Introduction
The first human genome draft (1) was based on Sanger sequencing
technology (2), cost $2.7 billion
and lasted over a period of 10 years (3). In comparison, the sequencing of the
human genome (~3 Gbp haploid genome size) in a next generation
sequencing (NGS) platform where millions of reads are efficiently
mapped to the reference genome, currently costs <$1,000 and it
can be performed in <2 days (4).
Short-read de novo genome assemblers have difficulty to
produce large and reliable contigs, particularly in low complexity
regions such as centromeres, telomeres and other repetitive regions
(5,6). To address this issue, third generation
sequencing (7) technologies have
been developed. Nanopore (https://nanoporetech.com/) (8,9) and
PacBio (https://www.pacb.com/) (10) sequencing platforms were launched
around 2010. Third generation sequencers are sequencing
single-molecules in real-time (10)
without the need of PCR amplification and thus, avoid PCR bias
(11,12). The main drawback of long reads is
lower accuracy compared to Illumina short-reads: Typical Nanopore
and PacBio Sequel I long-reads have an average accuracy of 90%
(13) compared to 99.9% of typical
Illumina short-reads (4). As a
consequence, assemblies produced only by long-reads were more
contiguous, but they also contained more errors, which made genome
annotation, variant calling and other genome analyses, challenging
tasks (6,12).
By following the hybrid assembly strategy (14,15),
the advantages of the two generations are combined, incorporating
the information contained in the two read types, overcoming their
drawbacks. Recent advantages in long-read sequencing by PacBio have
shown very promising results: Sequel System II was released in 2019
with an upgraded SMRT flow cell that was first introduced in 2013
(16), which was able to increase
the sequencing yield up to 8-fold. However, the greatest
breakthrough was the advance of circular consensus sequencing (CCS)
(17) which sequences the same
circular DNA molecule 10 times, to produce a highly accurate
(99.9%) high-fidelity (HiFi) consensus read, while increasing
unique molecular yield and insert size (up to 25 Kbp). At the same
time, recent advances in Nanopore's base identification algorithm,
Bonito (https://github.com/nanoporetech/bonito) (18), have led to greater than 97% base
accuracy.
Usually, the primary genome assembly is very
fragmented and some contigs are misassembled. For this reason, the
completion of the assembly requires the construction of scaffolds
(19). To this end, Hi-C sequencing
method provides chromosomal conformation information necessary to
assemble chromosome-level scaffolds. The general principle of this
method is based on the proximity and contacts of chromosomal
regions in the cell nucleus. The frequency of contacts is higher
between regions of the same chromosome; thus, different chromosomes
can be distinguished during the assembly (20). The result of this method is a
collection of pairs of reads of chimeric fragments that can be
mapped to the assembly, joining very remote areas.
Using the recent sequencing and scaffolding
technologies, it is now possible to construct new reference genomes
and finish the assembly of existing ones, by closing gaps in the
centromeres, telomeres and other low complexity regions. For this
reason, new projects have been launched and new consortia have been
formed (21–23). The telomere to telomere (T2T)
consortium (https://sites.google.com/ucsc.edu/t2tworkinggroup/)
(24,25) aims to finish the entire human genome
by producing chromosomes without gaps. Almost two decades after the
first draft of the human genome by the International Human Genome
Sequencing Consortium, T2T published a completed human genome with
the exception of five known gaps withing the rDNA arrays
(https://genomeinformatics.github.io/CHM13v1/).
The development of sequencing technologies and
assembly and scaffolding algorithms, as well as the sharp increase
of publicly available data (https://www.ncbi.nlm.nih.gov/genbank/statistics/),
democratised de novo genome assembly projects by making them
more approachable to smaller labs. The present study aimed to
compare genome assembly pipelines, which use different assembly
strategies, evaluating them in terms of accuracy, speed and
computational power needed. Finally, the need for scaffold
construction, incorporating Hi-C sequencing data was also
evaluated.
Materials and methods
Data acquisition and experimental
overview
Primary sequencing data were downloaded from 3
organisms, Drosophila virilis, Drosophila melanogaster and
Homo sapiens (Table I). Some
FASTQ files were subsampled using Reformat tool from BBtools
(https://sourceforge.net/projects/bbmap/). Following
the hybrid assembly strategy, using short paired-end Illumina reads
in combination with long Nanopore reads, the low complexity genome
of Drosophila virilis and the high complexity genome of
Homo sapiens were constructed, downloading read data from
the European Nucleotide Archive (ENA) (26) and the T2T Consortium, respectively.
Drosophila melanogaster genome was assembled following the
long-read assembly strategy using only HiFi reads retrieved from
ENA. Finally, Hi-C reads were used to create the scaffolds of our
assemblies. It is important to note that the sequencing data used
to assemble Homo sapiens genome, derives from CHM13hTERT,
which is a female haploid cell line; thus, there will be no Y
chromosome in the final assemblies. The experiments were performed
on the Biomedical Research Foundation, Academy of Athens (BRFAA)
computer cluster that consists of 24 nodes of 128 GB RAM each. Each
node consists of 2 Intel® Xeon® Silver 4116
processors with 12 cores per processor and 2 threads per core (i.e.
48 CPUs per node). Additionally, Homo sapiens assembly by
Wengan was performed on an Aristotle University of Thessaloniki
(AUTh) computational system on a single node which consists of 4
AMD Opteron™ 6274 processors with 16 cores per processor and 1
thread per core (i.e., 64 CPUs) and 256 GB RAM.
 | Table I.ENA accessions and T2T links of
primary sequencing data. |
Table I.
ENA accessions and T2T links of
primary sequencing data.
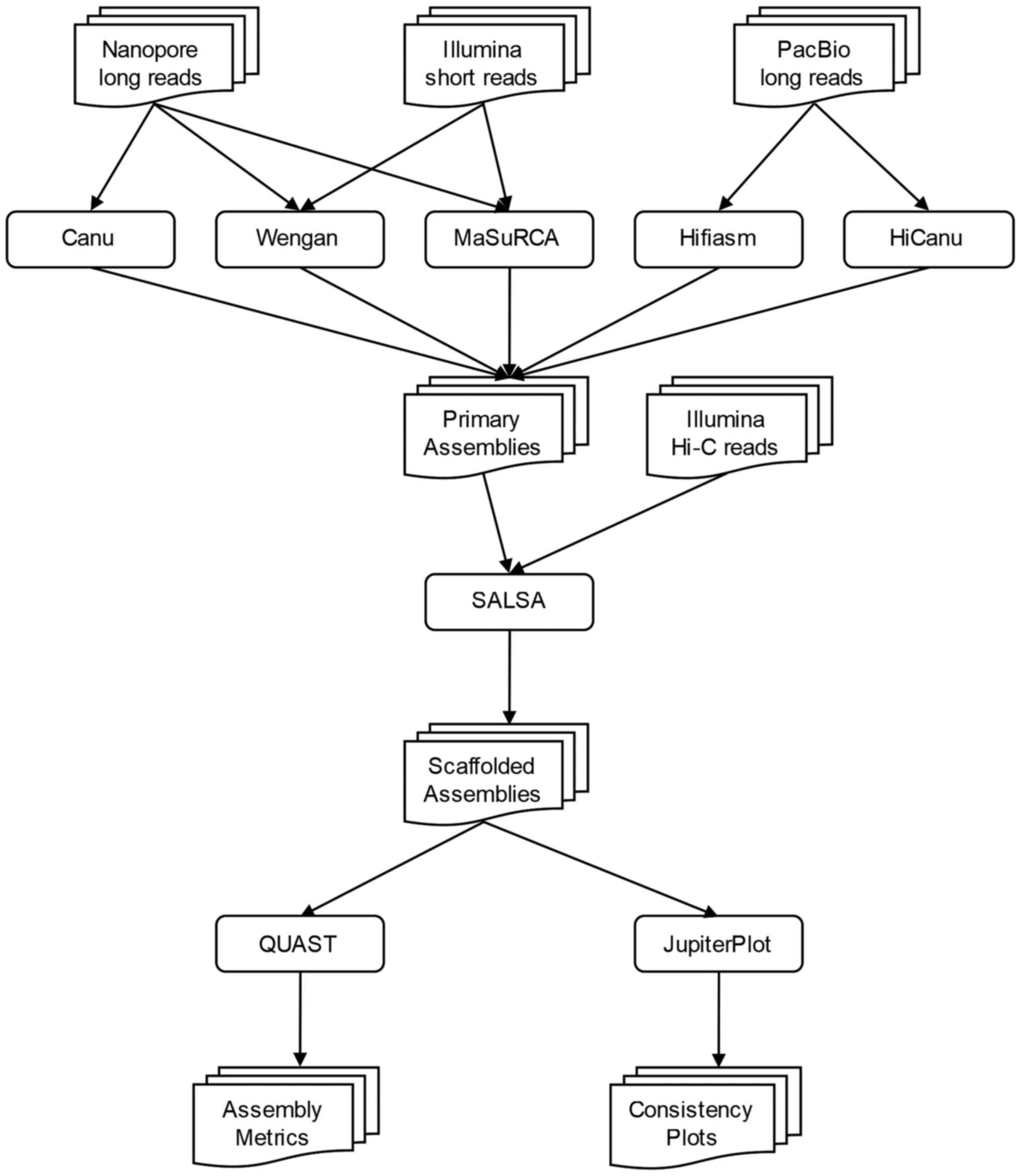
The pipeline is divided into 3 parts: In the first
stage of the current workflow (Fig.
1), different assemblers were used for the genome construction.
In the second stage, the scaffolding, Hi-C data were combined with
the initial assembly, in order to increase its continuity and
accuracy. In the last stage, the final assembly was assessed and
evaluated with the use of various tools.
Genome assembly
In order to assess the hybrid assembly strategy, the
present study chose to evaluate two pipelines, MaSuRCA (version
3.3.5) (27,28) and Wengan (version 0.1) (29). MaSuRCA workflow offers three
different assemblers, CABOG (30),
SOAPdenovo (31) and Flye (32). The pipeline was tested using CABOG
and Flye assemblers, which are designed for long-read assembly.
Wengan pipeline is based on DiscovarDenovo assembler (33).
Canu (version 2.0) (34) is a long-read assembler, designed to
use long high-noise single-molecule sequencing data, such as
Nanopore and PacBio reads. Its workflow is based on the Celera
assembler (35) which was used in
the Human Genome Project to produce the first draft of the human
genome. Hifiasm (version 0.13) (36) and HiCanu (Canu version 2.1.1)
(37) are long-read assemblers
exclusively for HiFi reads. The main difference between HiFi
assemblers and the ones mentioned previously, is that Hifiasm and
HiCanu produce phased assemblies. A phased assembly is a
haplotype-resolved assembly, where high complexity regions, such as
genes, will be separated into two different alleles (36,38).
HiCanu is a modified version of Canu, adapted to take advantage of
the characteristics of HiFi reads. Hifiasm produces two different
files for the primary and alternative assembly, whereas HiCanu
combines the primary and the alternative assembly in the same FASTA
file.
Scaffolding
In order to test the necessity of scaffolding, a
scaffolder was used to improve the assembly continuity and
completeness, as follows: Hi-C data are mapped to the primary
assembly by Arima mapping pipeline (39), to produce a BAM file which is
consequently converted to a BED file. SALSA (version 2.2) (40) uses this BED file which contains the
mapping information of Hi-C reads on the assembly, to scaffold the
primary assembly.
Quality control metrics
For the quality control of the assemblies produced,
different evaluation tools were used. These tools produce and
present the qualitative and quantitative characteristics of the
assemblies in a comprehensible way. QUAST (version 5.0.2) (41), a genome assembly evaluation tool,
produces various metrics for our assemblies, using a reference
genome (Table II). The standard
assembly statistics include the calculation of N50/NG50 and
L50/LG50 values (42), as follows:
N50 (or NG50) is the size of the contig, where at least 50% of the
genome assembly size (or the reference genome size), is contained
in contigs of equal or larger size than this contig. Higher
N50/NG50 values signify more contiguous assemblies. L50 (or LG50)
is the smallest number of contigs whose length sum makes up for at
least 50% of the genome assembly length (or reference genome
length). Lower L50/LG50 values signify more contiguous assemblies.
Furthermore, QUAST makes use of BUSCO (Quast version 5.0.2)
(43), to assess genome assembly
and annotation completeness, based on evolutionarily-informed
expectations of gene content of near-universal single-copy
orthologs.
| Table II.Reference genomes used for the
evaluation of the assemblies. |
Table II.
Reference genomes used for the
evaluation of the assemblies.
Genome consistency plots
JupiterPlot (version 1.0) (44) is a workflow that uses Circos
(45) to generate a genome assembly
consistency plot between a reference genome and a genome assembly.
The chromosomes of the reference genome are represented as coloured
arcs on the left half circle of the plot, whereas the
contigs/scaffolds of the assembled genome are represented as
outlined white arcs on the right half circle. The number and size
of white arcs is indicative of the genome contiguity. JupiterPlot
represents synteny between the reference and the assembled genome,
indicating corresponding contiguous regions as ribbons whose width
is proportional to their sequence length. In this manner, assembly
errors and chromosomal misassemblies can be visually identified: A
ribbon in twisted position represents an inversion, a ribbon which
crosses over other ribbons represents a translocation, a lack of a
ribbon connecting a region of the reference genome represents a
deletion and the overlap of two ribbons connecting the same
reference genome region represents a duplication. Although in other
cases these misassemblies may represent genuine chromosomal
aberrations, in our case they represent assembly errors due to low
sequence complexity of repetitive regions such as centromeres,
telomeres, etc., low sequencing coverage and weaknesses of each
assembly algorithm.
Results
Drosophila genome assemblies
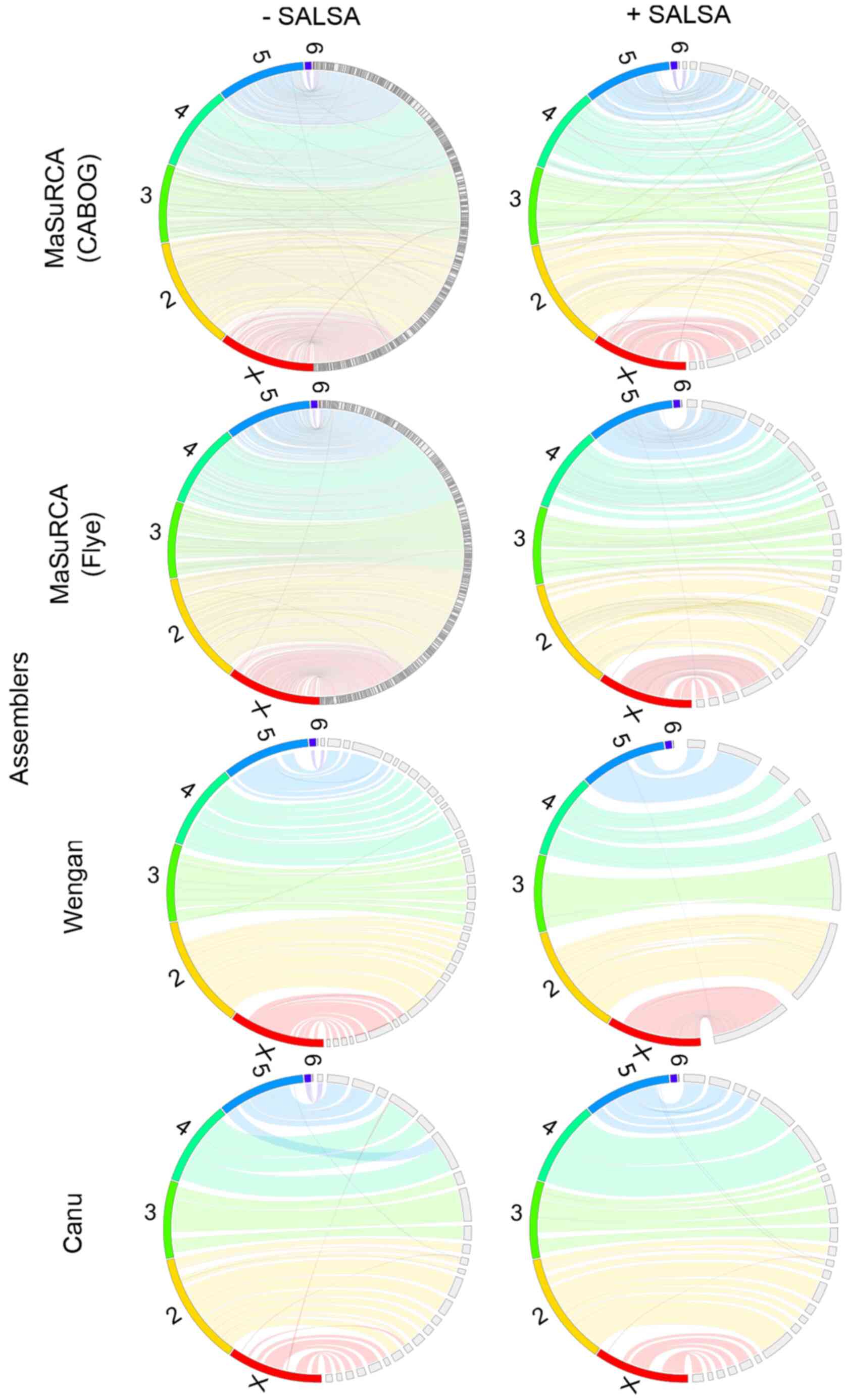
Primary (unscaffolded) MaSuRCA (CABOG or Flye)
hybrid assemblies are by far the most fragmented of all
Drosophila virilis assemblies, based on N50/NG50 and
L50/LG50 values (Table III) and
manual inspection of genome assembly consistency plots (Fig. 2). Canu, based exclusively on long
Nanopore data, produced the most contiguous primary assembly.
MaSuRCA/CABOG produced the most misassembled contigs, while Wengan
hybrid assembler created the least misassembled ones. All but Canu
assemblies present very high rates of preserved gene completeness,
similar to the rates of the reference genomes (Table IV). The sizes of all Drosophila
virilis primary assemblies are comparable to each other and
very similar to that of the reference genome. Wengan is the fastest
hybrid assembler and produced the Drosophila virilis genome
71 times faster than Canu, while the average CPU usage of Wengan is
smaller than the rest of these assemblers (Table V). Hi-C-based scaffolding
ameliorated the contiguity and it limited the misassemblies of all
assemblies, but it did not improve the gene completeness and it did
not alter the final assembly size.
| Table III.Metrics of Drosophila
assemblies. |
Table III.
Metrics of Drosophila
assemblies.
| Assemblers |
Contigs/scaffolds | Genome assembly
size (bp) | N50 | NG50 | L50 | LG50 |
|---|
| MaSuRCA
(CABOG) | 1,016 | 167,374,624 | 366,859 | 359,873 | 127 | 131 |
| MaSuRCA
(CABOG)/SALSA (Arima) | 532 | 167,617,624 | 3,400,369 | 3,400,369 | 15 | 15 |
| MaSuRCA (Flye) | 689 | 163,000,738 | 419,467 | 406,899 | 113 | 121 |
| MaSuRCA
(Flye)/SALSA (Arima) | 230 | 163,230,238 | 5,261,864 | 5,258,634 | 9 | 10 |
| Wengan | 329 | 153,989,049 | 3,232,846 | 3,013,042 | 13 | 16 |
| Wengan/SALSA
(Arima) | 229 | 154,046,842 | 21,036,706 | 16,232,289 | 3 | 4 |
| Canu | 425 | 169,315,961 | 4,435,749 | 4,435,749 | 10 | 10 |
| Canu/SALSA
(Arima) | 488 | 176,029,265 | 25,182,285 | 25,182,285 | 4 | 4 |
| Hifiasm |
|
|
|
|
|
|
| Insert
size: 11 Kbp | 314 | 149,971,598 | 23,693,975 | 23,693,975 | 3 | 3 |
|
Coverage: 37× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 149 | 164,010,561 | 21,707,601 | 24,110,342 | 4 | 3 |
|
Coverage: 40× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 186 | 169,871,295 | 23,943,049 | 24,211,538 | 4 | 3 |
|
Coverage: 92× |
|
|
|
|
|
|
| Hifiasm/SALSA |
|
|
|
|
|
|
| Insert
size: 11 Kbp | 308 | 149,976,098 | 23,693,975 | 23,693,975 | 3 | 3 |
|
Coverage: 37× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 141 | 164,015,561 | 24,110,342 | 24,620,248 | 4 | 3 |
|
Coverage: 40× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 183 | 169,876,757 | 23,943,049 | 24,211,538 | 4 | 3 |
|
Coverage: 92× |
|
|
|
|
|
|
| HiCanu |
|
|
|
|
|
|
| Insert
size: 11 Kbp | 1,792 | 295,986,869 | 2,513,964 | 6,791,534 | 24 | 7 |
|
Coverage: 37× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 1,024 | 322,211,690 | 6,752,429 | 17,694,921 | 12 | 4 |
|
Coverage: 40× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 1,269 | 337,795,659 | 11,255,983 | 26,987,095 | 8 | 2 |
|
Coverage: 92× |
|
|
|
|
|
|
| HiCanu/SALSA |
|
|
|
|
|
|
| Insert
size: 11 Kbp | 1,747 | 296,025,369 | 5,836,825 | 10,646,076 | 14 | 4 |
|
Coverage: 37× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 1,023 | 322,224,690 | 12,833,112 | 30,402,815 | 7 | 2 |
|
Coverage: 40× |
|
|
|
|
|
|
| Insert
size: 24 Kbp | 1,281 | 337,778,159 | 6,830,725 | 16,844,691 | 12 | 4 |
|
Coverage: 92× |
|
|
|
|
|
|
| Table IV.BUSCO values of Drosophila
assemblies. |
Table IV.
BUSCO values of Drosophila
assemblies.
| Assemblers | Completed and
single-copy BUSCOs (S) | Completed and
duplicated BUSCOs (D) | Fragmented BUSCOs
(F) | Missing BUSCOs
(M) |
|---|
| Drosophila
virilis reference genome | 98.0% | 0.5% | 0.7% | 0.8% |
| MaSuRCA
(CABOG) | 96.1% | 1.5% | 0.8% | 1.6% |
| MaSuRCA
(CABOG)/SALSA (Arima) | 96.1% | 1.4% | 0.8% | 1.7% |
| MaSuRCA (Flye) | 98.2% | 0.5% | 0.8% | 0.5% |
| MaSuRCA
(Flye)/SALSA (Arima) | 98.0% | 0.5% | 0.8% | 0.7% |
| Wengan | 98.0% | 0.4% | 0.7% | 0.9% |
| Wengan/SALSA
(Arima) | 97.9% | 0.3% | 0.8% | 1.0% |
| Canu | 62.7% | 0.2% | 21.3% | 15.8% |
| Canu/SALSA
(Arima) | 64.0% | 0.3% | 20.7% | 15.0% |
| Drosophila
melanogaster reference genome | 97.9% | 0.7% | 0.9% | 0.5% |
| Hifiasmx |
|
|
|
|
| Insert
size: 11 Kbp |
|
|
|
|
|
Coverage: 37× | 98.1% | 0.6% | 0.7% | 0.6% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 40× | 98.2% | 0.4% | 0.7% | 0.7% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 90× | 98.1% | 0.5% | 0.7% | 0.7% |
| Hifiasm/SALSA |
|
|
|
|
| Insert
size: 11 Kbp |
|
|
|
|
|
Coverage: 37× | 98.1% | 0.6% | 0.7% | 0.6% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 40× | 98.2% | 0.4% | 0.7% | 0.7% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 90× | 98.2% | 0.5% | 0.7% | 0.6% |
| HiCanu |
|
|
|
|
| Insert
size: 11 Kbp |
|
|
|
|
|
Coverage: 37× | 4.8% | 94.1% | 0.6% | 0.5% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 40× | 3.8% | 95.2% | 0.5% | 0.5% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 90× | 3.2% | 95.5% | 0.7% | 0.6% |
| HiCanu/SALSA |
|
|
|
|
| Insert
size: 11 Kbp |
|
|
|
|
|
Coverage: 37× | 42.3% | 56.7% | 0.5% | 0.5% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 40× | 37.3% | 61.6% | 0.5% | 0.6% |
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 90× | 39.0% | 59.9% | 0.5% | 0.6% |
| Table V.Assembly time and CPU usage
comparison. |
Table V.
Assembly time and CPU usage
comparison.
| Organism | Assemblers | CPU time (sec) | CPU usage | Elapsed (wall
clock) time (h:mm:ss) |
|---|
| Drosophila
virilis | MaSuRCA
(CABOG) | 1,638,637.72 | 3,954% | 11:30:39 |
|
| MaSuRCA (Flye) | 1,344,633.10 | 3,961% | 9:25:44 |
|
| Canu | 993,441,898 | 3,532% | 78:07:27 |
|
| Wengan | 198,241.94 | 2,831% | 1:56:42 |
| Drosophila
melanogaster | Hifiasm |
|
|
|
|
| Insert
size: 11 Kbp |
|
|
|
|
|
Coverage: 37× | 163,816.92 | 4,098% | 1:06:37 |
|
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 40× | 215,855.05 | 4,287% | 1:23:54 |
|
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 90× | 4,271,030.94 | 4,313% | 25:40:58 |
|
| HiCanu |
|
|
|
|
| Insert
size: 11 Kbp |
|
|
|
|
|
Coverage: 37× | 85,224.85 | 1,752% | 1:21:03 |
|
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 40× | 107,146.65 | 2,235% | 1:19:53 |
|
| Insert
size: 24 Kbp |
|
|
|
|
|
Coverage: 90× | 176,649.77 | 1,646% | 2:58:46 |
| Homo
sapiens | Hifiasm | 1,272,271.15 | 4,113% | 8:35:29 |
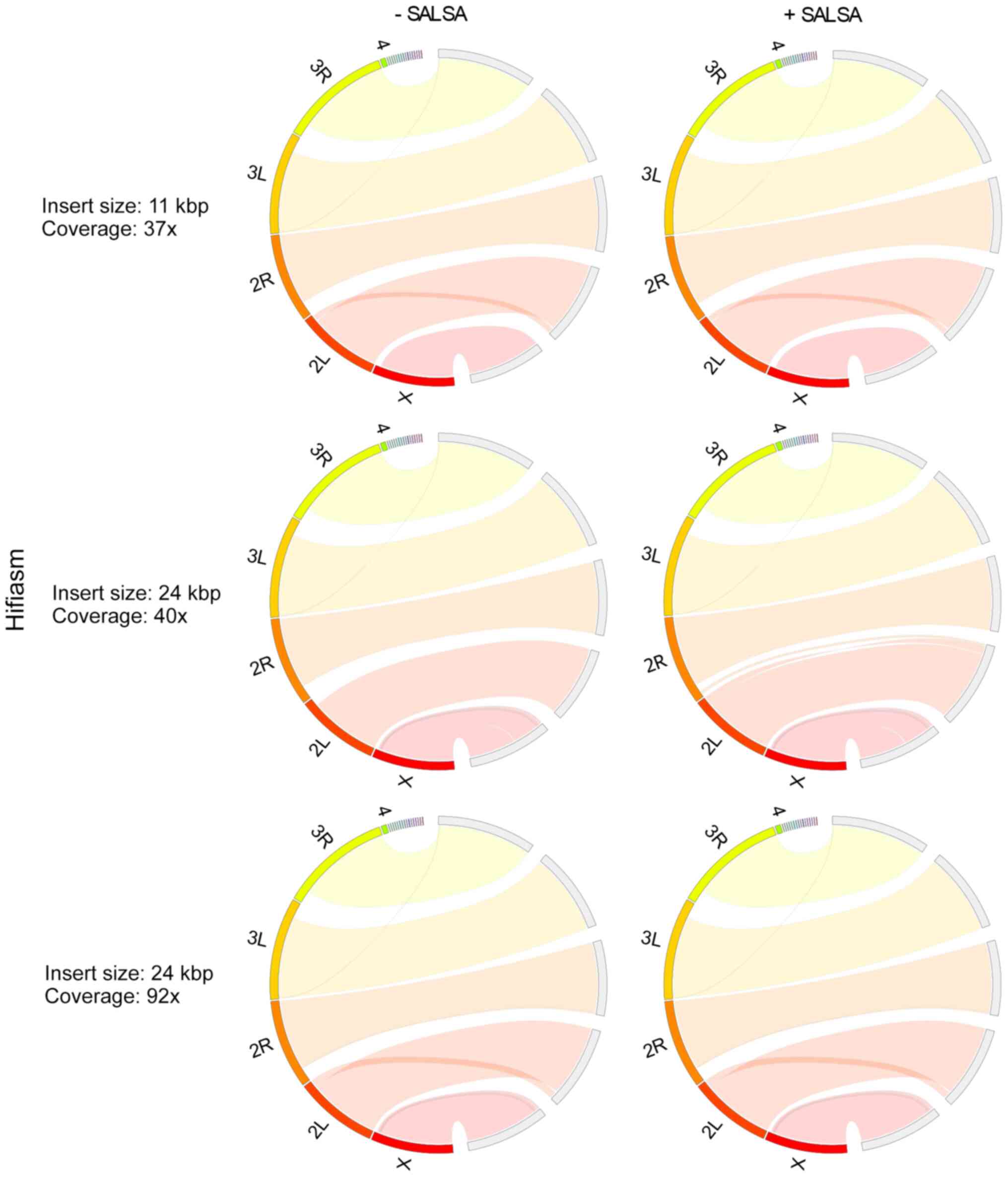
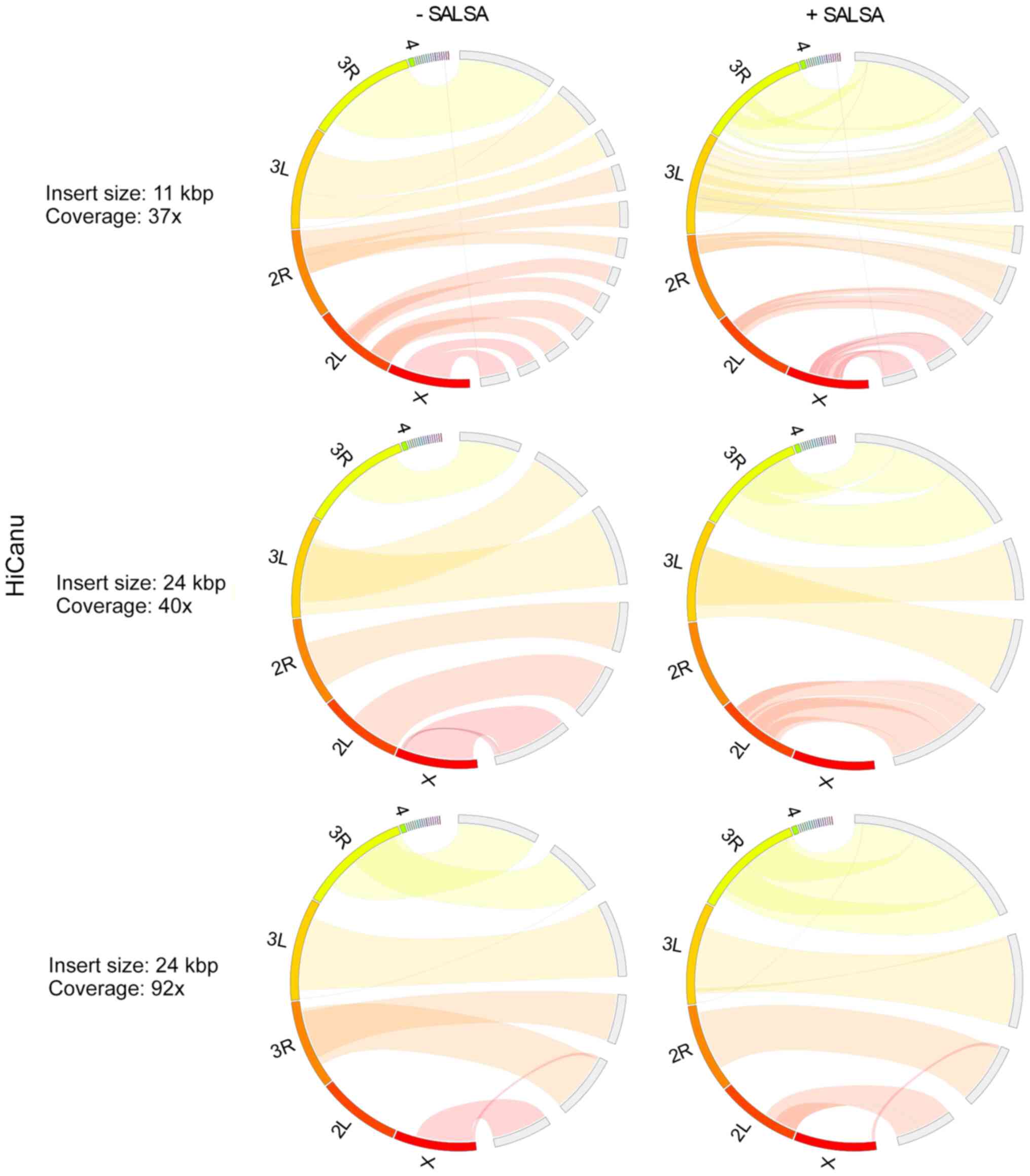
In Drosophila melanogaster primary
assemblies, Hifiasm outperformed HiCanu, producing less fragmented
and misassembled contigs (Figs. 3
and 4). As HiCanu produces phased
assemblies, the vast majority of single-copy genes appeared as
completed and duplicated in BUSCO analysis (Table IV). Nevertheless, the sum of
completed single and duplicated BUSCOs in Hifiasm and HiCanu was
practically identical to that of the reference genome. While using
the 11 Kbp insert size and 37× coverage data, Hifiasm produced
Drosophila melanogaster genome faster than HiCanu. However,
as the coverage was increased, the assembly time of Hifiasm
increased more rapidly than that of HiCanu: The assembly time of
Hifiasm and HiCanu using 24 Kbp insert size and 40× coverage data
was approximately the same, while HiCanu was 12× faster than
Hifiasm, when 24 Kbp insert size and 92× coverage was used. The
average CPU usage of HiCanu was also smaller than that of Hifiasm
(Table V). SALSA scaffolding based
on Hi-C data, slightly improved Hifiasm assemblies, while it
ameliorated the contiguity of HiCanu ones. It also slightly limited
the misassemblies of HiCanu outputs. It did not influence the gene
completeness of any assembly. Insert size (11 and 24 Kbp) and
coverage (37×, 40× and 92×) did not influence the outcome of
Hifiasm; however, a small deterioration in assembly contiguity at
the 92× coverage was noted. On the other hand, a higher insert size
and coverage improved HiCanu performance.
Overall, Hifiasm performed most effectively in the
primary assembly of Drosophila melanogaster genome (which is
comparable to that of Drosophila virilis), in terms of
genome contiguity, accuracy and completeness. At 37× and 40×
coverages, Hifiasm was also the fastest assembler; however, the CPU
usage of Wengan and HiCanu was half of that of Hifiasm. The
combination of Hi-C data had a minimal effect on the improvement of
Hifiasm assembly. Among hybrid assemblers, Wengan performed best
when combined with SALSA.
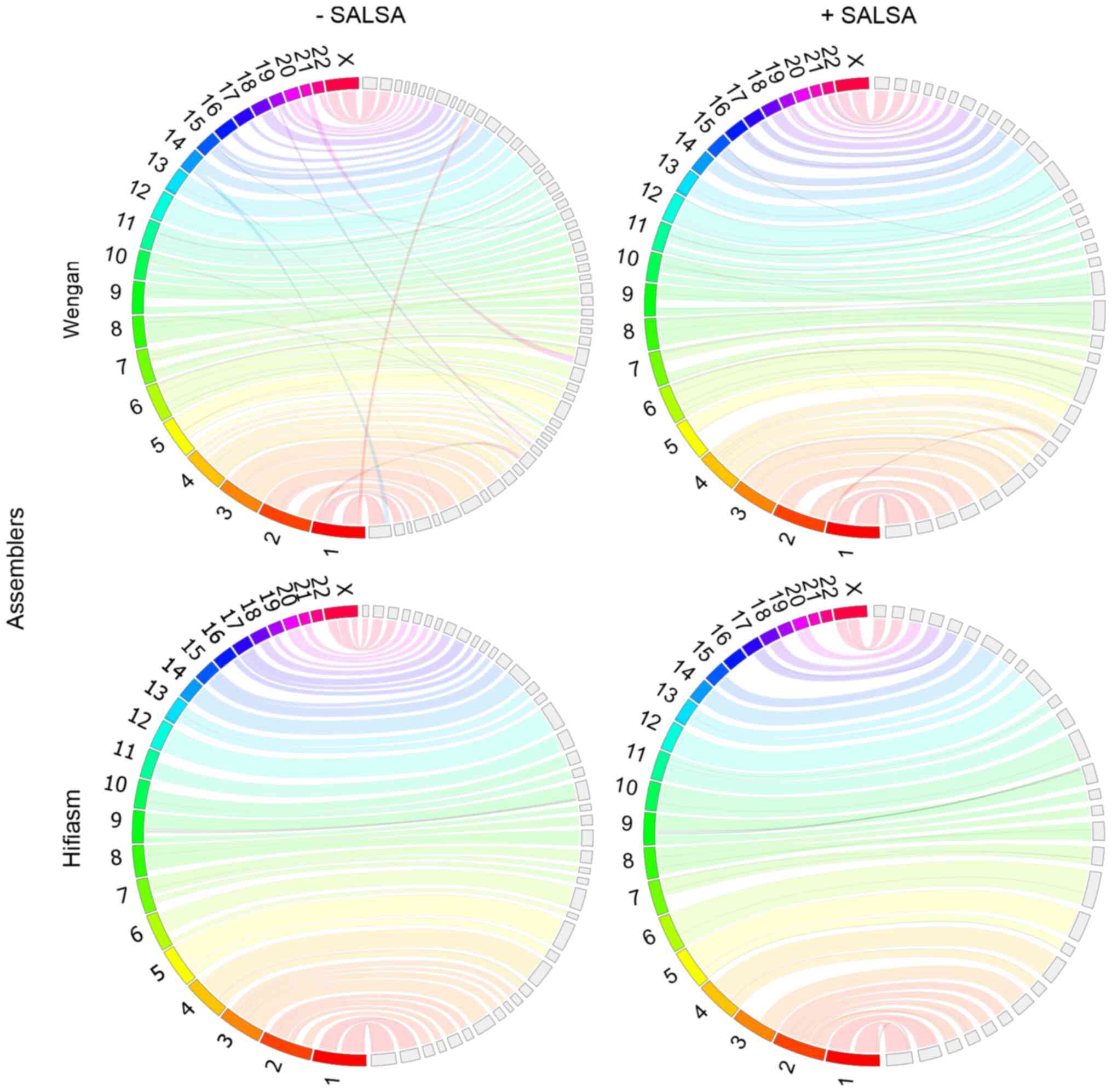
Homo sapiens genome assemblies
The human genome is much more complex than that of
Drosophila; thus, its assembly is a more demanding task
which requires much more computational resources. MaSuRCA and
Wengan hybrid assemblers and Canu long-read assembler, were not
able to complete the assembly of the human genome, even in half of
the original Illumina and Nanopore coverage, on the BRFAA cluster
with 128 GB RAM. Wengan, though, was able to produce a human genome
assembly on AUTh computational system with 256 GB RAM, when FASTQ
files were subsampled by half (Fig.
5). The incorporation of Hi-C data improved the genome
continuity and completeness, while reducing misassemblies (Table VI).
| Table VI.Homo sapiens assembly
metrics. |
Table VI.
Homo sapiens assembly
metrics.
| Assemblers |
Contigs/scaffolds | Genome assembly
size (bp) | N50 | NG50 | L50 | LG50 |
|---|
| Reference | 24 | 3,056,916,522 | 154,259,625 |
| 8 |
|
| Wengan | 2,000 | 2,845,883,522 | 39,733,923 | 36,783,291 | 23 | 26 |
| Wengan/SALSA
(Arima) | 1,689 | 2,845,883,522 | 59,573,195 | 56,310,190 | 15 | 17 |
| Hifiasm | 498 | 3,045,796,332 | 45,256,540 | 45,256,540 | 20 | 20 |
| Hifiasm/SALSA
(Arima) | 431 | 3,045,840,332 | 61,206,687 | 61,206,687 | 15 | 15 |
Hifiasm was unable to assemble the human genome on
the BRFAA cluster when the original 30× coverage of HiFi data was
used. Nevertheless, it succeeded to produce a notable assembly on
the same computational system with subsampled data (16× coverage),
in contrast to HiCanu, which failed to run because of low memory
resources, even with the subsampled data. Hifiasm failed to produce
a contig for chromosome 22. SALSA improved the contiguity, accuracy
and completeness of Hifiasm assembly (Table VI). The longest chromosomes of the
genome are well assembled, however, four of the smallest autosomal
chromosomes (chr 16, 19, 21, 22) are missing (Fig. 5).
Hifiasm outperformed HiCanu, Canu, Wengan and
MaSuRCA, as it managed to run in low resources and low coverage,
producing superior primary and scaffolded assemblies to those of
Wengan.
Discussion
The use of a reference genome in the study of
medical genetics, with the help of novel tools and methods, can
help the identification of novel drug-sequence variant interactions
(46) and the identification of
variants which may be related to mutations with a genetic base of a
variety of genetic diseases, such as cancer (47) and produce further analysis (48). By studying these variants, we are
able to analyse the differences and the heterogeneity of different
populations in order to understand their differences (49).
To propose an optimised de novo genome
assembly workflow, in the present study, factors such as the
maximum assembly contiguity, accuracy and completeness were taken
into account, without ignoring other parameters crucial for the
execution of the sequencing experiments and the production of the
assemblies, such as financial, computational power and time
limitations.
These findings suggest that the assembly exclusively
based on long highly accurate PacBio Hifi reads outperforms
Illumina-Nanopore hybrid and Nanopore assembly. de novo
genome assemblers which use HiFi reads, require lower amounts of
data compared to other strategies. It has been reported that a 30×
genome coverage, using HiFi data, is sufficient in order to produce
high quality assemblies (18,50).
The present study revealed that even a 16× coverage of the human
genome was adequate for that purpose. Thus, subsampling in Hifiasm
assembly strategy allows the adaptation of sequencing data to the
computational resources available as follows: Sequencing data with
a coverage of no higher than 40× can be produced as the current
findings and previous experience from other Hifiasm users
(https://downloads.pacbcloud.com/public/dataset/redwood2020/hifiasm/v12/)
suggest, and if the computational system fails to run, the data can
be subsampled using the divide and conquer approach, until the
computational resources are adequate for the analysis. However, if
the subsampled data correspond to <30× coverage, the final
assembly can be deteriorated, as we notice on Homo sapiens
assembly, where chromosome 22 is missing from the primary assembly
and chromosomes 16, 19, 21 and 22 from the final assembly, after
the scaffolding and correction process. On the other hand, it has
been reported that a hybrid assembly would need 50× Illumina
short-read coverage and 30× Nanopore long-read coverage of the
genome (15,51,52).
In the case of the human genome, notable results with a 34×
Illumina and 30× Nanopore coverage were able to be produced.
Therefore, the volume of data used for HiFi assemblies is much
smaller. As the volume of data decreases, so do the computational
requirements for CPU power and particularly memory. In addition,
the use of highly accurate long reads, bypasses several
computationally demanding, time consuming steps of the assembly
workflow.
In hybrid assembly strategy, Wengan performed most
effectively in terms of accuracy and speed. Wengan produced the
most contiguous Drosophila virilis assemblies. Although no
hybrid assembler produced a human genome assembly in BRFAA cluster,
Wengan was the only assembler that managed to construct a primary
assembly in AUTh computational system.
The assembler we recommend for HiFi reads is
Hifiasm, as it outperformed HiCanu in a small genome and it
succeeded to produce a notable assembly of a large genome whereas
HiCanu failed to run. Hifiasm performed equally well in respect of
insert size and coverage, while HiCanu output improves with the
increase in insert size and coverage. We recommend the use of
Hifiasm or HiCanu assemblers, depending on the available
computational resources as well as the organism's genome size and
complexity. Hifiasm produced the most contiguous assemblies and its
assembly strategy is highly efficient in terms of computational
power and time on a single node of the cluster. For this reason,
Hifiasm is also used by the Human Pangenome Project (https://humanpangenome.org/). On the other hand,
HiCanu gives the possibility to run the assembly on grid when using
a computational cluster. Distributing the tasks on different nodes
allows the use of more computational resources than running on a
central resource and jobs can be executed in parallel speeding
performance. Although running on grid, HiCanu was unable to produce
a human genome assembly, as the main bottleneck of all assemblers
is RAM size. Finally, by following PacBio HiFi assembly strategy
for small genomes, we utilise only one sample preparation and one
sequencing technology, in contrast to the Illumina/Nanopore hybrid
strategy where we need to make three sample preparations (Illumina,
Nanopore and Hi-C) and utilise two sequencing technologies
(Illumina sequencing for short genomic and Hi-C reads and Nanopore
for long genomic reads). For larger genomes, similar to the human
one, PacBio HiFi assembly strategy relies on two sample
preparations and two sequencing technologies (Illumina sequencing
for short Hi-C reads and PacBio long genomic reads).
Our analysis suggests that the use of additional
information for scaffolding is not necessary in small genomes (such
as insect genomes); however, it offers a noticeable improvement in
larger and more complex genomes (such as the human genome and
higher plant genomes). The computational resources required for
scaffolding, even for the most complex genomes, are far less than
those for the assembly step. Ideally, the use of multiple types of
data, seems to exploit different genome features. The successive
use of 10× (https://www.10×genomics.com/) (53,54),
Bionano (https://bionanogenomics.com/)
(55) and Hi-C data will generate
the most accurate scaffolds (25,56).
Although the use of 10× and Bionano data is not imperative, Hi-C
sequencing reads are highly recommended for complex genomes, in
order to increase the continuity of the assembly, while improving
the accuracy by reducing major misassemblies and
translocations.
The development of sequencing technologies led to a
great reduction on sequencing cost. The purchase of a sequencer is
no longer compulsory for genome assembly projects, as different
institutes provide a variety of sequencing services at affordable,
by many labs, prices. Each of PacBio, Illumina and Nanopore, offers
a network of certified sequencing service providers. Some of these
providers are certified for more than one of those sequencing
technologies. Moreover, the purchase of a computational cluster is
no longer necessary, as bioinformatics infrastructures, such as
ELIXIR (57), can offer researchers
the computational recourses necessary for the accomplishment of
demanding tasks, such as a de novo genome assembly.
The major bottlenecks in genome assembly projects
were the computationally demanding assembly algorithms and the
large cost of sequencing. The development of new assembly
algorithms, which require much less computational power and memory,
is the result of major improvements in long-read accuracy by
PacBio. The future of genomics relies on long-reads in order to
resolve low complexity regions of the genomes and perform
telomere-to-telomere assemblies. Alongside to the advances of read
accuracy, third generation sequencing led to the reduction of
sequencing cost. Furthermore, the increase of genomic data
availability in public databases (58), such as Sequence Read Archive (SRA)
(59), allows researchers to find
and use a variety of raw sequencing data from the same species of
interest, already produced by others, for the primary assembly
and/or the scaffolding process. Finally, it is important to note
that all assembly algorithms and methods we utilised during this
work, are being constantly updated in order to improve in terms of
performance and computational efficiency, allowing even the
reanalysis of older data and the discovery of novel information. In
addition, as basecallers are also constantly updated, reusing raw
signal files (for example, fast5-formatted files in Nanopore) can
produce more accurate reads.
In conclusion, continuous advancements in all fields
mentioned above, lead towards the democratisation of de novo
genome assembly projects, by enabling scientific laboratories with
limited technical and financial resources to perform a great
variety of genomic studies, without the need for expensive
sequencing equipment and computational infrastructure.
Acknowledgements
The analyses of this work have been performed using
the computing cluster of the Greek Genome Centre of the Biomedical
Research Foundation, Academy of Athens and the Aristotle University
of Thessaloniki (AUTh) High Performance Computing Infrastructure
and Resources.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MG and KK analysed and interpreted the data. IM
conceived and coordinated the current study. DAS was also involved
in the conception of the study. MG and IM assessed the authenticity
of all the raw data to ensure its legitimacy. All authors
contributed to the writing and revision of the work and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
DAS is the Editor-in-Chief for the journal, but had
no personal involvement in the reviewing process, or any influence
in terms of adjudicating on the final decision, for this article.
The other authors declare that they have no competing
interests.
References
|
1
|
Lander ES, Linton LM, Birren B, Nusbaum C,
Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al
International Human Genome Sequencing Consortium, : Initial
sequencing and analysis of the human genome. Nature. 409:860–921.
2001. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanger F, Nicklen S and Coulson AR: DNA
sequencing with chain-terminating inhibitors. Proc Natl Acad Sci
USA. 74:5463–5467. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kent WJ and Haussler D: Assembly of the
working draft of the human genome with GigAssembler. Genome Res.
11:1541–1548. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shendure J, Balasubramanian S, Church GM,
Gilbert W, Rogers J, Schloss JA and Waterston RH: DNA sequencing at
40: Past, present and future. Nature. 550:345–353. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Salzberg SL and Yorke JA: Beware of
mis-assembled genomes. Bioinformatics. 21:4320–4321. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chaisson MJ, Wilson RK and Eichler EE:
Genetic variation and the de novo assembly of human genomes. Nat
Rev Genet. 16:627–640. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Dijk EL, Jaszczyszyn Y, Naquin D and
Thermes C: The Third Revolution in sequencing technology. Trends
Genet. 34:666–681. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kasianowicz JJ, Brandin E, Branton D and
Deamer DW: Characterization of individual polynucleotide molecules
using a membrane channel. Proc Natl Acad Sci USA. 93:13770–13773.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haque F, Li J, Wu HC, Liang XJ and Guo P:
Solid-state and biological nanopore for real-time sensing of single
chemical and sequencing of DNA. Nano Today. 8:56–74. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eid J, Fehr A, Gray J, Luong K, Lyle J,
Otto G, Peluso P, Rank D, Baybayan P, Bettman B, et al: Real-time
DNA sequencing from single polymerase molecules. Science.
323:133–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Aird D, Ross MG, Chen W-S, Danielsson M,
Fennell T, Russ C, Jaffe DB, Nusbaum C and Gnirke A: Analyzing and
minimizing PCR amplification bias in Illumina sequencing libraries.
Genome Biol. 12:R182011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jain M, Koren S, Miga KH, Quick J, Rand
AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, et al:
Nanopore sequencing and assembly of a human genome with ultra-long
reads. Nat Biotechnol. 36:338–345. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin Y, Yuan J, Kolmogorov M, Shen MW,
Chaisson M and Pevzner PA: Assembly of long error-prone reads using
de Bruijn graphs. Proc Natl Acad Sci USA. 113:E8396–E8405. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tan MH, Austin CM, Hammer MP, Lee YP,
Croft LJ and Gan HM: Finding Nemo: Hybrid assembly with Oxford
Nanopore and Illumina reads greatly improves the clownfish
(Amphiprion ocellaris) genome assembly. Gigascience. 7:1–6.
2018. View Article : Google Scholar
|
|
15
|
Nowak RM, Jastrzębski JP, Kuśmirek W,
Sałamatin R, Rydzanicz M, Sobczyk-Kopcioł A, Sulima-Celińska A,
Paukszto Ł, Makowczenko KG, Płoski R, et al: Hybrid de novo
whole-genome assembly and annotation of the model tapeworm
Hymenolepis diminuta. Sci Data. 6:3022019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Korlach J and Turner SW: Zero-Mode
Waveguides. Encyclopedia of Biophysics. Roberts GC: Springer;
Heidelberg: pp. 2793–2795. 2013, View Article : Google Scholar
|
|
17
|
Wenger AM, Peluso P, Rowell WJ, Chang PC,
Hall RJ, Concepcion GT, Ebler J, Fungtammasan A, Kolesnikov A,
Olson ND, et al: Accurate circular consensus long-read sequencing
improves variant detection and assembly of a human genome. Nat
Biotechnol. 37:1155–1162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Silvestre-Ryan J and Holmes I: Pair
consensus decoding improves accuracy of neural network basecallers
for nanopore sequencing. Genome Biol. 22:382021. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ghurye J and Pop M: Modern technologies
and algorithms for scaffolding assembled genomes. PLoS Comput Biol.
15:e10069942019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lieberman-Aiden E, van Berkum NL, Williams
L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ,
Dorschner MO, et al: Comprehensive mapping of long-range
interactions reveals folding principles of the human genome.
Science. 326:289–293. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Auton A, Brooks LD, Durbin RM, Garrison
EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA and
Abecasis GR; 1000 Genomes Project Consortium, : A global reference
for human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Koepfli KP, Paten B and O'Brien SJ; Genome
10K Community of Scientists, : The Genome 10K Project: A way
forward. Annu Rev Anim Biosci. 3:57–111. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
ICGC/TCGA Pan-Cancer Analysis of Whole
Genomes Consortium: Pan-cancer analysis of whole genomes. Nature.
578:82–93. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Logsdon GA, Vollger MR, Hsieh P, Mao Y,
Liskovykh MA, Koren S, Nurk S, Mercuri L, Dishuck PC and Rhie A:
The structure, function, and evolution of a complete human
chromosome 8. bioRxiv. Sep 8–2020.(Epub ahead of print).
https://doi.org/10.1101/2020.09.08.285395.
|
|
25
|
Miga KH, Koren S, Rhie A, Vollger MR,
Gershman A, Bzikadze A, Brooks S, Howe E, Porubsky D, Logsdon GA,
et al: Telomere-to-telomere assembly of a complete human X
chromosome. Nature. 585:79–84. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Amid C, Alako BTF, Balavenkataraman
Kadhirvelu V, Burdett T, Burgin J, Fan J, Harrison PW, Holt S,
Hussein A, Ivanov E, et al: The European nucleotide archive in
2019. Nucleic Acids Res. 48:D70–D76. 2020.PubMed/NCBI
|
|
27
|
Zimin AV, Marçais G, Puiu D, Roberts M,
Salzberg SL and Yorke JA: The MaSuRCA genome assembler.
Bioinformatics. 29:2669–2677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zimin AV, Puiu D, Luo MC, Zhu T, Koren S,
Marçais G, Yorke JA, Dvořák J and Salzberg SL: Hybrid assembly of
the large and highly repetitive genome of Aegilops tauschii,
a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm.
Genome Res. 27:787–792. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Di Genova A, Buena-Atienza E, Ossowski S
and Sagot MF: Wengan: Efficient and high quality hybrid de novo
assembly of human genomes. bioRxiv. Nov 25–2019.(Epub ahead of
print). doi: https://doi.org/10.1101/840447.
|
|
30
|
Miller JR, Delcher AL, Koren S, Venter E,
Walenz BP, Brownley A, Johnson J, Li K, Mobarry C and Sutton G:
Aggressive assembly of pyrosequencing reads with mates.
Bioinformatics. 24:2818–2824. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan
J, He G, Chen Y, Pan Q, Liu Y, et al: SOAPdenovo2: An empirically
improved memory-efficient short-read de novo assembler.
Gigascience. 1:182012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kolmogorov M, Yuan J, Lin Y and Pevzner
PA: Assembly of long, error-prone reads using repeat graphs. Nat
Biotechnol. 37:540–546. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weisenfeld NI, Yin S, Sharpe T, Lau B,
Hegarty R, Holmes L, Sogoloff B, Tabbaa D, Williams L, Russ C, et
al: Comprehensive variation discovery in single human genomes. Nat
Genet. 46:1350–1355. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Koren S, Walenz BP, Berlin K, Miller JR,
Bergman NH and Phillippy AM: Canu: Scalable and accurate long-read
assembly via adaptive k-mer weighting and repeat separation. Genome
Res. 27:722–736. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Myers EW, Sutton GG, Delcher AL, Dew IM,
Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH,
Remington KA, et al: A whole-genome assembly of Drosophila.
Science. 287:2196–2204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cheng H, Concepcion GT, Feng X, Zhang H
and Li H: Haplotype-resolved de novo assembly with phased assembly
graphs. arXiv. Aug 3–2020.(Epub ahead of print).
|
|
37
|
Nurk S, Walenz BP, Rhie A, Vollger MR,
Logsdon GA, Grothe R, Miga KH, Eichler EE, Phillippy AM and Koren
S: HiCanu: Accurate assembly of segmental duplications, satellites,
and allelic variants from high-fidelity long reads. Genome Res.
30:1291–1305. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chin C-S, Peluso P, Sedlazeck FJ,
Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R,
Figueroa-Balderas R, Morales-Cruz A, et al: Phased diploid genome
assembly with single-molecule real-time sequencing. Nat Methods.
13:1050–1054. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Arima Genomics, Inc., . Arima-HiC Mapping
Pipeline. San Diego. 2019, GitHub. https://github.com/ArimaGenomics/mapping_pipeline/blob/master/Arima_Mapping_UserGuide_A160156_v02.pdf
|
|
40
|
Ghurye J, Rhie A, Walenz BP, Schmitt A,
Selvaraj S, Pop M, Phillippy AM and Koren S: Integrating Hi-C links
with assembly graphs for chromosome-scale assembly. PLoS Comput
Biol. 15:e10072732019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gurevich A, Saveliev V, Vyahhi N and
Tesler G: QUAST: Quality assessment tool for genome assemblies.
Bioinformatics. 29:1072–1075. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Earl D, Bradnam K, St John J, Darling A,
Lin D, Fass J, Yu HO, Buffalo V, Zerbino DR, Diekhans M, et al:
Assemblathon 1: A competitive assessment of de novo short read
assembly methods. Genome Res. 21:2224–2241. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Seppey M, Manni M and Zdobnov EM: BUSCO:
Assessing genome assembly and annotation completeness. Methods Mol
Biol. 1962:227–245. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chu J: Jupiter Plot: A circos-based tool
to visualize genome assembly consistency. 2018, GitHub. https://github.com/JustinChu/JupiterPlot/find/master
|
|
45
|
Krzywinski M, Schein J, Birol I, Connors
J, Gascoyne R, Horsman D, Jones SJ and Marra MA: Circos: An
information aesthetic for comparative genomics. Genome Res.
19:1639–1645. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kyriakidis K, Charalampidou A, Natsiavas
P, Vizirianakis IS and Malousi A: Linking exome sequencing data
with drug response aberrations. Stud Health Technol Inform.
264:1845–1846. 2019.PubMed/NCBI
|
|
47
|
Wei X, Ju X, Yi X, Zhu Q, Qu N, Liu T,
Chen Y, Jiang H, Yang G, Zhen R, et al: Identification of sequence
variants in genetic disease-causing genes using targeted
next-generation sequencing. PLoS One. 6:e295002011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kanakoglou DS, Michalettou TD, Vasileiou
C, Gioukakis E, Maneta D, Kyriakidis KV, Georgakilas AG and
Michalopoulos I: Effects of high-dose ionizing radiation in human
gene expression: A meta-analysis. Int J Mol Sci. 21:212020.
View Article : Google Scholar
|
|
49
|
DePristo MA, Banks E, Poplin R, Garimella
KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA,
Hanna M, et al: A framework for variation discovery and genotyping
using next-generation DNA sequencing data. Nat Genet. 43:491–498.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Vollger MR, Logsdon GA, Audano PA,
Sulovari A, Porubsky D, Peluso P, Wenger AM, Concepcion GT,
Kronenberg ZN, Munson KM, et al: Improved assembly and variant
detection of a haploid human genome using single-molecule,
high-fidelity long reads. Ann Hum Genet. 84:125–140. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Chen Z, Erickson DL and Meng J:
Benchmarking hybrid assembly approaches for genomic analyses of
bacterial pathogens using Illumina and Oxford Nanopore sequencing.
BMC Genomics. 21:6312020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Johnson LK, Sahasrabudhe R, Gill JA, Roach
JL, Froenicke L, Brown CT and Whitehead A: Draft genome assemblies
using sequencing reads from Oxford Nanopore Technology and Illumina
platforms for four species of North American Fundulus killifish.
Gigascience. 9:92020. View Article : Google Scholar
|
|
53
|
Coombe L, Zhang J, Vandervalk BP, Chu J,
Jackman SD, Birol I and Warren RL: ARKS: Chromosome-scale
scaffolding of human genome drafts with linked read kmers. BMC
Bioinformatics. 19:2342018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yeo S, Coombe L, Warren RL, Chu J and
Birol I: ARCS: Scaffolding genome drafts with linked reads.
Bioinformatics. 34:725–731. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lam ET, Hastie A, Lin C, Ehrlich D, Das
SK, Austin MD, Deshpande P, Cao H, Nagarajan N, Xiao M, et al:
Genome mapping on nanochannel arrays for structural variation
analysis and sequence assembly. Nat Biotechnol. 30:771–776. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wallberg A, Bunikis I, Pettersson OV,
Mosbech MB, Childers AK, Evans JD, Mikheyev AS, Robertson HM,
Robinson GE and Webster MT: A hybrid de novo genome assembly of the
honeybee, Apis mellifera, with chromosome-length scaffolds.
BMC Genomics. 20:2752019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Crosswell LC and Thornton JM: ELIXIR: A
distributed infrastructure for European biological data. Trends
Biotechnol. 30:241–242. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kodama Y, Shumway M and Leinonen R;
International Nucleotide Sequence Database Collaboration, : The
sequence read archive: Explosive growth of sequencing data. Nucleic
Acids Res. 40:D54–D56. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Leinonen R, Sugawara H and Shumway M;
International Nucleotide Sequence Database Collaboration, : The
sequence read archive. Nucleic Acids Res. 39:D19–D21. 2011.
View Article : Google Scholar : PubMed/NCBI
|