Introduction
Congenital ectopia lentis (CEL) is characterized by
displacement of the lens from its normal location (posterior to the
iris and anterior to the vitreous body) as a result of congenital
dysplasia of the suspensory ligament. It can lead to blindness due
to complications, such as high myopia, irregular astigmatism,
glaucoma, and retinal detachment (1,2). The
estimated prevalence rate of CEL was 6.4/100,000 in Denmark in 1998
(3,4). CEL can occur in isolation; however, it
is more common as a feature of disorders involving multiple systems
(e.g., cardiovascular, skeletal, and nervous systems) and syndromic
diseases, including Marfan syndrome [MFS; Online Mendelian
Inheritance in Man (OMIM) no. 154700], homocystinuria (OMIM no.
236200), Weill-Marchesani syndrome (WMS; OMIM no. 277600), and
Ehlers-Danlos syndrome (OMIM no. 225310) (5).
In a Danish study involving patients diagnosed with
CEL, only 29.2% were diagnosed with isolated ectopia lentis (IEL)
(3). Several studies have reported
that CEL can not only be a monogenic disease but also an autosomal
recessive inheritance (such as the ADAMTSL4 gene) (6–8). To
our knowledge, IEL is most commonly caused by variants in the
fillinbrin-1 (FBN1) gene. This gene is located on human
chromosome 15q21 and contains 66 exons spanning 235 kb (9,10). In
1994, Lönnqvist et al first described the p.Glu2447Lys FBN1
variant in a family with autosomal dominant EL inheritance and no
cardiovascular disorders characteristic of MFS (11). To date, there have been reports of
only 16 FBN1 variants causing IEL, of which 12 involved a
cysteine substitution causing a missense variant and eight involved
the EGF domain (12–18). Faivre et al (19) demonstrated that the cysteine
substitution in the FBN1 protein was strongly associated with EL
and that a missense variant in the 5′region of the FBN1 gene
may be more likely to lead to EL. Yang et al (18) found that a cysteine insertion in the
LTBP motif of the FBN1 protein could cause IEL. Liang et al
(20) suggested that the
p.Cys587Arg variant could affect the function of ciliary zonules,
causing IEL; however, it may not affect the conformation of FBN1 or
elastin fibers, nor cause any syndromic features.
Although >2,500 variants have been identified in
FBN1, few have been associated with IEL (21). Variant spectrum and potential
genotype-phenotype correlation analyses of the FBN1 gene in
patients with IEL would be valuable for understanding the mechanism
of high phenotypic heterogeneity in this gene. Therefore, in this
study, variant screening of FBN1 was carried out in twelve
unrelated probands to investigate the genotype-phenotype
association in patients with IEL.
Materials and methods
Study participants
A total of twelve unrelated probands with IEL
participated in this study. Detailed medical histories were
obtained, and physical examinations were performed on each proband,
including evaluation of the ophthalmic, cardiovascular, and
skeletal systems. The ophthalmic examination included vision,
intraocular pressure, dilated optometry results, slit lamp
inspection, Pentacam, and Ultrasound Biomicroscope. In addition, a
cardiac color Doppler ultrasound was performed to assess the aortic
diameter and heart valve. Height, fingers and sternum were examined
to evaluate the skeletal system. Patients were included if they had
EL in one or both eyes and if their clinical features satisfied the
2010 Ghent criteria (22). Patients
with a history of trauma (such as contusion of the eyeball) were
excluded. This study was performed in accordance with The
Declaration of Helsinki and was approved by The Institutional
Review Board of the Zhongshan Ophthalmic Center, Sun Yat-sen
University of Medicine. Written informed consent was obtained from
all 12 participants (including the probands, their parents and the
legal guardians of participants <18 years old) in accordance
with The Declaration of Helsinki.
Variant detection
Genomic DNA was extracted from peripheral blood
leucocytes of the 12 probands with IEL and their relatives using
Maxwell 16 DNA Purification kit (Promega Corporation) according to
the manufacturer's protocol. Targeted exome sequencing (TES)
involving 126 genes associated with inherited eye diseases,
including FBN1, was performed on DNA samples from all
probands, as described previously (23). Briefly, coding sequences of the 126
genes were captured using the SeqCap EZ Library SR V5 kit (cat. no.
7145594001; Roche NimbleGen, Inc.). A paired library was generated
using a KAPA HTP Library Preparation kit (cat. no. 07961901001,
Roche), then sequenced on the Illumina Miseq platform (Illumina,
Inc.) using an Illumina Miseq v2 kit (cat. no. MS-102-2002, Roche;
300 cycles; paired-end). The average sequencing coverage was ≥125×.
The raw sequencing data in fastq format were exported to the Strand
NGS software (v2.6; Strand Life Sciences Pvt. Ltd.) to call single
nucleotide variants and insertion-deletion variants. All small
insertions, deletions indels and single-nucleotide variants (SNVs)
were annotated and filtered using dbSNP138 InDels, NCBI dbSNP 146,
NCBI RefSeq Gene (https://www.ncbi.nlm.nih.gov/refseq/rsg/), Human
Genome Mutation Database (HGMD; Professional Version; Qiagen GmbH),
and 1000 Genomes Project, ExAC database (https://console.cloud.google.com/storage/browser/gnomad-public/legacy).
Pathogenic variants in FBN1 gene (GRCh37, accession
number NC_000015.9). Variants in FBN1 were selected with a position
ranging from 48700503 to 48937985 at chromosome 15 (Human
GRCh37/hg19) from the TES data from the twelve probands. Common
variants with an allelic frequency of ≥0.01 according to the
non-coding region or synonymous variations not affecting splicing
were excluded. The pathogenicity of missense variants was predicted
using several online tools, including Polyphen-2 (score 0–1,
http://genetics.bwh.harvard.edu/pph2/), Combined
Annotation Dependent Depletion (CADD; http://cadd.gs.washington.edu/), Rare Exome Variant
Ensemble learner (REVEL score 0–1; http://sites.google.com/site/revelgenomics/), and
ClinPred (score 0–1; release 2018.10; http://sites.google.com/site/clinpred/). The PhyloP
software (https://ccg.epfl.ch//mga/hg19/phylop/phylop.html)
and GERP++_RS software (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html)
were used to analyze the conservation of variations. A missense
variant would be classified as potentially pathogenic if it was
predicted to be deleterious by at least two of the four prediction
tools. The sequences of potential pathogenic variants were
validated using Sanger sequencing. Protein structures were
predicted using wild-type and mutant amino acid sequences using
Swiss-model software (https://swissmodel.expasy.org/). All potential
variants were classified manually according to the guidelines of
the American College of Medical Genetics and Genomics (24). Comparison between the results of the
present study with the main available variants in FBN1 reported by
previous literatures was also performed (25,26).
Statistical analysis
The differences in the genomic position of the
FBN1 variants and in the age at diagnosis between patients
with MFS or IEL were analyzed using Mann-Whitney U test. Data
analysis was performed using the SPSS statistical package (version
26.0 for Windows; IBM Corp.) P<0.05 was considered to indicate a
statistically significant difference.
Results
Clinical findings
A total of twelve probands participated in the
study, including nine male and three female patients. Clinical
information of all patients is displayed in Table I. The age at examination ranged from
3 to 27 years old, with a mean age of diagnosis of 9.91 years
(±8.85 years). All subjects had symptoms of poor vision and were
diagnosed with IEL. Eleven were first diagnosed with IEL before six
years of age, while the remaining proband was first diagnosed at 20
years of age. All patients with IEL in the present study presented
lens subluxation or luxation in both eyes and none showed
extraocular (cardiovascular or skeletal) features. High myopia was
a common feature of these probands (10/12 cases; 83.33%). The other
two patients had hyperopia. Except for probands 4 and 9, who were
not eligible for surgery due to their young age, all other probands
underwent transscleral-fixated intraocular lens implantation, which
effectively improved their vision.
 | Table I.Characteristics of patients with IEL
in the present study. |
Table I.
Characteristics of patients with IEL
in the present study.
|
|
|
|
| Ocular
examination | Extraocular
system |
|---|
|
|
|
|
|
|
|
|---|
| Patient | Age, years | Sex | Age at, diagnosis
years | VA, (logMAR)
OD/OS | Preoperative
Refraction, OD/OS (D) | Surgery for
IEL | Postoperative BCVA,
(logMAR) OD/OS | Skeletal
anomaly | AO diameter,
mm | Cardiovascular
symptom |
|---|
| 1 | 6 | M | 2 | 0.5/1.3 | −13.5/−9.5 | Yes | 0.09/0.3 | None | 22 | None |
| 2 | 5 | M | 4 | 1.3/1.0 | +14.0/+14.75 | Yes | 0.3/0.0 | None | 18 | None |
| 3 | 4 | M | 3 | 1.2/1.0 | −10.0/−12.0 | Yes | 0.15/0.0 | None | 23 | None |
| 4 | 3 | M | 3 | NA | −7.75/−8.0 | No | NA | None | 18 | None |
| 5 | 3 | M | 3 | 1.3/1.5 | −24.0/−23.0 | Yes | 0.15/0.3 | None | 23 | None |
| 6 | 4 | M | 3 | 1.3/0.6 | −14.0/−18.0 | Yes | 0.15/0.3 | None | 17 | None |
| 7 | 11 | F | 5 | 1.3/1.5 | −8.0/−7.0 | Yes | 0.15/0.09 | None | 29 | None |
| 8 | 27 | F | 5 | 0.7/0.9 | −10.0/−14.0 | Yes | 0.04/0.00 | None | 29 | None |
| 9 | 4 | M | 3 | 0.6/1.0 | +2/+2 | No | NA | None | 18 | None |
| 10 | 26 | F | 6 | 2.0/1.3 | −18.0/−14.0 | Yes | NA/0.00 | None | 27 | None |
| 11 | 21 | M | 20 | 0.3/1.3 | −17.0/−8.5 | Yes | 0.15/0.00 | None | NA | None |
| 12 | 5 | M | 3 | 1.1/0.9 | −7.5/−5.5 | Yes | 0.15/0.00 | None | 16 | None |
Variants in FBN1 gene
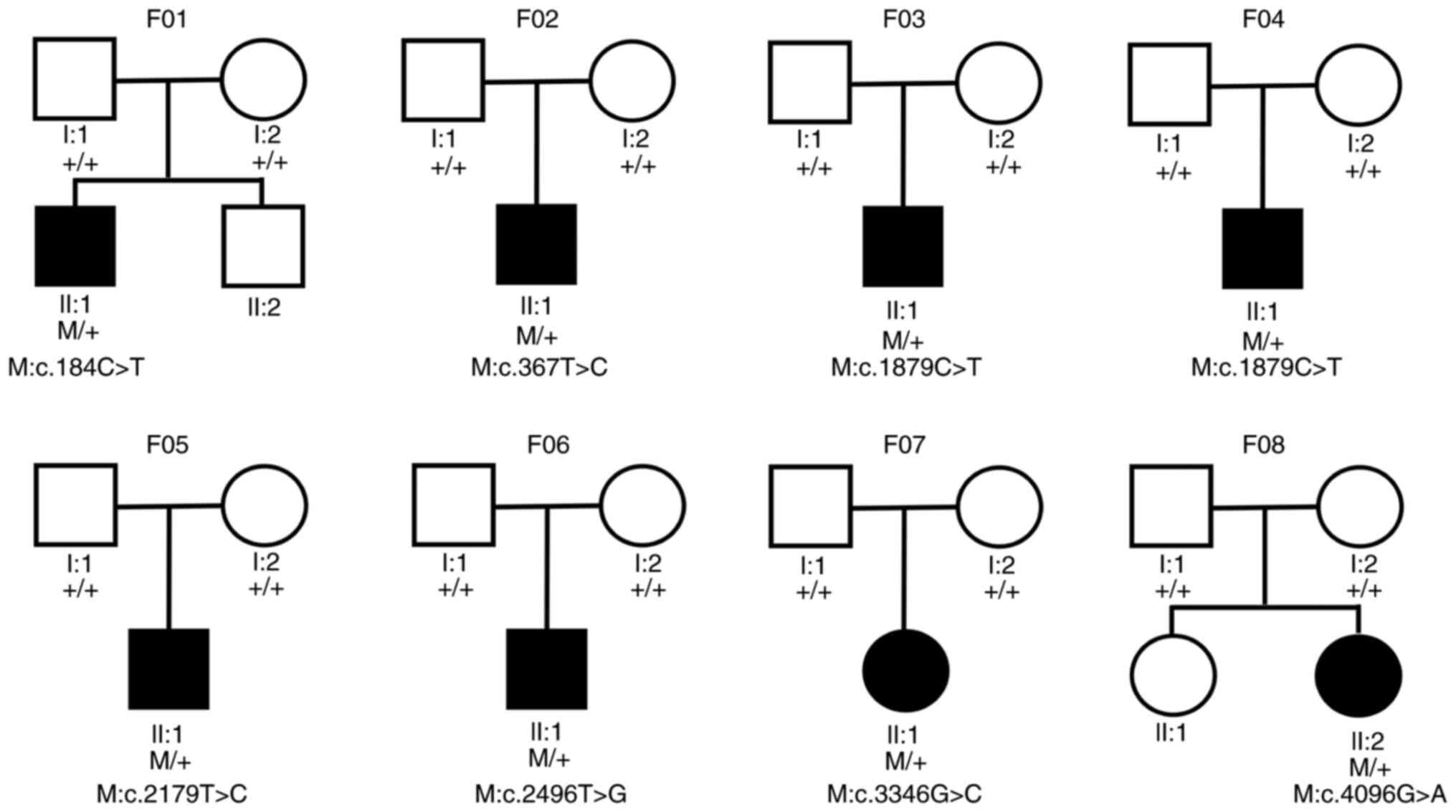
In total, seven missense variants in FBN1
were identified in eight of the 12 probands with EL, and no variant
was detected in the other four probands. The variant frequency was
66.67%. All eight variants of FBN1 were de novo,
since they were not detected in the parents of the eight probands.
The pedigrees of eight families are displayed in Fig. 1. In total, three (c.2179T>C,
c.2496T>G and c.3346G>C) of the eight variants were novel
variants. The other four (c.184C>T, c.367T>C, c.1879C>T
and c.4096G>A) had been reported previously (Table II; Fig.
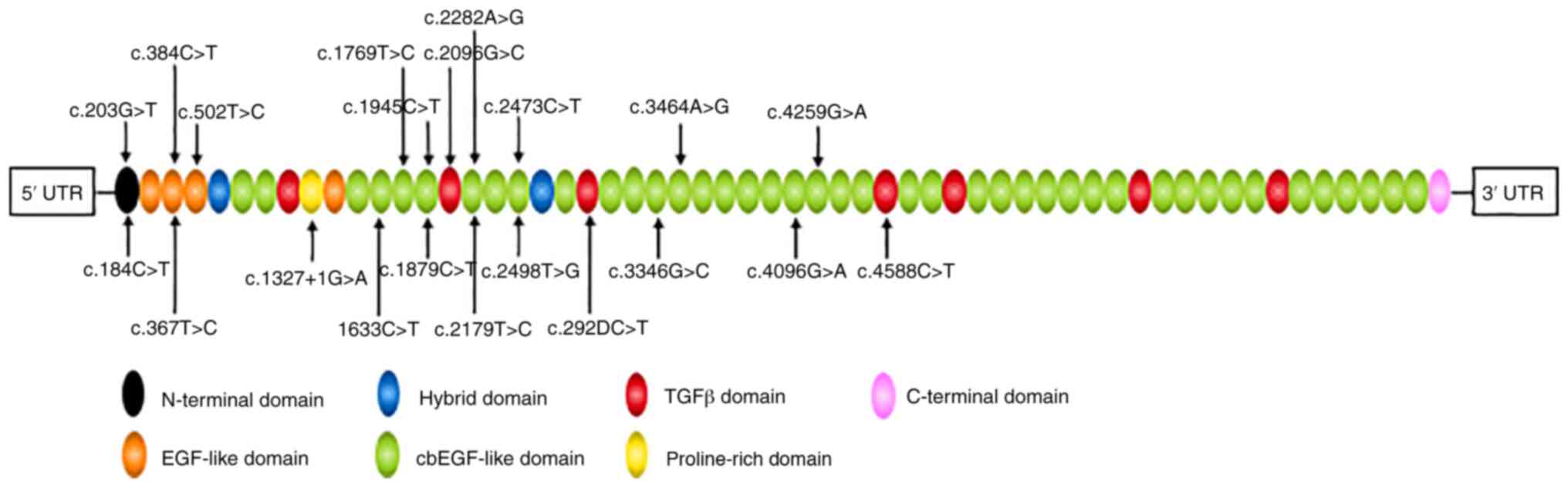
2) (27–29). All eight variants were located at
the calcium-binding epithermal growth factor (cbEGF)-like domain
(Fig. 3) and were predicted to be
deleterious by all four prediction tools (Table II).
| Table II.The seven FBN1 variants
identified in patients with isolated ectopia lentis. |
Table II.
The seven FBN1 variants
identified in patients with isolated ectopia lentis.
| First author,
year | Patient | Exon | Nucleotide
variant | Protein
variant | Domain | PolyPhen-2 | REVEL | CADD | ClinPred | HGMD | ACMG | (Refs.) |
|---|
| Körkkö et
al, 2002 | 1 | 2 | c.184C>T | p.Arg62Cys | 4-Cys motif
LTBP-like | 1.0 | 0.601 | 28.6 | 0.994 | DM | PS1, PS2, PM2,
PP3 | (27) |
| Arbustini et
al, 2005 | 2 | 4 | c.367T>C | p.Cys123Arg | ECF-like #02 | 0.998 | 0.991 | 28.0 | 0.999 | DM | PS1, PS2, PM2,
PP3 | (28) |
| Hayward et
al, 1994 | 3 and 4 | 15 | c.1879C>T | p.Arg627Cys | cbEGF-like #06 | 1.0 | 0.684 | 22.9 | 0.768 | DM | PS1, PS2, PM2,
PP3 | (44) |
| Present study | 5 | 18 | c.2179T>C | p.Cys727Arg | cbEGF-like #07 | 0.997 | 0.977 | 27.6 | 0.999 | NA | PS2, PM2, PP3 | – |
| Present study | 6 | 20 | c.2496T>G | p.Cys832Trp | cbEGF-like #09 | 0.999 | 0.914 | 12.56 | 0.999 | NA | PS2, PM2, PP3 | – |
| Present study | 7 | 27 | c.3346G>C | p.Glu1116Gln | cbEGF-like #13 | 0.997 | 0.899 | 28.0 | 0.979 | NA | PS2, PM2, PP3 | – |
| Uyeda et al,
2004 | 8 | 33 | c.4096G>A | p.Glu1366Lys | cbEGF-like #19 | 0.999 | 0.950 | 41 | 0.999 | DM | PS1, PM2, PP3 | (46) |
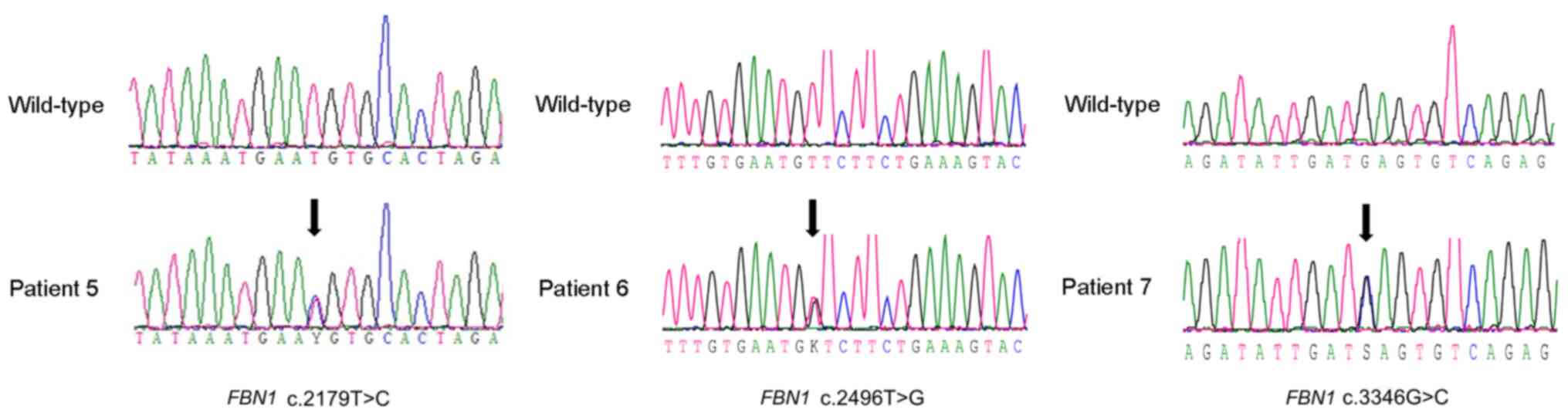
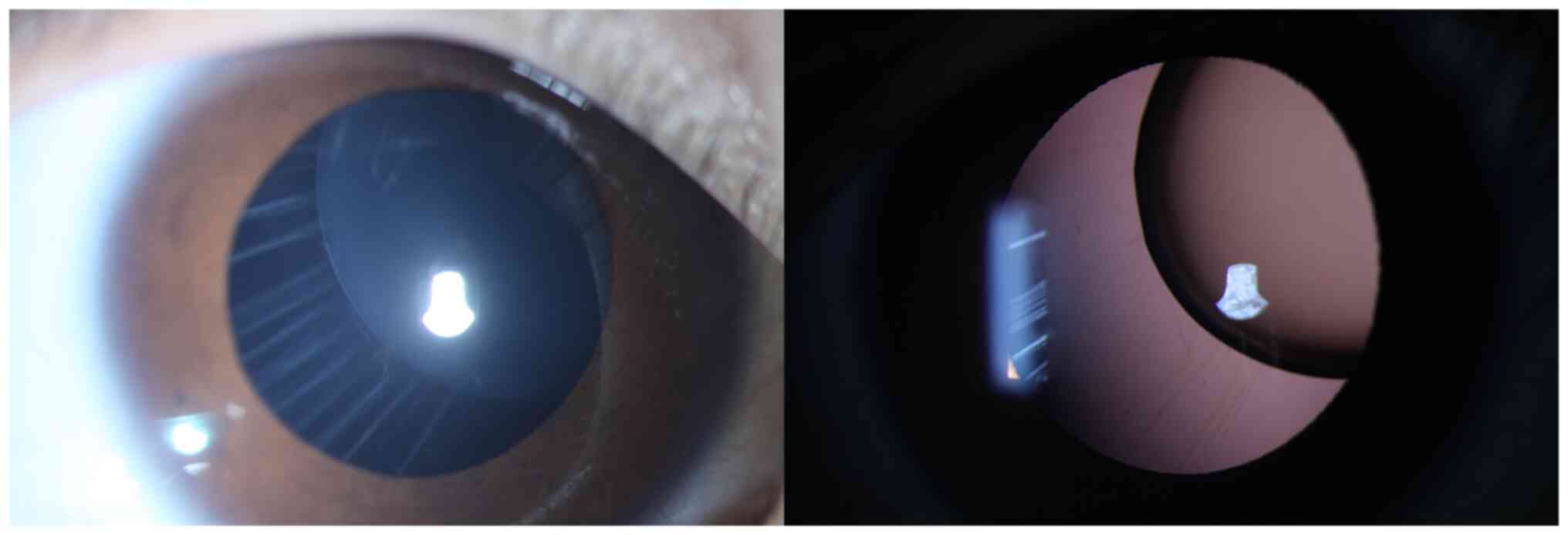
The lens dislocation of a patient with a
heterozygous c. 367T>C (p.Cys123Arg) variant in FBN1 is
shown in Fig. 4. The main reported
variants in FBN1 gene were collected and shown in Table SI, the average age of patients is
24.6 years old, and there is no obvious trend in the mutation sites
of the FBN1 gene. Compared with patients with MFS, patients with
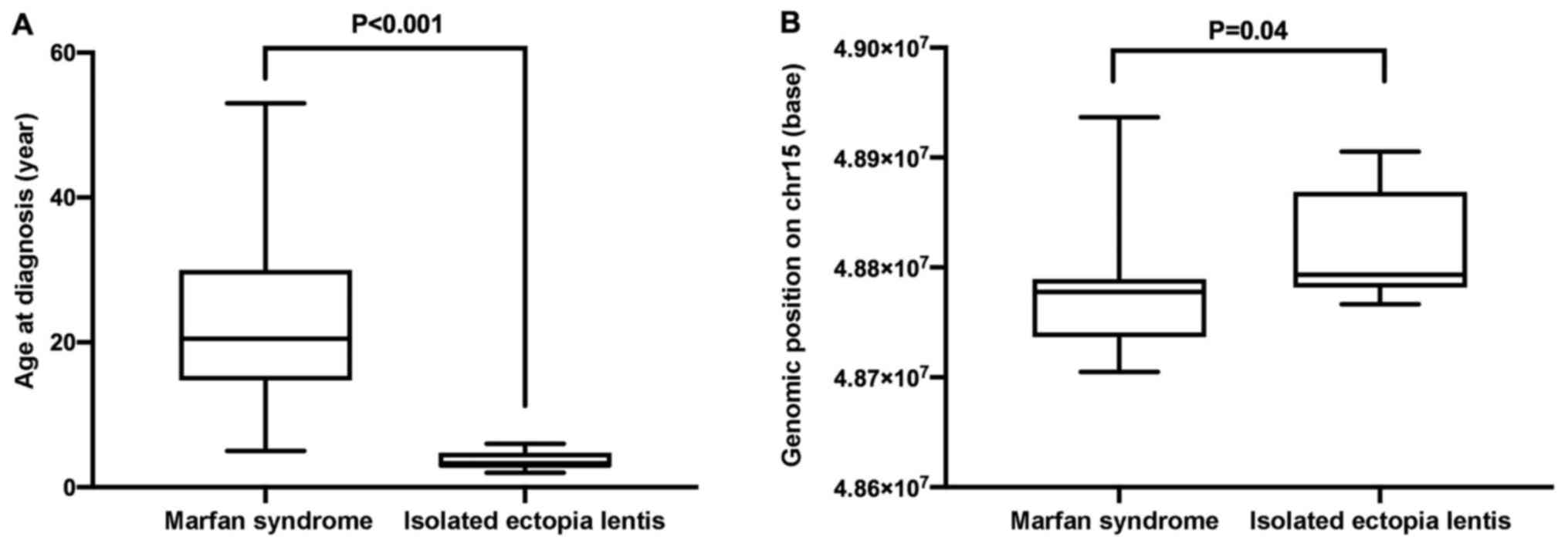
IEL were diagnosed at younger ages (Fig. 5A; P<0.001). Moreover, the
variants identified in the probands with IEL in the present study
were located in upstream coding regions of the gene (from exon 2 to
exon 33; Table II), whereas
variants associated with MFS were located throughout the whole
region of the gene as reported by previous studies (25,26).
The genomic positions of the FBN1 gene variants were in the
upstream regions of chromosome 15 (P=0.04; Fig. 5B).
Discussion
CEL is an autosomal dominant connective tissue
disorder. Several FBN1 variants have been reported to be
pathogenic for CEL, the majority of which are associated with
syndromic CEL (including MFS) (19). However, only a few studies have
examined FBN1 variants in patients with IEL (30). In the present study, seven
FBN1 variants in eight patients with IEL, including three
novel variants and four previously reported variants.
The FBN1 gene encodes the fibrillin 1
protein, which is an essential element of microfibrils found in
elastic and nonelastic tissues, including ciliary bodies, blood
vessels, lungs, and skin (5). In
patients with MFS, WMS or IEL (31), >2,500 variants have been
identified in this gene, most of which were identified in patients
with MFS. It has been reported that variants in FBN1
accounted for ~90% of patients with MFS (32). However, only 16 identified variants
in the FBN1 are associated with IEL (30), consistent with the variant frequency
in the present study. Missense variants account for two-thirds of
the 2,500 known variants in FBN1 (19). Most missense variants are located at
the cbEGF domain, and ~70% of these are conserved cysteine
substitutions (33). Cysteine plays
an important role in the EGF-like domain of the FBN1 protein.
Indeed, substitutions of cysteine residues can disrupt one of three
disulfide bridges that covalently connect three pairs of cysteine
residues that are highly conserved in EGF-like domains, thereby
affecting the structural stability of the domain itself and
neighboring domains as well (34,35).
Previous studies have suggested that missense variants in
FBN1 are associated with EL phenotype (19,36).
EL is more likely when the missense variants involve cysteine
residues (37). In the present
study, missense variants were detected in eight probands, of which
six were located in the cbEGF domain, and five involved a cysteine
residue. The present findings are consistent with those of the
previous study in which EL was associated with missense mutations
and cysteine residue change (37).
A total of three novel heterozygous variants
(c.2179T>C/p.Cys727Arg; c.2496T>G/p. Cys832Trp;
c.3346G>C/p.Glu1116Gln) in FBN1 were identified in three
probands with IEL. The initial symptoms experienced by the three
probands included blurred vision. Only high myopia and EL were
present in these three probands without extraocular features. All
three novel variants are de novo and were predicted to have
a possible pathogenic effect according to PolyPhen-2 and SIFT. In
addition, the two novel variants, p.Cys727Arg and p.Cys832Trp,
occurred at the same positions as the known p.Cys727Tyr and
p.Cys832Tyr respectively. These two variants may abolish a
disulfide bond and affect the first (C1) and sixth (C6) conserved
cysteine of the cbEGF domain that are essential for correct
EGF-like domain structure (38,39).
The variant c.3346G>C is located in cbEGF domain and changes the
conserved glutamic acid residue. It is suggested that the change of
glutamic acid may affect Ca2+ binding of cbEGF domain
and destructs the structure of FBN1 gene (35).
The variant c.184C>T carried by proband 1 has
previously been associated with late-onset IEL (38,40).
In a Chinese family, the age of diagnosis ranged between 41 and 65
years (41). In contrast with these
previous findings, in the present study, the patient with this
variant developed EL symptoms at age 3, however in this study it
was found that it could develop at 2 years old. The c.367T>C
variant is associated with classical MSF, was identified in a
Frenchman in 2005 (28). The
phenotype of the patients in this study only involved the ocular
system. The c.1879C>T variant was reported in 1994 and causes
manifestations in the ocular and skeletal systems, with mild aortic
involvement (29), but the present
study did not find any system disorder in probands. The
c.4096G>A variant was identified in 2004 in two patients, one
with cardiovascular manifestations but no ocular signs, and the
other with MFS (36) and no
cardiovascular system signs were found in any of the probands in
the present study.
Mutations in the FBN1 gene are commonly
associated with MFS. However, in the present study, the eight
probands with FBN1 variants, did not have any systemic
disorders except for IEL. The examination of aortic diameter
indicated that these patients did not meet the standards for
abnormal aortic roots, nor did they meet the revised Ghent criteria
for MFS (22). Nevertheless, it
remains possible that the patients could develop aortic root
dilation or other manifestations of MFS in the future. The present
findings also suggested that ocular disorders might occur in
isolation or may be the first sign of the syndrome. Currently, it
remains difficult to diagnose MFS in young patients with EL; as a
result, these pediatric patients still require long-term annual
cardiovascular imaging.
In the present study, the FBN1 variant
identified in the eight probands were located in the first 33
exons, and some involved arginine-to-cysteine substitutions. Faivre
et al (19) also noted that
EL was more likely when missense changes occurred in the exon 1–21.
Compared with patients with MFS, the average genomic positions of
the variants carried by the eight probands with IEL were located
more upstream of the FBN1 gene. Moreover, IEL was diagnosed
at a younger age, compared with MFS. This result is consistent with
those of previous studies describing EL as the first symptom of
MFS, occurring before the age of 10 (42). Most probands with FBN1 gene
variant suffered from poor visual acuity, significant EL, and high
myopia. Most also underwent surgical treatment, except for proband
11 who was older than 20 years, and proband 4, who was not eligible
for surgery due to parents' disagreement. Therefore, it may be
hypothesized that these variants lead to earlier ocular
symptoms.
The limitations of the current study were that the
patients were young; patients may not have developed other clinical
manifestations at the time of the present study. In addition to MFS
and EL, the FBN1 gene has also been implicated in conditions
such as WMS, isolated ascending aorta, mitral valve prolapse,
aortic root dilatation without dissection (43), and familial arachnodactyly (44). Associations between genotype and
phenotype have not been determined. In addition to the FBN1
gene, IEL can also be inherited in an autosomal recessive fashion
because of ADAMTS4, identified in 2009; variants in this
gene can cause earlier ocular phenotypes than those associated with
FBN1 in Caucasian populations (16,45).
Altogether, the evidence suggests a need for the identification of
other target genes and examination of larger patient cohorts in
future studies. Further study is required to examine associations
between genotype and phenotype in the FBN1 gene. Early
genetic screening contributes to early disease prediction and
management for patients and their families.
In summary, three novel variants and four reported
variants were identified in the FBN1 gene from eight Chinese
probands with IEL. These findings further enrich the FBN1
variant spectrum and may provide insight into the clinical
diagnosis and management of IEL.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by The National Natural
Science Foundation of China (grant nos. 81873673 and 81900841) and
The Fundamental Research Funds of the State Key Laboratory of
Ophthalmology (grant no. 30306020240020212); Dr Guangming Jin
receives support from The Young Teachers Training Program of Sun
Yat-sen University (grant no. 20ykpy143).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The sequencing datasets are also available from the
Sequence Read Archive under BioProject no. PRJNA668437.
Authors' contributions
YZ, GJ and DZ conceived and designed the
experiments. YZ performed the experiments. DG, QAC and XZ analyzed
the data. YZ, DG, QAC and XZ contributed
reagents/materials/analysis tools. YZ, GJ and DG wrote the
manuscript. GJ and DZ confirmed the authenticity of the data in the
present study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was performed in accordance with The
Declaration of Helsinki and was approved by The Institutional
Review Board at the Zhongshan Ophthalmic Center, Sun Yat-sen
University of Medicine. Written informed consent was obtained from
all participants and from the legal guardians of participants
<18 years old, in accordance with The Declaration of
Helsinki.
Patient consent for publication
All participants provided informed consent
(including the probands, their parents and the legal guardians of
participants under 18 years old) for the publication of their data,
including images and examination results.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Maumenee IH: The eye in the marfan
syndrome. Trans Am Ophthalmol Soc. 79:684–733. 1981.PubMed/NCBI
|
|
2
|
Chandra A and Charteris D: Molecular
pathogenesis and management strategies of ectopia lentis. Eye
(Lond). 28:162–168. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fuchs J and Rosenberg T: Congenital
ectopia lentis. A Danish national survey. Acta Ophthalmol Scand.
76:20–26. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sadiq MA and Vanderveen D: Genetics of
ectopia lentis. Semin Ophthalmol. 28:313–320. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nelson LB and Maumenee IH: Ectopia lentis.
Surv Ophthalmol. 27:143–160. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ruiz C, Rivas F, Villar-Calvo VM,
Serrano-Lucas JI and Cantú JM: Familial simple ectopia lentis. A
probable autosomal recessive form. Ophthalmic Paediatr Genet.
7:81–84. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sinha A and Rahman A: Ectopia lentis in a
family. Indian J Ophthalmol. 28:33–35. 1980.PubMed/NCBI
|
|
8
|
Casper DS, Simon JW, Nelson LB, Porter IH
and Lichtenstein SB: Familial simple ectopia lentis: A case study.
J Pediatr Ophthalmol Strabismus. 22:227–230. 1985.PubMed/NCBI
|
|
9
|
Sakai LY, Keene DR and Engvall E:
Fibrillin, a new 350-kD glycoprotein, is a component of
extracellular microfibrils. J Cell Biol. 103:2499–2509. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tynan K, Comeau K, Pearson M, Wilgenbus P,
Levitt D, Gasner C, Berg MA, Miller DC and Francke U: Mutation
screening of complete fibrillin-1 coding sequence: Report of five
new mutations, including two in 8-cysteine domains. Hum Mol Genet.
2:1813–1821. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lönnqvist L, Child A, Kainulainen K,
Davidson R, Puhakka L and Peltonen L: A novel mutation of the
fibrillin gene causing ectopia lentis. Genomics. 19:573–576. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin C, Yao K, Jiang J, Tang X, Shentu X
and Wu R: Novel FBN1 mutations associated with predominant ectopia
lentis and marfanoid habitus in Chinese patients. Mol Vis.
13:1280–1284. 2007.PubMed/NCBI
|
|
13
|
Deng T, Dong B, Zhang X, Dai H and Li Y:
Late-Onset bilateral lens dislocation and glaucoma associated with
a novel mutation in FBN1. Mol Vis. 14:1229–1233. 2008.PubMed/NCBI
|
|
14
|
Turner CL, Emery H, Collins AL, Howarth
RJ, Yearwood CM, Cross E, Duncan PJ, Bunyan DJ, Harvey JF and
Foulds NC: Detection of 53 FBN1 mutations (41 novel and 12
recurrent) and genotype-phenotype correlations in 113 unrelated
probands referred with Marfan syndrome, or a related
fibrillinopathy. Am J Med Genet A. 149A:161–170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zadeh N, Bernstein JA, Niemi AK, Dugan S,
Kwan A, Liang D, Hyland JC, Hoyme HE, Hudgins L and Manning MA:
Ectopia lentis as the presenting and primary feature in marfan
syndrome. Am J Med Genet A. 155A:2661–2668. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chandra A, Aragon-Martin JA, Hughes K,
Gati S, Reddy MA, Deshpande C, Cormack G, Child AH, Charteris DG
and Arno G: A genotype-phenotype comparison of ADAMTSL4 and FBN1 in
isolated ectopia lentis. Invest Ophthalmol Vis Sci. 53:4889–4896.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H, Qu W, Meng B, Zhang S, Yang T, Huang
S and Yuan H: Identification and study of a FBN1 gene mutation in a
Chinese family with ectopia lentis. Mol Vis. 18:504–511.
2012.PubMed/NCBI
|
|
18
|
Yang G, Chu M, Zhai X and Zhao J: A novel
FBN1 mutation in a Chinese family with isolated ectopia lentis. Mol
Vis. 18:945–950. 2012.PubMed/NCBI
|
|
19
|
Faivre L, Collod-Beroud G, Loeys BL, Child
A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, et al:
Effect of mutation type and location on clinical outcome in 1,013
probands with Marfan syndrome or related phenotypes and FBN1
mutations: an international study. Am J Hum Genet. 81:454–466.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liang C, Fan W, Wu S and Liu Y:
Identification of a novel FBN1 mutation in a Chinese family with
isolated ectopia lentis. Mol Vis. 17:3481–3485. 2011.PubMed/NCBI
|
|
21
|
Yang H, Ma Y, Luo M, Zhao K, Zhang Y, Zhu
G, Sun X, Luo F, Wang L, et al: Identification of gross deletions
in FBN1 gene by MLPA. Hum Genomics. 12:462018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Loeys BL, Dietz HC, Braverman AC,
Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y,
Jondeau G, Faivre L, et al: The revised Ghent nosology for the
Marfan syndrome. J Med Genet. 47:476–485. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang P, Li S, Sun W, Xiao X, Jia X, Liu M,
Xu L, Long Y and Zhang Q: An ophthalmic targeted exome sequencing
panel as a powerful tool to identify causative mutations in
patients suspected of hereditary eye diseases. Transl Vis Sci
Technol. 8:212019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chung BH, Lam ST, Tong TM, Li SY, Lun KS,
Chan DH, Fok SF, Or JS, Smith DK, Yang W and Lau YL: Identification
of novel FBN1 and TGFBR2 mutations in 65 probands with marfan
syndrome or marfan-like phenotypes. Am J Med Genet A.
149A:1452–1459. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang H, Luo M, Chen Q, Fu Y, Zhang J, Qian
X, Sun X, Fan Y, Zhou Z and Chang Q: Genetic testing of the FBN1
gene in Chinese patients with marfan/marfan-like syndrome. Clin
Chim Acta. 459:30–35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Körkkö J, Kaitila I, Körkkö L, Peltonen L
and Ala-Kokko L: Sensitivity of conformation sensitive gel
electrophoresis in detecting mutations in marfan syndrome and
related conditions. J Med Genet. 39:34–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arbustini E, Grasso M, Ansaldi S, Malattia
C, Pilotto A, Porcu E, Disabella E, Marziliano N, Pisani A,
Lanzarini L, et al: Identification of sixty-two novel and twelve
known FBN1 mutations in eighty-one unrelated probands with marfan
syndrome and other fibrillinopathies. Hum Mutat. 26:4942005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hayward C, Rae AL, Porteous ME, Logie LJ
and Brock DJ: Two novel mutations and a neutral polymorphism in
EGF-like domains of the fibrillin gene (FBN1): SSCP screening of
exons 15–21 in Marfan syndrome patients. Hum Mol Genet. 3:373–375.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang L, Lai YH, Capasso JE, Han S and
Levin AV: Early onset ectopia lentis due to a FBN1 mutation with
non-penetrance. Am J Med Genet A. 167:1365–1368. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sakai LY, Keene DR, Renard M and De Backer
J: FBN1: The disease-causing gene for marfan syndrome and other
genetic disorders. Gene. 591:279–291. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Baetens M, Van Laer L, De Leeneer K,
Hellemans J, De Schrijver J, Van De Voorde H, Renard M, Dietz H,
Lacro RV, Menten B, et al: Applying massive parallel sequencing to
molecular diagnosis of marfan and loeys-dietz syndromes. Hum Mutat.
32:1053–1062. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Seo GH, Kim YM, Kang E, Kim GH, Seo EJ,
Lee BH, Choi JH and Yoo HW: The phenotypic heterogeneity of
patients with Marfan-related disorders and their variant spectrums.
Medicine (Baltimore). 97:e107672018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aoyama T, Tynan K, Dietz HC, Francke U and
Furthmayr H: Missense mutations impair intracellular processing of
fibrillin and microfibril assembly in marfan syndrome. Hum Mol
Genet. 2:2135–2140. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Downing AK, Knott V, Werner JM, Cardy CM,
Campbell ID and Handford PA: Solution structure of a pair of
calcium-binding epidermal growth factor-like domains: Implications
for the marfan syndrome and other genetic disorders. Cell.
85:597–605. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Comeglio P, Johnson P, Arno G, Brice G,
Evans A, Aragon-Martin J, da Silva FP, Kiotsekoglou A and Child A:
The importance of mutation detection in marfan syndrome and
marfan-related disorders: Report of 193 FBN1 mutations. Hum Mutat.
28:9282007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schrijver I, Liu W, Brenn T, Furthmayr H
and Francke U: Cysteine substitutions in epidermal growth
factor-like domains of fibrillin-1: Distinct effects on biochemical
and clinical phenotypes. Am J Hum Genet. 65:1007–1020. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Stheneur C, Collod-Béroud G, Faivre L,
Buyck JF, Gouya L, Le Parc JM, Moura B, Muti C, Grandchamp B,
Sultan G, et al: Identification of the minimal combination of
clinical features in probands for efficient mutation detection in
the FBN1 gene. Eur J Hum Genet. 17:1121–1128. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu WO, Oefner PJ, Qian C, Odom RS and
Francke U: Denaturing HPLC-identified novel FBN1 mutations,
polymorphisms, and sequence variants in marfan syndrome and related
connective tissue disorders. Genet Test. 1:237–242. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao JH, Jin TB, Liu QB, Chen C and Hu HT:
Ophthalmic findings in a family with early-onset isolated ectopia
lentis and the p.Arg62Cys mutation of the fibrillin-1 gene (FBN1).
Ophthalmic Genet. 34:21–26. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu R, Lai Z, Zhou W, Ti DD and Zhang XN:
Recurrent FBN1 mutation (R62C) in a Chinese family with isolated
ectopia lentis. Am J Ophthalmol. 141:1136–1138. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tinkle BT and Saal HM; Committee on
Genetics, : Health supervision for children with marfan syndrome.
Pediatrics. 132:e1059–e1072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Milewicz DM, Michael K, Fisher N, Coselli
JS, Markello T and Biddinger A: Fibrillin-1 (FBN1) mutations in
patients with thoracic aortic aneurysms. Circulation. 94:2708–2711.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hayward C, Porteous ME and Brock DJ: A
novel mutation in the fibrillin gene (FBN1) in familial
arachnodactyly. Mol Cell Probes. 8:325–327. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ahram D, Sato TS, Kohilan A, Tayeh M, Chen
S, Leal S, Al-Salem M and El-Shanti H: A homozygous mutation in
ADAMTSL4 causes autosomal-recessive isolated ectopia lentis. Am J
Hum Genet. 84:274–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Uyeda T, Takahashi T, Eto S, Sato T, Xu G,
Kanezaki R, Toki T, Yonesaka S and Ito E: Three novel mutations of
the fibrillin-1 gene and ten single nucleotide polymorphisms of the
fibrillin-3 gene in marfan syndrome patients. J Hum Genet.
49:404–407. 2004. View Article : Google Scholar : PubMed/NCBI
|