Introduction
Alzheimer's disease (AD) is the most common type of
dementia; the two disease-defining pathological features of AD are
senile plaques (SPs) predominantly consisting of β-amyloid protein
(Aβ), and neurofibrillary tangles (NFTs) comprising p-Tau (1,2). The
excessive production and accumulation of Aβ are essential steps in
the formation of SPs. The amyloid hypothesis, which states that the
accumulation of Aβ induces neuronal loss and cognitive impairment
(3), holds a dominant position in
AD pathogenesis. Aβs are a group of polypeptides containing 39–43
amino acids produced by the proteolysis of amyloid precursor
protein (APP) by β-secretase and γ-secretase (4). The tau theory is another hypothesis of
AD, which states that the hyperphosphorylation of tau leads to the
formation of NFTs and degeneration of neurons. The apolipoprotein
(APO)E4 variant is considered a genetic risk factor for sporadic
AD; in a previous study, converting APOE4 to APOE3 in brain cell
types, including neurons, astrocytes and microglia-like cells, was
sufficient to attenuate numerous AD-related pathologies (5). In addition, there are several possible
novel etiologies of AD, including defective mitophagy (6), lack of nicotinamide adenine
dinucleotide (7) and Aβ-induced
oligodendrocyte progenitor cell senescence (8).
Nogo-A is one of three subtypes of the Nogo protein,
which is a member of the reticulon (RTN) family; Nogo-A consists of
a long N-terminal segment and a C-terminal reticulon homology
domain (RHD) (9). There is a
special domain between the two hydrophobic segments of the RHD
domain of Nogo-A, known as Nogo-66, which is the major inhibitory
region of Nogo-A (9). Nogo-A has
been reported to be expressed by oligodendrocytes and is considered
a major component of myelin in the central nervous system (CNS)
(10). In addition, Nogo-A has been
identified as a key molecule that limits axon regeneration and is
considered to be a major obstacle to nerve regeneration following
injury in the adult mammalian CNS (11). Nogo-A interacts with the specific
receptor complex on neurons to prevent neurite outgrowth; this
receptor complex is composed of Nogo-66 receptor 1 (NgR1), and two
complementary co-receptors p75 and Ig-like domain-containing
NgR1-interacting protein 1 (LINGO-1) (12).
It has been suggested that Nogo-A may be involved in
the pathology of AD. Nogo-A is normally expressed in the
hippocampus of healthy elderly individuals, but it is overexpressed
in the brain tissues of patients with AD and is associated with Aβ
deposits in SPs (13). It has
previously been demonstrated that inhibition of the Nogo-A/NgR
pathway by Nogo-66 antagonist peptide (NEP1-40) attenuated
amyloidogenic processing of APP and reduced Aβ plaque deposition in
APP/PS1 mice (14). It has also
been hypothesized that Nogo-A may alter neuronal APP metabolism and
Aβ production. Our previous study reported that Nogo-P4, the 31–55
amino acid of the Nogo-66 peptide, promoted Aβ secretion in
cortical neurons (15). However,
whether the full-length Nogo-66 protein has the same effect, as
well as the underlying mechanisms of Nogo-66 in APP regulation, are
not clear. Therefore, the aim of the present study was to
investigate whether Nogo-66 promotes Aβ secretion in cortical
neurons, Neuro2a (N2a) cells and Sprague-Dawley rats. Furthermore,
the present study explored the possible underlying molecular
mechanism of Nogo-66 on Aβ secretion, which in turn may provide
insight into possible effects and mechanisms of the Nogo-A/NgR1
pathway in AD pathogenesis.
Materials and methods
Reagents
Soluble Nogo-66 protein was obtained via SUMO fusion
in Escherichia coli from our laboratory (16). Y-27632 and the NEP1-40 were obtained
from Merck KGaA and Tocris Bioscience, respectively. The
Cy3-conjugated anti-rabbit secondary antibody was obtained from
Abcam (cat. no. ab6939). Rabbit monoclonal IgG anti-APP was
purchased from LifeSpan BioSciences, Inc. In addition, the
following primary antibodies were obtained: Anti-β-secretase 1
(BACE1; cat. no. 5606; Cell Signaling Technology, Inc.), anti-ROCK2
(cat. no. ab125025; Abcam), anti-phosphorylated (p)-collapsin
response mediator protein-2 (CRMP2; Thr514; cat. no. 9397; Cell
Signaling Technology, Inc.), anti-CRMP2 (cat. no. 35672; Cell
Signaling Technology, Inc.), anti-GAPDH (cat. no. BS72410; Bioworld
Technology, Inc.) and anti-microtubule-associated protein 2 (MAP2;
cat. no. 05-346; Merck KGaA). HRP-conjugated goat anti-rabbit IgG
antibody (cat. no. E030120-01; Earthox Life Sciences) was used. All
reagents and drugs used were of analytical grade.
Animals
Sprague-Dawley rats (n=24; age, 3 months; weight,
330±20 g) were provided by the Experimental Animal Center of
Guangdong Province. Animals were randomly divided into the
following groups (n=6 per group): i) Control; ii) low dose; iii)
middle dose; and iv) high dose. Rats were housed under standard
temperature (24±1°C) with 60±10% humidity, diurnal conditions
(lights on, 08:00-20:00), and ad libitum access to food and
water. The present study was approved by the Animal Research
Committee of the School of Medicine, Jinan University (Guangzhou,
China; approval no. IACUC-20180120-05).
Cell culture
Cortical neurons were prepared from postnatal rats
(n=20; age, 1–3 days; weight, 6–8 g) (17), which were obtained from the
Guangdong Medical Laboratory Animal Center. Animals were housed at
a standard temperature (24±1°C) with 60±10% humidity, diurnal
conditions (lights on, 08:00-20:00), and ad libitum access
to food and water. Briefly, the pups were sacrificed by cervical
dislocation without anesthesia, in accordance with the procedures
approved by the local ethics review board, and the cerebral tissues
were dissociated by combined trituration and 0.25% trypsin (Thermo
Fisher Scientific, Inc.) at 37°C for 30 min. The debris was removed
and the supernatant was centrifuged at 1,000 × g for 5 min at 4°C
to collect the cell pellet. Cortical neurons were plated at a
density of 2.1×105/well into 24-well plates on
poly-L-lysine (PLL; Merck KGaA)-coated borosilicate glass
coverslips (16-mm diameter; VWR International, LLC) or at a density
of 1×106/well in 6-well plates coated with 100 µg/ml PLL
in neurobasal medium (Thermo Fisher Scientific, Inc.) containing 25
mM KCl, 2% B27 (Thermo Fisher Scientific, Inc.), 1.5 mM glutamine
(Thermo Fisher Scientific, Inc.) and 0.01% penicillin/streptomycin
(Merck KGaA).
N2a mouse neuroblastoma cells stably expressing the
human Swedish mutant APP695 (APP695 N2a cells) (18) were used in the present study, and
were kindly provided by Dr Cuizan Cai (Macau University of Science
and Technology, Macao, China). The cells were maintained in
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Shanghai Shuangwei
Biological Technology Co., Ltd.), 200 µg/ml G418 (Thermo Fisher
Scientific, Inc.) and 1% penicillin/streptomycin. Cells were
cultured at 37°C in a humidified atmosphere containing 5%
CO2 before the experiments.
Following treatment with different concentrations of
Nogo-66 (0.25, 0.5 and 1 µM Nogo-66) or a buffer control (PBS) for
48 h at 37°C, cortical neurons and N2a mouse neuroblastoma cells
were cultured for 48 h. Cells were prepared in RIPA lysis buffer
containing a cocktail of complete protease inhibitors (Merck KGaA).
The lysates were clarified by centrifugation at 12,000 × g for 5
min at 4°C to remove the insoluble components.
Following culture for 48 h, cortical neurons and N2a
mouse neuroblastoma cells were treated with Y-27632 (100 µM) and
NEP1-40 (2 µM) for 1 h of pre-protection at 37°C. Subsequently,
cells were treated with different concentrations of Nogo-66 (1 µM)
for 48 h at 37°C. Cells were then prepared in RIPA lysis buffer
containing a cocktail of complete protease inhibitors (Merck KGaA).
The lysates were clarified by centrifugation at 12,000 × g for 5
min at 4°C to remove the insoluble components.
Neurite outgrowth assay
Cortical neuron cells were plated
(2.1×105/well) onto 0.01% PLL-coated coverslips in
24-well culture dishes for 48 h following treatment with different
concentrations of Nogo-66 (0.25, 0.5 and 1 µM) and a buffer control
(PBS) for 48 h at 37°C. The cells were fixed for 20 min in 4%
paraformaldehyde at room temperature and washed with PBS. After
rinsing several times with PBS, the cells were blocked and
permeabilized for 30 min at room temperature with PBS containing 5%
goat serum (Gibco; Thermo Fisher Scientific, Inc.), 1% (w/v) BSA
(GenView Scientific, Inc.) and 0.05% Triton X-100. The cells were
then rinsed and incubated with anti-MAP2 antibody (1:500; Merck
KGaA) at 4°C overnight. The cells were then rinsed again and
incubated for 1 h with goat anti-rabbit Cy3 secondary antibody
(1:1,000; cat. no. ab6939; Abcam) at room temperature. All slides
were visualized under a laser scanning confocal microscope (LSM
700; Carl Zeiss AG). Each group was assessed in triplicate and the
experiments were repeated four times. The average length of each
neuron was measured with Image-Pro Plus software (version 6.0;
Media Cybernetics, Inc.). Neurites were measured only if they were
>20 µm in length. A total of 100 neurons were measured from each
group in each experiment. These counts were averaged and the SD
values were calculated using Graph Pad Prism v5.02 (GraphPad
Software, Inc.). Mean values were obtained by averaging values from
the measurement of ~100 neurons per well in three separate wells
per group.
Intra-hippocampal injection of
Nogo-66
Male Sprague-Dawley rats (age, 12 weeks; weight,
330±20 g; Guangdong Medical Laboratory Animal Center) were
anesthetized via an intravenous injection of 30 mg/kg pentobarbital
sodium (Merck KGaA) and were placed in a stereotaxic frame.
Unilateral injections of 2.5 µl physiological saline, or 0.5, 1 and
2 mg/kg Nogo-66, were administered at a rate of 0.5 µl/min into the
left and right hippocampus (3.0 mm posterior to the bregma, 1.8 mm
lateral to the midline and 2.6 mm below the dura mater). Following
the intra-hippocampal injection, the scalp was sutured and
sterilized. The rats were kept warm for 2 h before being returned
to their cages. After 4 days, the rats were euthanized by
CO2 asphyxiation; the filling rate of CO2 was
20% chamber volume per minute. Subsequently, hippocampal tissues
from the rats were rapidly harvested and stored at −80°C until
further processing for biochemical analysis.
After 4 days, hippocampal tissue lysates were
extracted for biochemical assays. Hippocampal tissues were
dissected and homogenized in RIPA buffer (Beyotime Institute of
Biotechnology) containing a cocktail of complete protease
inhibitors (Merck KGaA). The lysate was centrifuged at 12,000 × g
for 15 min at 4°C. Protein concentrations were determined using a
BCA assay kit (Thermo Fisher Scientific, Inc.).
Western blotting
The cortical neurons and APP-overexpression (induced
by the human Swedish mutant APP695) N2a cells were prepared in RIPA
lysis buffer containing a cocktail of complete protease inhibitors
(Merck KGaA). The lysates were clarified by centrifugation at
12,000 × g for 5 min at 4°C to remove the insoluble components. The
protein concentration in the supernatant was detected using a BCA
assay kit (Thermo Fisher Scientific, Inc.). Equal amounts of
proteins (0.125–2 mg/ml) were separated by SDS-PAGE on 10 or 12%
gels and were transferred to PVDF membranes. The membranes were
blocked with TBS-Tween-20 (0.1%) containing 5% non-fat dry milk at
room temperature for 1 h, and incubated overnight at 4°C with the
following primary antibodies: Anti-APP (1:1,000; Lifespan
Biosciences, Inc.), anti-APP-C99 (1:2,000; Merck KGaA),
anti-secreted APP-α (sAPPα; 1:1,000; BioLegend, Inc.), anti-BACE1
(1:1,000; Cell Signaling Technology, Inc.), anti-ROCK2 (1:1,000;
Abcam), anti-CRMP2 (1:1,000; Cell Signaling Technology, Inc.),
anti-p-CRMP2 Thr 509/514 (1:1,000; Cell Signaling Technology, Inc.)
and anti-GAPDH (1:10,000; Bioworld Technology, Inc.). The membranes
were then incubated with HRP-conjugated goat anti-rabbit IgG
antibody (1:2,000; cat. no. E030120-01; Earthox Life Sciences) for
1 h at room temperature. The blots were detected using an ECL
detection kit (Tanon Science and Technology Co., Ltd.), according
to the manufacturer's instructions. The results were analyzed using
Quantity-One software (version 4.6.6; Bio-Rad Laboratories, Inc.)
to determine the relative ratio. The hippocampal tissue was
prepared and analyzed in the same way.
ELISA for Aβ40 and
Aβ42
The total levels of Aβ40 and
Aβ42 in the culture medium of cultured neurons and the
hippocampal homogenate of rats were detected using ELISA kits (cat.
nos. 294-62501 and 292-64501; Wako Chemicals GmbH), according to
the manufacturer's instructions. For human or rat Aβ40
and Aβ42 ELISA, the monoclonal BAN50 antibody, which
specifically detected the N-terminal portion of human
Aβ1-16, was used as the capturing antibody, and the
monoclonal BA27 and BC05 antibodies, which detect the C-terminal
portion of Aβ1-40 and Aβ1-42, respectively,
were used as detector antibodies. The amount of Aβ was calculated
by comparing these absorbance values with the control
solutions.
Enzymatic activity assay
The β-Secretase Activity Assay Kit (cat. no. 565785;
Merck KGaA) was used to measure the BACE1 activity of cells and
hippocampal tissue according to manufacturer's protocol. Protein
lysates (20–30 µl; 5–7 mg/ml) were used in vitro assays. All
fluorescent values were detected on a plate reader with excitation
(Ex) at 320 nm and emission (Em) at 405 nm (BioTek China). The
ADAM10 Activity Detection kit (AS-72226; AnaSpec) was used to
measure ADAM10 activity in cells and hippocampal tissue according
to the manufacturer's protocol. Samples (50 µl) were used for in
vitro assays. Fluorescence intensity was measured at
Ex/Em=490/520 nm after incubating the reaction at 37°C for 30 min
in the dark.
Statistical analysis
All experiments were repeated 3–4 times. All values
are expressed as the mean ± SD. Analyses were performed using SPSS
16.0 statistical software (SPSS, Inc.). Comparison of parameters
among more than two groups was made by one-way ANOVA followed by
the post hoc Tukey test. P<0.05 was considered to indicate a
statistically significant difference.
Results
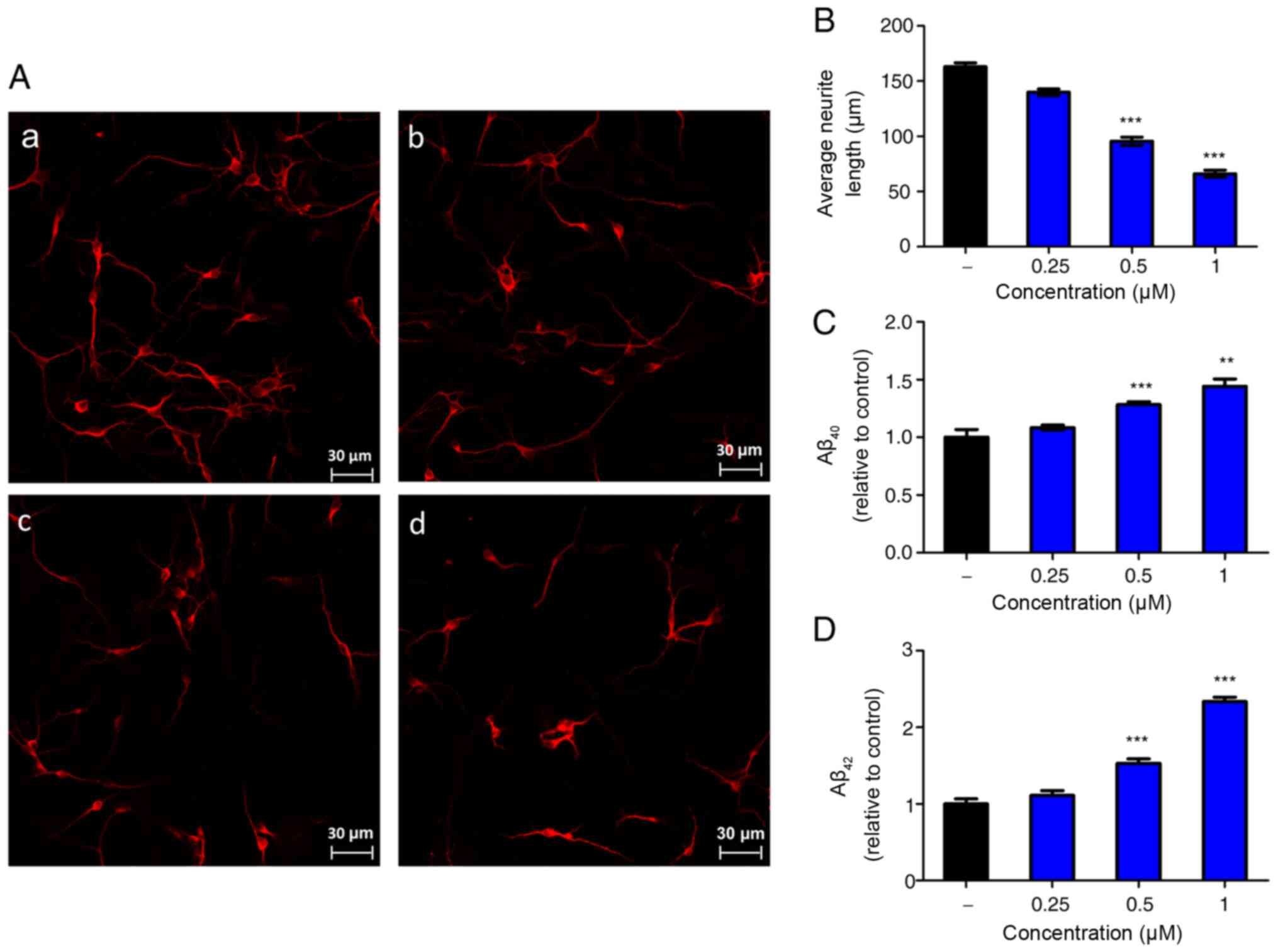
Nogo-66 inhibits neurite outgrowth and
promotes Aβ secretion in a dose-dependent manner
To observe the effect of Nogo-66 on neurite growth,
neurons from the cerebral cortex of newborn rats were analyzed
in vitro. A laser scanning confocal microscope was used to
analyze the average neurite length. Notably, Nogo-66 had a
dose-dependent inhibitory effect on neurite outgrowth (Fig. 1A and B). Quantification of the
neurite length demonstrated that 0.25, 0.5 and 1 µM Nogo-66
markedly decreased neurite outgrowth compared with that in the
control group; the neurite length was ~87, 62 and 37% of the
control values respectively (Fig.
1B).
To determine whether Nogo-66 had an effect on the
production of Aβ, the cortical neurons were exposed to various
concentrations of Nogo-66 (0.25, 0.5 and 1 µM) and a buffer control
(PBS), and secreted Aβ40 and Aβ42 levels were
measured by ELISA. Treatment with 0.5 or 1 µM Nogo-66 significantly
increased the levels of Aβ40 and Aβ42
compared with those in the control group (Fig. 1C and D). ELISA analysis indicated
that the levels of Aβ40 and Aβ42 in the
Nogo-66 group (0.5 and 1 µM) were higher than those detected in the
PBS groups.
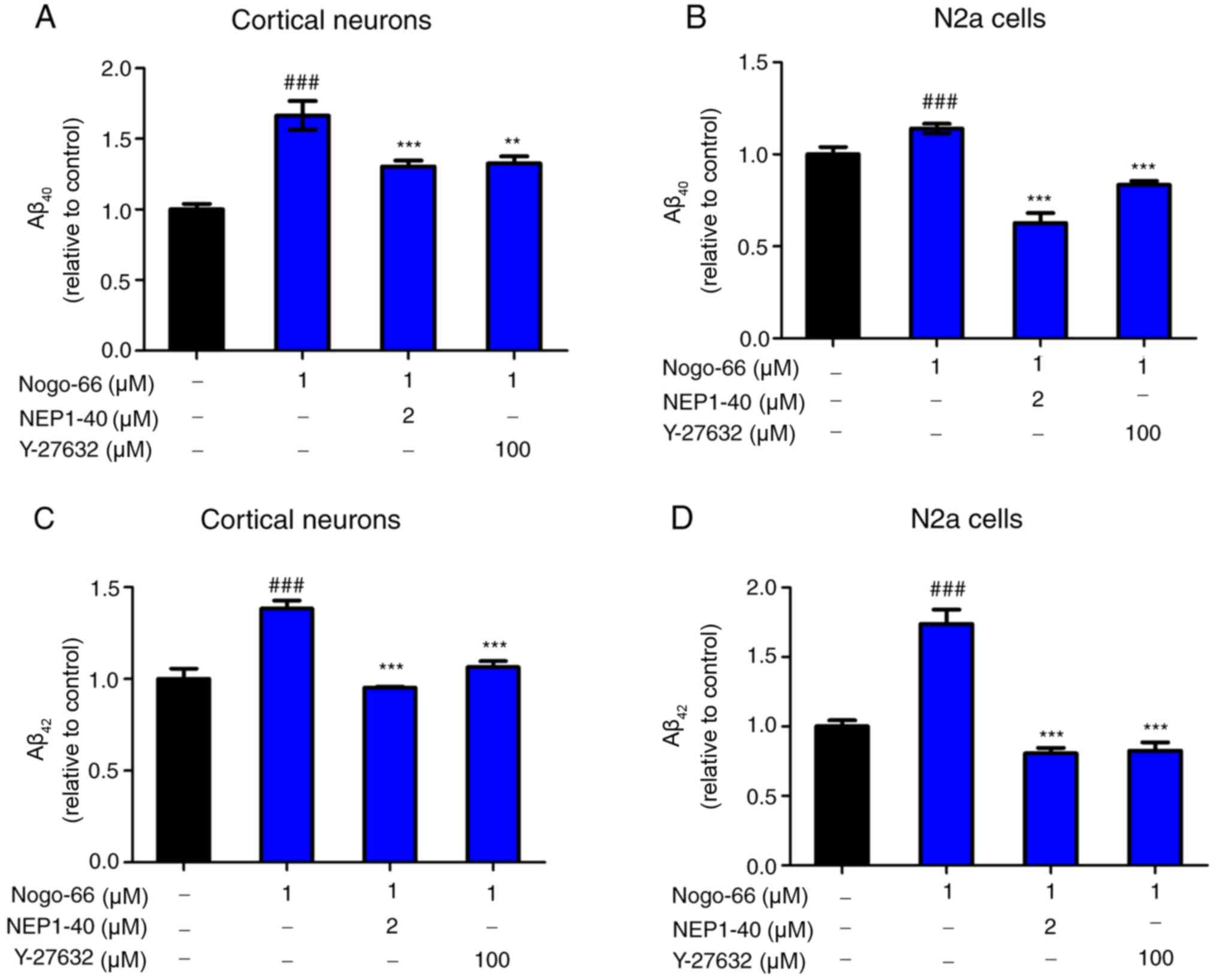
Inhibition of NgR1/ROCK pathway
reduces Aβ secretion
Primary cortical neurons and N2a cells were used,
which were transduced with lentivirus expressing human APP695, to
determine whether the pharmacological inhibition of ROCK and NgR1
reduced Aβ production. To determine how Nogo-66 influenced APP
processing to Aβ, secreted Aβ40 and Aβ42
levels were measured in primary cortical neurons and
APP-overexpressing N2a cells by ELISA, following treatment with
NEP1-40 or Y-27632 prior to treatment with 1 µM Nogo-66. The
optimum concentrations of NEP1-40 and Y-27632 had already been
explored by adding a series of concentrations of NEP1-40 (0, 0.5, 1
and 2 µM) or Y-27632 (0, 12.5, 25, 50 and 100 µM) with Nogo-66 to
the cortical neurons; the levels of Aβ40 and
Aβ42 were decreased in a dose-dependent manner (data not
shown). To explore whether NEP1-40 and Y-27632 could reduce Aβ
secretion, the cortical neurons were exposed to NEP1-40 and
Y-27632. Following treatment with 2 µM NEP1-40, secreted
Aβ40 and Aβ42 levels were lower compared with
those detected in the Nogo-66-only group (Fig. 2A and C). In addition, the effects of
treatment with 100 µM Y-27632 were similar to those detected
following treatment with NEP1-40 (Fig.
2A and C). In addition, compared with in the PBS group, Nogo-66
(1 µM) significantly increased the levels of secreted
Aβ40 and Aβ42. The results of
APP-overexpressing N2a cells were consistent with those for
cortical neurons (Fig. 2B and
D).
Effect of Nogo-66 on processing of
APP
To further characterize the effects of Nogo-66 on
APP processing to Aβ, the primary neurons were exposed to Nogo-66,
NEP1-40 and Y-27632. Subsequently, the protein expression levels of
cell-associated full-length APP, C99 and sAPPα were measured by
western blot analysis. Densitometric analysis indicated that
treatment with 1 µM Nogo-66 significantly increased the protein
expression levels of APP and C99 levels, whereas treatment with
NEP1-40 and Y-27632 decreased the expression levels of these
proteins (Fig. 3A, C and E).
Conversely, treatment with Nogo-66, NEP1-40 or Y-27632 had little
effect on the protein expression levels of sAPPα and BACE1
(Fig. 3A, G and I). To determine
whether Nogo-66 had a similar effect on APP-overexpressing N2a
cells, these cells were treated in the same manner. Consistent with
the aforementioned findings, treatment with 1 µM Nogo-66
significantly increased the protein expression levels of APP and
C99, whereas the effects of Nogo-66 were reversed following
treatment with NEP1-40 and Y-27632 (Fig. 3B, D and F). Furthermore, the protein
expression levels of sAPPα and BACE1 were not affected by the same
treatment (Fig. 3B, H and J). In
combination, these findings suggested that Nogo-66 may increase the
production of Aβ by augmenting the amyloidogenic processing of APP.
Notably, the opposing outcomes of NEP1-40 or Y-27632 on Aβ levels
suggested that inhibition of NgR1 and the receptor downstream
signaling molecule ROCK may reduce the amyloidogenic processing of
APP.
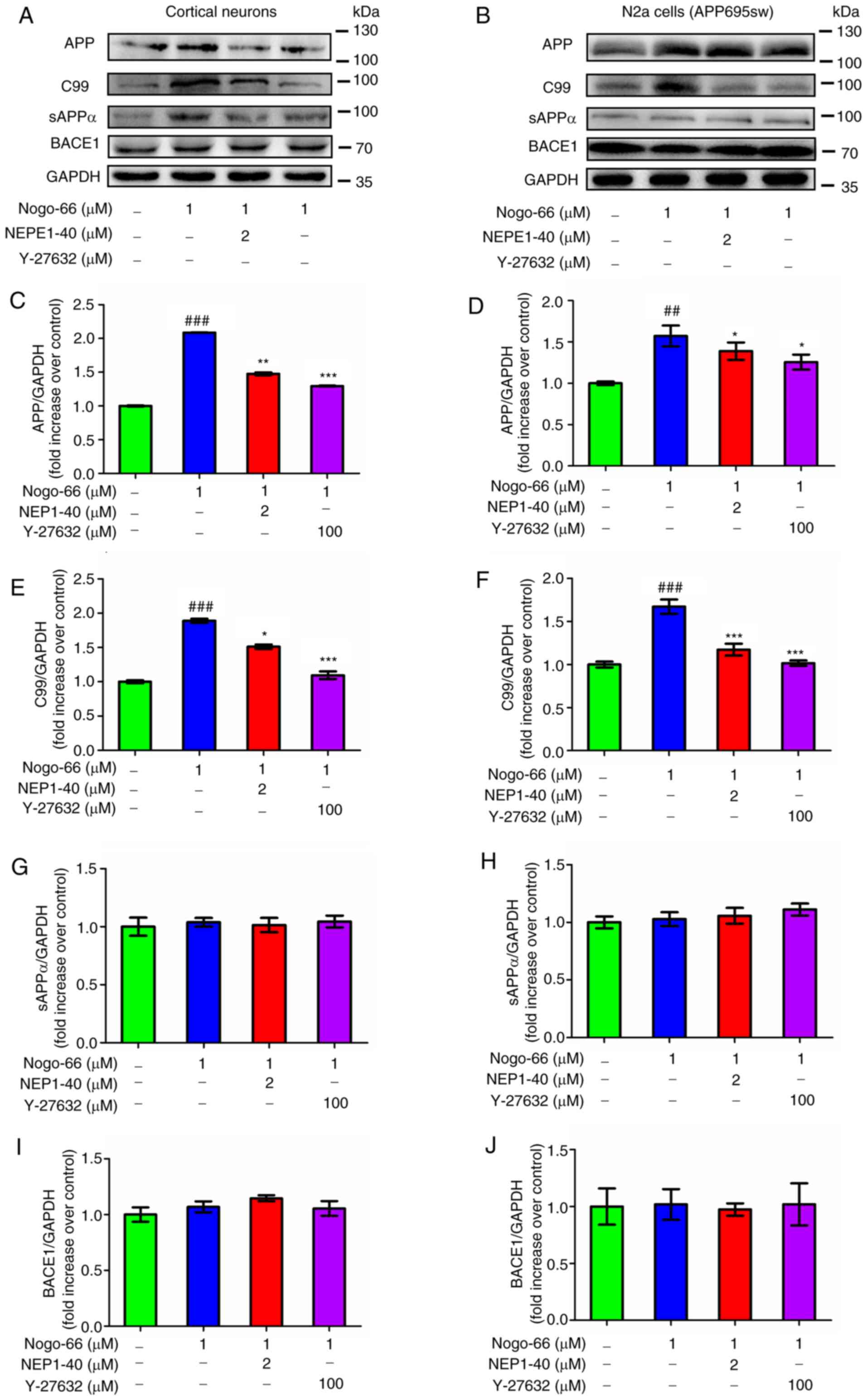 | Figure 3.Effects of Nogo-66, NEP1-40 and
Y-27632 on the protein expression levels of APP, C99, sAPPα and
BACE1 in (A, C, E, G and I) primary cortical neurons and (B, D, F,
H and J) N2a cells. Data are presented as the mean ± SD from three
biological repeats. ##P<0.01,
###P<0.001 vs. control; *P<0.05, **P<0.01,
***P<0.001 vs. Nogo-66 group. NEP1-40, Nogo-66 antagonist
peptide; APP, amyloid precursor protein; sAPPα, secreted APP-α;
BACE1, β-secretase 1; N2a, Neuro2a. |
Effect of Nogo-66 on the NgR/ROCK
pathway in cortical neurons and N2a cells
Western blot analysis was performed to explore
whether Nogo-66 increased the secretion of Aβ and inhibited neurite
regeneration by activating RhoA/ROCK2 signaling. As shown in
Fig. 4, the expression levels of
ROCK2 and p-CRMP2 were significantly enhanced by Nogo-66 in
cortical neurons (Fig. 4A, C and
E). Therefore, the results indicated that treatment with
Nogo-66 led to a significant dose-dependent increase in the
activation of the relevant signaling molecules, including ROCK2 and
p-CRMP2. The results in APP-overexpressing N2a cells were
consistent with the findings in cortical neurons (Fig. 4B, D and F). These results suggested
that the Rho/ROCK2 pathway may have an important role in the effect
of Nogo-66, including the increase in Aβ generation and the
inhibition of neurite outgrowth in cortical neurons. These findings
suggested that Nogo-66 regulated the expression of ROCK2 and
activated the phosphorylation of CRMP2, which was associated with
increasing APP levels.
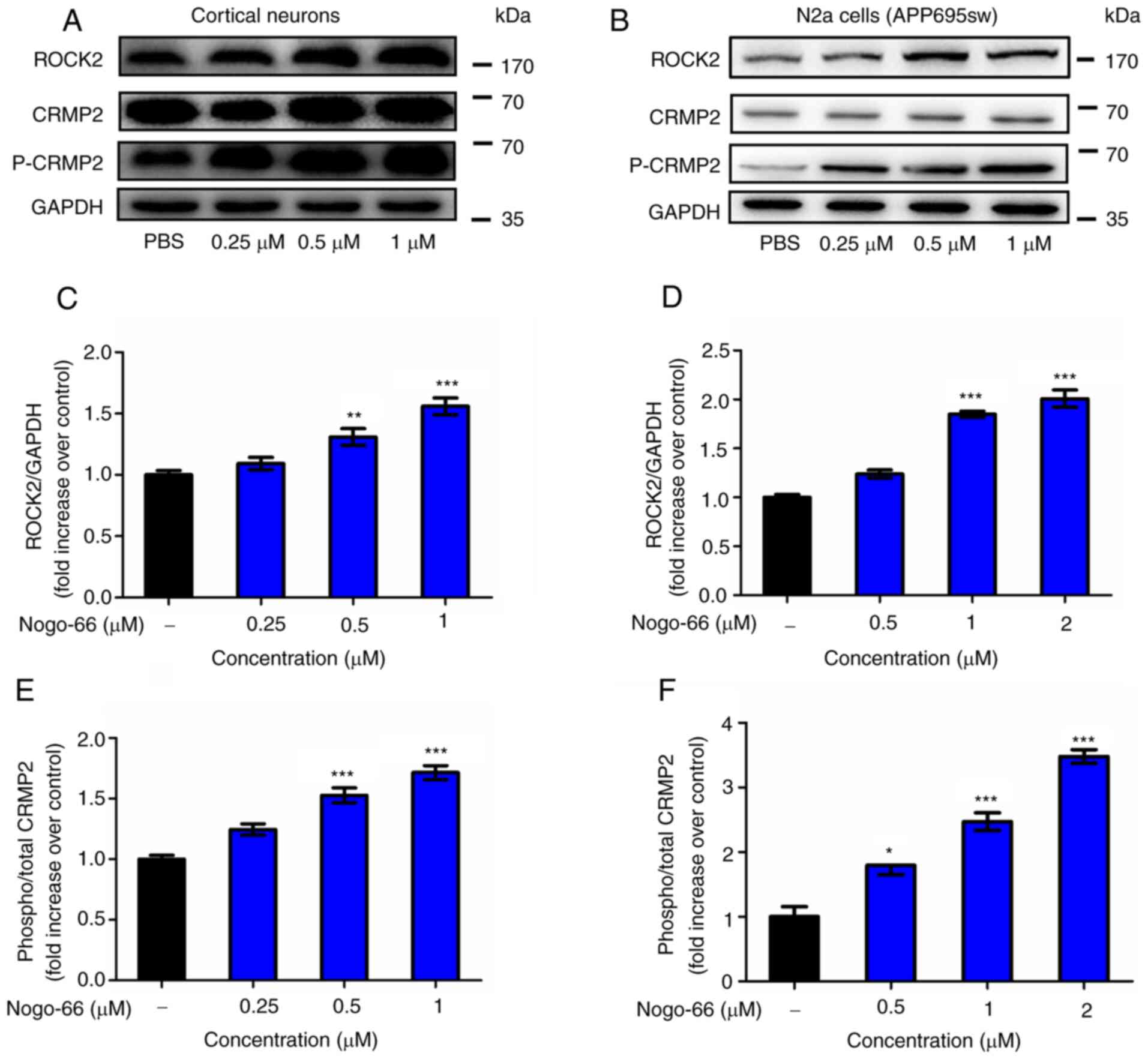 | Figure 4.Nogo-66 inhibits neurite outgrowth
and induces Aβ production by regulating ROCK2 protein expression
and CRMP2 phosphorylation. (A, C and E) Cortical neurons were
treated with various concentrations of Nogo-66. Western blot
analysis results showing the protein expression levels of ROCK2,
p-CRMP2 and CRMP2. Equal protein loading was confirmed using GAPDH.
Data are presented as the mean OD values ± SD of triplicate
samples. **P<0.01, ***P<0.001 vs. control. (B, D and F) N2a
cells were treated with various concentrations of Nogo-66. Western
blot analysis results showing the protein expression levels of
ROCK2, p-CRMP2 and CRMP2. Equal protein loading was confirmed using
GAPDH. Data are presented as the mean OD values ± SD of triplicate
samples. *P<0.05, ***P<0.001 vs. control. Aβ, β-amyloid
protein; N2a, Neuro2a; ROCK2, Rho-associated coiled-coil containing
kinase 2; CRMP2, collapsin response mediator protein-2; p-,
phosphorylated; OD, optical density. |
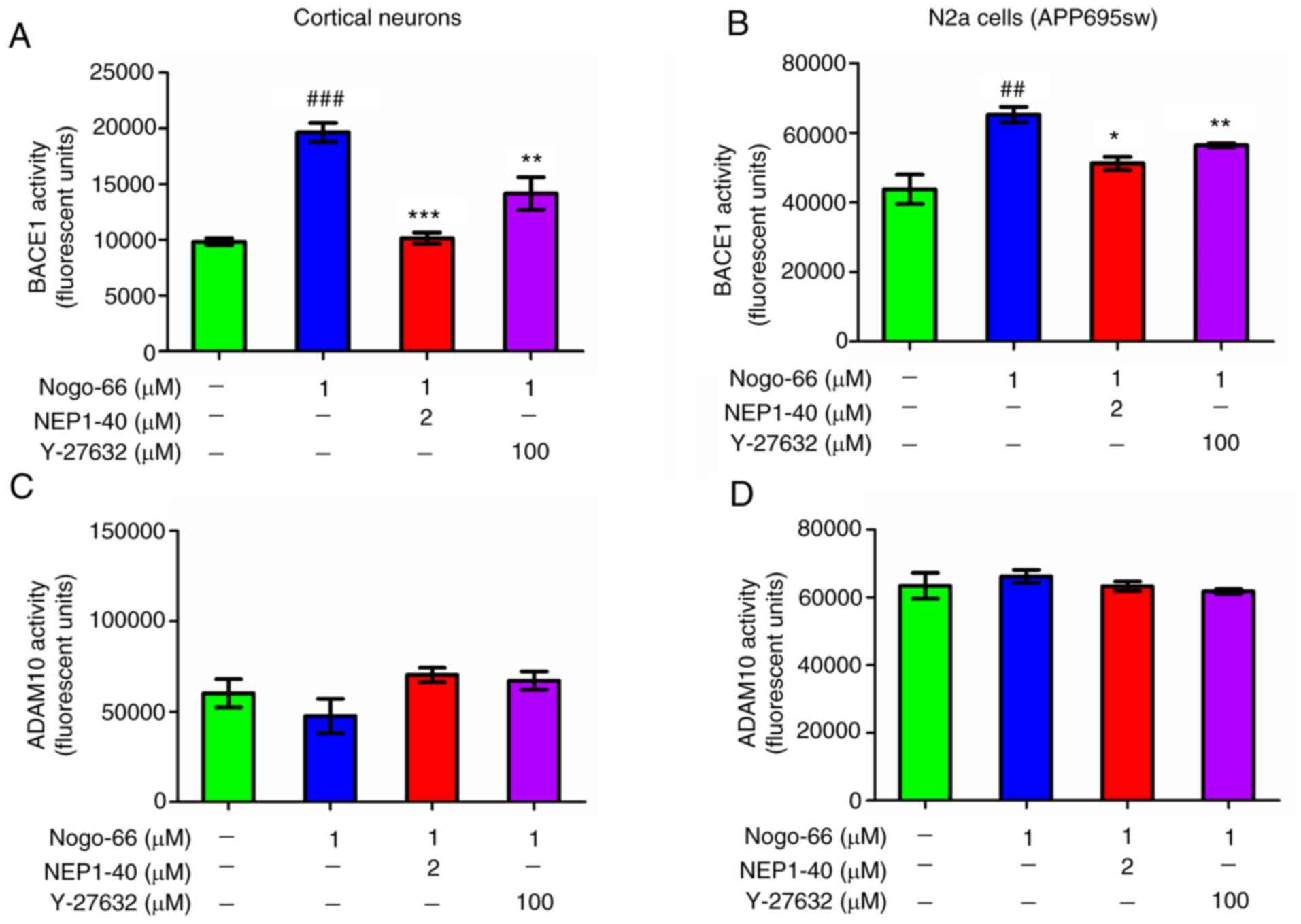
Nogo-66 promotes APP processing by
activating BACE1
Based on the considerable increase in APP and C99
expression levels following exposure to Nogo-66, it was speculated
that Nogo-66 may stimulate APP processing by activating BACE1. To
test this speculation, BACE1 activity in cortical neurons was
assessed using an Activity Detection kit. The results revealed that
BACE1 activity was almost twice as high in the Nogo-66 group
compared with that in the control group (Fig. 5A); however, the results of western
blotting indicated that the protein expression levels of BACE1 were
not altered in the presence of Nogo-66 (Fig. 3I and J). Furthermore, NEP1-40 and
Y-27632 prevented BACE1 activity from being increased by Nogo-66 in
the primary cultured cortical cells (Fig. 5A). ADAM10 activity was also detected
following exposure to Nogo-66, and inhibitors NEP1-40 and Y-27632.
The results revealed that ADAM10 activity was almost constant in
Nogo-66-treated cells, and ADAM10 activity remained almost
unchanged following treatment with the inhibitors (Fig. 5C). To determine whether Nogo-66 had
a comparable effect on APP-overexpressing cells, N2a cells were
treated in the same manner. Consistent with the aforementioned
findings, 1 µM Nogo-66 significantly increased BACE1 activity
compared with that in the control group, whereas the effects of
Nogo-66 were reversed after the addition of NEP1-40 and Y-27632 to
the cells (Fig. 5B). Furthermore,
ADAM10 activity remained the same after treatment with Nogo-66 or
the two inhibitors (Fig. 5D).
These results indicated that Nogo-66 increased Aβ
secretion by activating the enzymatic activity of BACE1. Based on
these findings, it was proposed that the hydrolysis of APP may be
increased through upregulation of BACE1 activity via the downstream
signaling pathway of Nogo-66 interacting with NgR, which could
result in increased Aβ.
Nogo-66 promotes Aβ secretion via the
NgR/ROCK-dependent activation of BACE1 in rats
The present study aimed to examine whether exposure
to Nogo-66 could influence Aβ secretion in Sprague-Dawley rats
(Fig. 6A).
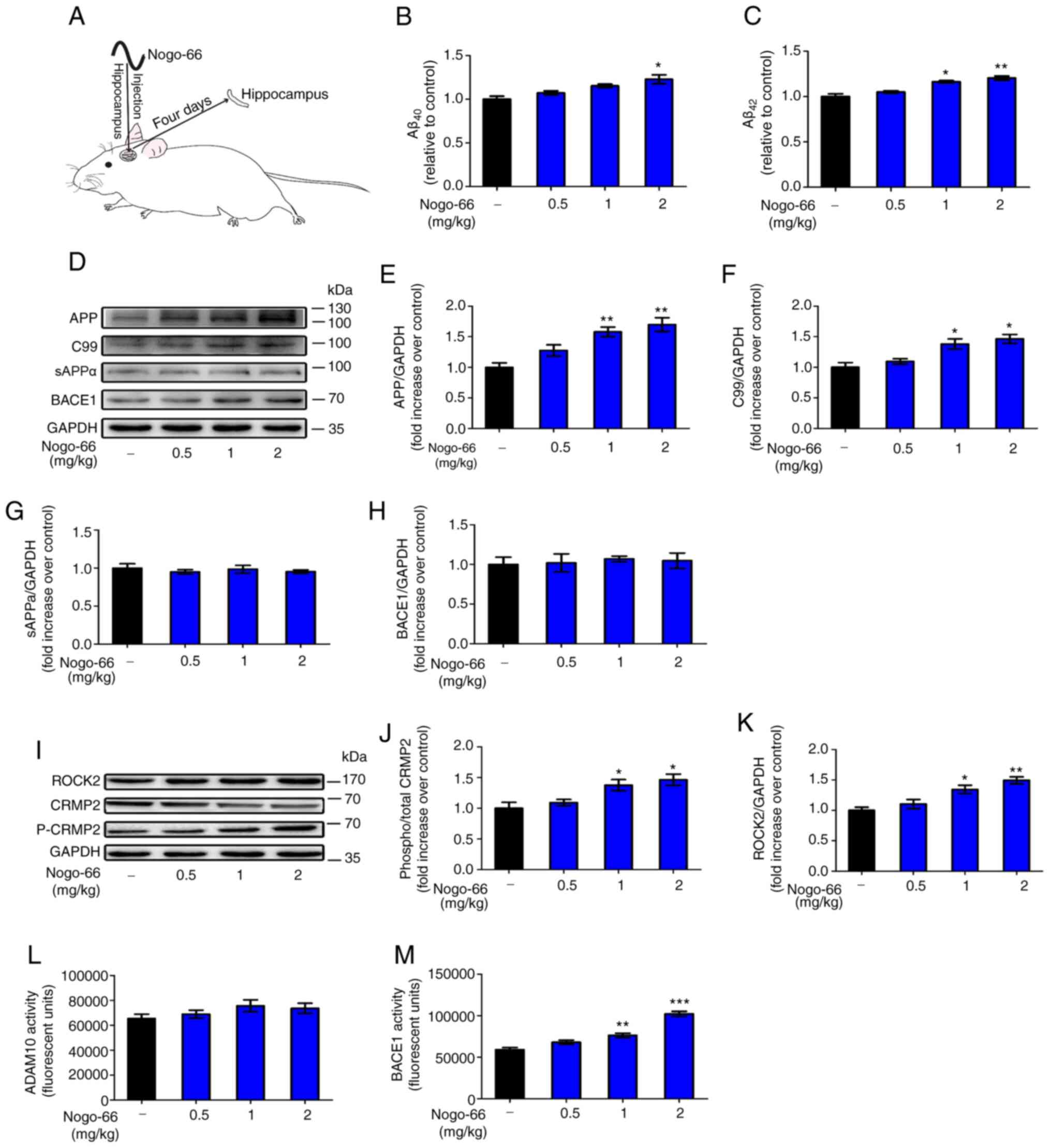 | Figure 6.Effects of Nogo-66 on the expression
levels and enzymatic activity of proteins involved in the Aβ
pathway in the hippocampus of Sprague-Dawley rats. (A) Schematic
diagram of treatment of rats with Nogo-66. Nogo-66 induced (B)
Aβ40 and (C) Aβ42 production in a
dose-dependent manner in the brains of rats. Data are presented as
the mean ± SD (n=6). *P<0.05, **P<0.01 vs. control. (D)
Western blotting results showing the protein expression levels of
APP, C99, sAPPα and BACE1 in the hippocampus. (E-H)
Semi-quantification of western blotting. (I) Western blot analysis
of the expression levels of ROCK2, p-CRMP2 and CRMP2 in the
hippocampus. Semi-quantification of (J) p-CRMP2 and (K) ROCK2
protein. Equal protein loading was confirmed using GAPDH. (L)
ADAM10 and (M) BACE1 activity in the hippocampus of Sprague-Dawley
rats was measured. Data are presented as the mean OD values ± SD of
triplicate samples (n=3). *P<0.05, **P<0.01, ***P<0.001
vs. control. APP, amyloid precursor protein; sAPPα, secreted APP-α;
BACE1, β-secretase 1; Aβ, β-amyloid protein; CRMP2, collapsin
response mediator protein-2; p-, phosphorylated; ROCK2,
Rho-associated coiled-coil containing kinase 2; OD, optical
density. |
Hippocampal homogenates were prepared for western
blotting, in order to examine the expression levels of proteins
associated with APP processing. Densitometric analysis revealed
that the protein expression levels of APP and C99 were
significantly increased following treatment with 1 and 2 mg/kg
Nogo-66 (Fig. 6D-F). Conversely,
treatment with various concentrations of Nogo-66 had little effect
on the protein expression levels of sAPPα and BACE1 (Fig. 6D, G and H). In addition, soluble Aβ
levels were measured by ELISA; Nogo-66-treated rats exhibited a
substantial increase in the levels of Aβ40 and
Aβ42 compared with those in the control groups (Fig. 6B and C). The results were consistent
with those obtained in vitro.
The protein expression levels of ROCK2 and p-CRMP2
were increased in the hippocampus of rats exposed to Nogo-66 in a
dose-dependent manner. High (2 mg/kg) and medium (1 mg/kg) doses of
Nogo-66 significantly increased the expression of phosphorylated
CRMP2 and ROCK2 in the hippocampus of rats (Fig. 6I-K). A low dose (0.5 mg/kg) of
Nogo-66 also increased the protein expression levels of ROCK2 and
p-CRMP2 in the hippocampus, but without a significant effect
(Fig. 6I-K). These trends in the
hippocampus were consistent with those observed in vitro,
indicating that Nogo-66 may promote the secretion of Aβ by
increasing the expression of APP and activating NgR1 downstream
signal proteins, such as ROCK2 and p-CRMP2.
The activity of BACE1 in the hippocampus of rats was
increased in a dose-dependent manner in response to Nogo-66. High
(2 mg/kg) and medium (1 mg/kg) doses of Nogo-66 significantly
increased BACE1 activity in the hippocampus of rats (Fig. 6M). A low dose (0.5 mg/kg) of Nogo-66
also increased the enzymatic activity of in the hippocampus;
however, this was not significant. In addition, Nogo-66 treatment
had a slight effect on ADAM10 enzymatic activity, but there was no
significant difference (Fig. 6L).
The effects of Nogo-66 on APP metabolic enzymatic activity in the
hippocampus were consistent with those observed in vitro,
and indicated that Nogo-66 mainly promoted the cleavage of the APP
protein through the BACE1 pathway by increasing the activity of
BACE1, thereby increasing the secretion of Aβ.
Discussion
Nogo-A is a member of the Nogo family found in the
myelin sheath of the CNS, which inhibits axonal regeneration.
Nogo-A is considered a major obstacle to neurological regeneration
following traumatic injury in mammals (19). Nogo-66 is a 66-amino acid residue in
the extracellular domain of Nogo-A, which inhibits neurite
outgrowth by binding to the NgR1 in the CNS (20). In vitro, Nogo-66 was
previously shown to inhibit neurite outgrowth in a variety of nerve
cells, such as dorsal root ganglion cells (21,22),
PC12 cells (21) and cerebellar
granule cells (23). In the present
study, Nogo-66 was revealed to inhibit neurite outgrowth of
cortical neurons in a dose-dependent manner.
Previous studies have reported that Nogo-A and its
receptors are involved in the regulation of synaptic plasticity, as
well as the metabolism of Aβ, suggesting that it may serve an
important role in AD. In a previous study, the levels of Aβ
deposition were decreased in NgR2-knockout mice (24). However, whereas NgR1 has been shown
to bind to APP (24), contrasting
effects of this interaction have also been reported; Park et
al (25) observed decreased Aβ
production following NgR1-overexpression in neuroblastoma in
vitro. These previous studies have suggested a strong linkage
between Nogo-A and Aβ; however, the exact roles need to be
elucidated. The excess production of Aβ is considered the
initiating factor of AD, which is generated from the hydrolysis of
APP via BACE-1 and γ-secretase. NgR can cause parallel changes in
sAPPα and Aβ, indicating that NgR blocks APP hydrolysis (26). In contrast to these findings, Zhou
et al (24) reported that
the increased interaction between NgR and APP reduced the surface
expression of APP and favored the processing of APP via the BACE1
pathway.
The Nogo-A protein, also termed RTN4A, is a member
of the RTN family. RTNs are characterized by the highly conserved
C-terminal RHD, which is composed of two putative transmembrane
domains separated by one hydrophilic loop, plus a hydrophilic tail.
Notably, all four human RTN proteins appear to be involved in the
pathogenesis of AD. He et al (27) discovered that BACE1
co-immunoprecipitated with RTN1, RTN2, RTN3 and RTN4, and revealed
that RTN family members are binding partners of BACE1. Furthermore,
increased RTN3 expression caused a dose-dependent reduction in Aβ,
whereas suppressing the expression of RTN3 by RNA interference
increased the secretion of Aβ, thus suggesting that RTN3 may block
access of BACE1 to APP, reduce the cleavage of this protein and
inhibit the production of Aβ. RTN4-B/C has also been reported to
interact with BACE1 and inhibit its ability to produce Aβ (28). Certain studies have reported that
Nogo-A promotes the pathogenesis of AD. Masliah et al
(29) demonstrated that deleting
Nogo-A ameliorated learning and memory deficits of APP-transgenic
mice in the Morris water maze at an early/intermediate stage of the
disease, thus suggesting that Nogo-A may influence the metabolism
of APP; however, the detailed effect and mechanism are not clear.
In the present study, ELISA results revealed that Nogo-66 promoted
the levels of Aβ40 and Aβ42 in primary
cortical neurons in a dose-dependent manner. The present results
showed that increased Nogo-66 increased production of Aβ, which is
the opposite effect to other RTNs. Notably, Nogo-A may be
considered a very different protein compared with others from the
RTN family, as it not only has the unique function of inhibiting
neurite outgrowth but it may also increase Aβ.
When cortical neurons were treated with NEP1-40 in
the present study, the levels of Aβ40 and
Aβ42 were decreased. After treatment with Nogo-66, the
protein expression levels of APP and activation of BACE1 were
increased in cortical neurons and N2a cells, indicating that
Nogo-66 simultaneously increased the levels of APP and activated
BACE1, which may eventually lead to an increase in the secretion of
Aβ.
The Rho/ROCK signaling pathway was activated when
Nogo-A interacts with NgR1 (30).
The downstream event included activation of RhoA-GTP, ROCK, MAP and
CRMP2, which eventually exerted effects on the cytoskeleton
associated with neurite growth. The results of western blotting
revealed that higher ROCK2 expression and increased p-CRMP2
expression was induced by Nogo-66. It was therefore suggested that
Nogo-66-stimulated ROCK2-induced regulation of CRMP2
phosphorylation may contribute to neurite outgrowth inhibition.
The present results also revealed that the ROCK
inhibitor Y-27632 suppressed Nogo-66-stimulated Aβ42 and
Aβ40 secretion. These findings indicated that Y-27632
may reduce Aβ42 by inhibiting the activation of ROCK and
Nogo-66 may increase Aβ42 secretion by activating the
ROCK pathway. Subsequently, western blot analysis was performed to
explore whether Nogo-66 stimulated Aβ secretion by regulating ROCK2
expression. It was revealed that Nogo-66 significantly increased
the expression levels of ROCK2 in cortical neurons and N2a cells. A
previous study used a variety of non-steroidal anti-inflammatory
drugs to demonstrate that the activation of Rho/ROCK2 led to a
large production of Aβ42, whereas the inhibition of
ROCK2 activity reduced the APP amyloid enzymatic degradation
pathway and the levels of Aβ42 in the brain (31). Another study confirmed that
inhibition of RhoA or ROCK2 knockdown could interfere with the
β-cleavage of APP, and significantly reduced the levels of brain Aβ
in AD model mice (32). It was
therefore hypothesized that Nogo-66 bound to NgR, and potentially
exerted its regulatory effects on Aβ levels by increasing the
expression of ROCK2 and promoting the catalytic action of BACE1,
which may then facilitate amyloidogenic metabolism of APP, thus
resulting in increased Aβ42 secretion.
In the in vivo experiment conducted in the
present study, Nogo-66 promoted the levels of Aβ40 and
Aβ42 in rat tissues. Nogo-66 also enhanced the
expression levels of proteins associated with the APP processing
pathway and activated BACE1 in rats, which may eventually lead to
increased secretion of Aβ. Subsequently, western blot analysis was
used to explore the expression levels of ROCK2 and p-CRMP2 in the
hippocampus; it was revealed that Nogo-66 increased the expression
levels of ROCK2 and p-CRMP2 in vivo. The results regarding
the effects of Nogo-66 on the rat hippocampus and related mechanism
were consistent with the results obtained for neurons in
vitro.
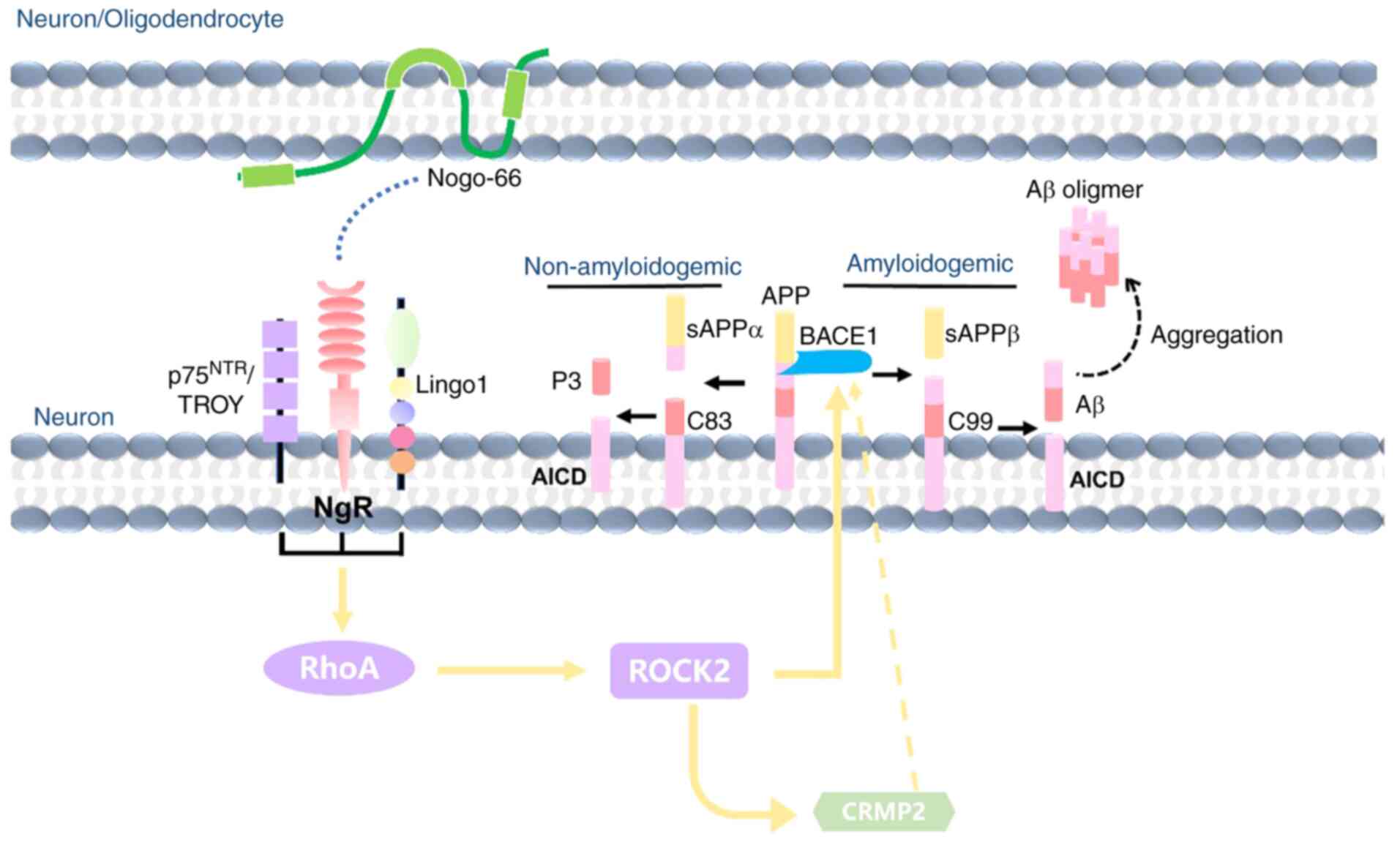
In conclusion, although Nogo-A has an important role
in AD pathology, its effects and mechanisms are not clear. The
present study demonstrated that Nogo-66 inhibited neurite
outgrowth, and increased the levels of Aβ42 and
Aβ40 in a dose-dependent manner. The underlying
mechanism may be as follows (Fig.
7): Nogo-66 increases Aβ generation by activating BACE1 and
increasing APP expression, which is associated with activation of
NgR1 and its downstream signaling molecule ROCK2, as well as the
phosphorylation of CRMP2. Therefore, Nogo-A may be a precipitating
factor that causes the onset and development of AD; however, the
exact mechanism still requires further exploration. From the
viewpoint of drug therapeutic targets, NgR and ROCK2 appear to be
potentially useful and effective therapeutic targets for AD
treatment, and targeting them may promote neurite outgrowth and
inhibit the neuronal production of Aβ.
Acknowledgements
The authors would like to thank Dr Fei Fang
(Department of Clinical Molecular Biology, Akershus University
Hospital, University of Oslo, Norway) for critical discussion and
assistance in the preparation of the manuscript.
Funding
The study was funded by the National Natural Science
Foundation of China (grant nos. 81202519, 81873739, 81572497 and
81703011), the Science and Technology Program of Guangzhou Province
(grant no. 201607010216) and the Natural Science Foundation of
Guangdong Province (grant nos. 2019A1515010936, 2014A030313362 and
2017A030313487).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
FX, QZ, HY and FCL were involved in revising the
manuscript critically for important intellectual content, making
substantial contributions to conception and design, and giving
final approval of the version to be published. QQX participated in
the entire process, drafting of the article and data analysis. XF,
YYH, NF, QYC, GFL, JPP, YH and ZJW fed the animals, performed the
experiments and collected samples from the rats. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were conducted according to
the Jinan University ethical guidelines and were approved by the
Animal Research Committee of the School of Medicine of Jinan
University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kerr JS, Adriaanse BA, Greig NH, Mattson
MP, Cader MZ, Bohr VA and Fang EF: Mitophagy and Alzheimer's
disease: Cellular and molecular mechanisms. Trends Neurosci.
40:151–166. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stephenson-Jones M, Yu K, Ahrens S,
Tucciarone JM, van Huijstee AN, Mejia LA, Penzo MA, Tai LH,
Wilbrecht L and Li B: A basal ganglia circuit for evaluating action
outcomes. Nature. 539:289–293. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pereira JB, Janelidze S, Ossenkoppele R,
Kvartsberg H, Brinkmalm A, Mattsson-Carlgren N, Stomrud E, Smith R,
Zetterberg H, Blennow K, et al: Untangling the association of
amyloid-β and tau with synaptic and axonal loss in Alzheimer's
disease. Brain awaa395. 2020. View Article : Google Scholar
|
|
4
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin Y-T, Seo J, Gao F, Feldman HM, Wen HL,
Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, et al: APOE4 causes
widespread molecular and cellular alterations associated with
Alzheimer's disease phenotypes in human iPSC-derived brain cell
types. Neuron. 98:1141–1154.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fang EF, Hou Y, Palikaras K, Adriaanse BA,
Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, et
al: Mitophagy inhibits amyloid-β and tau pathology and reverses
cognitive deficits in models of Alzheimer's disease. Nat Neurosci.
22:401–412. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lautrup S, Sinclair DA, Mattson MP and
Fang EF: NAD+ in brain aging and neurodegenerative
disorders. Cell Metab. 30:630–655. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang P, Kishimoto Y, Grammatikakis I,
Gottimukkala K, Cutler RG, Zhang S, Abdelmohsen K, Bohr VA, Misra
Sen J, Gorospe M, et al: Senolytic therapy alleviates Aβ-associated
oligodendrocyte progenitor cell senescence and cognitive deficits
in an Alzheimer's disease model. Nat Neurosci. 22:719–728. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oertle T, van der Haar ME, Bandtlow CE,
Robeva A, Burfeind P, Buss A, Huber AB, Simonen M, Schnell L,
Brösamle C, et al: Nogo-A inhibits neurite outgrowth and cell
spreading with three discrete regions. J Neurosci. 23:5393–5406.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huebner EA and Strittmatter SM: Axon
regeneration in the peripheral and central nervous systems. Results
Probl Cell Differ. 48:339–351. 2009.PubMed/NCBI
|
|
11
|
Grandpré T and Strittmatter SM: Nogo: A
molecular determinant of axonal growth and regeneration.
Neuroscientist. 7:377–386. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mosyak L, Wood A, Dwyer B, Buddha M,
Johnson M, Aulabaugh A, Zhong X, Presman E, Benard S, Kelleher K,
et al: The structure of the Lingo-1 ectodomain, a module implicated
in central nervous system repair inhibition. J Biol Chem.
281:36378–36390. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gil V, Nicolas O, Mingorance A, Ureña JM,
Tang BL, Hirata T, Sáez-Valero J, Ferrer I, Soriano E and del Río
JA: Nogo-A expression in the human hippocampus in normal aging and
in Alzheimer disease. J Neuropathol Exp Neurol. 65:433–444. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fang Y, Yao L, Li C, Wang J, Wang J, Chen
S, Zhou XF and Liao H: The blockage of the Nogo/NgR signal pathway
in microglia alleviates the formation of Aβ plaques and tau
phosphorylation in APP/PS1 transgenic mice. J Neuroinflammation.
13:562016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xiao F, Lin LF, Cheng X, Gao Q and Luo HM:
Nogo-66 receptor activation inhibits neurite outgrowth and
increases β-amyloid protein secretion of cortical neurons. Mol Med
Rep. 5:619–624. 2012.PubMed/NCBI
|
|
16
|
Dai X, Sun Z, Liang R, Li Y, Luo H, Huang
Y, Chen M, Su Z and Xiao F: Recombinant Nogo-66 via soluble
expression with SUMO fusion in Escherichia coli inhibits neurite
outgrowth in vitro. Appl Microbiol Biotechnol. 99:5997–6007. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
zur Nedden S, Doney AS and Frenguelli BG:
Modulation of intracellular ATP determines adenosine release and
functional outcome in response to metabolic stress in rat
hippocampal slices and cerebellar granule cells. J Neurochem.
128:111–124. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gao R, Wang Y, Pan Q, Huang G, Li N, Mou J
and Wang D: Fuzhisan, a chinese herbal medicine, suppresses
beta-secretase gene transcription via upregulation of SIRT1
expression in N2a-APP695 cells. Int J Clin Exp Med. 8:7231–7240.
2015.PubMed/NCBI
|
|
19
|
Jin SG, Ryu HH, Li SY, Li CH, Lim SH, Jang
WY and Jung S: Nogo-A inhibits the migration and invasion of human
malignant glioma U87MG cells. Oncol Rep. 35:3395–3402. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yan J, Zhou X, Guo JJ, Mao L, Wang YJ, Sun
J, Sun LX, Zhang LY, Zhou XF and Liao H: Nogo-66 inhibits adhesion
and migration of microglia via GTPase Rho pathway in vitro. J
Neurochem. 120:721–731. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
GrandPré T, Nakamura F, Vartanian T and
Strittmatter SM: Identification of the Nogo inhibitor of axon
regeneration as a Reticulon protein. Nature. 403:439–444. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen MS, Huber AB, van der Haar ME, Frank
M, Schnell L, Spillmann AA, Christ F and Schwab ME: Nogo-A is a
myelin-associated neurite outgrowth inhibitor and an antigen for
monoclonal antibody IN-1. Nature. 403:434–439. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Niederöst B, Oertle T, Fritsche J,
McKinney RA and Bandtlow CE: Nogo-A and myelin-associated
glycoprotein mediate neurite growth inhibition by antagonistic
regulation of RhoA and Rac1. J Neurosci. 22:10368–10376. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhou X, Hu X, He W, Tang X, Shi Q, Zhang Z
and Yan R: Interaction between amyloid precursor protein and Nogo
receptors regulates amyloid deposition. FASEB J. 25:3146–3156.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Park JH, Gimbel DA, GrandPre T, Lee JK,
Kim JE, Li W, Lee DH and Strittmatter SM: Alzheimer precursor
protein interaction with the Nogo-66 receptor reduces amyloid-beta
plaque deposition. J Neurosci. 26:1386–1395. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang R, Wu XF, Wang B, Guan RX, Lv LM, Li
AP, Lei L, Ma Y, Li N, Li QF, et al: Reduction of NgR in perforant
path decreases amyloid-β peptide production and ameliorates
synaptic and cognitive deficits in APP/PS1 mice. Alzheimers Res
Ther. 12:472020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He W, Lu Y, Qahwash I, Hu X-Y, Chang A and
Yan R: Reticulon family members modulate BACE1 activity and
amyloid-β peptide generation. Nat Med. 10:959–965. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Murayama KS, Kametani F, Saito S, Kume H,
Akiyama H and Araki W: Reticulons RTN3 and RTN4-B/C interact with
BACE1 and inhibit its ability to produce amyloid β-protein. Eur J
Neurosci. 24:1237–1244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Masliah E, Xie F, Dayan S, Rockenstein E,
Mante M, Adame A, Patrick CM, Chan AF and Zheng B: Genetic deletion
of Nogo/Rtn4 ameliorates behavioral and neuropathological outcomes
in amyloid precursor protein transgenic mice. Neuroscience.
169:488–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu W, Xiao P, Fan S, Chen Y, Huang W, Chen
X, Liu G, Dang C, Zeng J and Xing S: Blockade of Nogo-A/Nogo-66
receptor 1 (NgR1) inhibits autophagic activation and prevents
secondary neuronal damage in the thalamus after focal cerebral
infarction in hypertensive rats. Neuroscience. 431:103–114. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhou Y, Su Y, Li B, Liu F, Ryder JW, Wu X,
Gonzalez-DeWhitt PA, Gelfanova V, Hale JE, May PC, et al:
Nonsteroidal anti-inflammatory drugs can lower amyloidogenic
Abeta42 by inhibiting Rho. Science. 302:1215–1217. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Herskowitz JH, Feng Y, Mattheyses AL,
Hales CM, Higginbotham LA, Duong DM, Montine TJ, Troncoso JC,
Thambisetty M, Seyfried NT, et al: Pharmacologic inhibition of
ROCK2 suppresses amyloid-β production in an Alzheimer's disease
mouse model. J Neurosci. 33:19086–19098. 2013. View Article : Google Scholar : PubMed/NCBI
|