Introduction
Acute myocardial infarction (AMI) is a subset of
acute coronary syndrome that displays high morbidity and mortality
(1). In the United States, the
estimated annual incidence of AMI is 605,000 new cases and 200,000
recurrent cases, and the mortality is ~14% (2). At present, the treatment strategies
for AMI primarily include thrombolytic therapy and primary
percutaneous coronary interventions (3). Although blood perfusion in the
ischemic region is often rapidly recovered following immediate
therapy, recanalization can aggravate myocardial injury, which is
known as ischemia/reperfusion (I/R) injury (4). It has been suggested that myocardial
I/R injury may decrease the benefits of myocardial reperfusion and
worsen clinical outcomes (5).
However, to the best of our knowledge, no effective treatment
strategy for myocardial I/R injury has been developed to date
(6). Therefore, the discovery of
novel effective therapeutic strategies for myocardial I/R injury is
important.
Tissue kallikrein 1 (TK1), a glycoprotein that
belongs to the serine proteinase superfamily, can cleave
low-molecular-weight kininogen into a number of bioactive kinin
peptides, including kininogens and kallikreins (7). Vasoactive kinins serve a beneficial
role in a number of cardiovascular, cerebrovascular and renal
diseases (8). A previous study
revealed that hTK1 gene-modified mesenchymal stem cells display
protective effects against cardiac injury following MI via
suppressing apoptosis and inflammation, and promoting
neovascularization (9). However,
the functional role of TK1 in myocardial I/R injury requires
further investigation.
Matrix metallopeptidases (MMPs) are a family of
proteinases that possess the ability to degrade and remodel the
extracellular matrix (ECM) (10).
Adverse ECM remodeling is one of the key pathological processes in
myocardial I/R injury (11). MMP
activities are inhibited by tissue inhibitors of matrix
metalloproteinases (TIMPs), of which there are four subtypes,
TIMP1-4 (12). TIMP1 may serve as a
plasma biomarker for predicting prognosis following MI (13). However, to the best of our
knowledge, the potential role of TIMP1 in myocardial I/R injury has
not been previously reported. Our previous study successfully
constructed an adenovirus (Ad) vector containing hTK1 and hTIMP1
genes (14). In the present study,
the beneficial effects of the co-expression of hTK1 and hTIMP1
genes on myocardial I/R injury in vivo and in vitro
were assessed using the aforementioned Ad vector.
Materials and methods
Animals and treatments
A total of 30 male Sprague-Dawley rats (weight,
200–220 g; age, 4–6 weeks) were purchased from Beijing Vital River
Laboratory Animal Technology Co., Ltd.. Rats were randomly divided
into the following three groups (n=10 per group): i) Sham; ii) I/R;
and iii) I/R + Ad-hTK1/hTIMP1. All rats were housed in an
environment of the constant temperature (22±2°C) and humidity
(55±5%) with 12-h light/dark cycles, and ad libitum access
to food and water. At 1 week prior to myocardial I/R injury, rats
in the I/R + Ad-hTK1/hTIMP1 group were intravenously injected with
recombinant Ad vector containing hTK1 and hTIMP1 genes
(Ad-hTK1/hTIMP1; 5×1012 gc/kg). At the same time point,
rats in the I/R and sham groups were intravenously injected with
the control vector (Ad-EGFP; 5×1012 gc/kg). The
Ad-hTK1/hTIMP1 and Ad-EGFP were constructed and maintained in our
laboratory (Fujian Provincial Hospital Key Laboratory of
Geriatrics, Fuzhou, China). All animal experiments were conducted
in accordance with the National Institutes of Health Guide for the
Care and Use of Laboratory Animals (15), and approved by the Institutional
Ethics Committee for Laboratory Animal Care of Fujian Provincial
Hospital (approval no. K2019-01-038).
Cell culture and transfection
Rat cardiac microvascular endothelial cells (CMVECs;
LifeLine Cell Technology, LLC) were cultured in Dulbecco's modified
Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 20% fetal bovine serum (FBS; Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin (Gibco; Thermo Fisher
Scientific, Inc.) and 100 µg/ml streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in an air sealed chamber. CMVECs were
transfected with Ad-hTK1/hTIMP1 or the control vector (Ad-EGFP),
according to a previous study (16).
Myocardial I/R injury protocol
The induction of myocardial I/R injury in the
present study was performed as previously described (17), with a few modifications. Briefly,
rats were anesthetized by the intraperitoneal injection of
pentobarbital sodium (40 mg/kg) and were mechanically ventilated.
The heart was exposed via a thoracotomy and subsequent opening of
the pericardium. The left anterior descending coronary artery was
ligated with a slipknot using a 7-0 silk suture. Ischemia of the
anterior wall of the left ventricle was confirmed by observation of
ST-segment elevation on the electrocardiogram. The heart was
immediately placed back into the thoracic cavity and the chest was
closed using a 4-0 silk suture. Following ischemia for 30 min, the
slipknot was released to allow for myocardial reperfusion. Rats in
the sham group underwent an identical procedure, but ligation of
the left anterior descending coronary artery was not performed. At
24 h post-surgery, echocardiographic measurements were conducted
and blood samples (2 ml) were collected from the carotid artery of
each rat. All rats were euthanized by the intraperitoneal injection
of an overdose of pentobarbital sodium (120 mg/kg). Death was
verified by cessation of spontaneous breathing and the heartbeat.
Subsequently, cardiac tissues were collected for use in subsequent
experiments.
Establishment of the cell model with
hypoxia/reoxygenation (H/R) treatment
The cell model with H/R treatment was established as
previously described (18).
Briefly, CMVECs were cultured in serum- and glucose-free DMEM in a
hypoxic chamber (1% O2) at 37°C for 6 h. Subsequently,
CMVECs were maintained in DMEM containing 20% FBS in normoxic
conditions (21% O2) at 37°C for 12 h for
reoxygenation.
Histological staining
Rat cardiac tissues were fixed in 4%
paraformaldehyde (Beyotime Institute of Biotechnology) at 4°C for
24 h, embedded in paraffin and cut into 4 µm-thick sections. For
haematoxylin and eosin (H&E) staining, sections were stained
with haematoxylin for 3–5 min and 1% eosin for 5 min at room
temperature. For Masson and Sirius red staining, sections were
stained using the Masson's Trichrome Stain kit and the Sirius Red
Stain kit (both Beijing Solarbio Science & Technology Co.,
Ltd.) according to the manufacturer's protocol, respectively.
Stained sections were viewed and imaged at ×200 magnification using
an inverted optical light microscope (Olympus Corporation).
Triphenyl-tetrazolium-chloride (TTC)
staining
TTC staining was performed to measure the myocardial
infarct size as previously described (19). Briefly, hearts were harvested and
perfused with ice-cold PBS. Subsequently, hearts were cut into
2-mm-thick slices and incubated with 1% TTC (Sigma-Aldrich; Merck
KGaA) diluted in 0.9% sodium chloride for 30 min at 37°C, followed
by fixation with 4% paraformaldehyde at room temperature for 30
min. Then, stained slices were placed under natural light and
imaged using a Canon EOS 2000D digital camera. The infarct area was
quantified using ImageJ v1.8.0 software (National Institutes of
Health).
Echocardiographic measurement
Rats were anaesthetized by the intraperitoneal
injection of pentobarbital sodium (40 mg/kg) and fixed in the
supine position. Subsequently, two cardiac function parameters,
left ventricular fractional shortening (LVFS) and left ventricular
ejection fraction (LVEF), were assessed using a transthoracic
echocardiography system (VisualSonics, Inc.).
Immunofluorescence staining
CD31, a marker of endothelial cells,
immunofluorescence staining was performed to detect microvessels.
Rat cardiac tissues were rapidly frozen at −20°C and cut into 6
µm-thick sections using a freezing microtome (Leica Microsystems
GmbH). Sections were fixed in 4% paraformaldehyde at room
temperature for 30 min and washed with PBS three times. Following
blocking in Immunol Staining Blocking Buffer (Beyotime Institute of
Biotechnology) at room temperature for 15 min, sections were
incubated with an anti-CD31 primary antibody (cat. no. ab222783;
1:100; Abcam) overnight at 37°C. Then, sections were incubated with
a FITC-labelled goat anti-rabbit IgG secondary antibody (cat. no.
A0562; 1:200; Beyotime Institute of Biotechnology) at room
temperature for 1 h. Stained sections were observed and imaged at
×200 magnification using a confocal laser-scanning microscope
(Leica Microsystems GmbH). The fluorescent density of microvessels
was analysed using ImageJ v1.8.0 software.
Immunohistochemistry
Rat cardiac tissues were fixed in 4%
paraformaldehyde (Beyotime Institute of Biotechnology) at 4°C for
24 h, embedded in paraffin and cut into 4 µm-thick sections.
Following blocking in Immunol Staining Blocking Buffer at room
temperature for 15 min, sections were incubated with an anti-von
Willebrand Factor (vWF) primary antibody (cat. no. ab6994; 1:200;
Abcam) overnight at 4°C. Subsequently, sections were incubated with
a biotinylated mouse anti-rabbit IgG secondary antibody (cat. no.
bs-0295M; 1:100; BIOSS) at room temperature for 30 min. Sections
were stained with dyaminobenzidine (Beyotime Institute of
Biotechnology) for 10 min and haematoxylin for 3–5 min at room
temperature. Stained sections were visualized and imaged at ×200
magnification using an inverted optical light microscope (Olympus
Corporation).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from rat cardiac tissues
using TRIzol® reagent (Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. Total RNA was reverse
transcribed into cDNA using the ReverTra Ace qPCR RT kit (Toyobo
Life Science), according to the manufacturer's protocol.
Subsequently, qPCR was performed using a SYBR Green PCR master mix
(Takara Biotechnology Co., Ltd.) on a LightCycler 480 instrument
(Roche Diagnostics). The following thermocycling conditions were
used for qPCR: Initial denaturation at 95°C for 30 sec; and 40
cycles of amplification at 95°C for 5 sec, 60°C for 20 sec and 72°C
for 10 sec. The following primers were used for qPCR: MMP2 forward,
5′-GGGAATGAGTACTGGGTCTATT-3′ and reverse,
5′-CCAGTTAAAGGCAGCGTCTA-3′; MMP9 forward,
5′-GATCAGCCGGGAACGTATCT-3′ and reverse, 5′-AACTACAACGCCAGAAGTAT-3′;
GAPDH forward, 5′-AACTTGGCATCGTGGAAGG-3′ and reverse,
5′-GTGGATGCAGGGATGATGTTC-3′. mRNA expression levels were quantified
using the 2−∆∆Cq method (20) and normalized to the internal
reference gene GAPDH.
Western blotting
Total protein was isolated from rat cardiac tissues
and CMVECs using RIPA buffer (Beyotime Institute of Biotechnology)
containing 1% protease inhibitor. Protein concentrations were
determined using the BCA kit (Beyotime Institute of Biotechnology).
Proteins (40 µg) were separated via 8 or 15% SDS-PAGE and
subsequently transferred to PVDF membranes (MilliporeSigma).
Following blocking in 5% skimmed milk at room temperature for 2 h,
the membranes were incubated overnight at 4°C with the following
primary antibodies (all purchased from Abcam): Anti-TK1 (cat. no.
ab28289; 1:1,000), anti-TIMP1 (cat. no. ab109125; 1:1,000),
anti-MMP2 (cat. no. ab92536; 1:1,000), anti-MMP9 (cat. no. ab76003;
1:1,000), anti-GAPDH (cat. no. ab181602; 1:10,000), anti-γ-H2A.X
(cat. no. ab81299; 1:5,000), anti-H2A.X (cat. no. ab11175; 1:1,000)
and anti-TATA binding protein (TBP; cat. no. ab63766; 1:1,000).
Subsequently, the membranes were incubated with a HRP-conjugated
goat anti-rabbit IgG secondary antibody (cat. no. BA1054; 1:5,000;
Wuhan Boster Biological Technology, Ltd.) at room temperature for 2
h. Protein bands were visualized using an enhanced
chemiluminescence kit (Thermo Fisher Scientific, Inc.) and a
chemiluminescence imaging system (Bio-Rad Laboratories, Inc.).
Protein expression levels were semi-quantified using ImageJ v1.8.0
software with GAPDH and TBP as the loading controls.
Assessment of oxidative stress
Following reperfusion for 24 h, blood samples were
collected from the carotid artery of anesthetized rats. To obtain
serum samples, blood samples were maintained at room temperature
for 2 h prior to centrifugation at 1,000 × g for 20 min at 4°C.
Serum levels of superoxide dismutase (SOD), malondialdehyde (MDA),
glutathione (GSH), catalase (CAT) and glutathione-S-transferase
(GST) were measured using corresponding kits (SOD, cat. no. BC0175;
MDA, cat. no. BC0025; GSH, cat. no. BC1175; CAT, cat. no. BC0205;
GST, cat. no. BC0350; all purchased from Beijing Solarbio Science
& Technology Co., Ltd.) according to the manufacturer's
protocols.
Following H/R treatment, intracellular ROS
production in CMVECs was detected using CellROX Green reagent
(Thermo Fisher Scientific, Inc.). Cells were incubated with 5 µM
CellROX Green reagent at 37°C for 30 min and washed three times
with PBS. Subsequently, cells were incubated with DAPI (Beyotime
Institute of Biotechnology) at room temperature for 10 min. Stained
cells were observed and imaged at ×200 magnification using a
confocal laser-scanning microscope (Leica Microsystems GmbH). The
ratio of green fluorescence/nuclei staining in each image was
calculated.
Cell viability assay
CMVECs were seeded (1×103 cells/well)
into 96-well plates and treated with H/R. Cells were washed with
PBS and incubated with 0.5 mg/ml MTT solution at 37°C for 4 h. The
incubation solution was discarded and the formazan crystals were
dissolved in 150 µl DMSO. The absorbance was measured at a
wavelength of 570 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
Wound healing assay
CMVECs were seeded (5×105 cells/well)
into 6-well plates and cultured in FBS-free medium until cells
reached 90% confluence. The cell monolayer was scraped using a
sterile 200 µl pipette tip and washed twice with sterile PBS. The
remaining adherent cells were treated with H/R and then cultured
for 48 h in FBS-free medium. Images of the wound at ×100
magnification were captured at 0 and 48 h using an inverted optical
light microscope (Olympus Corporation).
Transwell assay
CMVECs (5×104) were seeded into the upper
chambers of the Transwell inserts (pore size, 8 µm; EMD Millipore)
with FBS-free medium. DMEM supplemented with 20% FBS was plated
into the lower chambers. Following treatment with H/R and cell
culture for 48 h, migratory cells were fixed in 4% paraformaldehyde
at room temperature for 30 min and stained with 0.1% crystal violet
(Beyotime Institute of Biotechnology) at room temperature for 15
min. Following washing twice with PBS, migratory cells were
observed and imaged at ×200 magnification using an inverted optical
light microscope (Olympus Corporation) and the number of cells was
quantified for analysis.
Tube formation assay
For the tube formation assay, 96-well plates were
coated with Matrigel (BD Biosciences) and incubated at 37°C for 30
min. CMVECs were seeded (1×104 cells/well) into the
96-well plates and treated with H/R. After culturing at 37°C for 24
h, capillary-like tubes were observed and imaged at ×200
magnification using an inverted optical light microscope (Olympus
Corporation). Tube length was quantified using ImageJ v1.8.0
software.
Statistical analysis
Data are presented as the mean ± SD. All experiments
were conducted with at least three independent repeats. Comparisons
among multiple groups were analysed using one-way ANOVA followed by
Bonferroni's post hoc test. Statistical analyses were performed
using SPSS software (version 22.0; IBM Corp.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Ad-hTK1/hTIMP1 significantly
ameliorates myocardial injury and improves cardiac function in
myocardial I/R model rats
Compared with the control vector, Ad-hTK1/hTIMP1
injection notably increased the protein expression levels of hTK1
and hTIMP1 in rat hearts (Fig.
S1A), which demonstrated that hTK1 and hTIMP1 were expressed in
rat hearts after gene delivery. Moreover, the H&E staining
results demonstrated that myocardial cells in sham rats were
well-arranged and displayed intact muscle fibres, whereas
myocardial cells were irregularly arranged and obvious necrosis was
observed in the myocardium following myocardial I/R injury
(Fig. 1A). Injection with
Ad-hTK1/hTIMP1 partly ameliorated histopathological alterations in
myocardial I/R model rats. Furthermore, the Masson and Sirius red
staining results displayed obvious collagen deposition in the
myocardium following myocardial I/R, which was notably reduced by
Ad-hTK1/hTIMP1 (Fig. 1A).
Furthermore, the results demonstrated that I/R treatment
significantly increased myocardial infarct size compared with sham
rats (Fig. 1B), whereas
Ad-hTK1/hTIMP1 significantly decreased myocardial infarct size in
myocardial I/R model rats. The results also demonstrated that LVFS
and LVEF in myocardial I/R model rats were significantly reduced
compared with sham rats, but these alterations were significantly
reversed by Ad-hTK1/hTIMP1 (Fig. 1C and
D). The aforementioned results indicated that Ad-hTK1/hTIMP1
significantly alleviated myocardial injury and improved cardiac
function in myocardial I/R model rats.
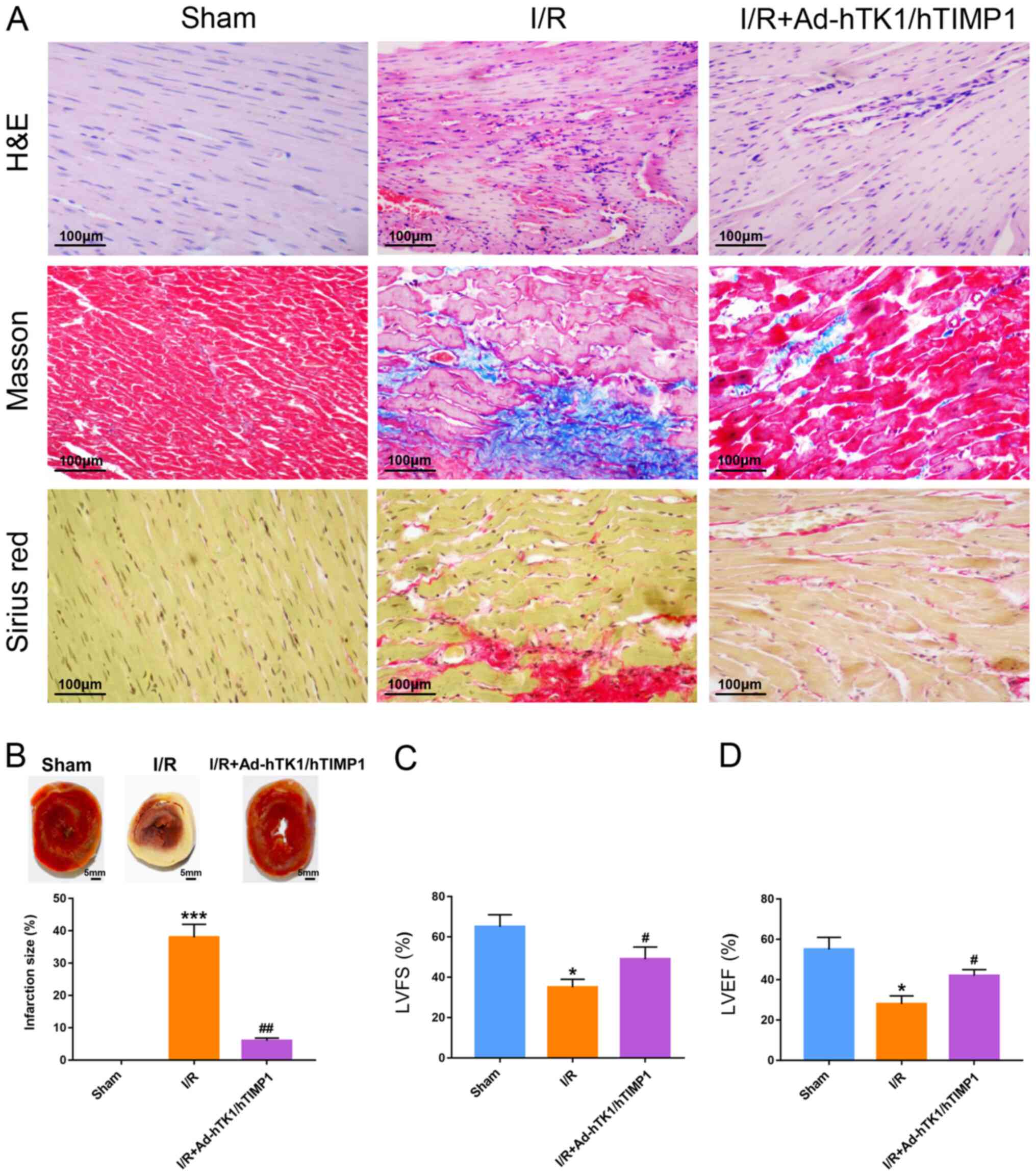 | Figure 1.Ad-hTK1/hTIMP1 significantly
ameliorates myocardial injury and improves cardiac function in
myocardial I/R model rats. (A) H&E, Masson and Sirius red
staining were performed on cardiac tissue sections of rats in the
sham, I/R and I/R + Ad-hTK1/hTIMP1 groups. Blue in Masson-stained
sections and red in Sirius red-stained sections represent collagen
deposition. Scale bar, 100 µm. (B) Myocardial infarction size was
measured via triphenyl-tetrazolium-chloride staining. Scale bar, 5
mm. Echocardiographic measurement of the two cardiac function
parameters (C) LVFS and (D) LVEF. *P<0.05 and ***P<0.001 vs.
Sham; #P<0.05 and ##P<0.01 vs. I/R. Ad,
adenovirus; hTK1, human tissue kallikrein 1; hTIMP1, human tissue
inhibitors of matrix metalloproteinase 1; I/R,
ischemia/reperfusion; LVFS, left ventricular fractional shortening;
LVEF, left ventricular ejection fraction. |
Ad-hTK1/hTIMP1 promotes microvessel
formation, and reduces MMP2 and MMP9 expression levels in
myocardial I/R model rats
The microvessels in rat hearts were detected via
CD31 immunofluorescence staining (Fig.
2A). The fluorescence density of microvessels in the myocardium
of myocardial I/R model rats was significantly lower compared with
sham rats, and was significantly enhanced following Ad-hTK1/hTIMP1
treatment (Fig. 2B). Furthermore,
Ad-hTK1/hTIMP1 markedly increased the number of vWF-positive cells
in the myocardium of myocardial I/R model rats (Fig. 2C). The results also indicated that
compared with the sham group, I/R treatment significantly increased
MMP2 and MMP9 mRNA and protein expression levels, and
Ad-hTK1/hTIMP1 significantly downregulated MMP2 and MMP9 expression
levels in myocardial I/R model rats (Fig. 2D-F).
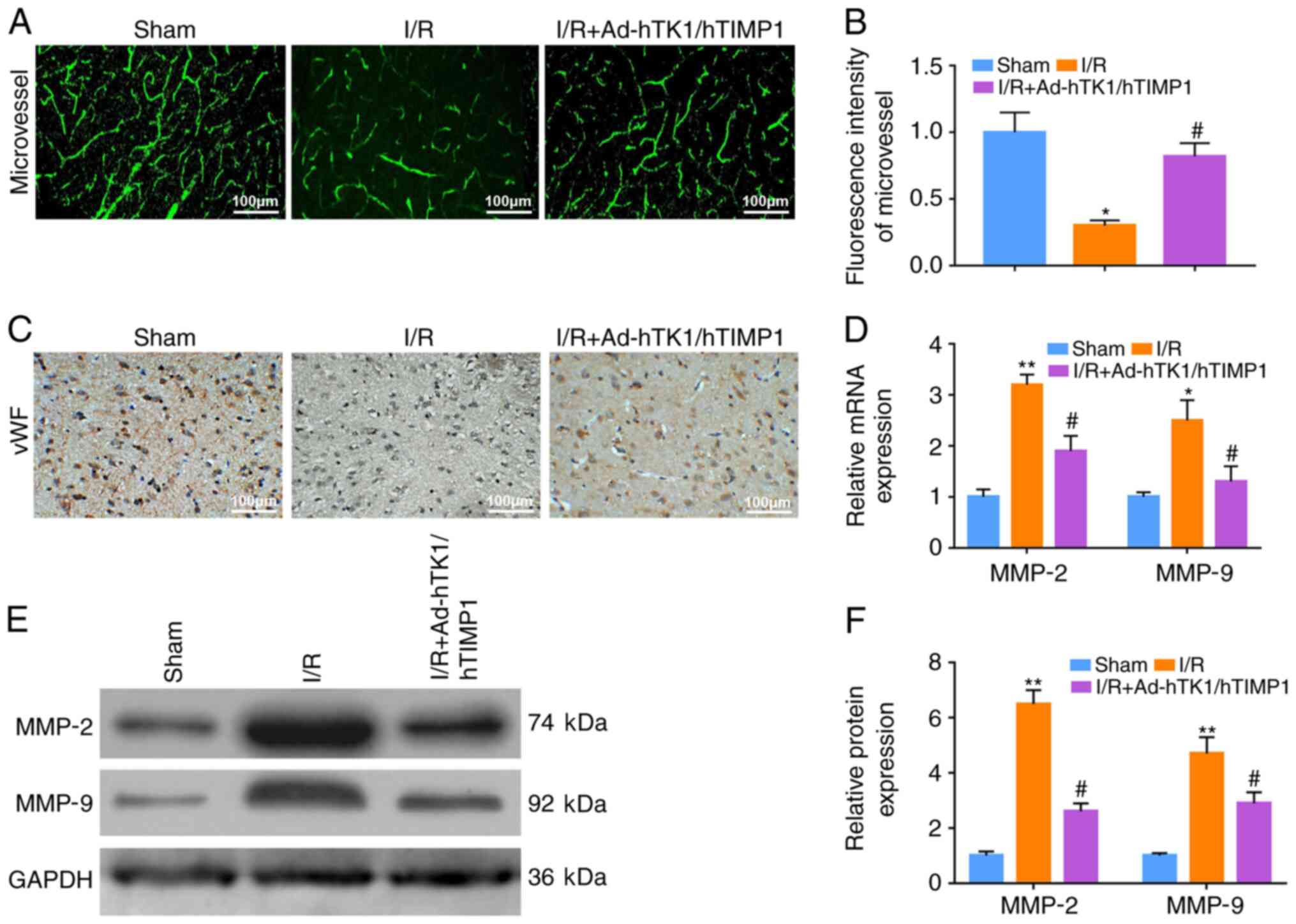 | Figure 2.Ad-hTK1/hTIMP1 promotes microvessel
formation, and reduces MMP2 and MMP9 expression levels in
myocardial I/R model rats. Microvessels in the myocardium of rats
in the sham, I/R and I/R + Ad-hTK1/hTIMP1 groups were (A) detected
via immunofluorescence staining using an anti-CD31 primary antibody
and (B) quantified. Scale bar, 100 µm. (C) Immunohistochemical
staining of vWF-positive cells in the myocardium of rats in the
sham, I/R and I/R + Ad-hTK1/hTIMP1 groups. Scale bar, 100 µm. (D)
MMP2 and MMP9 mRNA expression levels in rat hearts. MMP2 and MMP9
protein expression levels in rat hearts were (E) determined via
western blotting and (F) semi-quantified. *P<0.05 and
**P<0.01 vs. Sham; #P<0.05 vs. I/R. Ad,
adenovirus; hTK1, human tissue kallikrein 1; hTIMP1, human tissue
inhibitors of matrix metalloproteinase 1; MMP, matrix
metallopeptidase; I/R, ischemia/reperfusion; vWF, von Willebrand
Factor. |
Ad-hTK1/hTIMP1 significantly reduces
oxidative stress in myocardial I/R model rats
Subsequently, serum levels of oxidative stress
biomarkers were measured. The results indicated that GSH serum
levels, and SOD, CAT and GST activities in myocardial I/R model
rats were significantly decreased compared with sham rats. However,
Ad-hTK1/hTIMP1 significantly reversed I/R-induced effects on
oxidative stress in myocardial I/R model rats (Fig. 3A-D). Compared with sham rats, the
serum level of MDA in myocardial I/R model rats was significantly
increased, which was significantly reduced by Ad-hTK1/hTIMP1
(Fig. 3E). The results indicated
that Ad-hTK1/hTIMP1 significantly reduced oxidative stress in
myocardial I/R model rats.
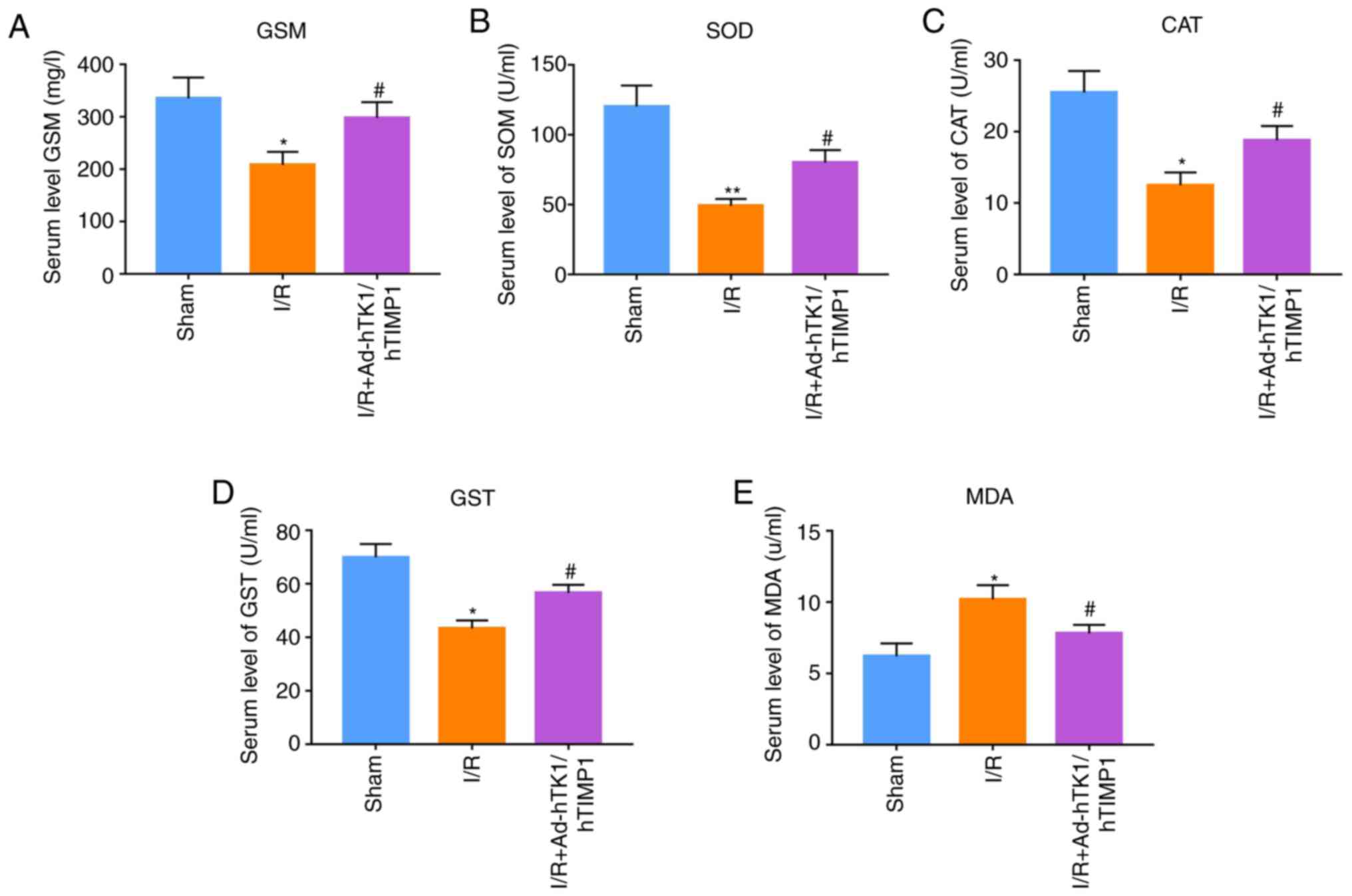 | Figure 3.Ad-hTK1/hTIMP1 significantly reduces
oxidative stress in myocardial I/R model rats. Measurement of serum
levels of oxidative stress biomarkers, including (A) GSH, (B) SOD,
(C) CAT, (D) GST and (E) MDA, in rats in the sham, I/R and I/R +
Ad-hTK1/hTIMP1 groups. *P<0.05 and **P<0.01 vs. Sham;
#P<0.05 vs. I/R. Ad, adenovirus; hTK1, human tissue
kallikrein 1; hTIMP1, human tissue inhibitors of matrix
metalloproteinase 1; I/R, ischemia/reperfusion; GSH, glutathione;
SOD, superoxide dismutase; CAT, catalase; GST,
glutathione-S-transferase; MDA, malondialdehyde. |
Ad-hTK1/hTIMP1 significantly enhances
H/R-treated CMVEC proliferation, migration and tube formation
Following transfection with Ad-hTK1/hTIMP1, the
protein expression levels of hTK1 and hTIMP1 in CMVECs were notably
increased compared with the control vector (Fig. S1B), demonstrating the successful
transfection of Ad-hTK1/hTIMP1 in CMVECs. Subsequently, the cell
model with H/R treatment was established in CMVECs. Compared with
the control group, CMVEC proliferation in the H/R group was
significantly suppressed, whereas Ad-hTK1/hTIMP1 significantly
enhanced proliferation in H/R-treated CMVECs (Fig. 4A). Furthermore, H/R treatment
notably inhibited CMVEC migration at 48 h compared with the control
group, and Ad-hTK1/hTIMP1 considerably attenuated the inhibitory
effect of H/R treatment (Fig. 4B).
Furthermore, the Transwell assay results demonstrated that compared
with the control group, the number of migratory CMVECs was
significantly decreased following treatment with H/R, which was
significantly increased by Ad-hTK1/hTIMP1 (Fig. 4C and D). Additionally, compared with
the control group, tube lengths in the H/R group were significantly
reduced, which was significantly ameliorated by Ad-hTK1/hTIMP1
(Fig. 4E and F).
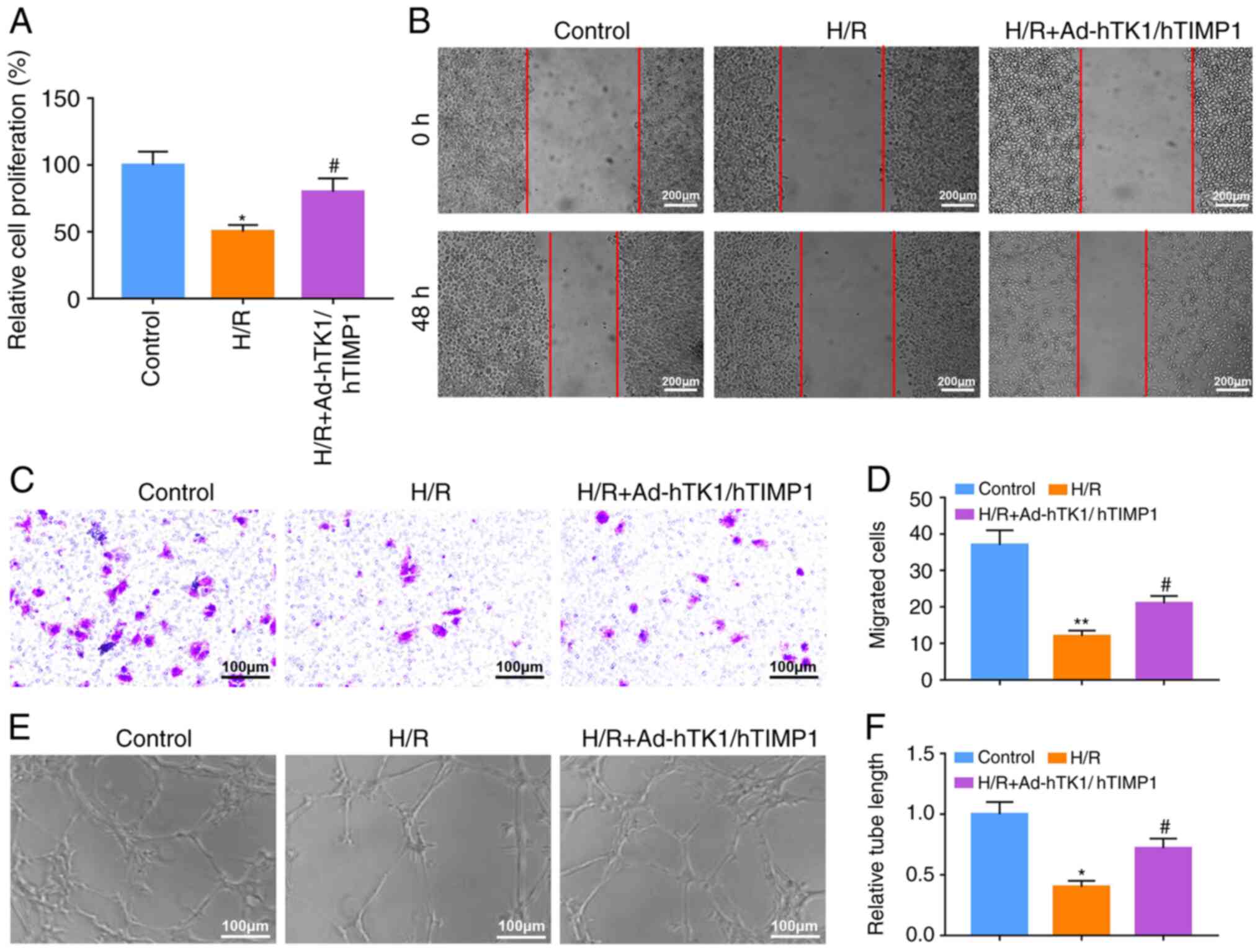 | Figure 4.Ad-hTK1/hTIMP1 significantly enhances
H/R-treated CMVEC proliferation, migration and tube formation. The
cell model with H/R treatment was established in CMVECs. (A) CMVEC
proliferation was detected by performing the MTT assay. (B) CMVEC
migration was assessed by conducting wound healing assays. The area
between the two red lines indicates the wound area. Scale bar, 200
µm. CMVEC migration was also (C) measured by performing a Transwell
assay and (D) quantified. Scale bar, 100 µm. CMVEC tube formation
was (E) assessed by conducting a tube formation assay and (F)
quantified. Scale bar, 100 µm. *P<0.05 and **P<0.01 vs.
Control; #P<0.05 vs. H/R. Ad, adenovirus; hTK1, human
tissue kallikrein 1; hTIMP1, human tissue inhibitors of matrix
metalloproteinase 1; H/R, hypoxia/reoxygenation; CMVEC, cardiac
microvascular endothelial cells; I/R, ischemia/reperfusion. |
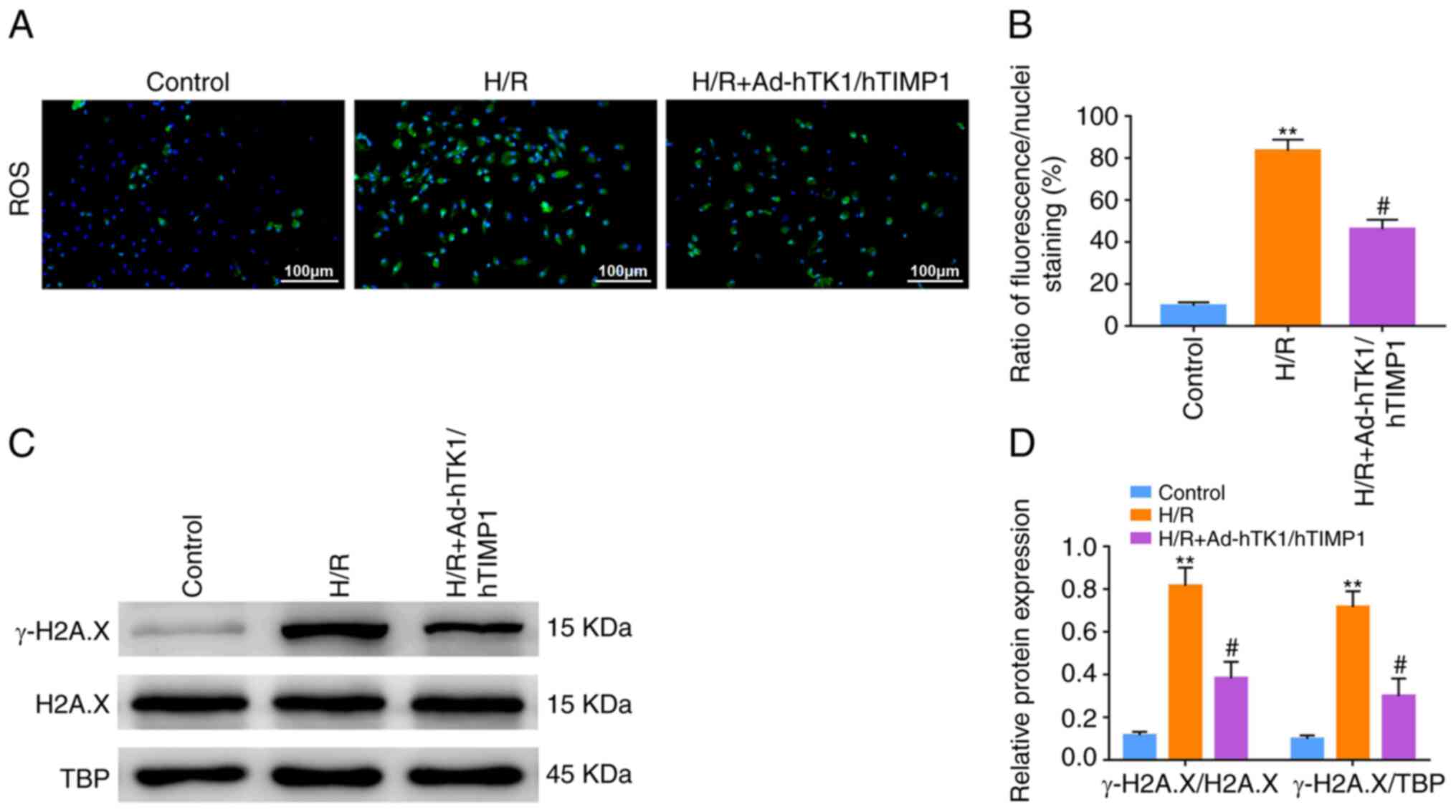
Ad-hTK1/hTIMP1 significantly decreases
intracellular ROS production and γ-H2A.X expression in H/R-treated
CMVECs
The ratio of green fluorescence/nuclei staining of
the H/R group was significantly higher compared with the control
group, indicating that intracellular ROS production in CMVECs was
significantly enhanced following H/R treatment (Fig. 5A). However, Ad-hTK1/hTIMP1
significantly reduced intracellular ROS production in H/R-treated
CMVECs (Fig. 5B). Furthermore, the
relative protein expression levels of γ-H2A.X in H/R-treated CMVECs
were significantly increased compared with the control group. By
contrast, γ-H2A.X expression levels in H/R-treated CMVECs were
significantly decreased by Ad-hTK1/hTIMP1 (Fig. 5C and D).
Discussion
Collagen deposition due to an imbalance of
myocardial ECM deposition and degradation following myocardial I/R
injury may lead to myocardial fibrosis, which may induce myocardial
pathological remodeling and decrease cardiac function (21). MMPs and their inhibitors, TIMPs,
have been reported to be vital regulators in ECM remodelling
(22). A previous study reported
that a substantial increase in MMP9 and MMP2 expression levels was
positively correlated with the degree of left ventricular fibrosis
in a mouse model of pressure overload (23). Additionally, a previous study
demonstrated that MMP9 and MMP2 expression levels were
significantly increased in myocardial I/R model mice compared with
sham mice (24). In the present
study, compared with sham rats, MMP9 and MMP2 mRNA and protein
expression levels were significantly increased following myocardial
I/R injury, but significantly reduced following Ad-hTK1/hTIMP1
injection. Meanwhile, a notable decrease in collagen deposition in
the myocardium of myocardial I/R model rats following
Ad-hTK1/hTIMP1 treatment was also observed in the present study,
which may be attributed to exogenous TIMP1. In addition, a previous
study reported that exogenous hTK1 gene delivery decreases infarct
size and improves cardiac function following myocardial I/R injury
by limiting post-infarct ventricular remodeling via inhibition of
NF-κB activation (25).
Furthermore, hTK1-modified mesenchymal stem cells have been
demonstrated to exhibit enhanced beneficial effects on cardiac
injury following MI, which are primarily manifested as promoting
cardiomyocyte survival, inhibiting ventricular remodeling and
improving cardiac function (9). The
aforementioned results indicated the protective role of hTK1 gene
against cardiac injury in ischemic heart diseases. Consistently,
the present study indicated that Ad-hTK1/hTIMP1 significantly
ameliorated myocardial injury, decreased infarct size and improved
cardiac function following myocardial I/R injury. Therefore, the
results suggested that Ad-hTK1/hTIMP1 displayed its protective
effects of alleviating cardiac injury and preventing cardiac
remodeling after myocardial I/R injury via the co-expression of
hTK1 and hTIMP1 genes.
I/R can cause vascular endothelial injury via a
number of different mechanisms, including pH change induced
cytotoxicity, oxidative stress and inhibition of endothelial nitric
oxide synthase (26). According to
the American College of Cardiology/American Heart Association
guidelines, clinical treatments that minimize microvascular damage
should be prioritized when attempting to protect the injured
myocardium (27). Therefore, any
treatment that can promote angiogenesis may be an effective
therapeutic strategy for myocardial I/R injury. It has been
previously reported that exogenous TK1 administration can
significantly enhance vascular density in the peri-infarction
region following myocardial infarction (28) or cerebral cortex infarction
(29). TIMP1 has also been reported
to facilitate blood vessel formation via inhibiting MMP9 in
vitro (30). Consistently, the
present study demonstrated that co-treatment of hTK1 and hTIMP1
genes significantly increased the microvessel density in the
myocardium of myocardial I/R model rats. It was also demonstrated
that the number of vWF [a biomarker of angiogenesis (31)]-positive cells was significantly
increased by Ad-hTK1/hTIMP1 in the myocardium of myocardial I/R
model rats. Furthermore, in vitro experiments demonstrated
that Ad-hTK1/hTIMP1 significantly ameliorated H/R-induced
suppression of CMVEC proliferation and migration. Ad-hTK1/hTIMP1
also significantly increased tube formation in H/R-treated CMVECs.
The results of the present study indicated the prominent effects of
co-expression of hTK1 and hTIMP1 genes on promoting angiogenesis
following I/R injury, which may serve a key role in improving
myocardial I/R injury.
Oxidative stress is an important pathological
process that contributes to myocardial I/R injury (32) and can damage all components of the
cell, including DNA, proteins and lipids (33). Previous research has reported that
any treatment that can reduce oxidative stress may attenuate
myocardial I/R injury (34–36). It has been previously reported that
hTK1 can improve erectile dysfunction of streptozotocin-induced
diabetic model rats and attenuate salt-induced renal fibrosis via
inhibition of oxidative stress (8,37).
Furthermore, a previous study demonstrated that free TIMP1 and
magnetic nanoparticle-bound TIMP1 significantly reduced ROS
production in HIV-infected neuroblastoma cells (38). In the present study, Ad-hTK1/hTIMP1
significantly reduced oxidative stress levels in myocardial I/R
model rats and H/R-treated CMVECs. γ-H2A.X is H2A.X (a specialized
histone H2A variant) phosphorylated at Ser 139 via ATM
serine/threonine kinase and ATR serine/threonine kinase (39). γ-H2A.X is widely considered as a
marker of DNA damage (40,41). The results of the present study
demonstrated that Ad-hTK1/hTIMP1 significantly decreased γ-H2A.X
expression in H/R-treated CMVECs, indicating reduced DNA damage in
H/R-treated CMVECs following transfection with Ad-hTK1/hTIMP1,
which may be attributed to decreased oxidative stress. The
aforementioned results demonstrated that co-expression of hTK1 and
hTIMP1 genes may ameliorate myocardial I/R injury via suppressing
oxidative stress.
The present study demonstrated the protective
effects of co-expression of hTK1 and hTIMP1 genes against
myocardial I/R injury. However, a limitation of the present study
was that only the preliminary protective effects of Ad-hTK1/hTIMP1,
including promoting angiogenesis and suppressing oxidative stress,
were investigated; therefore, the in-depth mechanisms underlying
hTK1 and hTIMP1 expression in myocardial I/R injury require further
investigation.
In conclusion, the present study demonstrated that
co-expression of hTK1 and hTIMP1 genes displayed significant
protective effects in myocardial I/R injury via promoting
angiogenesis and suppressing oxidative stress. Therefore,
co-expression of hTK1 and hTIMP1 may serve as a potential
therapeutic strategy for myocardial I/R injury.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Sailing Fund
of Fujian Medical University (grant no. 2018QH1136).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SH performed the in vivo experiments and
drafted the manuscript. MC performed the in vitro
experiments. HY and KL analysed the data. YG and PZ designed the
study and revised the manuscript. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
All animal experiments were conducted in accordance
with the National Institutes of Health Guide for the Care and Use
of Laboratory Animals (15), and
approved by the Institutional Ethics Committee for Laboratory
Animal Care of Fujian Provincial Hospital (approval no.
K2019-01-038).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AMI
|
acute myocardial infarction
|
|
CAT
|
catalase
|
|
CMVECs
|
cardiac microvascular endothelial
cells
|
|
ECM
|
extracellular matrix
|
|
GSH
|
glutathione
|
|
GST
|
glutathione-S-transferase
|
|
H&E
|
haematoxylin and eosin
|
|
H/R
|
hypoxia/reoxygenation
|
|
I/R
|
ischemia/reperfusion
|
|
LVEF
|
left ventricular ejection fraction
|
|
LVFS
|
left ventricular fractional
shortening
|
|
MDA
|
malondialdehyde
|
|
MMPs
|
matrix metallopeptidases
|
|
ROS
|
reactive oxygen species
|
|
SOD
|
superoxide dismutase
|
|
TIMPs
|
tissue inhibitors of matrix
metalloproteinases
|
|
TK1
|
tissue kallikrein 1
|
|
TTC
|
triphenyl-tetrazolium-chloride
|
|
vWF
|
von Willebrand Factor
|
References
|
1
|
Boateng S and Sanborn T: Acute myocardial
infarction. Dis Mon. 59:83–96. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Benjamin EJ, Muntner P, Alonso A,
Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR,
Cheng S, Das SR, et al: Heart disease and stroke statistics-2019
update: A report from the american heart association. Circulation.
139:e56–e528. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mehta LS, Beckie TM, DeVon HA, Grines CL,
Krumholz HM, Johnson MN, Lindley KJ, Vaccarino V, Wang TY, Watson
KE, et al: Acute myocardial infarction in women: A scientific
statement from the American heart association. Circulation.
133:916–947. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ni XQ and Hu ZY: Remifentanil improves
myocardial ischemia-reperfusion injury in rats through inhibiting
IL-18 signaling pathway. Eur Rev Med Pharmacol Sci. 24:3915–3922.
2020.PubMed/NCBI
|
|
5
|
Chen Q, Zhou Y, Richards AM and Wang P:
Up-regulation of miRNA-221 inhibits hypoxia/reoxygenation-induced
autophagy through the DDIT4/mTORC1 and Tp53inp1/p62 pathways.
Biochem Biophys Res Commun. 474:168–174. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hausenloy DJ and Yellon DM: Myocardial
ischemia-reperfusion injury: A neglected therapeutic target. J Clin
Invest. 123:92–100. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Luan Y, Ruan Y, Wang T, Zhuan L, Wen Z,
Chen R, Zhang Y, Cui K, Yang J, Wang S, et al: Preserved erectile
function in the aged transgenic rat harboring human tissue
kallikrein 1. J Sex Med. 13:1311–1322. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luan Y, Cui K, Tang Z, Ruan Y, Liu K, Wang
T, Chen Z, Wang S and Liu J: Human tissue kallikrein 1 improves
erectile dysfunction of streptozotocin-induced diabetic rats by
inhibition of excessive oxidative stress and activation of the
PI3K/AKT/eNOS pathway. Oxid Med Cell Longev. 2020:68342362020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao L, Bledsoe G, Yin H, Shen B, Chao L
and Chao J: Tissue kallikrein-modified mesenchymal stem cells
provide enhanced protection against ischemic cardiac injury after
myocardial infarction. Circ J. 77:2134–2144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lorente L, Martín MM, Ramos L, Argueso M,
Cáceres JJ, Solé-Violán J, Jiménez A, Borreguero-León JM,
González-Rivero AF, Orbe J, et al: Persistently high circulating
tissue inhibitor of matrix metalloproteinase-1 levels in
non-survivor brain trauma injury patients. J Crit Care. 51:117–121.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Takawale A, Fan D, Basu R, Shen M,
Parajuli N, Wang W, Wang X, Oudit GY and Kassiri Z: Myocardial
recovery from ischemia-reperfusion is compromised in the absence of
tissue inhibitor of metalloproteinase 4. Circ Heart Fail.
7:652–662. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Su YY, Li HM, Yan ZX, Li MC, Wei JP, Zheng
WX, Liu SQ, Deng YT, Xie HF and Li CG: Renin-angiotensin system
activation and imbalance of matrix metalloproteinase-9/tissue
inhibitor of matrix metalloproteinase-1 in cold-induced stroke.
Life Sci. 231:1165632019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kelly D, Squire IB, Khan SQ, Dhillon O,
Narayan H, Ng KH, Quinn P, Davies JE and Ng LL: Usefulness of
plasma tissue inhibitors of metalloproteinases as markers of
prognosis after acute myocardial infarction. Am J Cardiol.
106:477–482. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu P, Yu H, Huang S, Xiang H, Li F and
Zheng W: Synergistic effect of a tissue kallikrein 1 and tissue
inhibitor of matrix metalloproteinase 1 co-expression vector on the
proliferation of rat vascular smooth muscle cells. Mol Med Rep.
12:5671–5678. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington, DC: 2011
|
|
16
|
Yu HZ, Xie LD, Zhu PL, Xu CS and Wang HJ:
Human tissue kallikrein 1 gene delivery inhibits PDGF-BB-induced
vascular smooth muscle cells proliferation and upregulates the
expressions of p27Kip1 and p2lCip1. Mol Cell Biochem. 360:363–371.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhu L, Xu C, Huo X, Hao H, Wan Q, Chen H,
Zhang X, Breyer RM, Huang Y, Cao X, et al: The
cyclooxygenase-1/mPGES-1/endothelial prostaglandin EP4 receptor
pathway constrains myocardial ischemia-reperfusion injury. Nat
Commun. 10:18882019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu S, Cao S, Tong Z and Liu J: FGF21
protects myocardial ischemia-reperfusion injury through reduction
of miR-145-mediated autophagy. Am J Transl Res. 10:3677–3688.
2018.PubMed/NCBI
|
|
19
|
Li W, Feng G, Gauthier JM, Lokshina I,
Higashikubo R, Evans S, Liu X, Hassan A, Tanaka S, Cicka M, et al:
Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil
recruitment after heart transplantation. J Clin Invest.
129:2293–2304. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zheng QN, Wei XH, Pan CS, Li Q, Liu YY,
Fan JY and Han JY: QiShenYiQi Pills® ameliorates
ischemia/reperfusion-induced myocardial fibrosis involving RP
S19-mediated TGFβ1/Smads signaling pathway. Pharmacol Res.
146:1042722019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lan TH, Huang XQ and Tan HM: Vascular
fibrosis in atherosclerosis. Cardiovasc Pathol. 22:401–407. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Podesser BK, Kreibich M, Dzilic E, Santer
D, Förster L, Trojanek S, Abraham D, Krššák M, Klein KU, Tretter
EV, et al: Tenascin-C promotes chronic pressure overload-induced
cardiac dysfunction, hypertrophy and myocardial fibrosis. J
Hypertens. 36:847–856. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Scofield SLC, Dalal S, Lim KA, Thrasher
PR, Daniels CR, Peterson JM, Singh M and Singh K: Exogenous
ubiquitin reduces inflammatory response and preserves myocardial
function 3 days post-ischemia-reperfusion injury. Am J Physiol
Heart Circ Physiol. 316:H617–H628. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yin H, Chao L and Chao J: Nitric oxide
mediates cardiac protection of tissue kallikrein by reducing
inflammation and ventricular remodeling after myocardial
ischemia/reperfusion. Life Sci. 82:156–165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang Q, He GW, Underwood MJ and Yu CM:
Cellular and molecular mechanisms of endothelial
ischemia/reperfusion injury: Perspectives and implications for
postischemic myocardial protection. Am J Transl Res. 8:765–777.
2016.PubMed/NCBI
|
|
27
|
Galaup A, Gomez E, Souktani R, Durand M,
Cazes A, Monnot C, Teillon J, Le Jan S, Bouleti C, Briois G, et al:
Protection against myocardial infarction and no-reflow through
preservation of vascular integrity by angiopoietin-like 4.
Circulation. 125:140–149. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yao YY, Yin H, Shen B, Chao L and Chao J:
Tissue kallikrein and kinin infusion rescues failing myocardium
after myocardial infarction. J Card Fail. 13:588–596. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ling L, Hou Q, Xing S, Yu J, Pei Z and
Zeng J: Exogenous kallikrein enhances neurogenesis and angiogenesis
in the subventricular zone and the peri-infarction region and
improves neurological function after focal cortical infarction in
hypertensive rats. Brain Res. 1206:89–97. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu H, Chen B and Lilly B: Fibroblasts
potentiate blood vessel formation partially through secreted factor
TIMP-1. Angiogenesis. 11:223–234. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin KC, Yip HK, Shao PL, Wu SC, Chen KH,
Chen YT, Yang CC, Sun CK, Kao GS, Chen SY, et al: Combination of
adipose-derived mesenchymal stem cells (ADMSC) and ADMSC-derived
exosomes for protecting kidney from acute ischemia-reperfusion
injury. Int J Cardiol. 216:173–185. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mokhtari-Zaer A, Marefati N, Atkin SL,
Butler AE and Sahebkar A: The protective role of curcumin in
myocardial ischemia-reperfusion injury. J Cell Physiol.
234:214–222. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li P, Stetler RA, Leak RK, Shi Y, Li Y, Yu
W, Bennett MVL and Chen J: Oxidative stress and DNA damage after
cerebral ischemia: Potential therapeutic targets to repair the
genome and improve stroke recovery. Neuropharmacology. 134:208–217.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rocca C, Boukhzar L, Granieri MC, Alsharif
I, Mazza R, Lefranc B, Tota B, Leprince J, Cerra MC, Anouar Y and
Angelone T: A selenoprotein T-derived peptide protects the heart
against ischaemia/reperfusion injury through inhibition of
apoptosis and oxidative stress. Acta Physiol (Oxf). 223:e130672018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang M, Ni J, Cao Y, Xing X, Wu Q and Fan
G: Astragaloside IV attenuates myocardial ischemia-reperfusion
injury from oxidative stress by regulating succinate,
lysophospholipid metabolism, and ROS scavenging system. Oxid Med
Cell Longev. 2019:91376542019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yao BJ, He XQ, Lin YH and Dai WJ:
Cardioprotective effects of anisodamine against myocardial
ischemia/reperfusion injury through the inhibition of oxidative
stress, inflammation and apoptosis. Mol Med Rep. 17:1253–1260.
2018.PubMed/NCBI
|
|
37
|
Zhang JJ, Bledsoe G, Kato K, Chao L and
Chao J: Tissue kallikrein attenuates salt-induced renal fibrosis by
inhibition of oxidative stress. Kidney Int. 66:722–732. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Atluri VS, Jayant RD, Pilakka-Kanthikeel
S, Garcia G, Samikkannu T, Yndart A, Kaushik A and Nair M:
Development of TIMP1 magnetic nanoformulation for regulation of
synaptic plasticity in HIV-1 infection. Int J Nanomedicine.
11:4287–4298. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xiao A, Li H, Shechter D, Ahn SH, Fabrizio
LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P,
Hofmann K, et al: WSTF regulates the H2A.X DNA damage response via
a novel tyrosine kinase activity. Nature. 457:57–62. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang X, Mei Y, Wang T, Liu F, Jiang N,
Zhou W and Zhang Y: Early oxidative stress, DNA damage and
inflammation resulting from subcutaneous injection of sulfur
mustard into mice. Environ Toxicol Pharmacol. 55:68–73. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Liu Y, Long YH, Wang SQ, Li YF and Zhang
JH: Phosphorylation of H2A.XTyr39 positively regulates
DNA damage response and is linked to cancer progression. FEBS J.
283:4462–4473. 2016. View Article : Google Scholar : PubMed/NCBI
|