Introduction
Osteoarthritis (OA), characterized by progressive
cartilage degradation, is the most common joint disorder worldwide
(1). OA accounts for major pain,
and loss of function and disability in adults. As OA is an
age-related disease, the rapid increase in the number of older-aged
adults in the population has led to increased incidence. Effective
disease-modifying drugs are still limited for the treatment of OA
(2). The last option for late-stage
OA is the surgical approach of total joint replacement (3). The etiology and underlying mechanisms
responsible for the development of OA remain largely unknown.
Recent findings have suggested that the overproduction of reactive
oxygen species (ROS) and oxidative stress induces chondrocyte
senescence/apoptosis, the degradation of the extracellular matrix
or synovial inflammation in OA (4,5).
Nuclear factor erythroid 2-related factor 2 (Nrf2) has been
identified as one of the transcription factors that binds to
antioxidant response elements (AREs) to induce the expression of a
number of detoxication enzymes, including heme oxygenase-1 (HO-1),
which exerts anti-inflammatory effects during OA (6,7).
Previously published research from our laboratory demonstrated that
deficiency of Nrf2 leads to an increased severity of OA in an
inflammatory model and a post-traumatic model of OA (8). Thus, the activation of the Nrf2/ARE
signaling pathway offers a novel perspective for the exploration of
potential treatment strategies for OA. Coniferaldehyde (CFA), a
food flavoring, has been demonstrated to potently activate Nrf2 in
RAW264.7 macrophages, exerting cytoprotective effects against
lipopolysaccharide-induced cell death (9). The aim of the present study was to
investigate whether CFA can exert protective effects against OA by
activating the Nrf-2/HO-1 signaling pathway.
Materials and methods
Chemicals and reagents
CFA (purity, 98%) was purchased from Sigma-Aldrich
(Merck KGaA; cat. no. 382051). Anti-HO-1 (cat. no. BS6626; 1:2,000)
and secondary HRP-conjugated goat anti-rabbit (cat. no. BS13278;
1:5,000) were obtained from Bioworld Technology, Inc. Anti-Nrf2
(cat. no. 12721; 1:2,000), anti-lamin B (cat. no. 13435; 1:1,000),
Alexa Fluor 488 anti-rabbit IgG (cat. no. 4412; 1:200) and
anti-β-actin (cat. no. 8457; 1:5,000) antibodies were procured from
Cell Signaling Technology, Inc. The nuclear protein extraction kit
(cat. no. P0028) and Cell Counting Kit-8 (CCK-8; cat. no. C0037)
were supplied by Beyotime Institute of Biotechnology. The
malondialdehyde (MDA; cat. no. A003-4), catalase (CAT; cat. no.
A007-2) and superoxidase dismutase (SOD; cat. no. A001-3) assay
kits were purchased from Nanjing Jiancheng Bioengineering
Institute. The fluorescent probe, 2′,7′-dichlorodihydrofluorescein
diacetate (H2DCFDA; cat. no. D399), was supplied by Thermo Fisher
Scientific, Inc.
Cells and cell culture
Primary mouse chondrocytes were isolated from murine
costal cartilage obtained from 29 male neonatal B6 mice (age, 6
days old; weight, 2.92±0.46 g) purchased from the Comparative
Medical Center of Yangzhou University (Yangzhou, China) and were
housed under a 12 h light/dark cycle at a room temperature of
22±1°C and 50% relative humidity with food and water available
ad libitum, as previously described (10). The animal research was carried out
in April 2019 in accordance with Nanjing Medical University
Institutional Animal Care and Use Committee guidelines (approval
no. IACUC 1901062). Mice were euthanized using 100% CO2
anesthesia using an air displacement rate of 20% of the chamber
volume/min. Following digestion with collagenase D overnight at
37°C, the collected cells were cultured at a density of
5×105 cells/per dish (10 cm in diameter) in Ham's F-12
(DMEM/F12; Gibco; Thermo Fisher Scientific, Inc.) containing 2 mM
L-glutamine, 5% FBS (Gibco; Thermo Fisher Scientific, Inc.), 100
IU/ml penicillin and 100 µg/ml streptomycin at 37°C and 5%
CO2. Morphology observation of chondrocytes with
light-contrast microscopes and immunostaining of chondrocytes in
cultures with an anti-type II collagen antibody were used to
phenotype chondrocytes (data not shown). For immunostaining, the
cells was blocked with 3% goat serum (Invitrogen; Thermo Fisher
Scientific, Inc.) for 2 h at 22°C, and then incubated with primary
antibody against type II collagen (Bioworld Technology, Inc; cat.
no. BS1071; 1:200) for 2 h at room temperature, and were
subsequently incubated with fluorescein-conjugated antibody (Cell
Signaling Technology, Inc; cat. no. 4412; 1:200) for 1 h at 22°C.
Primary chondrocytes at 80% confluency were detached and plated in
6-well plates (2.4 cm in diameter) for use in further assays.
Cell viability assays
The viability of the treated and untreated primary
mouse chondrocytes was measured by a CCK-8 assay. Briefly,
chondrocytes at passage 1 were incubated in a 12-well plate at a
density of 5×103 cells/well. Following 24 h of
incubation with various concentrations of CFA (0, 30, 60, 90 and
120 µM) at 37°C, CCK-8 working solution was added at 10 µl/well
followed by further incubation for 1 h in the dark. The absorbance
was monitored at a wavelength of 450 nm using a microplate reader
(Bio-Rad Laboratories, Inc.) according to the manufacturer's
protocols.
OA model and histological
analysis
A total of 95 B6 male mice (weight, 26.40±3.92 g;
age, 8–10 weeks old) supplied by the Comparative Medical Center of
Yangzhou University (Yangzhou, China) were used in the present
study. The mice were anesthetized by administering intraperitoneal
ketamine hydrochloride (120 mg/kg) and xylazine hydrochloride (5
mg/kg). The levels of anesthesia were considered adequate when the
mice stayed still quietly, were unresponsive to external stimuli
and had constant heart and respiratory rates. Mice were euthanized
using 100% CO2 anesthesia using an air displacement rate
of 20% of the chamber volume/min. All animal research was executed
in accordance with the Nanjing Medical University Institutional
Animal Care and Use Committee guidelines. The mice were housed in a
normal atmosphere under a 12-h light-dark cycle at 24°C with 50%
humidity, and provided with standard laboratory food (RM3; Special
Dietary Systems) and water ad libitum. Mice were divided
into four groups [sham surgery + saline, sham surgery + CFA,
destabilization of the medial meniscus (DMM) surgery + saline and
DMM surgery + CFA). OA was induced by DMM surgery on the right knee
joints according to previously reported procedures (11). The delivery of CFA (0.05 mmol
kg/day) was performed via peritoneal injection at 2 weeks following
surgery. The mice were sacrificed at 8 weeks post-OA surgery for
histological assessment and WB analysis. Serial sections
(thickness, 5 µm) of knee joints were placed in 70% ethyl alcohol
for 15 min. Then, the sections were stained with 0.04% safranin
O/sodium acetate buffer for 10 min at room temperature. The
severity of cartilage deterioration was evaluated using the 0–6
scoring system recommended by the Osteoarthritis Research Society
International (OARSI) with an Olympus BX51 light microscope and
photographed by a computer-operated Olympus DP72 digital camera
(Olympus Corporation) (12). The
observers were blinded to the experimental conditions.
Protein expression analysis via
western blotting (WB)
The chondrocytes at passage 1 were treated with
various concentrations of CFA for 24 h. The collected cells were
lysed on ice for 15 min with pre-cooled RIPA lysis buffer (50 mM
Tris-HCl, 150 mM NaCl, pH 7.4, 1 mM EDTA, 0.5% sodium deoxycholate,
1% Nonidet P40 and 0.1% SDS). Mice were administered CFA (0.05 mmol
kg/day) or saline for 2 weeks or 6 weeks, and the harvested
cartilage samples were prepared as previously described (13). Briefly, knee cartilage obtained from
mice using a scalpel blade with a surgical microscope was stored in
liquid nitrogen. Following pulverization, samples were solubilized
at 4°C with lysis buffer for 30 min. For WB analysis, both cell and
tissue samples were centrifuged at 12,000 × g for 20 min at 4°C,
and the supernatant was collected to determine the protein content
with a BCA protein quantitation kit (Pierce; Thermo Fisher
Scientific, Inc.). Proteins of the same quality were loaded on 10%
gels and separated via SDS-PAGE, and subsequently separated
proteins were transferred onto PVDF membranes. The membranes were
blocked with 5% milk at room temperature for 1 h, and then probed
with the desired primary antibodies (anti-Nrf2, anti-HO-1,
anti-lamin B and anti-β-actin) at 4°C overnight. Following
incubation with primary antibodies, the membranes were washed three
times in Tris-buffered saline with 0.1% Tween-20 (TBST), then
detected with secondary HRP-conjugated goat anti-rabbit antibodies
at room temperature for 1 h. The membranes were finally washed
three times in TTBS, and were then visualized by ECL High-Signal
reagent (Thermo Fisher Scientific, Inc.) and analyzed with ImageJ
software (version 1.43, National Institutes of Health) for
semiquantification. To obtain a suitable amount of protein, the
pooling of the samples was performed; each experimental unit for
mice comprised a pool of two to three compartments; when pooling
was performed, the experimental unit was regarded as one.
Gene transcript analysis via reverse
transcription-quantitative PCR (RT-qPCR)
Cartilage was procured and snap-frozen in liquid
nitrogen prior to RNA extraction. Pooling was also performed to
obtain a suitable amount of cartilage and each experimental unit
comprised a pool of two compartments. Total RNA extracted from the
cartilage in the knee joints from the mice was isolated using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). cDNA was synthesized from total RNA using the PrimeScript RT
Reagent kit (Takara Bio, Inc.), according to the manufacturer's
protocol. PCR thermocycling conditions were as follows: Initial
denaturation at 96°C for 10 min, followed by 30 cycles of
denaturation at 95°C for 15 sec, annealing and elongation at 60°C
for 30 sec; and final extension at 72°C for 10 min. qPCR was
performed on a 7500 real-time PCR system using SYBR-Green PCR
Master Mix (Thermo Fisher Scientific, Inc.) and repeated in
triplicate for each sample. Primer pairs used in the present study
are listed in Table I. Relative
quantification was performed using the 2−ΔΔCq method
(14).
 | Table I.Gene specific primer sequences for
reverse transcription-quantitative PCR. |
Table I.
Gene specific primer sequences for
reverse transcription-quantitative PCR.
| Genes | Primer sequences
(5′→3′) |
|---|
| IL-1 | F:
ATGGCAGAAGTACCTAAGCTCGC |
|
| R:
ACACAAATTGCATGGTGAAGTCAGTT |
| IL-6 | F:
ACACACTGGTTCTGAGGGAC |
|
| R:
TACCACAAGGTTGGCAGGTG |
| TNF-α | F:
ATGAGCACAGAAAGCATGATCCGC |
|
| R:
CCAAAGTAGACCTGCCCGGACTC |
| MMP1 | F:
GCCACAAAGTTGATGCAGTT |
|
| R:
GCAGTTGAACCAGCTATTAG |
| MMP3 | F:
ATGAAAATGAAGGGTCTTCCGG |
|
| R:
GCAGAAGCTCCATACCAGCA |
| MMP13 | F:
ATGCATTCAGCTATCCTGGCCA |
|
| R:
AAGATTGCATTTCTCGGAGCCTG |
| ACTB | F:
TGACGGGGTCACCCACACTGTGCCCATCTA |
|
| R:
CTAGAAGCATTTGCGGTGGACGATGGAGGG |
Luciferase assays
The HO-1 promoter was amplified by general PCR and
the product was subsequently inserted into the pGL3 vector (Promega
Corporation) at the HindIII and Bg1II sites. A
non-specific oligonucleotide was used to construct a control
plasmid. The DNA was extracted from RAW cells using the TIANamp
Genomic DNA kit (cat. no. DP304; Tiangen Biotech Co., Ltd.) and
amplified with AmpliTaq Gold™ DNA Polymerase (Thermo Fisher
Scientific, Inc.) following the manufacturer's instructions. The
primer sequences were as follows: HO-1 forward,
5′-GGAAGATCTCTGCAGAGCCCCACTGGAG-3′ and reverse,
5′-CCCAAGCTTGGAACAGCAACGCTGT-3′. The thermocycling conditions were
as follows: 2 min at 94°C; 30 cycles of 30 sec at 94°C, 30 sec at
56°C and 30 sec at 72°C, and a final 5 min at 72°C. The 293 cells
(5×103 cells/well) obtained from the Cell Bank of the
Type Culture Collection of the Chinese Academy of Science (China)
were transfected with the plasmid (plasmid DNA 1 µg) using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Following 24 h of transfection, the cells were treated with 0, 50
and 100 µM CFA for 24 h at 37°C, and a luciferase assay system
(Promega Corporation) was used to measure the luciferase activity
by normalizing the Firefly luciferase activity to Renilla
luciferase activity.
Immunofluorescence staining
Primary chondrocytes were seeded on glass coverslips
in a 6-well plate (2.4 cm in diameter; 1×105 cells/well)
and treated with CFA (100 µM) for 24 h. Following the rinsing of
the glass coverslips with ice-cold PBS three times, the cells were
fixed in 4% paraformaldehyde for 15 min at 22°C and subsequently
permeabilized in PBS containing 2% Triton X-100 for 15 min at room
temperature. After the cells were blocked with 3% goat serum
(Invitrogen; Thermo Fisher Scientific, Inc.) for 90 min at 22°C,
the cells were incubated with primary antibody against Nrf2 in
blocking medium for 2 h at room temperature, and were subsequently
incubated with fluorescein-conjugated antibody (Cell Signaling
Technology, Inc; cat. no. 4412; 1:200) for 1 h at 22°C. After
washing with PBS, the cells were labeled with DAPI (Invitrogen;
Thermo Fisher Scientific, Inc.) for 15 min at room temperature. The
stained cells were observed under a confocal microscope (Zeiss LSM
710 META; Carl Zeiss AG).
Apoptosis detection by flow
cytometry
The apoptosis of murine primary chondrocytes was
measured by Annexin-V-FITC/PI staining assay (Thermo Fisher
Scientific, Inc.). Primary chondrocytes cultured in 12-well plates
(5×103 cells/well) subjected to different treatments
were stained with 5 µl Annexin V-FITC and 5 µl PI. The mixture was
incubated for 15 min at room temperature in the dark. The
percentage of apoptotic cells (Annexin
V-FITC+/PI−) was calculated using a flow
cytometer (BD Biosciences) according to the manufacturer's
instructions. Data were acquired via flow cytometry (Beckman
Coulter, Inc.). Fluorescence analyses were performed on COULTER
EPICS XL Flow Cytometer (Beckman Coulter, Inc.) using Expo32-ADC
software (version 1.2B; Beckman Coulter, Inc.).
Detection of intracellular ROS and
measurement of antioxidant enzyme activities
The intracellular ROS levels were determined with
the use of the fluorescent probe, H2DCFDA. The cells
were treated or not with CFA (100 µM) for 24 h, followed by
incubation with or without H2O2 (40 ng/ml)
for a further 24 h at room temperature. After discarding the
medium, the chondrocytes were incubated for 25 min with
H2DCFDA (10 µM) in the dark at room temperature. A flow
cytometer (BD Biosciences) was used to monitor the intracellular
accumulation of ROS. Data were acquired by flow cytometry (Beckman
Coulter, Inc.). Fluorescence analyses were performed on Coulter
Epics XL Flow Cytometer (Beckman Coulter, Inc.) using Expo32-ADC
software. In order to monitor the activities of antioxidant
enzymes, the treated cells were homogenized and centrifuged at
13,000 × g for 5 min at 4°C for supernatants. The levels of SOD,
MDA and CAT in the supernatants were measured according to the
instructions provided with the respective kits (Nanjing Jiancheng
Bioengineering Institute). All the analyses were performed in six
samples, each of them in triplicate.
Statistical analysis
All data are expressed as the mean ± SD. All assays
were repeated at least three times independently. The Mann-Whitney
U test was used to assess independent pairs, and one-way analysis
of variance followed by Tukey's post hoc test were used to assess
multiple samples. Statistical analyses were conducted using SPSS
software version 11.5 (SPSS, Inc.). P<0.05 was considered to
indicate a statistically significant difference.
Results
CFA activates Nrf2 in primary
chondrocytes and knee cartilage
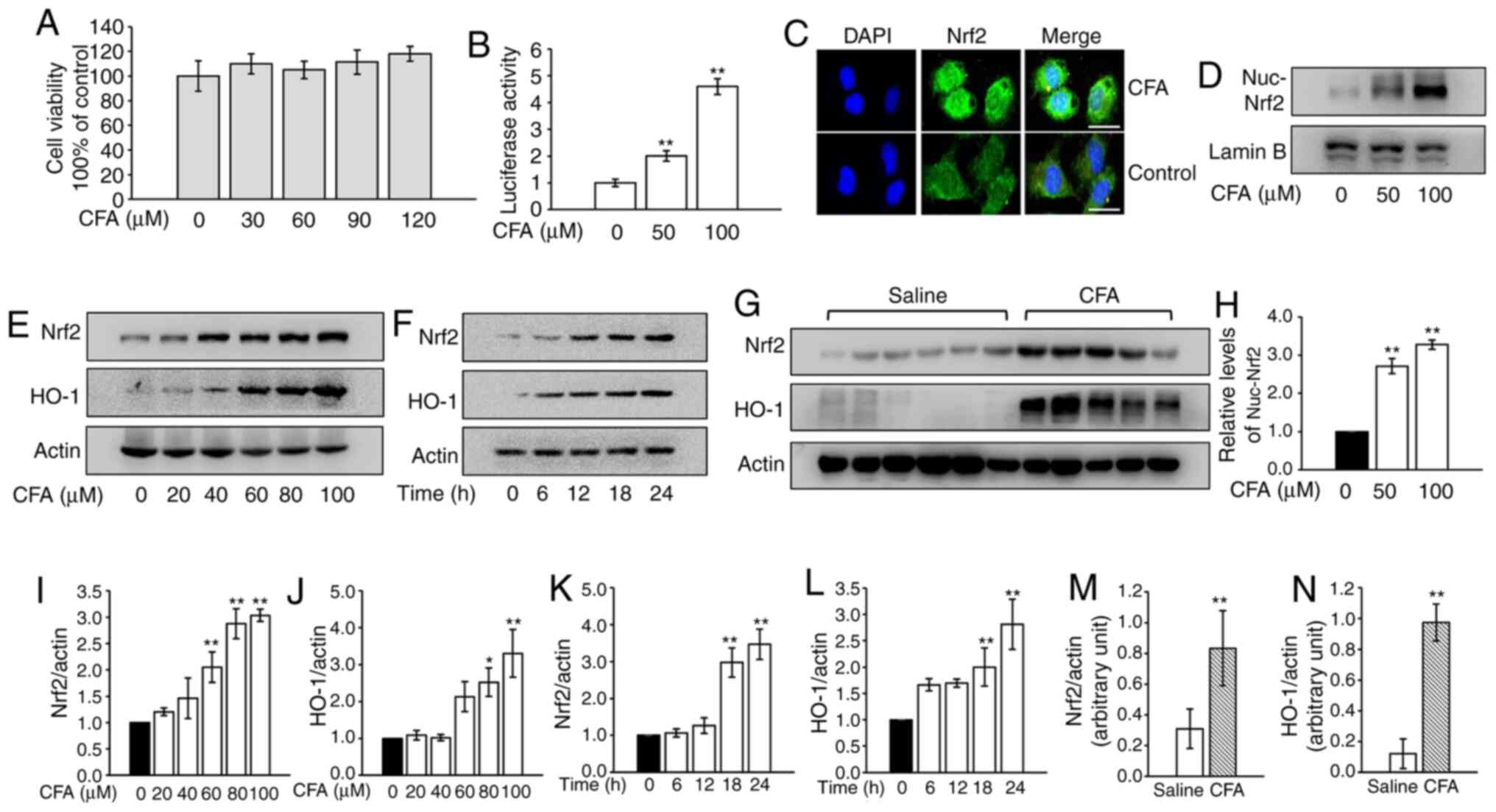
The cytotoxicity of CFA was analyzed in primary
murine chondrocytes and concentrations <120 µM were used in
subsequent assays (Fig. 1A). CFA
treatment led to a significant increase in luciferase activity in a
dose-dependent manner (Fig. 1B).
The results of the immunofluorescence assays revealed that CFA
treatment promoted the nuclear translocation of Nrf2 (Fig. 1C). Consistently, nuclear protein was
extracted and assayed for Nrf2 by WB analysis. CFA significantly
upregulated the expression of nuclear Nrf2 (Fig. 1D and H). The protein expression of
Nrf2 and its downstream protein, HO-1, in primary mouse
chondrocytes steadily increased following CFA treatment in a dose-
and time-dependent manner (Fig. 1E, F
and I-L). To investigate the effects of CFA on Nrf2 and HO-1
expression in the cartilage of knee joints, mice were treated with
CFA (0.2 mmol kg/day) via gastric gavage for 2 weeks and cartilage
samples were harvested for use in WB analysis. The results revealed
that expression of Nrf2 and HO-1 in cartilage were significantly
higher in the CFA-treated group (Fig.
1G, M and N).
CFA regulates the activities of
antioxidant markers and apoptosis in chondrocytes
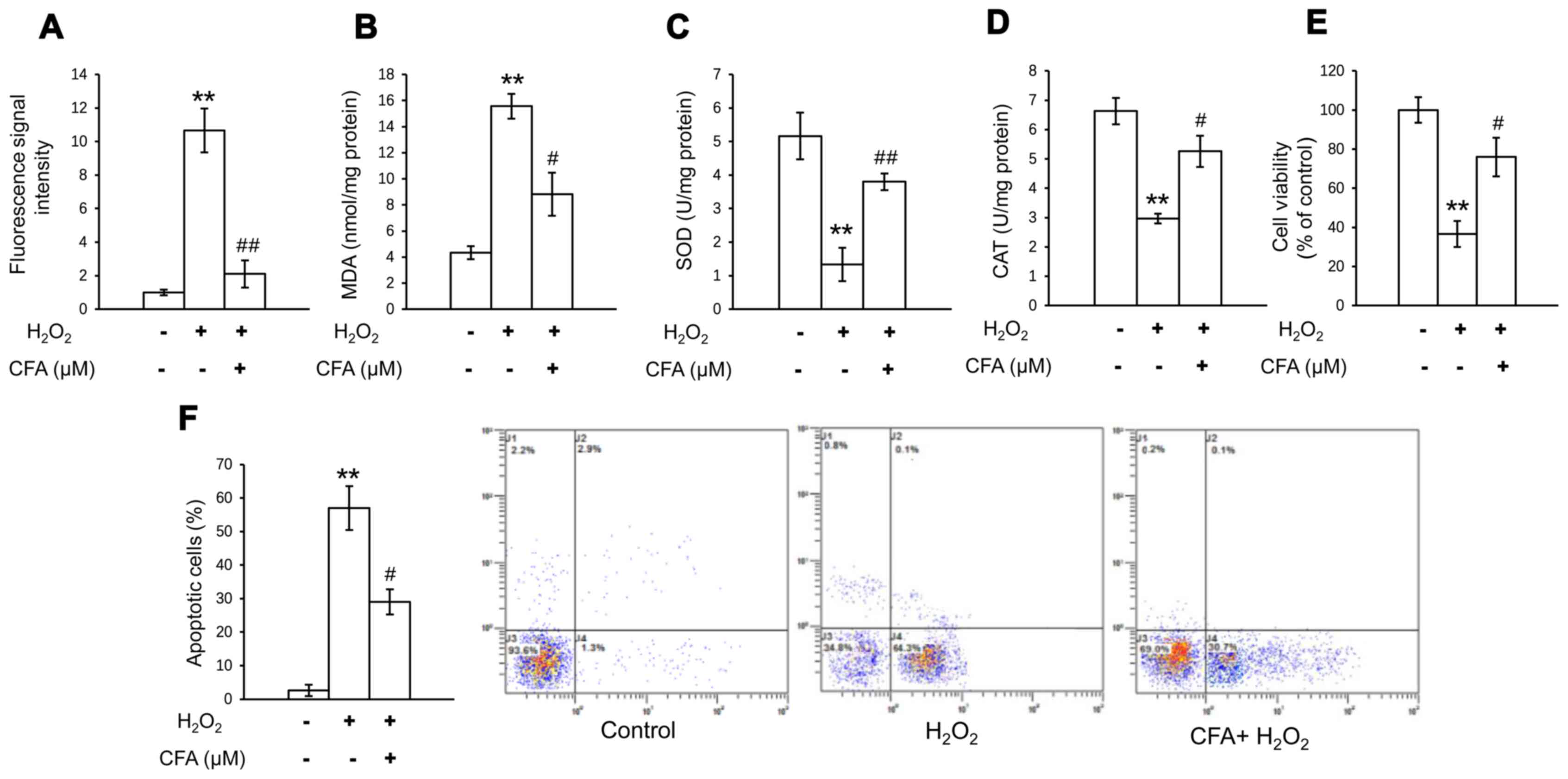
In order to investigate the antioxidative effects of
CFA in primary chondrocytes, oxidative stress was determined by
measuring the activities of CAT, MDA and SOD, and the level of
intracellular ROS. The H2O2-induced increase
in the MDA and ROS levels was significantly inhibited following 24
h of CFA treatment (Fig. 2A and B).
In addition, the suppressed activity of SOD and CAT was evidently
restored by treatment with CFA (Fig. 2C
and D). These results suggested that CFA exerted a potent
ROS-eliminating effect, protecting primary chondrocytes against
oxidative stress-induced damage. It was also found that CFA
reversed the decrease in cell viability and the increase in the
apoptotic rate, which were induced by exposure of the cells to
H2O2 (Fig. 2E and
F). This suggested that CFA exerted protective effects against
cartilage damage by scavenging the overproduction of ROS, which
causes the apoptosis of chondrocytes.
CFA alleviates cartilage destruction
in experimental OA
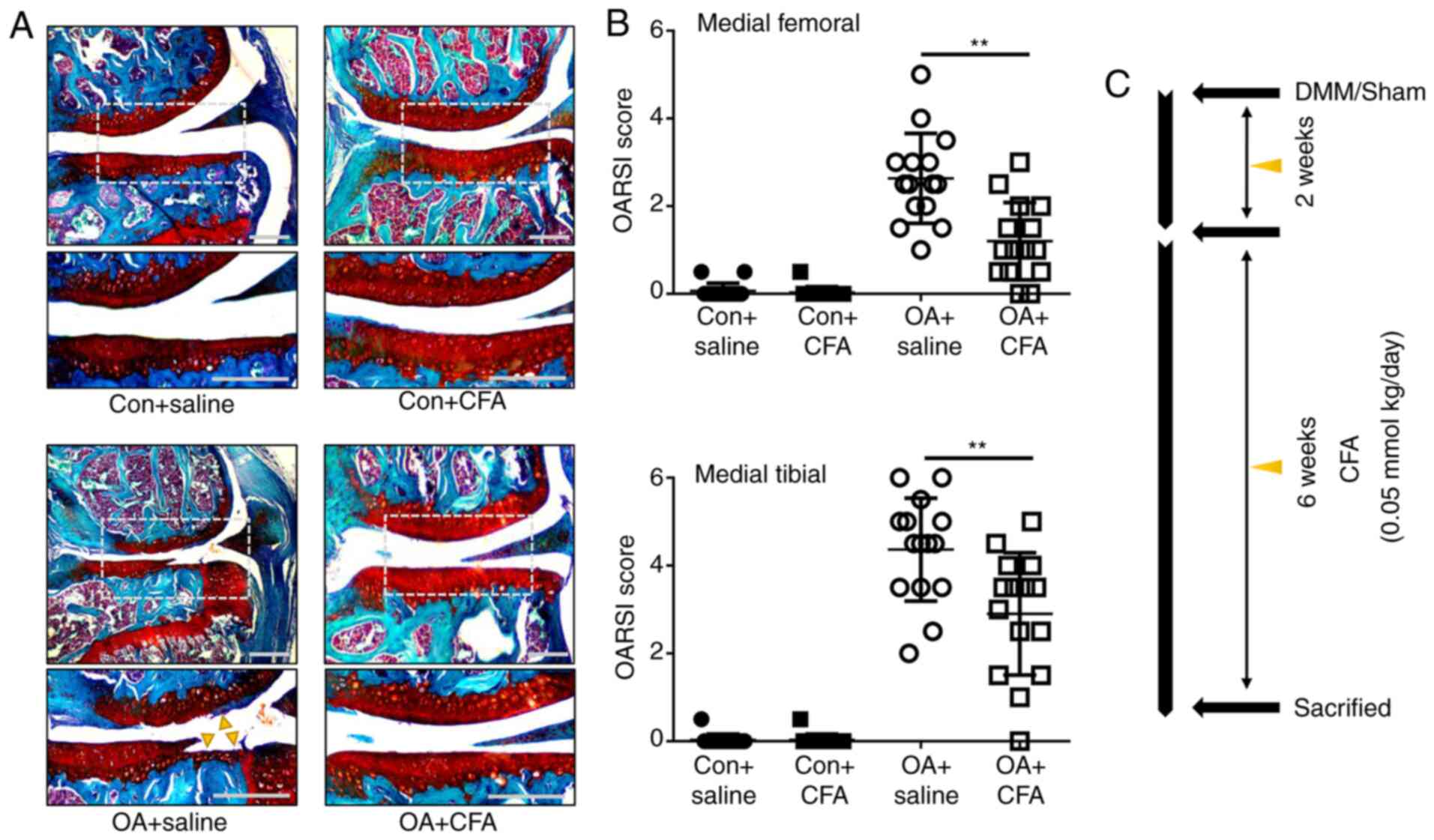
To determine the protective effects of CFA on OA, a
murine model of OA was established by transecting the medial
meniscotibial ligament of mice, followed by treatment with either
CFA (0.05 mmol/kg/day) or the same amount of the vehicle for 6
weeks (Fig. 3C). Histological
sections stained with Safranin O/Fast Green were assessed using
OARSI scores in a blinded manner. The vehicle-treated (OA + saline)
group displayed notable proteoglycan loss, and cartilage erosion or
cartilage damage at 8 weeks post-DMM surgery (Fig. 3A). The OARSI scores of the
vehicle-treated group reached as high as 2.63±0.10 for the femur
and 4.40±1.33 for the tibia, while the CFA-treated group exhibited
significantly improved scores, with 1.20±0.85 for the femur and
3.03±1.45 for the tibia (Fig. 3B).
These data demonstrated that CFA suppressed the observed
degradation of cartilage, thus attenuating the progression of
OA.
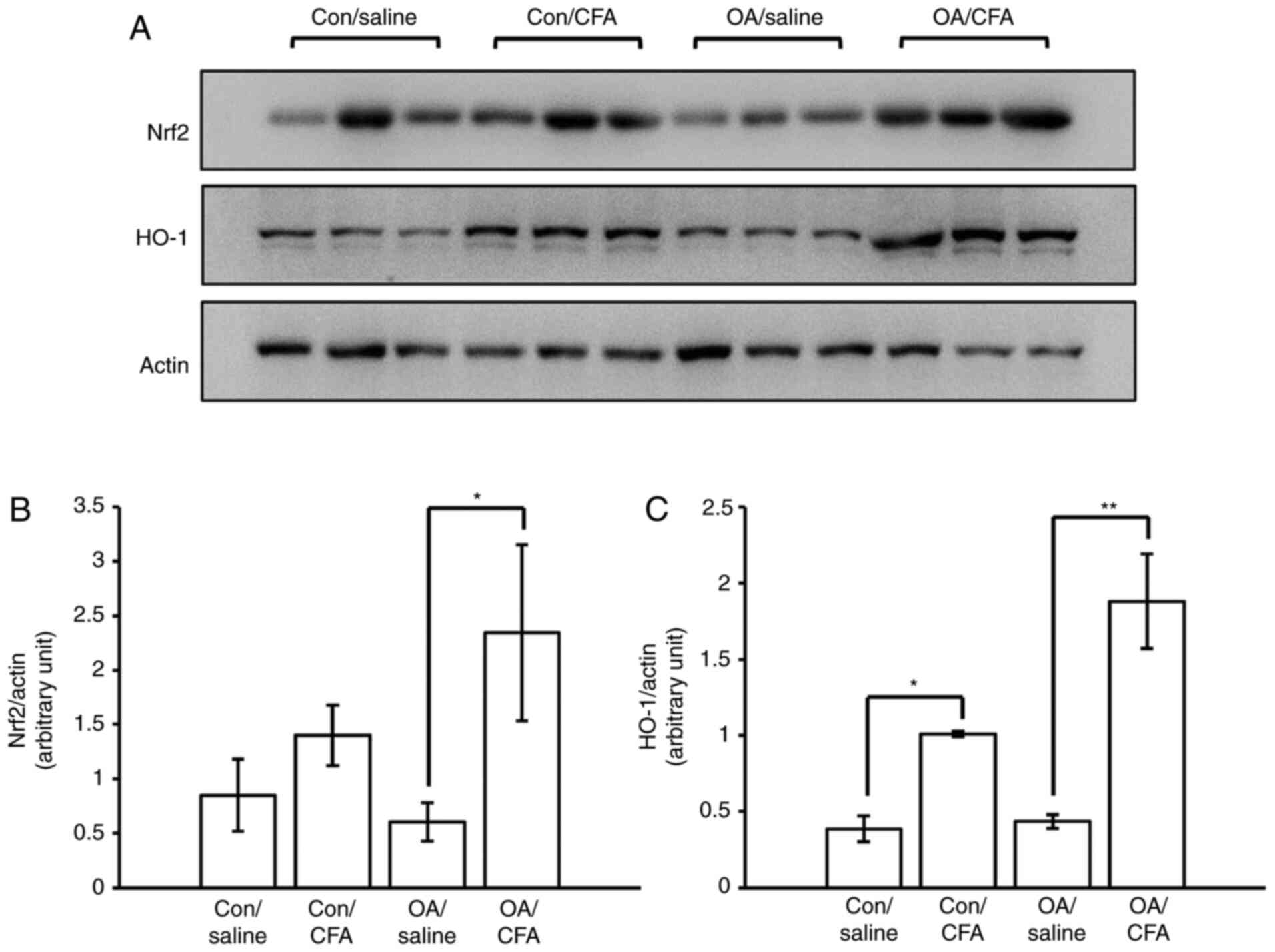
CFA promotes the expression of
Nrf2/HO-1 in experimental OA
The levels of Nrf2 and HO-1 in the cartilage from
knee joints were measured by WB analysis to ascertain whether the
regulation of Nrf2/HO-1 in response to CFA was the same as that
observed in vitro. To obtain sufficient proteins for WB
analysis, pooling from 32 joints was performed. The results
revealed that the expression levels of Nrf2/HO-1 were upregulated
in the cartilage from the knee joints of the murine model of OA
induced by surgery following the systematic oral administration of
CFA (Fig. 4A-C), suggesting that
CFA may confer protection against OA in mice via the activation of
the Nrf2/HO-1 pathway.
CFA regulates OA-associated mRNA
expression
To further elucidate the mechanisms underlying the
chondroprotective effects of CFA in vivo, the effects of CFA
on the expression of inflammatory and catabolic factors in
articular cartilage were further explored. The results revealed
that the protein expression levels of matrix metalloproteinase
(MMP)1, MMP13, TNF-α and interleukin (IL)-1 were significantly
increased in the OA group compared with the control group, and the
upregulated gene expression was suppressed, while the expression
levels of both MMP3 and IL-6 were not regulated by CFA (Fig. 5A-F).
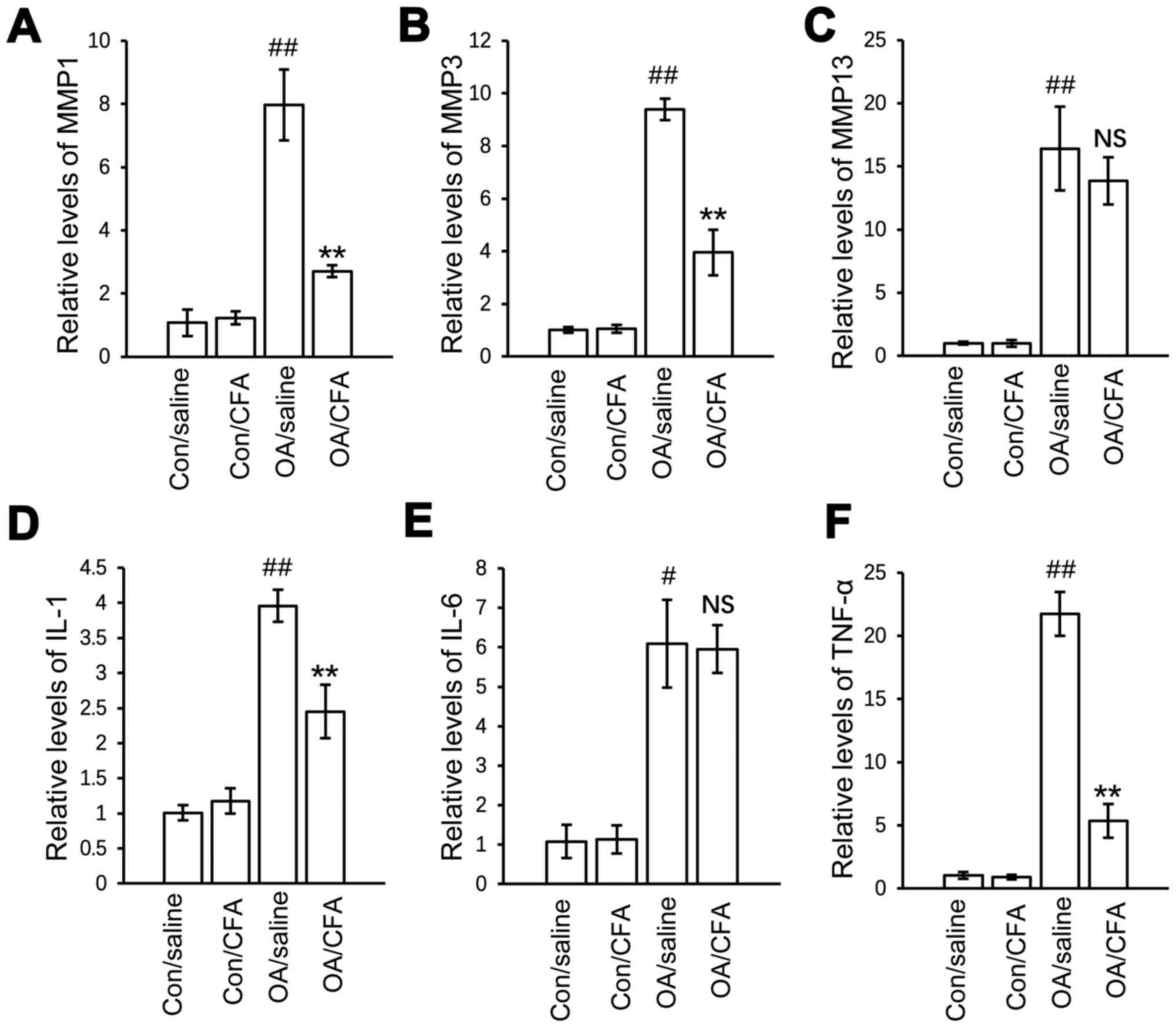 | Figure 5.Effects of CFA on the expression of
OA-related genes in experimental osteoarthritis. A total of 24
knees harvested from 12 mice were used for gene transcript
analysis. There were a total of four groups: Control knees from
saline-treated mice, control knees from CFA-treated mice, OA knees
from saline-treated mice, OA knees from CFA-treated mice. mRNA
expression of (A) MMP1, (B) MMP3, (C) MMP13, (D) IL-1, (E) IL-6 and
(F) TNF-α in the cartilage from the four groups of knee joints
quantified by reverse transcription-quantitative PCR.
#P<0.05, ##P<0.01 vs.
con/saline-treated mice; **P<0.01 vs. OA/saline-treated mice.
CFA, coniferaldehyde; OA, osteoarthritis; MMP, matrix
metalloproteinase; IL-, interleukin. |
Discussion
Nrf2 is a crucial transcriptional regulator
mediating protection against oxidative stress. In the resting
state, Nrf2 is tethered to its inhibitor in the cytoplasm,
Kelch-like ECH-associated protein 1 (Keap1), which regulates its
proteasomal degradation (15). When
exposed to oxidative stress, Nrf2 is released from Keap1 and
translocates to the nucleus, binding to the antioxidant response
elements (AREs) located in the promoter regions to regulate the
expression of a variety of antioxidant enzymes, thereby providing
inducible antioxidant defense. The CFA-induced nuclear
translocation of Nrf2, the activation of the HO-1 promoter
transactivation activity and the upregulation of its downstream
gene expression were all observed in the present study, which
indicated that CFA activated the Nrf2 signaling pathway in
vivo and in vitro.
Trichostatin A (a histone deacetylase inhibitor) was
previously used to induce Nrf2 activation and HO-1 expression,
subsequently resulting in the decreased severity of cartilage
destruction in mice with OA (8).
However, considering the side-effects caused by trichostatin A,
potential drugs targeting Nrf2 activation are required. Dong et
al (16) treated mice with CFA
(0.2 mmol/kg/day) orally for >6 months and the results of
immunohistochemistry indicated that CFA upregulated Nrf2 expression
in brain tissues. Since drugs given by oral route usually have low
bioavailability due to first pass metabolism, the dose of CFA was
adjusted for peritoneal route of administration in the present
study. The activation of Nrf2 was observed by WB analysis in the
cartilage from the knee joints of mice that were administered CFA
(0.05 mmol/kg/day) by peritoneal injection for 2 weeks. These
results demonstrated that Nrf2 activation serves as an important
mechanism for the chondroprotective effects of CFA.
Notably, CFA has been reported to induce the growth
of SH-SY5Y cells within the range of 30–160 µM (16). The present study evaluated cell
viability using a CCK-8 assay, and no increase in cell viability
was observed with concentrations between 0 and 120 µM CFA (Fig. 1A). These differing results may
depend on the cell type used. ROS are usually considered to be a
toxic by-product of cell metabolism. A related study found that
increased mitochondrial ROS production can trigger chondrocyte
death and promote the expression of pro-inflammatory cytokines,
leading to the onset and progression of OA (17). On the other hand, the inhibition of
ROS production can attenuate the degeneration of cartilage in OA
(18,19). H2O2 is one of
the main types of ROS, which are spontaneously converted to the
highly reactive hydroxyl radicals, inducing DNA damage and cell
death. In the present study, chondrocytes exposed to
H2O2 exhibited a significant increase in
apoptosis with a decreased cell viability. Treatment with CFA
alleviated H2O2-induced apoptosis and
increased the viability of primary chondrocytes in the present
study, improving mitochondrial function by eliminating the
overproduction of ROS induced by H2O2, and
thereby conferring protection against oxidative stress during
OA.
Although there was no significant statistical
difference in the expression of both Nrf2 and HO-1 between the
OA/CFA group and the Control/CFA group (may be due to the limited
number of samples), it was speculated that the level of HO-1, a
member of heat shock protein (HSP) family, which is produced by
cells in response to exposure to stressful conditions, may be much
higher in the OA/CFA group compared with the Control/CFA group. In
our previous study, it was found that the level of HO-1 in knee
cartilage significantly increased in both patients with OA and
DMM-induced OA mice relative to controls, which may be regarded as
a beneficial adaptive response to oxidative stress in chondrocytes
during the progression of OA (20).
These findings suggested that the development of OA also
contributed to the increased expression of HO-1 in the OA/CFA
group, and the very obvious upregulation of HO-1 induced by CFA may
play a key role in delaying the progression of OA.
HO degrades heme to free iron, biliverdin and carbon
monoxide (21). The authors
previously reported that HO-1 expression is increased in cartilage
from patients diagnosed with OA, which is regarded as an adaptive
response against stress during cartilage damage (20). The induction of HO-1 suppresses the
IL-1-induced expression of MMPs and inflammatory factors (22,23),
which are abnormally enhanced in OA-affected cartilage. In
agreement with these studies, the present study found that the
expression of several matrix-degrading enzyme genes, including
MMP1, MMP3, IL-1 and TNF-α, were significantly inhibited in
cartilage from the mice subjected to DMM surgery at 8 weeks
post-CFA treatment. Although a previous study stated that the
upregulation of HO-1 induced by cobalt protoporphyrin IX suppressed
the expression of IL-6 in human tracheal smooth muscle cells
(24), the present study did not
observe a significant effect of CFA induced-HO-1 on the regulation
of IL-6 in cartilage. On the whole, this evidence suggested that
CFA also plays an important regulatory role in anti-inflammation
and anti-catabolism during OA.
To the best of our knowledge, the present study is
the first to demonstrate the cartilage-protective role of CFA in an
animal model of OA. CFA activated the Nrf2/HO-1 signaling pathway
to exert potent anti-apoptotic and anti-inflammatory effects on
cartilage. The present study suggested that the use of CFA may
represent a promising treatment strategy for OA.
Acknowledgements
Not applicable.
Funding
The present study was subsidized by a grant from the
Natural Science Foundation of the Jiangsu Higher Education
Institutions of China (grant no. 18KJB320009) for financial
support.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DC, JW and JQ conceived and designed the study. DC,
JW, SC, LJ, JC and JW performed the experiments. DC and JW confirm
the authenticity of all the raw data. DC and JW wrote the
manuscript. JQ reviewed and edited the manuscript. All authors read
and approved the manuscript.
Ethics approval and consent to
participate
All experiments related to the use of animals were
approved (approval no. IACUC 1901062) by the Institutional Animal
Care and Use Committee of The Sir Run Run Hospital of Nanjing
Medical University (Nanjing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bihlet AR, Byrjalsen I, Bay-Jensen AC,
Andersen JR, Christiansen C, Riis BJ and Karsdal MA: Associations
between biomarkers of bone and cartilage turnover, gender, pain
categories and radiographic severity in knee osteoarthritis.
Arthritis Res Ther. 21:2032019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang Y, Hussain SM, Wluka AE, Lim YZ,
Abram F, Pelletier JP, Martel-Pelletier J and Cicuttini FM:
Association between metformin use and disease progression in obese
people with knee osteoarthritis: Data from the osteoarthritis
Initiative-a prospective cohort study. Arthritis Res Ther.
21:1272019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu H, Zhao G, Xia F, Liu X, Gong L and Wen
X: The diagnosis and treatment of knee osteoarthritis: A literature
review. Int J Clin Exp Med. 12:4589–4599. 2019.
|
|
4
|
Lepetsos P and Papavassiliou AG:
ROS/oxidative stress signaling in osteoarthritis. Biochim Biophys
Acta. 1862:576–591. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khan NM, Haseeb A, Ansari MY, Devarapalli
P, Haynie S and Haqqi TM: Wogonin, a plant derived small molecule,
exerts potent anti-inflammatory and chondroprotective effects
through the activation of ROS/ERK/Nrf2 signaling pathways in human
osteoarthritis chondrocytes. Free Radic Biol Med. 106:288–301.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Loboda A, Damulewicz M, Pyza E, Jozkowicz
A and Dulak J: Role of Nrf2/HO-1 system in development, oxidative
stress response and diseases: An evolutionarily conserved
mechanism. Cell Mol Life Sci. 73:3221–3247. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhao K, Li Y, Wang Z, Han N and Wang Y:
Carnosine protects mouse podocytes from high glucose induced
apoptosis through PI3K/AKT and Nrf2 pathways. Biomed Res Int.
2019:43489732019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cai D, Yin S, Yang J, Jiang Q and Cao W:
Histone deacetylase inhibition activates Nrf2 and protects against
osteoarthritis. Arthritis Res Ther. 17:2692015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kim KM, Heo DR, Kim YA, Lee J, Kim NS and
Bang OS: Coniferaldehyde inhibits LPS-induced apoptosis through the
PKC α/β II/Nrf-2/HO-1 dependent pathway in RAW264.7 macrophage
cells. Environ Toxicol Pharmacol. 48:85–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gosset M, Berenbaum F, Thirion S and
Jacques C: Primary culture and phenotyping of murine chondrocytes.
Nat Protoc. 3:1253–1260. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Glasson SS, Blanchet TJ and Morris EA: The
surgical destabilization of the medial meniscus (DMM) model of
osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage.
15:1061–1069. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Glasson SS, Chambers MG, Van Den Berg WB
and Little CB: The OARSI histopathology initiative-recommendations
for histological assessments of osteoarthritis in the mouse.
Osteoarthritis Cartilage. 18 (Suppl 3):S17–S23. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cai D, Feng W, Liu J, Jiang L, Chen S,
Yuan T, Yu C, Xie H, Geng D and Qin J: 7,8-Dihydroxyflavone
activates Nrf2/HO-1 signaling pathways and protects against
osteoarthritis. Exp Ther Med. 18:1677–1684. 2019.PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giudice A, Arra C and Turco MC: Review of
molecular mechanisms involved in the activation of the Nrf2-ARE
signaling pathway by chemopreventive agents. 647. Higgins P:
Transcription Factors. Methods in Molecular Biology (Methods and
Protocols). Springer, Humana Press; Totowa, NJ: pp. 37–74. 2010,
View Article : Google Scholar
|
|
16
|
Dong Y, Stewart T, Bai L, Li X, Xu T,
Iliff J, Shi M, Zheng D, Yuan L, Wei T, et al: Coniferaldehyde
attenuates Alzheimer's pathology via activation of Nrf2 and its
targets. Theranostics. 10:179–200. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim J, Xu M, Xo R, Mates A, Wilson GL,
Pearsall AW IV and Grishko V: Mitochondrial DNA damage is involved
in apoptosis caused by pro-inflammatory cytokines in human OA
chondrocytes. Osteoarthritis Cartilage. 18:424–432. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Henrotin YE, Bruckner P and Pujol JP: The
role of reactive oxygen species in homeostasis and degradation of
cartilage. Osteoarthritis Cartilage. 11:747–755. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Henrotin Y, Kurz B and Aigner T: Oxygen
and reactive oxygen species in cartilage degradation: Friends or
foes? Osteoarthritis Cartilage. 13:643–654. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cai D, Huff TW, Liu J, Yuan T, Wei Z and
Qin J: Alleviation of cartilage destruction by sinapic acid in
experimental osteoarthritis. Biomed Res Int. 2019:56896132019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fernández P, Guillén MI, Gomar F and
Alcaraz MJ: Expression of heme oxygenase-1 and regulation by
cytokines in human osteoarthritic chondrocytes. Biochem Pharmacol.
66:2049–2052. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rousset F, Nguyen MVC, Grange L, Morel F
and Lardy B: Heme oxygenase-1 regulates matrix metalloproteinase
MMP-1 secretion and chondrocyte cell death via Nox4 NADPH oxidase
activity in chondrocytes. PLoS One. 8:e664782013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee TS and Chau LY: Heme oxygenase-1
mediates the anti-inflammatory effect of interleukin-10 in mice.
Nat Med. 8:240–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee IT, Luo SF, Lee CW, Wang SW, Lin CC,
Chang CC, Chen YL, Chau LY and Yang CM: Overexpression of HO-1
protects against TNF-alpha-mediated airway inflammation by
down-regulation of TNFR1-dependent oxidative stress. Am J Pathol.
175:519–532. 2009. View Article : Google Scholar : PubMed/NCBI
|