Introduction
Adenoids are lymphatic tissues of the nasopharynx
located at the junction of the posterior wall of the nasopharynx
and between the pharyngeal recesses on both sides. Adenoids exist
from birth, and can gradually shrink after 8–10 years of age under
normal physiological conditions (1). Adenoid hypertrophy (AH) is caused when
the adenoid is stimulated by repeated inflammation, thus resulting
in its pathological hypertrophy (2). AH is a common pediatric disease that
may cause a series of local and systemic symptoms, such as nasal
congestion, snoring, mouth breathing and obstructive sleep apnea.
These symptoms may lead to hypoxia and carbon dioxide retention,
thereby affecting children's intellectual development (3). AH can also directly cause mandibular
dysplasia, leading to an adenoid facial appearance (4).
The main cause of AH is mainly bacterial virus
infection, but there are also other factors, such as allergen
stimulation. AH is the hypertrophy of lymphoid tissue (loose
connective tissue) and lymphatic follicles, or may also be
hypertrophy of mucosal epithelial cells (5). The hypertrophy process is also
accompanied by increased numbers of various lymphocytes, which
further aggravates the stimulation. The inflammatory stimulation of
epithelial cells on the surface of adenoids can directly or
indirectly cause rhinitis and sinusitis (6). Inflammatory products that invade the
eustachian tube on both sides can cause otitis media (7). However, the related underlying
mechanisms of adenoid epithelial cell hypertrophy have not been
fully elucidated.
IL-32 is a pro-inflammatory cytokine produced by T
lymphocytes, natural killer cells, monocytes and epithelial cells,
and its association with airway inflammation has demonstrated in a
number of studies (8). For example,
IL-32 was identified as an important cytokine involved in the
inflammation of allergic rhinitis (AR) (9). Inhibition of IL-32 signaling was shown
to attenuate AR in both cellular and animal models (10,11).
Elevated levels of IL-32 were also found to play a role in the
pathogenesis of rhinosinusitis through its role as a
pro-inflammatory cytokine (12).
However, whether IL-32 is involved in AH remains to be
elucidated.
IL-32 can exert its pro-inflammatory role via
numerous pathways or mediators, including p38, MAPK, NF-κB,
caspase-1 and caspase-3 (13). In
fact, these pathways are interrelated. For example, IL-32 can
activate the NF-κB pathway by increasing nucleotide-binding
oligomerization domain-containing protein (NOD) 1 and NOD2
expression, therefore promoting NACHT LRR and PYD
domains-containing protein 3 (NLRP3) activation and IL-1β
production (14). Exogenous
activation of NOD1 and NOD2 can also in turn promote the secretion
of IL-32, stimulate the activation and release of IL-1β through the
caspase-1 pathway and thus further mediate the progression of
inflammation (15). Notably,
NLRP3-mediated caspase-1 activation was recently confirmed to
trigger cell pyroptosis, a form of programmed cell death (16). Moreover, the NLRP3 inflammasome was
implicated to promote the development and progression of AR by
enhancing inflammatory responses and epithelium pyroptosis
(17).
The present study demonstrated IL-32 expression in
the adenoid tissues of patients with AH. IL-32 stimulation
significantly promoted the production of pro-inflammatory cytokines
and cell pyroptosis in human nasal epithelial cells. Meanwhile,
silencing of IL-32 significantly inhibited LPS-induced inflammation
and pyroptosis.
Materials and methods
Sample collection
The study cohort in the present investigation
consisted of 10 children (age range, 6–12 years; 4 female patients
and 6 male patients) diagnosed with AH in Tianjin Children's
Hospital (Tianjin, China) between February 2019 and June 2019. The
diagnosis of AH was based on patient history as well as physical
and endoscopic examinations. The normal group consisted of 10
healthy children without AH, but needed required operation on the
adenoid due to other reasons. Tissue was collected from the adenoid
during surgical operation and immediately stored in liquid nitrogen
before use. Parents of the children were informed about the study
and signed consent was obtained. The study was approved by the
Ethics Committee of Tianjin Children's Hospital (approval no.
IACUC-20190111-13).
Cell culture and treatment
Human nasal epithelial cells (HNEpC; RPMI 2650,
ATCC-CCL-30; http://www.atcc.org/products/all/CCL-30.aspx), which
exhibit epithelioid morphology and have been largely utilized in
previous studies as the nasal epithelial cell (18–21),
were purchased from American Type Culture Collection. Cells were
cultured in DMEM (Invitrogen; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 µg/ml streptomycin and 100 U/ml penicillin
in an incubator at 37°C.
Cells were cultured overnight to reach 80–90%
confluence before transfection. IL-32 small interfering RNA (siRNA)
(cat. no. sc-60841) and control siRNA-A (cat. no. sc-37007) were
purchased from Santa Cruz Biotechnology, Inc. Transfection was
performed using Lipofectamine® 2000 transfection reagent
(Invitrogen; Thermo Fisher Scientific, Inc.). All experiments were
performed in strict accordance with the manufacturer's
instructions. At 48 h post-transfection, the cells were selected
for subsequent experiments.
Cell Counting Kit-8 (CCK-8) assay
The cell proliferation of HNEpCs exposed to
different concentrations (0, 2, 10 and 50 ng/ml) of recombinant
human IL-32 (R&D Systems, Inc.) for 12, 24 and 48 h (22), as well as HNEpCs transfected with
IL-32 siRNA or siRNA-negative control (NC) in the presence of
lipopolysaccharide (LPS; 1 µg/ml; Beijing Solarbio Science &
Technology Co., Ltd.) stimulation for 12, 24 and 48 h, were
determined using a CCK-8 assay (cat. no. HY-K0301; MedChemExpress),
according to the manufacturer's instructions.
Hematoxylin and eosin (H&E)
staining
Adenoid tissues were fixed with 10% formalin
(Beyotime Institute of Biotechnology) at 4°C for 24h and embedded
in paraffin following alcohol dehydration. The tissues were sliced
at a thickness of 4 mm according to the manufacturer's protocol
(Leica Autostainer XL; Leica Microsystems, Inc.) and stained with
H&E at room temperature for 2 min (Thermo Fisher Scientific,
Inc.). Stained samples were observed under light microscopy
(magnification, ×200; Olympus FV-1mm; Olympus Corporation).
Immunohistochemical staining
For immunohistochemical analysis, the tissue slides
were dewaxed in xylene, rehydrated using graded ethanol, and then
heated to expose antigenic sites. Following antigenic retrieval,
the sections were incubated overnight at 4°C with diluted rabbit
polyclonal antibody against IL-32 (cat. no. ab37158; Abcam;
1:1,000) at a concentration of 10 µg/ml. An HRP-conjugated
anti-rabbit secondary antibody (cat. no. ab6721; Abcam; 1:1,000)
were used and detected with peroxidase-labelled streptavidin, both
incubated for 10 min at room temperature. Immunoreactivity was
visualized by incubating the sections for 2 min in 0.1%
3,3′-diaminobenzidine (Beyotime Institute of Biotechnology). The
sections were observed under a light microscope (Carl Zeiss AG) and
photographed with a digital camera (AxioCam MRc5; Zeiss AG). IL-32
expression is shown by a brown or dark brown stain, while blue
staining indicates negative expression. The darker the brown color,
the higher the expression of IL-32.
Flow cytometry
Cell apoptosis was measured using flow cytometry
analysis based on an Annexin V-FITC Apoptosis Detection kit
(Sigma-Aldrich; Merck KGaA) according to the manufacturer's
protocol. In brief, HNEpCs and cells transfected with siRNA-IL-32
were seeded at a density of 1×105 cells/well in a
six-well culture plate and treated with IL-32 or LPS. The cells
were then harvested with 0.25% trypsin, washed twice with cold PBS
and suspended in 500 µl binding buffer that was included in the
kit. The cells were then incubated with 500 µl Annexin V and 500 µl
propidium iodide reagent in the dark for 20 min. Analysis was
performed using a BD FACSAria flow cytometer (Becton, Dickinson and
Company) and iSort Automated Cell Sorter software (version A.0;
Thermo Fisher Scientific, Inc.).
Immunofluorescence (IF) staining
The expression of cleaved gasdermin D (GSDMD) was
detected by IF staining. In brief, cells were exposed to 4%
paraformaldehyde for 20 min and then blocked with 1% BSA (Gibco;
Thermo Fisher Scientific, Inc.) and 0.1% Triton-X for 2 h at room
temperature. Subsequently, the cells were incubated with primary
antibody against the N-terminal of GSDMD (GSDMD-N; cat. no.
ab215203; 1:1,000; Abcam) at 4°C overnight, followed by incubation
with goat anti-rabbit IgG-H&L secondary antibody (cat. no.
ab150077; 1:500; Abcam) for 2 h at room temperature. The nuclei
were stained with DAPI (Beyotime Institute of Biotechnology) at
room temperature for 5 min. Stained cells were observed using a
fluorescence microscope.
ELISA
ELISA kits were used to measure the concentrations
of IL-1β (cat. no. ab217608; Abcam), IL-6 (cat. no. ab46027; Abcam)
and TNF-α (cat. no. ab181421; Abcam) as per the manufacturer's
instructions. Briefly, 50 µl samples or standards were added to
appropriate wells. This was followed by the addition of antibody
cocktails from the ELISA kits into each well and incubation at room
temperature for 1 h. Subsequently, 3,3′,5,5′-tetramethylbenzidine
substrate from the kits was added to each well and incubated for 10
min in the dark with shaking at 400 rpm. Following incubation with
stop solution, the optical density values of each well were
measured at a wavelength of 450 nm using a microplate reader.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from HNEpCs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RT to synthesize cDNA was performed using SuperScript™ III
Reverse Transcriptase (Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol. The PCR reaction system was prepared
using SYBR® Green Real-Time PCR Master mix (Thermo
Fisher Scientific, Inc). The following primers for IL-32 were used:
Forward, 5′-GGCTGAGTATTTGTGCCAGG-3′ and reverse,
5′-TATGGCCTGGTGCATTCGG-3′. The following thermocycling conditions
were used for qPCR: 95°C for 2 min, followed by 40 cycles of 95°C
for 20 sec and 65°C for 40 sec. The expression levels of IL-32 were
normalized to the endogenous control GAPDH (forward,
5′-AATGGGCAGCCGTTAGGAAA-3′ and reverse, 5′-AATGGGCAGCCGTTAGGAAA-3′)
using the 2−∆∆Cq method (23).
Western blotting
Total protein was extracted from adenoid tissues and
HNEpC cells using RIPA lysis buffer (Beyotime Institute of
Biotechnology). Following mixing with SDS lysis buffer and boiling,
the protein content in cell lysates were measured using BCA Protein
Assay reagent (Pierce; Thermo Fisher Scientific, Inc.). Equal
amounts of protein (30 µg) were loaded onto 12% SDS-PAGE and
separated via electrophoresis, then separated proteins were
transferred to PVDF membranes. Following blocking in 5% skimmed
milk at room temperature for 2 h, membranes were incubated
overnight with primary antibodies against IL-32 (cat. no. ab37158;
1:1,000; Abcam), NLRP3 (cat. no. ab263899; 1:1,000; Abcam), IL-1β
(cat. no. ab216995; 1:1,000; Abcam), caspase-1 (cat. no. ab207802;
1:1,000; Abcam), GSDMD-N (cat. no. ab215203; 1:1,000; Abcam), NOD1
(cat. no. ab189435; 1:1,000; Abcam), NOD2 (cat. no. ab31488; 1:500;
Abcam), Toll-like receptor 4 (TLR4; cat. no. ab13556; 1:500; Abcam)
and GAPDH (cat. no. ab8245; 1:5,000; Abcam). Membranes were then
incubated with goat anti-rabbit IgG (cat. no. ab6721; Abcam;
1:2,000) or goat anti-mouse IgG (cat. no. ab6789; Abcam; 1:2,000)
secondary antibodies at room temperature for 2 h. Proteins were
then visualized using a gel imaging system (Amersham; Cytiva).
Protein expression levels were semi-quantified using Image-Pro Plus
software (version 6.0; Media Cybernetics, Inc.).
Statistical analysis
All generated data were analyzed using SPSS software
(version 22.0; IBM Corp.). All experiments were repeated at least
three times. Data are presented as the mean ± SD. An unpaired
Student's t-test was conducted for two-group comparisons. One-way
ANOVA followed by Tukey's post hoc test was performed to compare
between three or more groups. P<0.05 was considered to indicate
a statistically significant difference.
Results
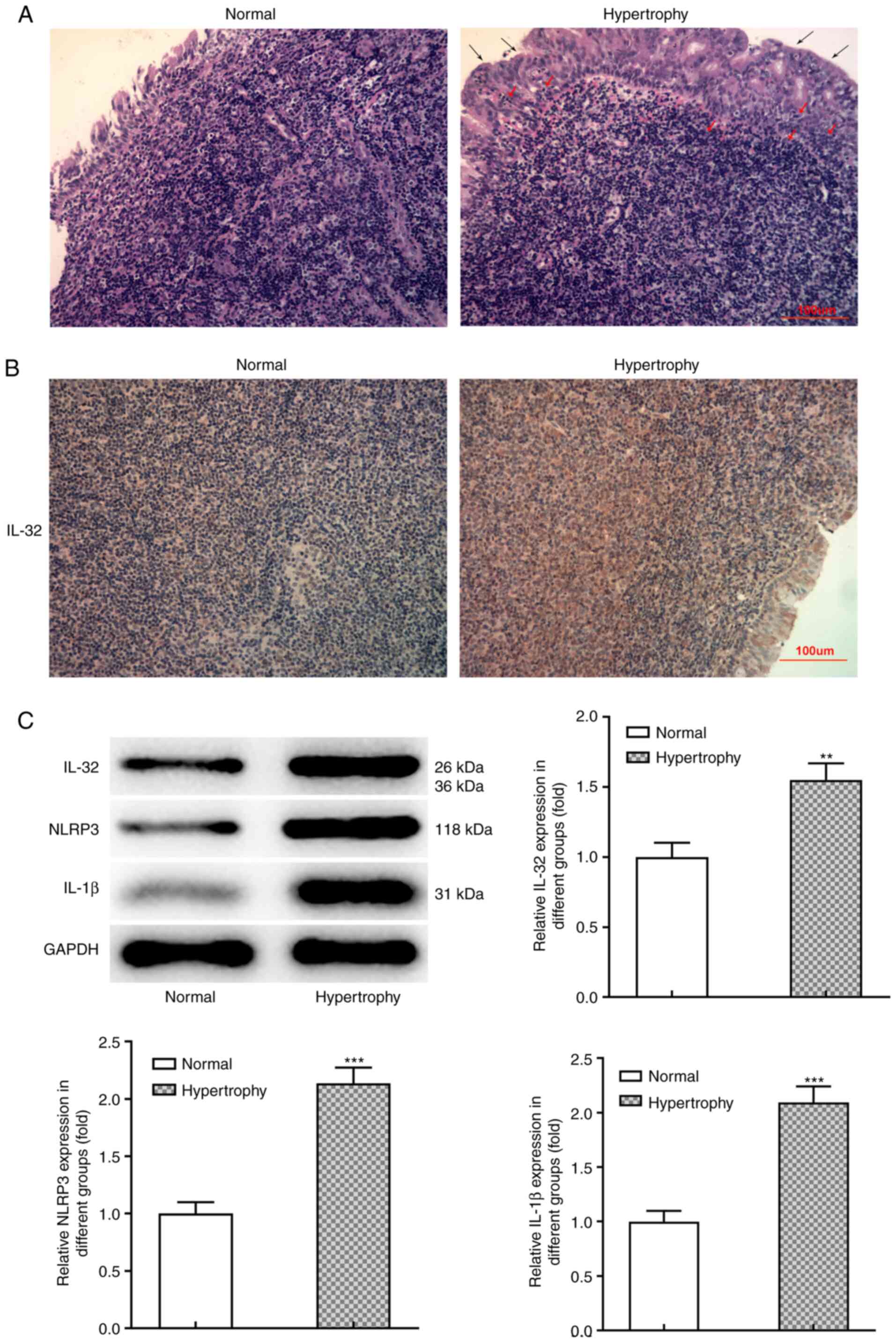
IL-32 is upregulated in adenoid
tissues with AH
Firstly, to determine whether IL-32 is involved in
AH, IL-32 expression in normal and hypertrophic adenoid tissues was
detected. Fig. 1A demonstrates the
typical pathological manifestation of hypertrophy and hyperplasia
in adenoid tissues from patients with AH. Compared with the normal
group, the obvious hypertrophy and lymphocyte infiltration can be
observed in the adenoid epithelial tissue of the hyperplasia group.
Immunohistochemical analysis (Fig.
1B) revealed that IL-32 expression was markedly upregulated in
hypertrophic adenoid tissues compared with normal tissues.
Consistently, western blot analysis confirmed higher protein
expression of IL-32 in hypertrophic adenoid tissues (Fig. 1C). Meanwhile, the protein expression
levels of NLRP3 and IL-1β, which both play important roles in
inflammation and pyroptosis, were also significantly increased in
hypertrophic adenoid tissues (Fig.
1C). These data indicated that IL-32, together with
inflammation and pyroptosis, may contribute to AH progression.
IL-32 induces apoptosis and
inflammation
Subsequently, HNEpCs were utilized as the cell model
and exposed to different concentrations (0, 2, 10 and 50 ng/ml) of
recombinant human IL-32. Fig. 2A
shows the cell proliferation rate at 12, 24 and 48 h post IL-32
treatment, revealing that 2 ng/ml IL-32 promoted cell proliferation
at 48 h post-treatment, while 10 ng/ml IL-32 significantly promoted
cell proliferation at 24 and 48 h post-treatment, owing to the
stressful cell proliferation caused by mild inflammation upon IL-1β
activation (24). Meanwhile, 50
ng/ml IL-32 significantly inhibited cell proliferation, which can
be explained by significant apoptosis or cell death (Fig. 2B and C) caused by 50 ng/ml IL-32
treatment. In the following experiments, cells were exposed to 2,
10 and 50 ng/ml for 48 h. The results shown in Fig. 2B and C revealed that 10 ng/ml IL-32
significantly increased the ratio of cell apoptosis, and 50 ng/ml
IL-32 resulted in an especially high ratio of apoptosis (nearly
6-fold of the control). As shown in Fig. 2D, IL-32 also increased the
production of pro-inflammatory cytokines, including TNF-α, IL-6 and
IL-1β, in a concentration-dependent manner. These results suggested
that IL-32 could induce apoptosis and inflammation in normal
HNEpCs.
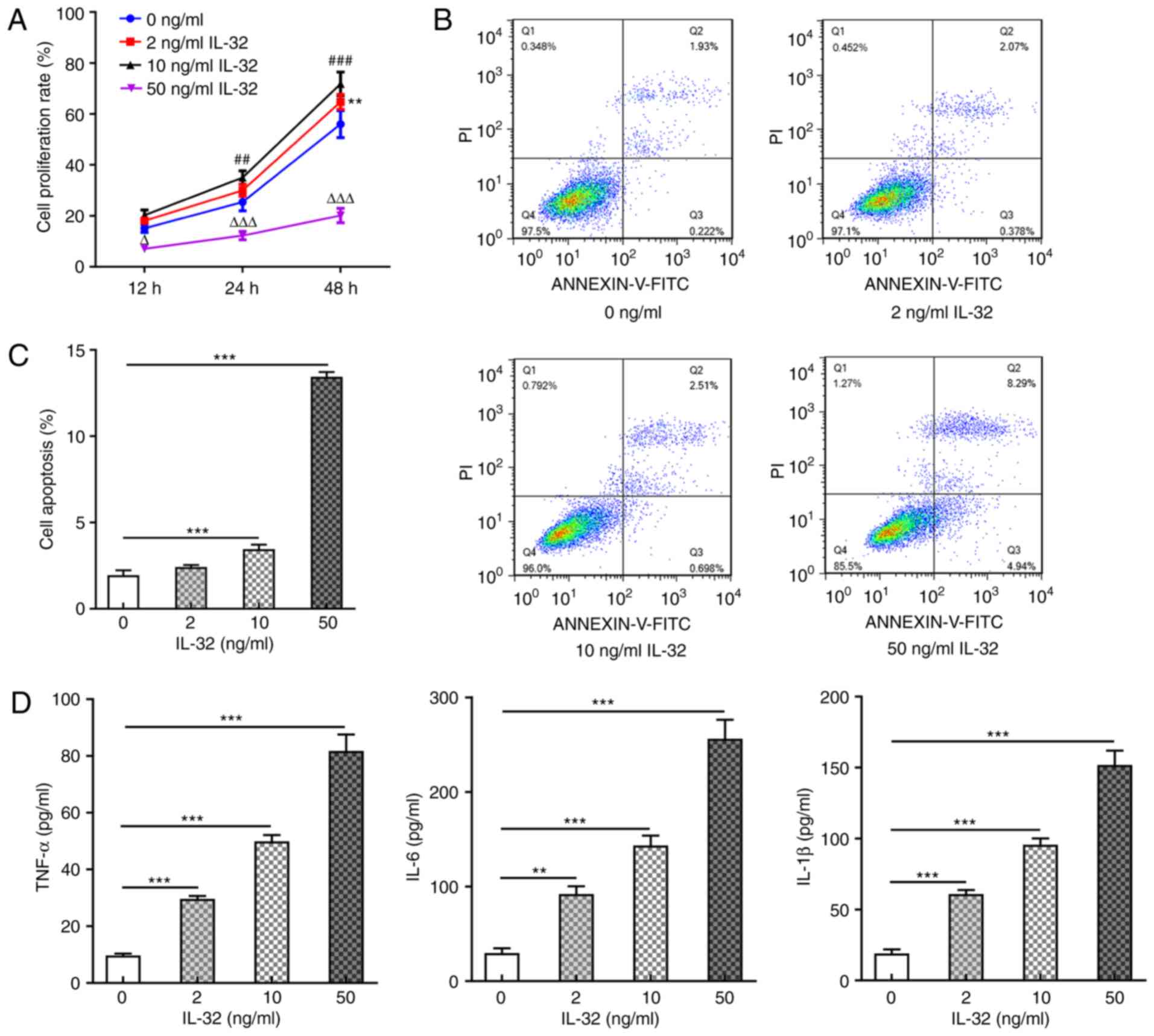 | Figure 2.Effects of IL-32 on HNEpC
proliferation, apoptosis and inflammation. (A) The cell
proliferation rate of HNEpCs exposed to 0, 2, 10 and 50 ng/ml IL-32
for 12, 24 and 48 h. **P<0.01, 2 vs. 0 ng/ml IL-32;
##P<0.01 and ###P<0.001, 10 vs. 0 ng/ml
IL-32; ∆P<0.05 and ∆∆∆P<0.001, 50 vs. 0
ng/ml IL-32. (B and C) The apoptosis ratio of HNEpCs exposed to 0,
2, 10 and 50 ng/ml IL-32 for 48 h was detected by flow cytometry.
***P<0.001 vs. 0 ng/ml IL-32. (D) The concentration of TNF-α,
IL-6 and IL-1β in the culture medium of HNEpCs exposed to 0, 2, 10
and 50 ng/ml IL-32 for 48 h was detected via ELISA. **P<0.01 and
***P<0.001, vs. 0 ng/ml IL-32. HNEpC, human nasal epithelial
cells. |
IL-32 promotes NLRP3-mediated
pyroptosis
Subsequently, the present study assessed whether
IL-32 could induce pyroptosis in HNEpCs. IF staining (Fig. 3A) illustrated that IL-32 stimulation
significantly enhanced the activation (cleavage) of GSDMD,
suggesting the occurrence of pyroptosis. Similar results were
observed in Fig. 3B, which show the
protein expression of NLRP3, cleaved-caspase-1 and GSDMD-N in
response to IL-32 stimulation. NLRP3, cleaved-caspase-1 and GSDMD-N
expression levels were also markedly increased upon IL-32
stimulation. In addition, following exposure to 2, 10 and 50 ng/ml
IL-32, the protein expression levels of NOD1, NOD2 and TLR4 were
also significantly enhanced (Fig.
3C). The aforementioned data indicated that IL-32 could induce
pyroptosis of normal HNEpCs.
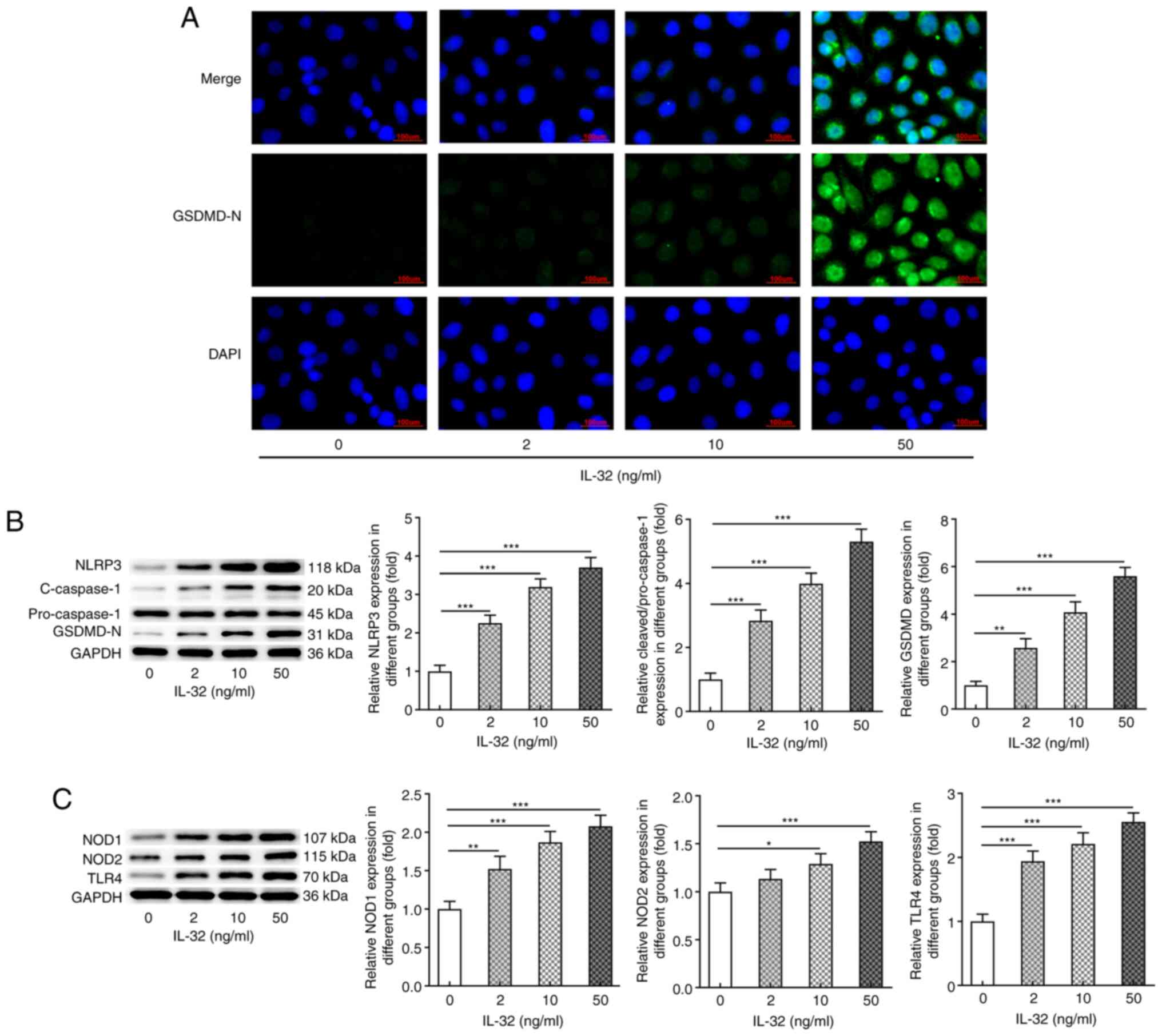 | Figure 3.Effects of IL-32 on the expression of
proteins involved in pyroptosis. (A) Representative
immunofluorescence staining for GSDMD-N in HNEpCs exposed to 0, 2,
10 and 50 ng/ml IL-32 for 48 h. Scale bar, 100 µm. (B) The
expression of NLRP3, cleaved-caspase-3/pro-caspase-3 and GSDMD-N in
HNEpCs exposed to 0, 2, 10 and 50 ng/ml IL-32 for 48 h was detected
via western blotting. (C) The expression of NOD1, NOD2 and TLR4 in
HNEpCs exposed to 0, 2, 10 and 50 ng/ml IL-32 for 48 h was detected
by western blotting. *P<0.05, **P<0.01 and ***P<0.001 vs.
0 ng/ml IL-32. GSDMD-N, N-terminal of gasdermin D; HNEpC, human
nasal epithelial cell; NLRP3, NACHT LRR and PYD domains-containing
protein 3; NOD, nucleotide-binding oligomerization
domain-containing protein; TLR, Toll-like receptor. |
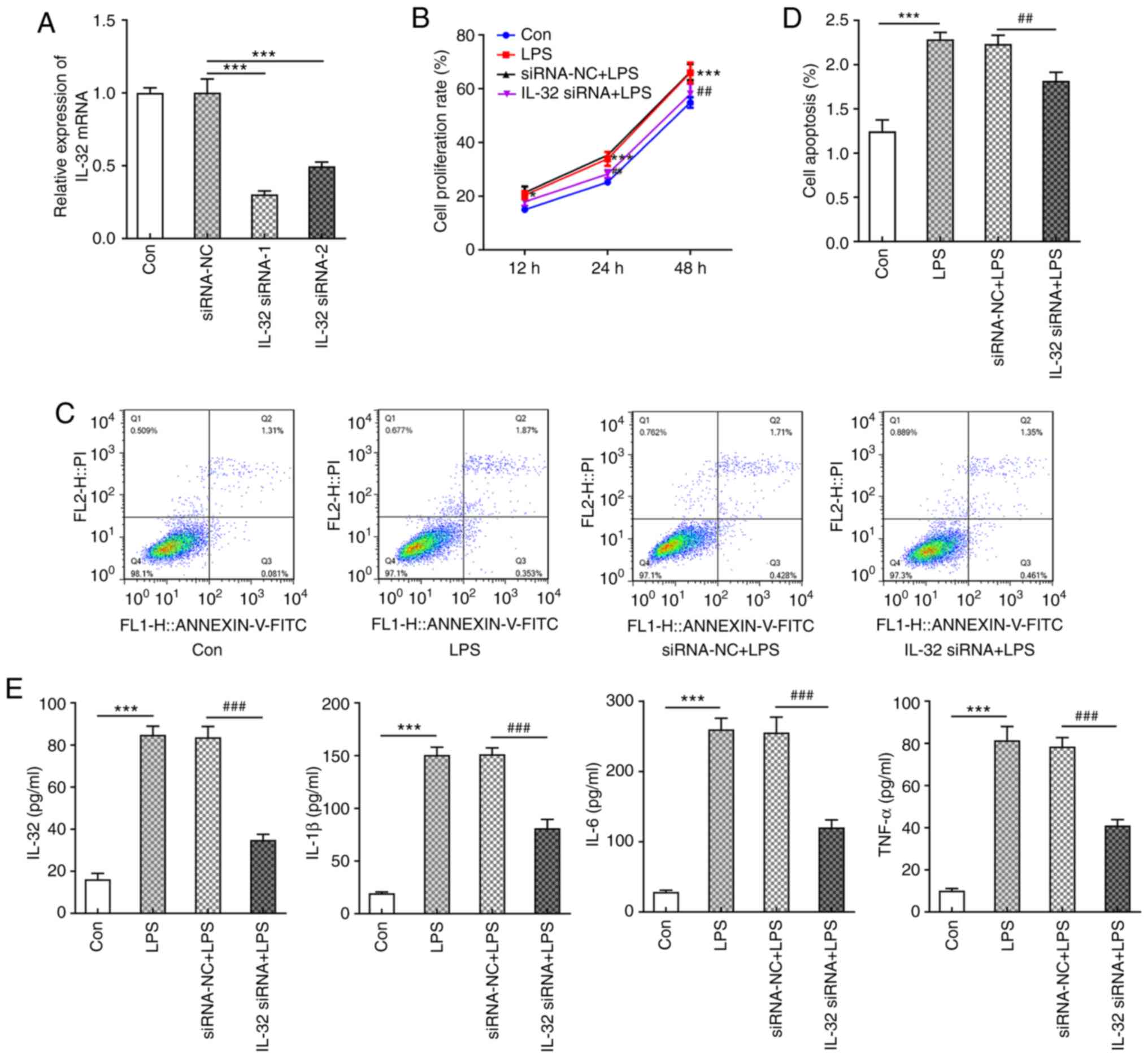
Knockdown of IL-32 inhibits
LPS-induced inflammation and pyroptosis
Next, to further examine the modulatory role of
IL-32 in AH, IL-32 expression levels in HNEpCs were knocked down
using siRNAs. IL-32 siRNA-1 was chosen for subsequent experiments
due to its enhanced silencing effects (Fig. 4A). LPS was used to stimulate control
HNEpC cells and IL-32 knockdown cells. Similar to low
concentrations of IL-32 (2 and 10 ng/ml), LPS also significantly
promoted cell proliferation compared with the control group
(Fig. 4B). However, cells silenced
with IL-32 siRNA-1 in the presence of LPS exerted a lower
proliferation rate compared with cells transfected with NC vectors
(Fig. 4B). LPS also enhanced the
ratio of cell apoptosis, which was reversed by IL-32 knockdown
(Fig. 4C and D). In addition, LPS
resulted in the production of a large amount of pro-inflammatory
cytokines, including IL-32, IL-1β, IL-6 and TNF-α, which was
significantly inhibited by IL-32 silencing (Fig. 4E). These results revealed that
silencing of IL-32 could inhibit LPS-induced acceleration of
proliferation, apoptosis and inflammation.
Finally, the expression levels of proteins involved
in NLRP3-mediated pyroptosis were evaluated. As shown in Fig. 5A, cells exerted significantly higher
expression of cleaved GSDMD in response to LPS stimulation, while
the knockdown of IL-32 markedly inhibited GSDMD activation. As
presented in Fig. 5B, LPS also
increased the expression of NLRP3, cleaved-caspase-1 and GSDMD-N,
which was reversed by IL-32 silencing. As illustrated in Fig. 5C, the LPS-induced enhanced
expression of NOD1, NOD2 and TLR4 was notably inhibited by IL-32
knockdown. These data indicated that IL-32 knockdown could
effectively inhibit LPS-induced pyroptosis.
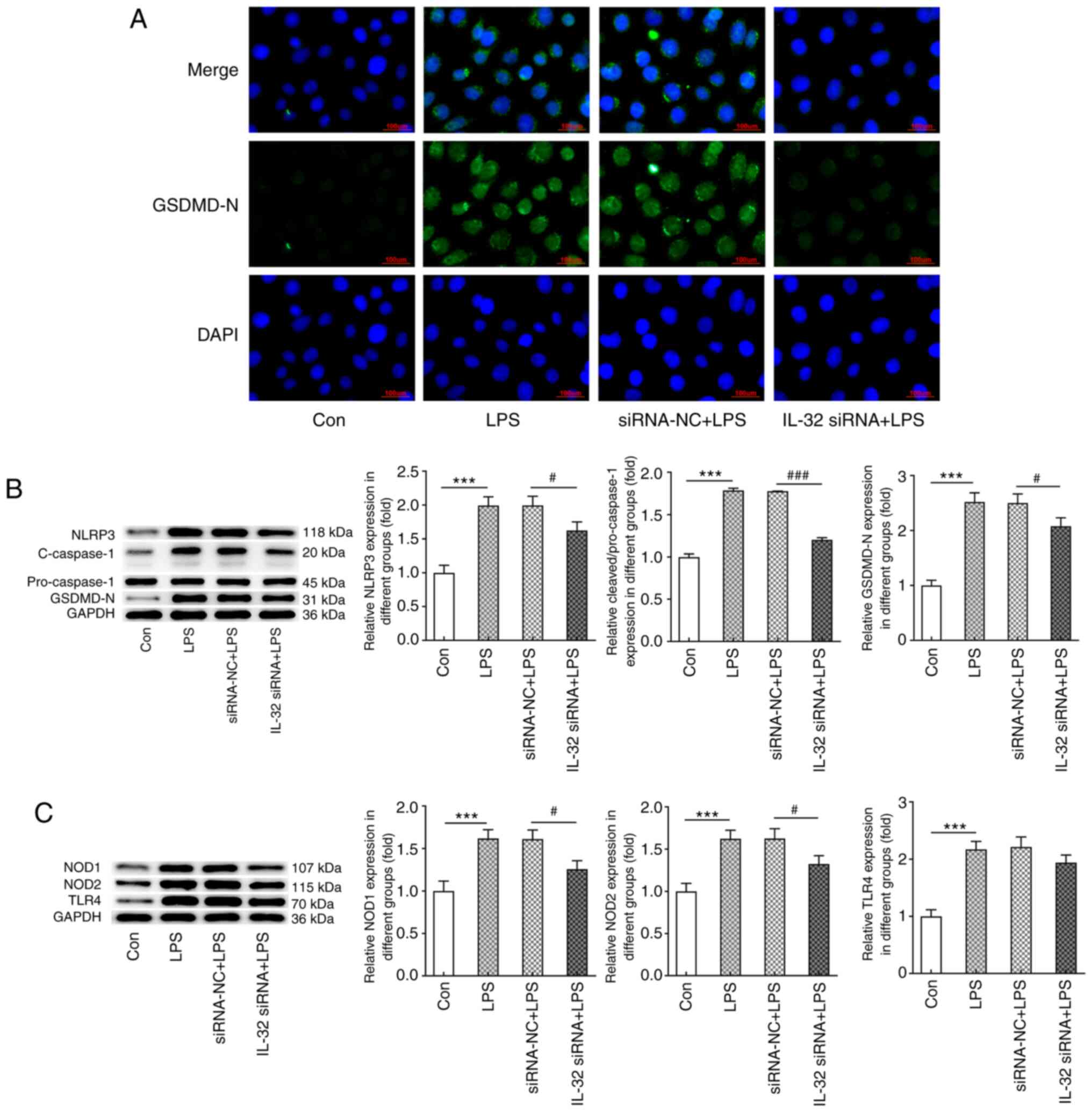 | Figure 5.Effects of IL-32 silencing on
LPS-induced HNEpC pyroptosis. (A) Immunofluorescence staining for
GSDMD-N in control HNEpCs and HNEpCs transfected with corresponding
siRNAs in the presence of LPS. Scale bar, 100 µm. (B) The
expression of NLRP3, cleaved-caspase-3/pro-caspase-3 and GSDMD-N in
control HNEpCs and HNEpCs transfected with corresponding siRNAs in
the presence of LPS was detected by western blotting. (C) The
expression of NOD1, NOD2 and TLR4 in control HNEpCs and HNEpCs
transfected with corresponding siRNAs in the presence of LPS was
detected by western blotting. ***P<0.001 vs. control;
#P<0.05 and ###P<0.001 vs. siRNA-NC +
LPS. HNEpC, human nasal epithelial cells; LPS, lipopolysaccharide;
siRNA, small interfering RNA; NC, negative control; GSDMD-N,
N-terminal of gasdermin D; NOD, nucleotide-binding oligomerization
domain-containing protein; TLR, Toll-like receptor; NLRP3, NACHT
LRR and PYD domains-containing protein 3. |
Discussion
The inflammatory response has been extensively
reported to contribute to the occurrence and progression of AH
(7,25). The present study aimed to analyze
the association between the pro-inflammatory cytokine IL-32 and AH.
IL-32 is a member of the IL family, which are lymphokines that
interact between leukocytes or immune cells (13). To the best of our knowledge, the
present study is the first to evaluate the association between
IL-32 and AH. The results from the present study suggested the
close association between IL-32 and AH pathogenesis as well as
LPS-induced HNEpC injury.
Adenoids are considered to be essential parts of the
system that protect organisms from pathogens. It is known that
human adenoids are immunoreactive lymphoid organs, which exhibit
specific antibodies together with B and T cell activities in
response to various antigens performing the functions of humoral
and cellular immunity (26). These
are extremely important for the growth and development of children
(26). At present, adenoidectomy is
a direct and preferred method to solve AH in children, but it may
bring risks and complications, including postoperative bleeding,
nasopharyngeal stenosis and velopharyngeal insufficiency (27). Another possible risk is that
removing adenoid tissue may result in a negative impact on immune
function (28). These factors
indicate the urgency to discover more suitable treatments.
ILs consist of a large family that can transmit
information, activate and regulate immune cells, mediate the
activation, proliferation and differentiation of T and B cells and
play an important role in inflammation. In addition to IL-32,
numerous other IL members have also been illustrated to regulate
airway inflammation (29–31). For example, IL-17A was found to be
upregulated in adenoid tissues from patients with AH and
pneumococcal carriage (32). IL-33
plays a role in the pathophysiology of chronic rhinosinusitis
(30). These data indicate the
modulatory role of ILs in AH and other types of airway
inflammation. The present study found that IL-32, together with
NLRP3 and IL-1β, were significantly upregulated in AH tissues,
indicating their role in AH. Subsequently, it was found that
treatment with low concentrations of IL-32 (2 and 10 ng/ml) could
promote HNEpC proliferation, while higher concentrations of IL-32
(50 ng/ml) inhibited cell proliferation. This can be explained by
the stressful cell proliferation caused by mild inflammation upon
low concentrations of IL-32 (2 and 10 ng/ml) stimulation (24), but predominant apoptosis or cell
death caused by high concentration of IL-32 (50 ng/ml) treatment.
The present study showed that IL-32 significantly enhanced
apoptosis, inflammation and the expression of proteins involved in
pyroptosis in a concentration-dependent manner.
Pyroptosis is a newly discovered programmed cell
death. It can cause excessive inflammation and immune response in
tissues, which in turn results in local and even systemic
inflammation and immunopathological damage (33). Pyroptosis can be divided into
classical and non-classical pathways according to its recognition
mechanisms, reactants and reaction pathways (16). Among both, GDSMD cleavage can expose
its N domain with pore-forming activities, which forms a large pore
in the membrane that induces pyroptosis (16). Meanwhile, the activation of
inflammasome NLRP3 induced by GSDMD-N and external stimuli can
trigger caspase-1 activation, thereby resulting in the release of a
large number of pro-inflammatory cytokines, ultimately aggravating
inflammation (17). The present
results showed that IL-32 increased GSDMD-N, NLRP3 and
cleaved-caspase-1 expression in HNEpCs, suggesting that IL-32
triggered pyroptosis in HNEpCs. TLRs and NOD-like receptors are two
major pattern recognition receptors that provide responses against
pathogenic invasion or tissue injury (34). It has been reported that TLR4, NOD1
and NOD2 are also associated with airway inflammation (35). The present study found that IL-32
also promoted TLR4, NOD1 and NOD2 expression in a
concentration-dependent manner. Notably, NOD1/2 and TLR4 are known
to activate NF-κB signaling, which is able to trigger NLRP3
activation (36–37). Therefore, the present results
indicated that IL-32 could also induce inflammation and
NLRP3-mediated pyroptosis by activating pattern recognition
receptors.
To further verify the present hypothesis, IL-32
expression was silenced in HNEpCs stimulated with LPS using siRNA.
Similar to IL-32, LPS significantly triggered high proliferation,
apoptosis, inflammation and activation of GSDMD, NLRP3, caspase-1,
NOD1/2 and TLR4. Besides, IL-32 knockdown significantly inhibited
all effects induced by LPS, revealing the protective effects of
IL-32 knockdown against LPS-induced injury in HNEpCs. To the best
of our knowledge, the present study is the first to identify IL-32
as an important inflammatory cytokine involved in AH inflammation.
However, this study were only carried out in a cell model, thus it
lacks the validation of in vivo models. Future research will
aim to verify these findings using in vivo experiments and
uncover the specific underlying mechanisms involved in the actions
of IL-32 in AH.
Taken together, the results of the present study
demonstrated that during AH and upon LPS exposure, adenoid tissues
and nasal epithelial cells released IL-32, which then regulated
apoptosis and excretion of inflammatory cytokines via activation of
the NOD1/2/TLR4/NLRP3 pathway, ultimately leading to pyroptosis.
Approaches targeting IL-32 to downregulate its expression may
provide novel therapeutic targets for AH. However, further in
vivo and clinical investigations need to be performed.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and XS conceived and designed the study. JZ, XS,
LZ and BS acquired and analyzed the data. JZ and XS confirmed the
authenticity of all the raw data. JZ prepared the draft of the
manuscript, including the figures. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Ethical Committee of the Tianjin Children's Hospital (Tianjin,
China). The parents of the children were informed about the study
and consent was obtained.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Coca-Pelaz A, Rodrigo JP, Bradley PJ,
Vander Poorten V, Triantafyllou A, Hunt JL, Strojan P, Rinaldo A,
Haigentz M Jr, Takes RP, et al: Adenoid cystic carcinoma of the
head and neck - An update. Oral Oncol. 51:652–661. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Buzatto GP, Tamashiro E, Proenca-Modena
JL, Saturn TH, Prates MC, Gagliardi TB, Carenzi LR, Massuda ET,
Hyppolito MA, Valera FC, et al: The pathogens profile in children
with otitis media with effusion and adenoid hypertrophy. PLoS One.
12:e01710492017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Durgut O and Dikici O: The effect of
adenoid hypertrophy on hearing thresholds in children with otitis
media with effusion. Int J Pediatr Otorhinolaryngol. 124:116–119.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Stupak HD and Park SY: Gravitational
forces, negative pressure and facial structure in the genesis of
airway dysfunction during sleep: A review of the paradigm. Sleep
Med. 51:125–132. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Scadding G: Non-surgical treatment of
adenoidal hypertrophy: the role of treating IgE-mediated
inflammation. Pediatr Allergy Immunol. 21:1095–1106. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gulotta G, Iannella G, Vicini C, Polimeni
A, Greco A, de Vincentiis M, Visconti IC, Meccariello G, Cammaroto
G, De Vito A, et al: Risk factors for obstructive sleep apnea
syndrome in children. Int J Environ Res Public Health. 16:32352019.
View Article : Google Scholar
|
|
7
|
Marazzato M, Zicari AM, Aleandri M, Conte
AL, Longhi C, Vitanza L, Bolognino V, Zagaglia C, De Castro G,
Brindisi G, et al: 16S metagenomics reveals dysbiosis of nasal core
microbiota in children with chronic nasal inflammation: Role of
adenoid hypertrophy and allergic rhinitis. Front Cell Infect
Microbiol. 10:4582020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hong JT, Son DJ, Lee CK, Yoon DY, Lee DH
and Park MH: Interleukin 32, inflammation and cancer. Pharmacol
Ther. 174:127–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jeong HJ, Shin SY, Oh HA, Kim MH, Cho JS
and Kim HM: IL-32 up-regulation is associated with inflammatory
cytokine production in allergic rhinitis. J Pathol. 224:553–563.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nam SY, Oh HA, Choi Y, Park KY, Kim HM and
Jeong HJ: Inhibition of IL-32 signaling by bamboo salt decreases
pro-inflammatory responses in cellular models of allergic rhinitis.
J Med Food. 17:939–948. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jeong HJ, Oh HA, Lee BJ and Kim HM:
Inhibition of IL-32 and TSLP production through the attenuation of
caspase-1 activation in an animal model of allergic rhinitis by
Naju Jjok (Polygonum tinctorium). Int J Mol Med. 33:142–150. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Keswani A, Kern RC, Schleimer RP and Kato
A: Role of interleukin-32 in chronic rhinosinusitis. Curr Opin
Allergy Clin Immunol. 13:13–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim SH, Han SY, Azam T, Yoon DY and
Dinarello CA: Interleukin-32: A cytokine and inducer of TNFalpha.
Immunity. 22:131–142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pan X, Cao H, Lu J, Shu X, Xiong X, Hong
X, Xu Q, Zhu H, Li G and Shen G: Interleukin-32 expression induced
by hepatitis B virus protein X is mediated through activation of
NF-κB. Mol Immunol. 48:1573–1577. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu J, Fang K, Wang S, Xiong L, Zhang C,
Liu Z, Guan X, Zheng R, Wang G, Zheng J, et al: Anti-inflammatory
effect of columbianetin on lipopolysaccharide-stimulated human
peripheral blood mononuclear cells. Mediators Inflamm.
2018:91917432018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang S, Yuan YH, Chen NH and Wang HB: The
mechanisms of NLRP3 inflammasome/pyroptosis activation and their
role in Parkinson's disease. Int Immunopharmacol. 67:458–464. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Z, Liang C, Wang T, Zou Q, Zhou M,
Cheng Y, Peng H, Ji Z, Deng Y, Liao J, et al: NLRP3 inflammasome
activation promotes the development of allergic rhinitis via
epithelium pyroptosis. Biochem Biophys Res Commun. 522:61–67. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hu H and Li H: Prunetin inhibits
lipopolysaccharide-induced inflammatory cytokine production and
MUC5AC expression by inactivating the TLR4/MyD88 pathway in human
nasal epithelial cells. Biomed Pharmacother. 106:1469–1477. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsou YA, Tung YT, Wu TF, Chang GRL, Chen
HC, Lin CD, Lai CH, Chen HL and Chen CM: Lactoferrin interacts with
SPLUNC1 to attenuate lipopolysaccharide-induced inflammation of
human nasal epithelial cells via down-regulated MEK1/2-MAPK
signaling. Biochem Cell Biol. 95:394–399. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li YQ, Zhong Y, Xiao XP, Li DD, Zhou Z and
Tian YY: IL-33/ST2 axis promotes the inflammatory response of nasal
mucosal epithelial cells through inducing the ERK1/2 pathway.
Innate Immun. 26:505–513. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nian JB, Zeng M, Zheng J, Zeng LY, Fu Z,
Huang QJ and Wei X: Epithelial cells expressed IL-33 to promote
degranulation of mast cells through inhibition on
ST2/PI3K/mTOR-mediated autophagy in allergic rhinitis. Cell Cycle.
19:1132–1142. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin J, Xu R, Hu L, You J, Jiang N, Li C,
Che C, Wang Q, Xu Q, Li J, et al: Interleukin-32 induced thymic
stromal lymphopoietin plays a critical role in the inflammatory
response in human corneal epithelium. Cell Signal. 49:39–45. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kiraly O, Gong G, Olipitz W, Muthupalani S
and Engelward BP: Inflammation-induced cell proliferation
potentiates DNA damage-induced mutations in vivo. PLoS Genet.
11:e10049012015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Anfuso A, Ramadan H, Terrell A, Demirdag
Y, Walton C, Skoner DP and Piedimonte G: Sinus and adenoid
inflammation in children with chronic rhinosinusitis and asthma.
Ann Allergy Asthma Immunol. 114:103–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun YL, Zheng HT, Tao JL, Jiang MC, Hu CC,
Li XM and Yuan B: Effectiveness and safety of Chinese herbal
medicine for pediatric adenoid hypertrophy: A meta-analysis. Int J
Pediatr Otorhinolaryngol. 119:79–85. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Richter GT and Bower CM: Cervical
complications following routine tonsillectomy and adenoidectomy.
Curr Opin Otolaryngol Head Neck Surg. 14:375–380. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van den Akker EH, Sanders EA, van Staaij
BK, Rijkers GT, Rovers MM, Hoes AW and Schilder AG: Long-term
effects of pediatric adenotonsillectomy on serum immunoglobulin
levels: Results of a randomized controlled trial. Ann Allergy
Asthma Immunol. 97:251–256. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rogala B and Glück J: The role of
interleukin-33 in rhinitis. Curr Allergy Asthma Rep. 13:196–202.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim DK, Jin HR, Eun KM, Mo JH, Cho SH, Oh
S, Cho D and Kim DW: The role of interleukin-33 in chronic
rhinosinusitis. Thorax. 72:635–645. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo XQ, Ma F, Wang S, Zhao MZ, Shao JB,
Geng XR, Liu JQ, Mo LH, Guan L, Liu ZG, et al: Interleukin-5
induces apoptotic defects in CD4+ T cells of patients
with allergic rhinitis. J Leukoc Biol. 105:719–727. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Huang CC, Wu PW, Chen CL, Wang CH, Lee TJ,
Tsai CN and Chiu CH: IL-17A expression in the adenoid tissue from
children with sleep disordered breathing and its association with
pneumococcal carriage. Sci Rep. 8:167702018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bergsbaken T, Fink SL and Cookson BT:
Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol.
7:99–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Keestra-Gounder AM, Byndloss MX, Seyffert
N, Young BM, Chávez-Arroyo A, Tsai AY, Cevallos SA, Winter MG, Pham
OH, Tiffany CR, et al: NOD1 and NOD2 signalling links ER stress
with inflammation. Nature. 532:394–397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Di Stefano A, Ricciardolo FLM, Caramori G,
Adcock IM, Chung KF, Barnes PJ, Brun P, Leonardi A, Andò F, Vallese
D, et al: Bronchial inflammation and bacterial load in stable COPD
is associated with TLR4 overexpression. Eur Respir J.
49:16020062017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Luo M, Yan D, Sun Q, Tao J, Xu L, Sun H
and Zhao H: Ginsenoside Rg1 attenuates cardiomyocyte apoptosis and
inflammation via the TLR4/NF-κB/NLRP3 pathway. J Cell Biochem.
121:2994–3004. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Aabdin ZU, Bilal MS, Dai H, Abaker JA, Liu
X, Benazir S, Yan J and Shen X: NOD1/NF-κB signaling pathway
inhibited by sodium butyrate in the mammary gland of lactating
goats during sub-acute ruminal acidosis. Microb Pathog. 122:58–62.
2018. View Article : Google Scholar : PubMed/NCBI
|