Introduction
Transient forebrain ischemia (tFI) can be induced by
temporary deprivation of arterial blood flow to the forebrain
through occlusion of bilateral common carotid arteries for five
minutes. A tFI can cause death (loss) of pyramidal cells (neurons)
only in the Conus Ammonis 1 region (CA1) of the hippocampus of the
gerbil (1). Pyramidal cells of the
CA1 are called CA1 pyramidal cells. They will die at 4–5 days after
tFI for 5 min. This phenomenon of CA1 pyramidal cell death is
called delayed neuronal death (DND) (1). Afterward, DND has been reported in the
hippocampal CA1 of rodents including rats and mice (2,3).
Possible mechanisms of DND after tFI have been
reported. Among them, transient increase in excitatory amino acid
transmitter efflux is probably associated with the degrees of
neuronal damage or death in ischemic brains (4). In detail, during ischemia, high
extracellular glutamate level can activate ionotropic glutamate
receptors (i.e., N-methyl-D-aspartate receptor (NMDAR) that
are ligand-gated ion channels leading to channel opening and
overflow of calcium ions into neurons. Finally, neuronal
damage/death occurs (5,6). Many researchers have reported that
brain diseases and dysfunctions are related to abnormal NMDAR
expressions (7,8). For example, it has been reported that
pathological activation of NMDAR1 is a major cause of neuronal
death following brain ischemia (9–12).
Brain or body temperature has been thought to be one
of major factors affecting neuronal survival or death after brain
ischemic injury (13–15). Preclinical studies have provided
evidence that hyperthermia has harmful effects on animal models of
transient global cerebral ischemia (16,17)
and transient focal cerebral ischemia models (18,19).
Clinical reports have confirmed that hyperthermia can accelerate
infarction and worsen outcomes of ischemic stroke patients
(20,21). Recently, we have reported that
hyperthermia can accelerate and worsen neuronal damage or death in
the gerbil hippocampus after tFI (13,22,23).
As described above, transient ischemic insults under
hyperthermic condition can lead to extensive brain damage. However,
molecular mechanisms involved in deleterious effects of
hyperthermic condition on results from tFI have not yet been fully
clarified. Thus, the objective of this study was to investigate
changes of NMDAR1 expressions in the hippocampus and relationships
between NMDAR1 changes and neuronal damage or death after tFI
induction under hypothermic condition in gerbils.
Materials and methods
Experimental animals, protocol and
groups
Male Mongolian gerbils (age, 6 months; body weight,
64–74 g) were used. They were obtained from the Experimental Animal
Center located in Kangwon National University (Chuncheon, Kangwon,
Korea). The protocol of this research was approved (approval
number, KW-200113-1) on January 13, 2020 by the Institutional
Animal Care and Use Committee (IACUC) located in Kangwon National
University. The gerbils used in this study were cared under
constant temperature (~23°C) and humidity (~55%) with a 12-h
light/dark cycle. The handling and caring animals conformed to the
guidelines in the ‘Current international laws and policies’ of the
‘NIH Guide for the Care and Use of Laboratory Animals’ (The
National Academies Press, 8th edition, 2011). Number of the gerbils
used for this study were minimized, and the suffering caused by our
procedures used in this experiment was minimized.
Experimental animals (total number=120) were
assigned to four groups: i) sham-operated animals with normothermia
(NT) (NT/sham group, n=12); ii) tFI-operated animals with NT
(NT/tFI group, n=12); iii) sham-operated animals with hyperthermia
(HT) (HT/sham group, n=48); and iv) tFI-operated animals with HT
(HT/tFI group, n=48).
Induction of tFI
As previously described (23), the gerbils to be used for tFI were
anesthetized with mixture of 2.5% isoflurane in 33% oxygen and 67%
nitrous oxide via inhalation. Under anesthesia, hyperthermia was
induced by heating them with a heating pad until rectal temperature
reached 39.5±0.2°C for 1 h before tFI induction. For normothermia,
rectal temperature was regulated at 37±0.2°C. tFI was induced as
follows. Both common carotid arteries were isolated from the
carotid sheath and occluded using aneurysm clips for 5 min.
Thereafter, the gerbils were kept in thermal incubator
(temperature, 23°C; humidity, 60%) to recover to normothermic
level. The gerbils of NT/sham and HT/sham groups received the same
surgical process of tFI without bilateral carotid artery
occlusion.
Passive avoidance test (PAT)
Short term memory was evaluated by PAT used by
previous researchers (24,25) with modifications. In short, we used
a Gemini Avoidance System from GEM 392 (San Diego Instruments)
consisting of two (light and dark) compartments which communicates
through a sliding door was used. The gerbils received training of
PAT for three days before tFI. Namely, the gerbils were allowed to
explore light and dark compartments for 1 min while the sliding
door was opened. And, when the gerbils enter the dark compartment,
the door was closed, and they received an electric foot-shock (0.5
mA for 5 sec) from a steel grid in the floor. Two and five days
after tFI, PAT was performed. The PAT was elevated as latency time.
Namely, the gerbils were put in the light compartment, while the
door was opened. The latency time was that the gerbils in the light
compartment went into the dark compartment after receiving the
electric foot-shock. Maximum latency time was 180 sec, which was to
stay in the light compartment with the an electric foot-shock.
Western blot analysis
The gerbils (n=7 at 6 h, 1 day, 2 days, and 5 days
after tFI) in each group were deeply anesthetized by
intraperitoneal injection of 60 mg/kg pentobarbital sodium and
sacrificed to analyze the level of NMDAR1 protein in the
hippocampal CA1 region after tFI. As previously described (26), the hippocampi were removed, and the
CA1 regions were obtained. The tissues of the CA1 regions were
homogenized in PBS (5 mM, pH 7.4), which contained 0.2% Nonidet
P-40, 0.1 mM ethylene glycol bis (2-aminoethyl Ether)-N,N,N′,N′
tetraacetic acid (EGTA) (pH 8.0), 10 mM ethylendiamine tetraacetic
acid (EDTA) (pH 8.0), 15 mM sodium pyrophosphate, 150 mM NaCl, 50
mM NaF, 2 mM sodium orthovanadate, 100 mM β-glycerophosphate, 1 mM
dithiothreitol (DTT) and 1 mM phenylmethylsulfonyl fluoride (PMSF).
The homogenates were separated by centrifugation, and the protein
level was determined in the supernatants with Micro BCA protein
assay kit (Pierce Chemical Co.). Aliquots containing total protein
(20 µg) were boiled in loading buffer (150 mM Tris, pH 6.8)
containing 6% SDS, 0.3% bromophenol blue, 3 mM DTT, and 30%
glycerol and loaded onto 12.5% polyacrylamide gel. Subsequently,
they were received electrophoresis, and the gels were transferred
to nitrocellulose transfer membranes. To block non-specific
staining, the membranes were incubated with 5% defatted milk at
room temperature for 60 min. The membranes, thereafter, were
immunoreacted with rabbit anti-NMDAR1 (diluted 1:1,000, Abcam),
peroxidase-conjugated goat anti-rabbit IgG (diluted 1:250,
Sigma-Aldrich Co.; Merck KGaA) and ECL kit (Pierce Chemical
Co.).
The analysis of western blotting was done according
to a published method (26). In
brief, the western bands were scanned with computer scanner, and
the quantification of the analysis was done using a Scion Image
software from Scion Corp. The expression rate of NMDAR1 protein was
normalized through the corresponding expression rate of
β-actin.
Tissue processing for histology
Sections containing the hippocampus were prepared as
previously described (13). In
short, the gerbils in each group were deeply anesthetized by
intraperitoneal injection of 60 mg/kg pentobarbital sodium at 6 h,
1 day, 2 days, and 5 days after tFI. Under anesthesia, the brains
of the gerbils were perfused with 4% paraformaldehyde solution (in
0.1 M PB, pH 7.4). The brains were removed and more fixed in the
fixative for 4 h. The brain tissues were infiltrated with solution
of 30% sucrose to avoided tissue damage from freeze. The brain
tissues were frontally sectioned into 30-µm sections in a cryostat,
and they were stored in well plates containing PBS (pH 7.4).
Histofluorescence with Fluoro-Jade
B
Histofluorescence with Fluoro-Jade B (F-JB, a
fluorescent marker of cellular degeneration or death) was carried
out to examine damage/death of cells or neurons in gerbil
hippocampus after tFI. In short, as described in our published
papers (13,23). the prepared brain sections were
immersed in solution of 0.06% potassium permanganate and stained
with solution of 0.0004% F-JB (Histochem).
Eight sections with 90-µm interval were selected in
reference to anatomical landmarks of the gerbil brain atlas
(27). The samples were observed
using a fluorescence microscope from Carl Zeiss with blue
excitation fluorescence filter between 450–490 nm. Images of F-JB
positive cells, which underwent degeneration, as bright fluoresce,
when compared to the background (28), were captured and counted with NIH
ImageJ 1.59 software.
Immunohistochemistry
Immunohistochemistry was performed to examine
neurons using rabbit anti-neuronal nuclei (NeuN) (diluted 1:1,100,
Chemicon International) and NMDAR1 using rabbit anti-NMDAR1
(diluted 1:100, Abcam) according to method in our published papers
(13,23). Briefly, the prepared sections were
treated with solution of 0.3% hydrogen peroxide
(H2O2) and followed by solution of 10% normal
goat serum. Subsequently, these sections were incubated in each
solution of primary antibody for 12 h at 4°C. Thereafter these
sections were incubated in biotinylated goat anti-rabbit IgG
(diluted 1:250, Vector) and streptavidin peroxidase complex
(diluted 1:250, Vector). Finally, these immunoreacted sections were
visualized by soaking them in solution of 3,3′-diaminobenzidine
tetrahydrochloride.
For the analysis of NeuN-positive neurons, images of
NeuN-positive structures were taken from the CA1 with AxioM1 light
microscope from Carl Zeiss. For numbers of NeuN-positive neuron,
the images of NeuN-positive neurons were captured in a 200×200
µm2 at approximate middle of the CA1 region. The cell
count was done by averaging the total cell numbers from all of the
observed sections in each group.
The analysis of NMDAR1-immunoreactive structures in
the area of interest (strata pyramidale of hippocampal CA1 region;
Fig. S1) was evaluated as relative
optical density (ROD), as %. Images of NMDAR1-immunoreactive
structures were capture like above-mentioned method. The background
in the image was controlled, and ROD was calibrated with Adobe
Photoshop version 8.0 and NIH Image 1.59 software according to
method by (29).
Statistical analysis
Data presented here are expressed as the mean ± SEM.
The differences of the means between the groups were statistically
analyzed. Analysis of variance (ANOVA) with a post hoc Bonferroni's
multiple comparison tests with SPSS program was used to elucidate
tFI-related differences among the experimental groups. In addition,
two-way ANOVA was applied with the Bonferroni post hoc for
comparison of two independent variables between the groups of
normothermia and hyperthermia as well as their interaction.
P<0.05 was used for statistical significance.
Results
PAT
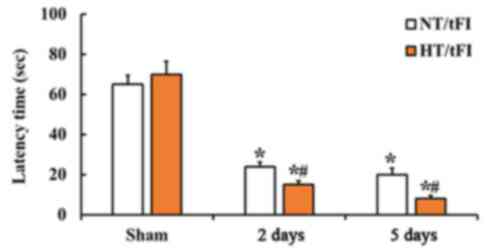
No significant difference in the latency time was
shown between the NT/sham and HT/sham groups (Fig. 1). At 2 days after tFI, the latency
time was significantly decreased in both NT/tFI and HT/tFI groups,
but, in the HT/tFI group, the latency time was significantly
shorter (about 62.5% of the NT/tFI group) than that in the NT/tFI
group (Fig. 1). At 5 days after
tFI, the latency time in both of the groups was much shorter than
that at 2 days after tFI, showing that the latency time in the
HT/tFI group was significantly shorter (about 40% of the NT/tFI
group) than that in the NT/tFI group (Fig. 1).
Neuronal damage/death
NeuN-immunoreactive neurons
NeuN-immunoreactive neurons or cells in the NT/sham
and HT/sham groups were easily shown in the stratum pyramidale (SP)
consisted of pyramidal neurons in the CA1 region (Fig. 2Aa, Ad, Ba, and Bd). In the NT/tFI
group, NeuN-positive cells in the SP was rarely observed 5 days
after tFI (Fig. 2Ac and Bc).
However, numbers of NeuN-positive cells in the SP of the HT/tFI
group were dramatically decreased (about 27.6% of the NT/tFI group)
from 2 days post-tFI (Fig. 2Ae, Be and
C), showing that the numbers of the NeuN+ neurons at
5 days after tFI was similar to that in the NT/tFI group (Fig. 2Af, Bf and C).
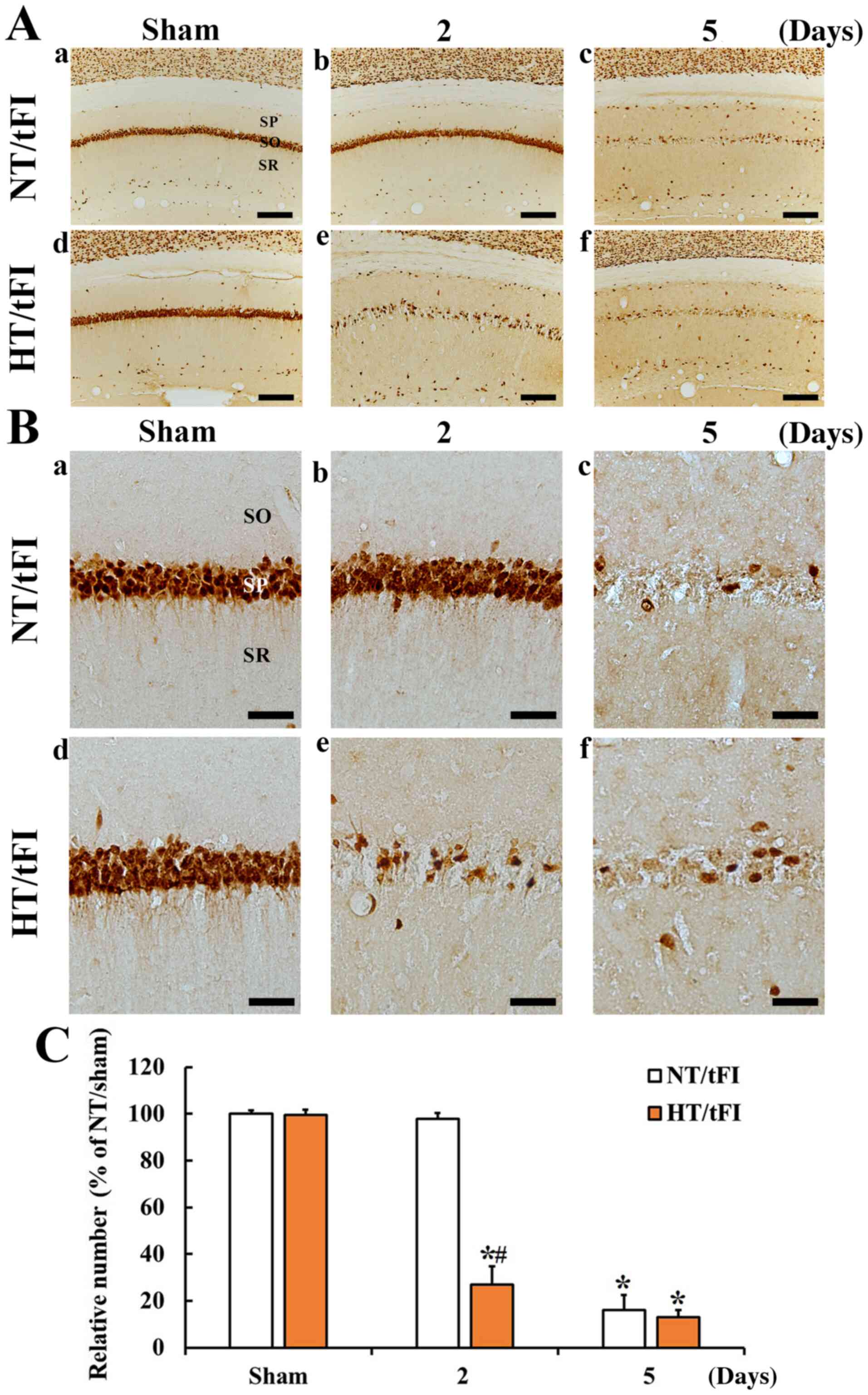 | Figure 2.(A) Low (×10; scale bar, 200 µm) and
(B) high (×40; scale bar, 50 µm) magnification of
immunohistochemical staining for NeuN in the CA1 region of the (a)
NT/sham, (b and c) NT/tFI at 2 and 5 days after tFI, (d) HT/sham
and (e and f) HT/tFI groups at 2 and 5 days after tFI. Five days
after tFI, NeuN+ neurons were rare in the SP. However,
in the HT/tFI group, NeuN+ neurons in the SP were
dramatically decreased from 2 days after tFI. (C) Numbers of
NeuN+ neurons in the CA1 region. The data are presented
as the mean ± SEM (n=7). *P<0.05 vs. respective sham group;
#P<0.05 vs. respective NT/tFI group. SP, stratum
pyramidale; SO, stratum oriens; SR, stratum radiatum; NT,
normothermia; HT, hyperthermia; tFI, transient forebrain ischemia;
NeuN, neuronal nuclei. |
F-JB positive cells
F-JB-positive cells, which are dead cells, were not
shown in the CA1 region of the NT/sham (Fig. 3Ac, Ba, C). In the NT/tFI group,
F-JB-positive cells were not found at 2 days after tFI (Fig. 3Ab, Bb and C), but many F-JB-positive
cells were shown in the SP at 5 days after tFI (Fig. 3Ac, Bc and C).
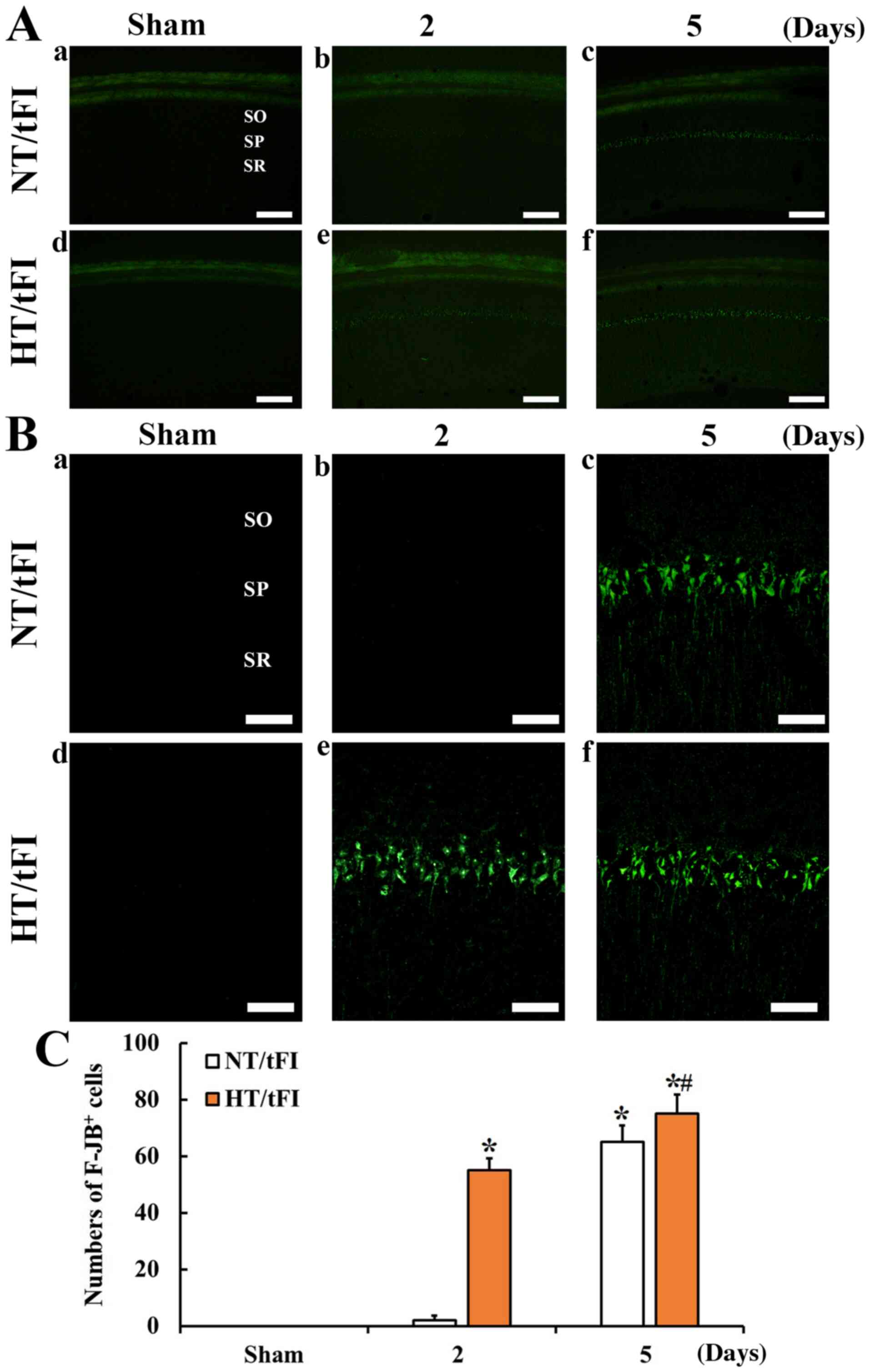 | Figure 3.(A) Low (×10; scale bar, 200 µm) and
(B) high (×40; scale bar, 50 µm) magnification of histofluorescence
with F-JB in the CA1 region of the (a) NT/sham, (b and c) NT/tFI at
2 and 5 days after tFI, (d) HT/sham and (e and f) HT/tFI groups at
2 and 5 days post-tFI. In the NT/tFI group, numerous
F-JB+ cells were detected in the SP 5 days after tFI.
However, in the HT/tFI group, numerous F-JB+ cells were
observed from 2 days post-tFI. (C) Numbers of F-JB+
cells in the CA1 region. The data are presented as the mean ± SEM
(n=7). *P<0.05 vs. respective sham group; #P<0.05
vs. respective NT/tFI group. SP, stratum pyramidale; SO, stratum
oriens; SR, stratum radiatum; NT, normothermia; HT, hyperthermia;
tFI, transient forebrain ischemia; F-JB, Fluoro-Jade B. |
In the HT/sham group, also F-JB-positive cells were
not shown in the CA1 region (Fig. 3Ad,
Bd and C). However, In the HT/tFI group, many F-JB-positive
cells were found in the SP at 2 days after tFI (Fig. 3Ae, Be and C). At 5 days after tFI in
this group, the distribution pattern of F-JB-positive cells was
similar to that at 2 days after tFI (Fig. 3Af, Bf and C).
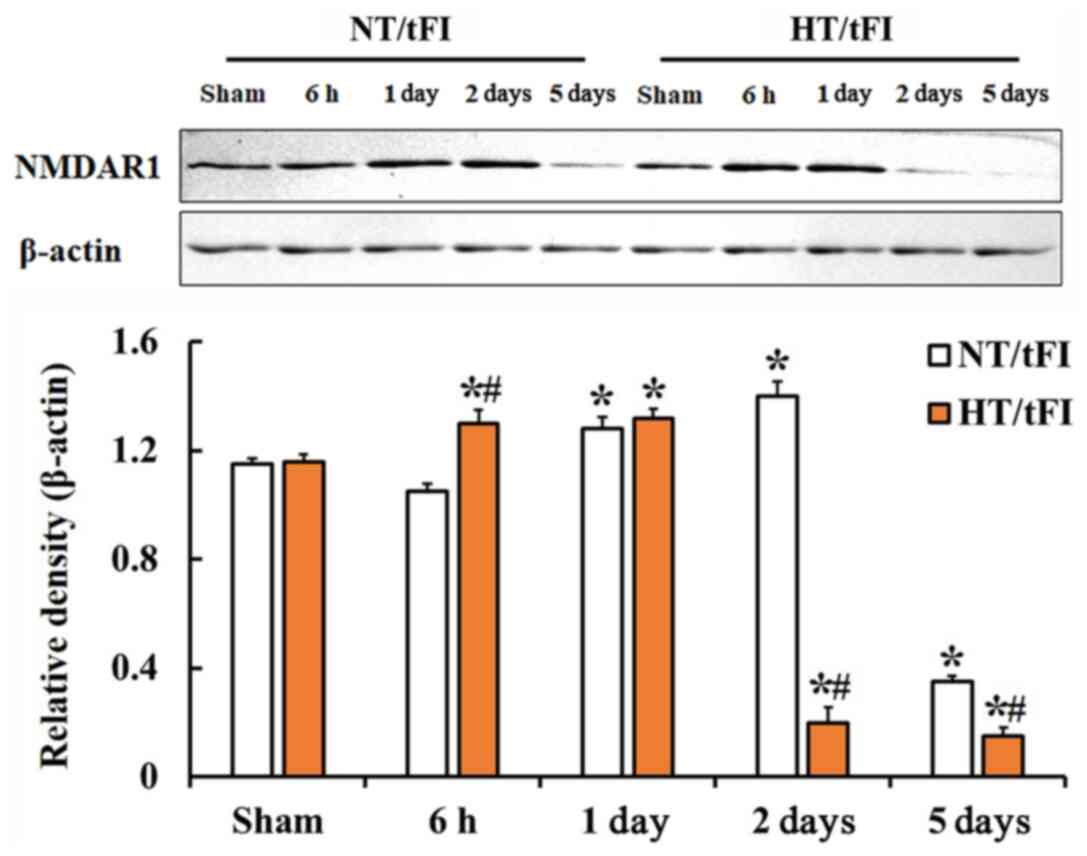
NMDAR1 protein level
NMDAR1 level in the CA1 region of the NT/tFI group
was changed after tFI, showing that the level began to increase
(111.3%) at 1 day, was highest (121.7%) at 2 days and very low
(30.4%) at 5 days after tFI when compared with that in the NT/sham
group (Fig. 4). NMDAR1 level in the
HT/sham group was similar to that in the NT/sham group (Fig. 4). However, in the HT/tFI group,
NMDAR1 level was differently altered after tFI compared to the
HT/tFI group, showing that the level was higher (113.1%) at 6 h,
similar at 1 day, very lower (17.4%) 2 days and low (13.2%) at 5
days after tFI compared with that in the NT/tFI group (Fig. 4).
NMDAR1 immunoreactivity
NMDAR1 immunoreactivity in the NT/sham group was
shown in the neuropil in the stratum oriens (SO) and radiatum (SR),
not in the SP of the CA1 region (Fig.
5Aa and Ba). In the NT/tFI group, NMDAR1 immunoreactivity at 6
h post-tFI was similar to that in the NT/sham group (Fig. 5Ab and Bb), but strong NMDAR1
immunoreactivity was shown in cells in the SP and processes
(dendrites) in the SR (Fig. 5Ab, Ac,
Bb, Bc and C). At 2 days post-tFI, NMDAR1 immunoreactivity in
the cells of the SP was increased compared with that at 1 day
post-tFI, showing that NMDAR1-immunoreactive processes were broken
(Fig. 5Ad, Bd and C). At 5 days
post-tFI, NMDAR1-immunoreactive cells and fibers were decreased
compared with those at 2 days post-tFI (Fig. 5Ae, Be and C).
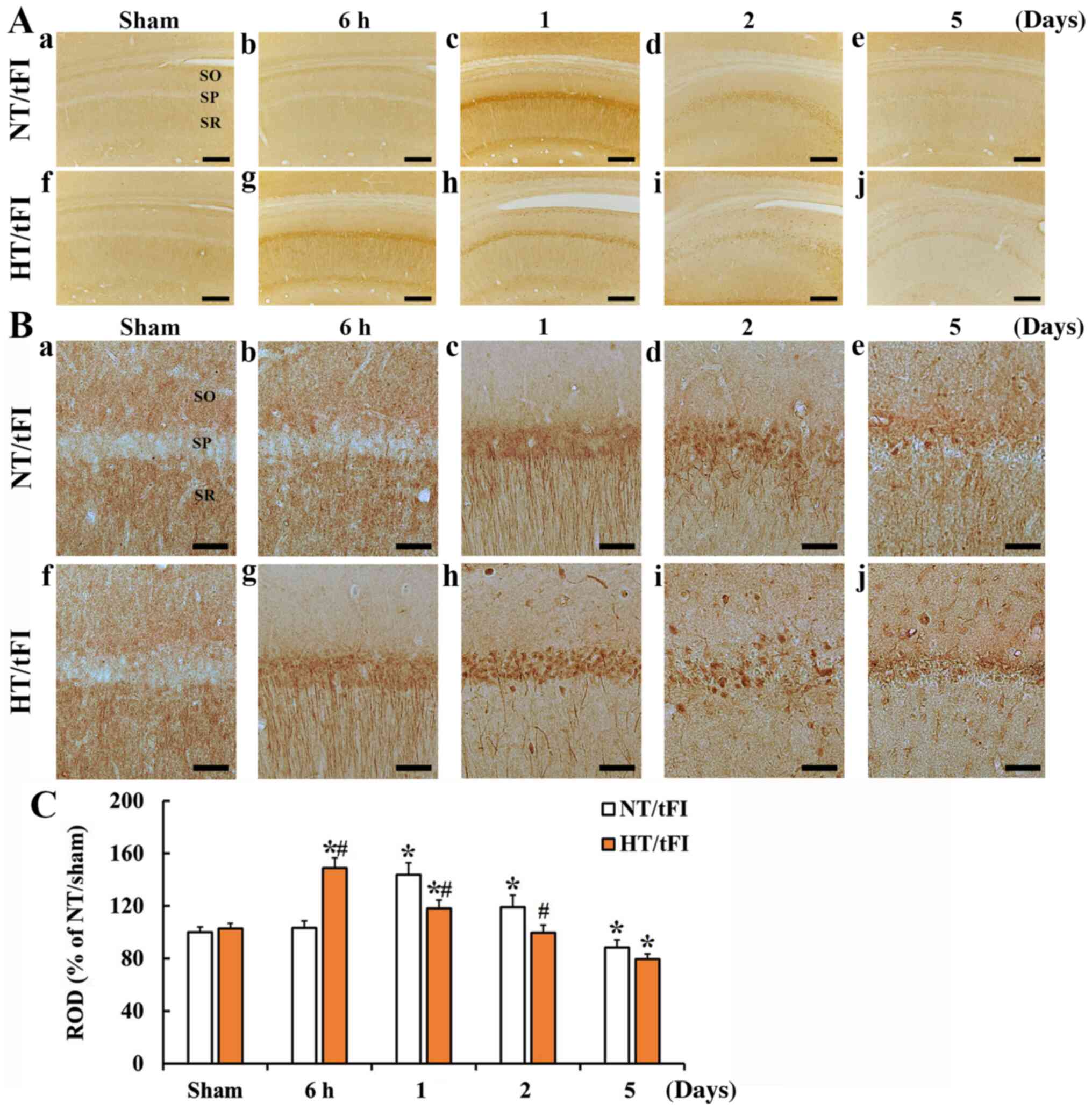 | Figure 5.(A) Low (×10; scale bar, 200 µm) and
(B) high (×40; scale bar, 50 µm) magnification of
immunohistochemical staining for NMDAR1 in the CA1 region of the
(a) NT/sham, (b-e) NT/tFI at 6 h, 1 day, 2 days and 5 days after
tFI, (f) HT/sham and (g-j) HT/tFI groups at 6 h, 1 day, 2 days and
5 days after tFI. NMDAR1 immunoreactivity was hardly observed in
the SP of the sham groups. In the NT/tFI group, strong NMDAR1
immunoreactivity was observed in the somata and processes at 1 and
2 days post-tFI. In the HT/tFI group, NMDAR1 immunoreactivity was
increased from 6 h post-tFI, indicating that the change pattern of
NMDAR1 immunoreactivity was different from that in the NT/tFI
group. (C) ROD of NMDAR1 immunoreactivity in the CA1 region. The
data are presented as the mean ± SEM (n=7). *P<0.05 vs.
respective sham group; #P<0.05 vs. respective NT/tFI
group. SP, stratum pyramidale; SO, stratum oriens; SR, stratum
radiatum; NT, normothermia; HT, hyperthermia; tFI, transient
forebrain ischemia; ROD, relative optical density; NMDAR1,
N-methyl-D-aspartate receptor 1. |
In the HT/sham group, there was no significant
difference in NMDAR1 immunoreactivity compared to that in the
NT/sham group (Fig. 5Af, Bf and C).
In the HT/tFI group, NMDAR1 immunoreactivity at 6 h was
significantly higher (143.9%) than that in the NT/tFI group,
showing that its distribution pattern was similar to that at 1 day
in the NT/tFI group (Fig. 5Ag, Bg and
C). At 1 day post-tFI, numerous cells in the SP showed strong
NMDAR1 immunoreactivity, thereafter, NMDAR1-immunoreactive
structures were gradually decreased with time after tFI, showing
that the ROD of NMDAR1 immunoreactivity was lower than that in the
NT/tFI group (Fig. 5Ah, Ai, Aj, Bh, Bi,
Bj and C).
Discussion
It has been reported that artificially elevated body
temperature is closely associated with deterioration of
neurological deficits, ischemic lesion (infarct), microvascular
injury, and neuronal damage in various rodent models of brain
ischemic insults (14,30–33).
In addition, clinical data have shown that hyperthermia can
increase infarction and worsen the outcome of ischemic injury in
ischemic stroke patients (20,21,34).
Namely, hyperthermic condition during ischemic insults can markedly
augment mortality and severity of ischemic damage than under a
normothermic condition.
Our current study showed that hyperthermic
preconditioning prior to tFI in the HT/tFI group could
significantly reduce short-term memory function evaluated by PAT
compared to the control (NT/tFI group). This finding agrees with
previous studies showing that cognitive impairment is accelerated
after brain injury under hyperthermia (35,36).
It has been reported that death of pyramidal cells
in the gerbil hippocampal CA1 region following tFI under
normothermic condition occurs at 4–5 days after tFI, and this
phenomenon is known as ‘delayed neuronal death’ (1). However, in the HT/tFI group with
memory deterioration, death (loss) of pyramidal neurons in the CA1
region was accelerated and deteriorated than that in the NT/tFI
group. In particular, neuronal loss in the HT/tFI group began at 2
days post-tFI, while neuronal loss in the NT/tFI group was found at
5 days post-tFI. Although the present study did not confirm
neuronal death at 6 h and 1day after tFI, this finding is similar
to our previously published results showing that, under
hyperthermic conditions before and during ischemia-reperfusion
(IR), neuronal death began at 1 day post-tFI and was accelerated at
2–3 days post-tFI, and that hyperthermia increased the extent and
severity of IR-induced neuronal death and glial activation in the
hippocampal CA1 region (13,23).
Similar findings have been reported in a canine model of transient
forebrain ischemia (37). Taken
together, these results suggest that hyperthermic conditioning in
ischemia can accelerate the extent and severity of neuronal death
in ischemic brains.
NMDAR1 is one of Ca2+-permeable receptors
that mediate glutamate excitotoxicity (38). In 1992, Choi reported that
ischemia-induced excessive glutamate release might result in a
pathological level of Ca2+ influx through NMDAR, leading
to subsequent neuronal death (39).
It has been reported that excessive glutamate release after
ischemic brain injury is accentuated by hyperthermia in animal
models of transient brain ischemia (40). Glutamate excitotoxicity is involved
in the vulnerability of neurons following transient ischemia
(41). It has been suggested that
neuroprotection is provided by late NMDA receptor blockade (i.e.,
blocking of presynaptic release of glutamate after excessive
activation of glutamate receptors and/or blocking of postsynaptic
sensitization of NMDA receptors) (41–43).
In our current study, the expression of NMDAR1 in pyramidal neurons
of the CA1 region in the NT/tFI group was diversely altered after
tFI. This finding is related to the process of damage and death of
pyramidal neurons for 4–5 days after tFI under normothermic
condition.
In this study, NMDAR1 expression in the HT/tFI group
was early augmented in the pyramidal neurons and differently
altered than that in the NT/tFI group. Castillo et al
(44) have reported a
glutamate-related relationship between increased body temperature
and deterioration of symptoms in ischemic stroke patients
clinically. In hyperthermic rats, significantly increased glutamate
release has been observed in the brain following global ischemia
(45). Therefore, the effect of
hyperthermic condition on glutamate release might be mediated by
overexpression of NMDARs under a hyperthermic condition. Results of
our current study and previous studies suggest that accumulation of
NMDAR1 in the early phase after tFI under hyperthermia plays a
critical role in the deterioration of neuronal function after
tFI.
In conclusion, NMDAR1 expression in the HT/tFI group
was earlier and more increased in pyramidal neurons located in the
CA1 stratum pyramidale than that in the NT/tFI group, suggesting
that hyperthermia-mediated overexpression of NMDAR1 might be
tightly associated with deterioration in memory decline and
neuronal damage or death following transient ischemic insults.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Cooperative
Research Program for Agriculture Science and Technology Development
(project no. PJ01329401) Rural Development Administration (Republic
of Korea) and by the Bio-Synergy Research Project (grant no.
NRF-2018M3A9C4076478) of the Ministry of Science and Information
and Communication Technologies through the National Research
Foundation.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
BK, JHA, JCL and MHW were responsible for
experimental design, data analysis and drafting and reviewing the
manuscript. DWK, TKL and YSK performed the experiments and data
collection. MCS, JHC, YMK, JHP and IJK performed the data analysis
and provided critical comments on the whole process of this work.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The protocol of the present study was approved
(approval no. KW-200113-1) on 13th January, 2020, by the
Institutional Animal Care and Use Committee of Kangwon National
University (Chuncheon, Republic of Korea).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kirino T: Delayed neuronal death in the
gerbil hippocampus following ischemia. Brain Res. 239:57–69. 1982.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kirino T, Tamura A and Sano K: Delayed
neuronal death in the rat hippocampus following transient forebrain
ischemia. Acta Neuropathol. 64:139–147. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen J, Nagayama T, Jin K, Stetler RA, Zhu
RL, Graham SH and Simon RP: Induction of caspase-3-like protease
may mediate delayed neuronal death in the hippocampus after
transient cerebral ischemia. J Neurosci. 18:4914–4928. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Benveniste H, Drejer J, Schousboe A and
Diemer NH: Elevation of the extracellular concentrations of
glutamate and aspartate in rat hippocampus during transient
cerebral ischemia monitored by intracerebral microdialysis. J
Neurochem. 43:1369–1374. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arundine M and Tymianski M: Molecular
mechanisms of calcium-dependent neurodegeneration in
excitotoxicity. Cell Calcium. 34:325–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kristian T and Siesjo BK: Calcium in
ischemic cell death. Stroke. 29:705–718. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Young AB, Greenamyre JT, Hollingsworth Z,
Albin R, D'Amato C, Shoulson I and Penney JB: NMDA receptor losses
in putamen from patients with Huntington's disease. Science.
241:981–983. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Stanika RI, Winters CA, Pivovarova NB and
Andrews SB: Differential NMDA receptor-dependent calcium loading
and mitochondrial dysfunction in CA1 vs. CA3 hippocampal neurons.
Neurobiol Dis. 37:403–411. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao S, Yu Y, Ma ZY, Sun H, Zhang YL, Wang
XT, Wang C, Fan WM, Zheng QY and Ma CL: NMDAR-mediated hippocampal
neuronal death is exacerbated by activities of ASIC1a. Neurotox
Res. 28:122–137. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Seo JY, Yan BC, Park JH, Ahn JH, Kim IH,
Lee JC, Kwon YG, Kim YM, Cho JH and Won MH: Comparison of the
immunoreactivities of NMDA receptors between the young and adult
hippocampal CA1 region induced by experimentally transient cerebral
ischemia. J Neurol Sci. 325:108–114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Benquet P, Gee CE and Gerber U: Transient
brain ischemia: NMDA receptor modulation and delayed neuronal
death. Med Sci (Paris). 24:185–190. 2008.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yanli L, Xizhou Z, Yan W, Bo Z, Yunhong Z,
Zicheng L, Lingling Y, Lingling Y, Zhangao C, Min Z and Zhi H:
Clonidine preconditioning alleviated focal cerebral ischemic insult
in rats via up-regulating p-NMDAR1 and down-regulating
NMDAR2A/p-NMDAR2B. Eur J Pharmacol. 793:89–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kim MJ, Cho JH, Cho JH, Park JH, Ahn JH,
Tae HJ, Cho GS, Yan BC, Hwang IK, Lee CH, et al: Impact of
hyperthermia before and during ischemia-reperfusion on neuronal
damage and gliosis in the gerbil hippocampus induced by transient
cerebral ischemia. J Neurol Sci. 348:101–110. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Busto R, Dietrich WD, Globus MY and
Ginsberg MD: The importance of brain temperature in cerebral
ischemic injury. Stroke. 20:1113–1114. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hsu SF, Niu KC, Lin CL and Lin MT: Brain
cooling causes attenuation of cerebral oxidative stress, systemic
inflammation, activated coagulation, and tissue ischemia/injury
during heatstroke. Shock. 26:210–220. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dietrich WD, Busto R, Valdes I and Loor Y:
Effects of normothermic versus mild hyperthermic forebrain ischemia
in rats. Stroke. 21:1318–1325. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Baena RC, Busto R, Dietrich WD, Globus MY
and Ginsberg MD: Hyperthermia delayed by 24 hours aggravates
neuronal damage in rat hippocampus following global ischemia.
Neurology. 48:768–773. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morikawa E, Ginsberg MD, Dietrich WD,
Duncan RC, Kraydieh S, Globus MY and Busto R: The significance of
brain temperature in focal cerebral ischemia: Histopathological
consequences of middle cerebral artery occlusion in the rat. J
Cereb Blood Flow Metab. 12:380–389. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
de Jonge JC, Wallet J and van der Worp HB:
Fever worsens outcomes in animal models of ischaemic stroke: A
systematic review and meta-analysis. Eur Stroke J. 4:29–38. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seo WK, Yu SW, Kim JH, Park KW and Koh SB:
The impact of hyperthermia and infection on acute ischemic stroke
patients in the intensive care unit. Neurocrit Care. 9:183–188.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Lim LL, Levi C, Heller RF and
Fisher J: Influence of admission body temperature on stroke
mortality. Stroke. 31:404–409. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim DW, Cho JH, Cho GS, Kim IH, Park JH,
Ahn JH, Chen BH, Shin BN, Tae HJ, Hong S, et al: Hyperthermic
preconditioning severely accelerates neuronal damage in the gerbil
ischemic hippocampal dentate gyrus via decreasing SODs expressions.
J Neurol Sci. 358:266–275. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lee JC, Cho JH, Lee TK, Kim IH, Won MH,
Cho GS, Shin BN, Hwang IK, Park JH, Ahn JH, et al: Effect of
hyperthermia on calbindin-D 28k immunoreactivity in the hippocampal
formation following transient global cerebral ischemia in gerbils.
Neural Regen Res. 12:1458–1464. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen BH, Park JH, Lee YL, Kang IJ, Kim DW,
Hwang IK, Lee CH, Yan BC, Kim YM, Lee TK, et al: Melatonin improves
vascular cognitive impairment induced by ischemic stroke by
remyelination via activation of ERK1/2 signaling and restoration of
glutamatergic synapses in the gerbil hippocampus. Biomed
Pharmacother. 108:687–697. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee JC, Park JH, Ahn JH, Kim IH, Cho JH,
Choi JH, Yoo KY, Lee CH, Hwang IK, Cho JH, et al: New GABAergic
neurogenesis in the hippocampal CA1 region of a gerbil model of
long-term survival after transient cerebral ischemic injury. Brain
Pathol. 26:581–592. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee JC, Kim IH, Park JH, Ahn JH, Cho JH,
Cho GS, Tae HJ, Chen BH, Yan BC, Yoo KY, et al: Ischemic
preconditioning protects hippocampal pyramidal neurons from
transient ischemic injury via the attenuation of oxidative damage
through upregulating heme oxygenase-1. Free Radic Biol Med.
79:78–90. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Radtke-Schuller S, Schuller G, Angenstein
F, Grosser OS, Goldschmidt J and Budinger E: Brain atlas of the
Mongolian gerbil (Meriones unguiculatus) in CT/MRI-aided
stereotaxic coordinates. Brain Struct Funct. 221 (Suppl 1):S1–S272.
2016. View Article : Google Scholar
|
|
28
|
Schmued LC and Hopkins KJ: Fluoro-Jade B:
A high affinity fluorescent marker for the localization of neuronal
degeneration. Brain Res. 874:123–130. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sugawara T, Lewen A, Noshita N, Gasche Y
and Chan PH: Effects of global ischemia duration on neuronal,
astroglial, oligodendroglial, and microglial reactions in the
vulnerable hippocampal CA1 subregion in rats. J Neurotrauma.
19:85–98. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Corbett D and Thornhill J: Temperature
modulation (hypothermic and hyperthermic conditions) and its
influence on histological and behavioral outcomes following
cerebral ischemia. Brain Pathol. 10:145–152. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dietrich WD, Halley M, Valdes I and Busto
R: Interrelationships between increased vascular permeability and
acute neuronal damage following temperature-controlled brain
ischemia in rats. Acta Neuropathol. 81:615–625. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barber PA, Hoyte L, Colbourne F and Buchan
AM: Temperature-regulated model of focal ischemia in the mouse: A
study with histopathological and behavioral outcomes. Stroke.
35:1720–1725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Campos F, Blanco M, Barral D, Agulla J,
Ramos-Cabrer P and Castillo J: Influence of temperature on ischemic
brain: Basic and clinical principles. Neurochem Int. 60:495–505.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Reith J, Jørgensen HS, Pedersen PM,
Nakayama H, Raaschou HO, Jeppesen LL and Olsen TS: Body temperature
in acute stroke: Relation to stroke severity, infarct size,
mortality, and outcome. Lancet. 347:422–425. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Titus DJ, Furones C, Atkins CM and
Dietrich WD: Emergence of cognitive deficits after mild traumatic
brain injury due to hyperthermia. Exp Neurol. 263:254–262. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Walter EJ and Carraretto M: The
neurological and cognitive consequences of hyperthermia. Crit Care.
20:1992016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wass CT, Lanier WL, Hofer RE, Scheithauer
BW and Andrews AG: Temperature changes of > or=1 degree C alter
functional neurologic outcome and histopathology in a canine model
of complete cerebral ischemia. Anesthesiology. 83:325–335. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moriyoshi K, Masu M, Ishii T, Shigemoto R,
Mizuno N and Nakanishi S: Molecular cloning and characterization of
the rat NMDA receptor. Nature. 354:31–37. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Choi DW: Excitotoxic cell death. J
Neurobiol. 23:1261–1276. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ginsberg MD and Busto R: Combating
hyperthermia in acute stroke: A significant clinical concern.
Stroke. 29:529–534. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Nishizawa Y: Glutamate release and
neuronal damage in ischemia. Life Sci. 69:369–381. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Traystman RJ: Animal models of focal and
global cerebral ischemia. ILAR J. 44:85–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dhawan J, Benveniste H, Luo Z, Nawrocky M,
Smith SD and Biegon A: A new look at glutamate and ischemia: NMDA
agonist improves long-term functional outcome in a rat model of
stroke. Future Neurol. 6:823–834. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Castillo J, Davalos A, Marrugat J and Noya
M: Timing for fever-related brain damage in acute ischemic stroke.
Stroke. 29:2455–2460. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Campos F, Pérez-Mato M, Agulla J, Blanco
M, Barral D, Almeida A, Brea D, Waeber C, Castillo J and
Ramos-Cabrer P: Glutamate excitoxicity is the key molecular
mechanism which is influenced by body temperature during the acute
phase of brain stroke. PLoS One. 7:e441912012. View Article : Google Scholar : PubMed/NCBI
|