and inflammatory
responses. Neuro-inflammation has been reported to be involved in
the progress of AD (32). Microglia
in the brain produce pro-inflammatory cytokines to activate the
inflammatory response. Moreover, NLR pyrin family domain containing
3 (NLRP3) has been reported to be activated by Aβ (33); activated NLRP3 can enhance AD
progression via the chronic inflammatory response. In addition, the
complement system is a family of key proteins that regulate the
immune response and is considered to be the first line of
homeostasis maintenance (34).
Members of the complement system have also been found to have high
expression levels in patients with AD. For example, C1q protein
expression levels, associated with the classical complement
pathway, are upregulated following activation by Aβ, and cause
neuronal atrophy (35). Another
study indicated that inhibiting C1q decrease synapse loss in mice
(36).
Following bioinformatics analysis, TYROBP was
predicted as a hub gene for AD. High expression levels of
TYROBP in AD have previously been reported (37), which is consistent with previous
results from second generation sequencing (15). However, previous research has
focused on Aβ42, τ and TREM2 instead of TYROBP (38). Consistent with the present GO
analysis, TYROBP serves as a core molecule in the immune system,
particularly as the adapter for TREM2 and CRs, which control signal
transduction (39). TREM2 and CR3
have been demonstrated to regulate the pathogenesis of AD (39). In addition, TYROBP inhibits
inflammatory responses via downregulation of microglia-mediated
cytokine production (40). Based on
the amyloid hypothesis, a number of drugs have been developed to
inhibit Aβ. TYROBP may downregulate Aβ and clear apoptotic neurons
by enhancing microglial phagocytic activity (41). To the best of our knowledge, the
present study is the first to use bioinformatics analysis to
predict TYROBP as a hub gene in AD. TYROBP may be a
novel AD diagnosis biomarker and treatment target.
Previous studies have reported the function of
TYROBP in AD (42,43). Although the detailed mechanism
underlying the cognitive decline of AD remains unknown, a partial
loss of the colony stimulating factor 1 receptor (CSF1R)/TYROBP
signaling pathway has been reported in neurological phenotypes
(44). The immune response is
reported to serve an important role during AD progression (45). TYROBP may mediate immune cell
phagocytosis by regulating CSF1R and by interfering with macrophage
proliferation, leading to white matter disease, which is clinically
characterized by behavioral, cognitive and motor abnormalities
(46). In addition, Haure-Mirande
et al (41) suggested that
TYROBP slows, arrests or prevents the development of sporadic
late-onset AD (LOAD). The complement system is activated in AD
(41). Deletion of TYROBP in a
tauopathy mouse model downregulates C1q expression levels, leading
to elevated learning behavior (retention tests) and synaptic
function (47). Therefore,
decreased expression levels of complement by TYROBP may be
beneficial during AD pathology by improving learning behavior and
synaptic function. Sekiya et al (38) found that glial expression of TYROBP
promotes τ-mediated neurodegeneration and affects LOAD;
furthermore, TYROBP was identified as a potential target
gene by comparing human AD profiles, which is similar to the
results of the present study.
miRNAs are a class of non-coding RNA molecule that
inhibit translation or cause degradation of target mRNAs (48). To date, miR-628-5p has not been
reported to participate in AD. Previously, miR-628-5p has been
reported to inhibit glioblastoma cell proliferation and migration
by targeting high mobility group box 3 (49); it can also decrease the
proliferation of prostate (50) and
epithelial ovarian cancer cells (51) by targeting fibroblast growth factor
receptor 2 and inhibiting glioma proliferation by binding DEAD-box
helicase 59 (52). Therefore,
miR-628-5p may serve as a suppressor miRNA targets TYROBP,
highlighting the potential of TYROBP as a novel therapeutic target
for AD.
To the best of our knowledge, the present study is
the first to use bioinformatics analysis and experimental
confirmation to predict that miR-628-5p may inhibit the expression
of AD-associated proteins. miR-628-5p/TYROBP may serve as a
novel molecular diagnostic marker and candidate therapeutic target
for AD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML and LY conceived and designed the study. SL, CM,
XW and YS performed the experiments and collected the data. ML, HL
and ZG analyzed and interpreted the data. ML performed experiments
and drafted the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
All experiments involving animals were approved by
The Changchun University of Chinese Medicine Animal Care and Use
Committee. RNA extraction, RNA reverse transcription and RT-qPCR
validation involving patients' blood was approved by the Ethics
Committee of Changchun University of Chinese Medicine.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
BP
|
biological processes
|
|
CC
|
cellular component
|
|
DEGs
|
differentially expressed genes
|
|
GO
|
Gene Ontology
|
|
KEGG
|
Kyoto Encyclopedia of Genes and
Genomes
|
|
MF
|
molecular function
|
|
PPI
|
protein-protein interaction
|
|
RT-q
|
reverse transcription-quantitative
|
References
|
1
|
Weiner MW, Veitch DP, Aisen PS, Beckett
LA, Cairns NJ, Green RC, Harvey D, Jack CR, Jagust W, Liu E, et al:
The Alzheimer's disease neuroimaging initiative: A review of papers
published since its inception. Alzheimers Dement. 9:e111–e194.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
De Strooper B: Lessons from a failed
γ-Secretase Alzheimer trial. Cell. 159:721–726. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mudher A and Lovestone S: Alzheimer's
disease-do tauists and baptists finally shake hands? Trends
Neurosci. 25:22–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Karran E and De Strooper B: The amyloid
cascade hypothesis: Are we poised for success or failure? J
Neurochem. 139 (Suppl 2):S237–S252. 2016. View Article : Google Scholar
|
|
5
|
Gervais F, Paquette J, Morissette C,
Krzywkowski P, Yu M, Azzi M, Lacombe D, Kong X, Aman A, Laurin J,
et al: Targeting soluble Abeta peptide with Tramiprosate for the
treatment of brain amyloidosis. Neurobiol Aging. 28:537–547. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Aisen PS, Gauthier S, Ferris SH, Saumier
D, Haine D, Garceau D, Duong A, Suhy J, Oh J, Lau WC and Sampalis
J: Tramiprosate in mild-to-moderate Alzheimer's disease - a
randomized, double-blind, placebo-controlled, multi-centre study
(the Alphase Study). Arch Med Sci. 7:102–111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tomasello E, Olcese L, Vely F, Geourgeon
C, Bléry M, Moqrich A, Gautheret D, Djabali M, Mattei MG and Vivier
E: Gene structure, expression pattern, and biological activity of
mouse killer cell activating receptor-associated protein
(KARAP)/DAP-12. J Biol Chem. 273:34115–34119. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hamerman JA, Ni M, Killebrew JR, Chu CL
and Lowell CA: The expanding roles of ITAM adapters FcRgamma and
DAP12 in myeloid cells. Immunol Rev. 232:42–58. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lanier LL, Corliss BC, Wu J, Leong C and
Phillips JH: Immunoreceptor DAP12 bearing a tyrosine-based
activation motif is involved in activating NK cells. Nature.
391:703–707. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paloneva J, Kestila M, Wu J, Salminen A,
Böhling T, Ruotsalainen V, Hakola P, Bakker AB, Phillips JH,
Pekkarinen P, et al: Loss-of-function mutations in TYROBP (DAP12)
result in a presenile dementia with bone cysts. Nat Genet.
25:357–361. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Varnum MM and Ikezu T: The classification
of microglial activation phenotypes on neurodegeneration and
regeneration in Alzheimer's disease brain. Arch Immunol Ther Exp
(Warsz). 60:251–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sessa G, Podini P, Mariani M, Meroni A,
Spreafico R, Sinigaglia F, Colonna M, Panina P and Meldolesi J:
Distribution and signaling of TREM2/DAP12, the receptor system
mutated in human polycystic lipomembraneous osteodysplasia with
sclerosing leukoencephalopathy dementia. Eur J Neurosci.
20:2617–2628. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Satoh J, Shimamura Y and Tabunoki H: Gene
expression profile of THP-1 monocytes following knockdown of DAP12,
a causative gene for Nasu-Hakola disease. Cell Mol Neurobiol.
32:337–343. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pareek CS, Smoczynski R and Tretyn A:
Sequencing technologies and genome sequencing. J Appl Genet.
52:413–435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Emilie F, Coelho JE, Zornbach K, Malik E,
Baqi Y, Schneider M, Cellai L, Carvalho K, Sebda S, Figeac M, et
al: Beneficial effect of a selective adenosine a2a
receptor antagonist in the APPswe/PS1dE9 mouse model of Alzheimer's
Disease. Front Mol Neurosci. 11:2352018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang W, Liu Q, Wang Y, Piao H, Li B, Zhu
Z, Li D, Wang T, Xu R and Liu K: Integration of gene expression
profile data of human epicardial adipose tissue from coronary
artery disease to verification of hub genes and pathways. Biomed
Res Int. 2019:85673062019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
You J, Qi S, Du Y, Wang C and Su G:
Multiple bioinformatics analyses of integrated gene expression
profiling data and verification of hub genes associated with
diabetic retinopathy. Med Sci Monit. 26:e9231462020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Faivre E, Coelho JE, Zornbach K, Malik E,
Baqi Y, Schneider M, Cellai L, Carvalho K, Sebda S, Figeac M, et
al: Beneficial effect of a selective adenosine A2A
receptor antagonist in the APPswe/PS1dE9 mouse model of Alzheimer's
disease. Front Mol Neurosci. 11:2352018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu W, Sun F, Wan M, Jiang F, Bo X, Lin L,
Tang H and Xu S: β-Sheet breaker peptide-HPYD for the treatment of
Alzheimer's disease: Primary studies on behavioral test and
transcriptional profiling. Front Pharmacol. 8:9692018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shim SR and Kim SJ: Intervention
meta-analysis: Application and practice using R software. Epidemiol
Health. 41:e20190082019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Otto R, Schirrmeister W, Majeed RW,
Greiner F, Lucas B, Röhrig R, Walcher F and Brammen D; AKTIN
Research Group, : Implementation of emergency department
performance benchmarking using R and LaTeX. Stud Health Technol
Inform. 267:238–246. 2019.PubMed/NCBI
|
|
22
|
Huang DW, Sherman BT, Tan Q, Collins JR,
Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC and Lempicki
RA: The DAVID gene functional classification tool: A novel
biological module-centric algorithm to functionally analyze large
gene lists. Genome Biol. 8:R1832007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McKhann G, Drachman D, Folstein M, Katzman
R, Price D and Stadlan EM: Clinical diagnosis of Alzheimer's
disease: Report of the NINCDS-ADRDA Work Group under the auspices
of the department of health and human services task force on
Alzheimer's disease. Neurology. 34:939–944. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zimmermann M: Ethical guidelines for
investigations of experimental pain in conscious animals. Pain.
16:109–110. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shen J and Kelleher RJ III: The presenilin
hypothesis of Alzheimer's disease: Evidence for a loss-of-function
pathogenic mechanism. Proc Natl Acad Sci USA. 104:403–409. 2017.
View Article : Google Scholar
|
|
28
|
Francis PT, Palmer AM, Snape M and Wilcock
GK: The cholinergic hypothesis of Alzheimer's disease: A review of
progress. J Neurol Neurosurg Psychiatry. 66:137–147. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hardy J and Allsop D: Amyloid deposition
as the central event in the aetiology of Alzheimer's disease.
Trends Pharmacol Sci. 12:383–388. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Goedert M, Spillantini MG and Crowther RA:
Tau proteins and neurofibrillary degeneration. Brain Pathol.
1:279–286. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen J, Qi Y, Liu CF, Lu JM, Shi J and Shi
Y: MicroRNA expression data analysis to identify key miRNAs
associated with Alzheimer's disease. J Gene Med. 20:e30142018.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sagy-Bross C, Hadad N and Levy R:
Cytosolic phospholipase A2α upregulation mediates apoptotic
neuronal death induced by aggregated amyloid-β peptide1-42.
Neurochem Int. 63:541–550. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heneka MT, Kummer MP, Stutz A, Delekate A,
Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et
al: NLRP3 is activated in Alzheimer's disease and contributes to
pathology in APP/PS1 mice. Nature. 493:674–678. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sarma JV and Ward PA: The compliment
system. Cell Tissue Res. 343:227–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rogers J, Cooper NR, Webster S, Schultz J,
McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, et al:
Complement activation by beta-amyloid in Alzheimer disease. Proc
Natl Acad Sci USA. 89:10016–10020. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schöll M, Lockhart SN, Schonhaut DR,
O'Neil JP, Janabi M, Ossenkoppele R, Baker SL, Vogel JW, Faria J,
Schwimmer HD, et al: PET imaging of tau deposition in the aging
human brain. Neuron. 89:971–982. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jiang Y, Li Z, Ma H, Cao X, Liu F, Tian A,
Sun X, Li X and Wang J: Upregulation of TREM2 ameliorates
neuroinflammatory responses and improves cognitive deficits
triggered by surgical trauma in Appswe/PS1dE9 mice. Cell Physiol
Biochem. 46:1398–1411. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sekiya M, Wang M, Fujisaki N, Sakakibara
Y, Quan X, Ehrlich ME, De Jager PL, Bennett DA, Schadt EE, Gandy S,
et al: Integrated biology approach reveals molecular and
pathological interactions among Alzheimer's Aβ42, Tau, TREM2, and
TYROBP in drosophila models. Genome Med. 10:262018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Haure-Mirande JV, Audrain M, Fanutza T,
Kim SH, Klein WL, Glabe C, Readhead B, Dudley JT, Blitzer RD, Wang
M, et al: Deficiency of TYROBP, an adapter protein for TREM2 and
CR3 receptors, is neuroprotective in a mouse model of early
Alzheimer's pathology. Acta Neuropathol. 134:769–788. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ma J, Jiang T, Tan L and Yu JT: TYROBP in
Alzheimer's disease. Mol Neurobiol. 51:820–826. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Haure-Mirande JV, Wang M, Audrain M,
Fanutza T, Kim SH, Heja S, Readhead B, Dudley JT, Blitzer RD,
Schadt EE, et al: Integrative approach to sporadic Alzheimer's
disease: Deficiency of Tyrobp in cerebral Abeta amyloidosis mouse
normalizes clinical phenotype and complement subnetwork molecular
pathology without reducing Abeta burden. Mol Psychiatry.
24:431–446. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pottier C, Ravenscroft TA, Brown PH, Finch
NA, Baker M, Parsons M, Asmann YW, Ren Y, Christopher E, Levitch D,
et al: TYROBP genetic variants in early-onset Alzheimer's disease.
Neurobiol Aging. 48:222.e9–222.e15. 2016. View Article : Google Scholar
|
|
43
|
Mori Y, Yoshino Y, Ochi S, Yamazaki K,
Kawabe K, Abe M, Kitano T, Ozaki Y, Yoshida T, Numata S, et al:
TREM2 mRNA expression in leukocytes is increased in Alzheimer's
disease and schizophrenia. PLoS One. 10:e01368352015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kempthorne L, Yoon H, Madore C, Smith S,
Wszolek ZK, Rademakers R, Kim J, Butovsky O and Dickson DW: Loss of
homeostatic microglial phenotype in CSF1R-related
Leukoencephalopathy. Acta Neuropathol Commun. 8:722020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Malik M, Parikh I, Vasquez JB, Smith C,
Tai L, Bu G, LaDu MJ, Fardo DW, Rebeck GW and Estus S: Genetics
ignite focus on microglial inflammation in Alzheimer's disease. Mol
Neurodegener. 10:522015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rademakers R, Baker M, Nicholson AM,
Rutherford NJ, Finch N, Soto-Ortolaza A, Lash J, Wider C, Wojtas A,
DeJesus-Hernandez M, et al: Mutations in the colony stimulating
factor 1 receptor (CSF1R) gene cause hereditary diffuse
leukoencephalopathy with spheroids. Nat Genet. 44:200–205. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Audrain M, Haure-Mirande JV, Wang M, Kim
SH, Fanutza T, Chakrabarty P, Fraser P, St George-Hyslop PH, Golde
TE, Blitzer RD, et al: Integrative approach to sporadic Alzheimer's
disease: Deficiency of TYROBP in a tauopathy mouse model reduces
C1q and normalizes clinical phenotype while increasing spread and
state of phosphorylation of tau. Mol Psychiatry. 24:1383–1397.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:e050052015. View Article : Google Scholar
|
|
49
|
Chen WL, Jiang L, Wang JS and Liao CX:
Circ-0001801 contributes to cell proliferation, migration, invasion
and epithelial to mesenchymal transition (EMT) in glioblastoma by
regulating miR-628-5p/HMGB3 axis. Eur Rev Med Pharmacol Sci.
23:10874–10885. 2019.PubMed/NCBI
|
|
50
|
Srivastava A, Goldberger H, Dimtchev A,
Marian C, Soldin O, Li X, Collins SP, Suy S and Kumar D:
Circulatory miR-628-5p is downregulated in prostate cancer
patients. Tumour Biol. 35:4867–4873. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li M, Qian Z, Ma X, Lin X, You Y, Li Y,
Chen T and Jiang H: miR-628-5p decreases the tumorigenicity of
epithelial ovarian cancer cells by targeting at FGFR2. Biochem
Biophys Res Commun. 495:2085–2091. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xie P, Wang Y, Liao Y, Han Q, Qiu Z, Chen
Y and Zuo X: MicroRNA-628-5p inhibits cell proliferation in glioma
by targeting DDX59. J Cell Biochem. 120:17293–17302. 2019.
View Article : Google Scholar : PubMed/NCBI
|