Introduction
Drug addiction, which is defined as the chronic and
compulsive use of drugs despite adverse consequences (1), is characterized by uncontrolled
drug-seeking behaviour (2),
accompanied by the functional alteration of specific neural
circuits caused by changes in neurotransmitters (3) and synaptic plasticity (4). As drug addiction is a cyclical,
chronic and recurrent disease (5),
the central problem in the treatment of drug addiction is
identifying the underlying mechanisms of relapse. According to
previous studies, a special type of memory, known as drug memory
(6,7), is formed by the association of
drug-induced euphoria with contextual cues, termed pathological
learning (8). Drug memory shapes
behaviours associated with drug addiction by stimulating the desire
or craving for drugs (9,10). Moreover, re-exposure to
drug-conditioned stimuli precipitates the recurrence of previously
extinguished drug-seeking behaviour (11) by recalling drug memory, while the
inhibition of drug memory prevents drug relapse (12). Drug addiction is therefore regarded
as a disease of learning and memory (13), and an overlap in the involved neural
circuitry and underlying molecular mechanisms between normal memory
and drug memory has been demonstrated (14).
A typical example of this overlap is long-term
potentiation (LTP) (15), described
as a lasting enhancement of synaptic transmission efficiency and
intensity induced by a transient high-frequency stimulus (HFS). LTP
was first considered to be a laboratory phenomenon; however, it has
been suggested that an LTP-like phenomenon is induced during memory
formation in an inhibitory avoidance model in rats (16), and LTP maintenance is also involved
in spatial information storage (17). Furthermore, drug-evoked plasticity,
which is a HFS-independent increase in synaptic strength and
connectivity, is observed in addiction (18) and is considered to be associated
with addictive behaviour (19).
Therefore, it was hypothesized that methods disrupting LTP
induction may be applied to inhibit drug memory and drug-seeking
behaviour.
Methamphetamine (MA), a globally abused
psychostimulant (20), induces
memory impairment (21). It has
been identified that endoplasmic reticulum (ER) stress, which
results from the accumulation of unfolded proteins (22), may be one of the underlying
mechanisms for MA-induced memory loss (data not shown). As ER
stress serves a role in the inhibition of normal memory, it was
hypothesized that ER stress may also serve a role in drug memory
inhibition. To validate this hypothesis, the present study first
tested whether MA-induced ER stress disrupted the formation of drug
memory using the conditioned place preference (CPP) test. Next, the
effects of MA-induced ER stress on hippocampal LTP induction were
investigated and the expression levels of several molecules
underlying both normal and addiction memory formation were
measured, which were used to elucidate the mechanisms underlying
drug memory disruption due to MA-induced ER stress.
Materials and methods
Animals
A total of 166 male C57BL/6 mice (age, 6–8 weeks;
weight, 25–30 g) were obtained from SPF (Beijing) Biotechnology
Co., Ltd. Animals were housed with free access to water and food in
a standard experiment room at 22–24°C and 50±5% humidity, with a
12-h light/dark cycle. All animal experimental procedures were
conducted following the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health (NIH) (23), and were approved by the Animal Care
and Use Committee of the Beijing Institute of Pharmacology &
Toxicology. Different mice were used for each experiment in the
present study.
Drugs
MA was obtained from Beijing Institute of
Pharmacology & Toxicology. The ER stress inhibitors 4-phenyl
butyric acid (PBA) and tauroursodeoxycholic acid (TUDCA) were
purchased from Sigma-Aldrich; Merck KGaA (cat. no. P21005) and
Shanghai Aladdin Bio-Chem Technology Co., Ltd. (cat. no. S101371),
respectively. All drugs were dissolved in saline and prepared at a
concentration of 20 mg/ml. The animals were divided into six
groups: i) Normal saline (S group; n=31 mice); ii) TUDCA (n=15
mice); iii) PBA (n=15 mice); iv) MA (n=69 mice); v) TUDCA+MA (n=18
mice); and vi) PBA+MA (n=18 mice). In the MA group, mice were
administered intraperitoneal (i.p.) injections of 1, 2 or 5 mg/kg
MA. In the TUDCA+MA or PBA+MA group, mice received i.p. injections
of 200 mg/kg TUDCA or 100 mg/kg PBA 60 min before receiving 5 mg/kg
MA injections. In the S, TUDCA and PBA groups, mice were
administered i.p. injections of saline, 200 mg/kg TUDCA and 100
mg/kg PBA, respectively.
CPP test
The protocol of the CPP test was based on a previous
report (24), with some necessary
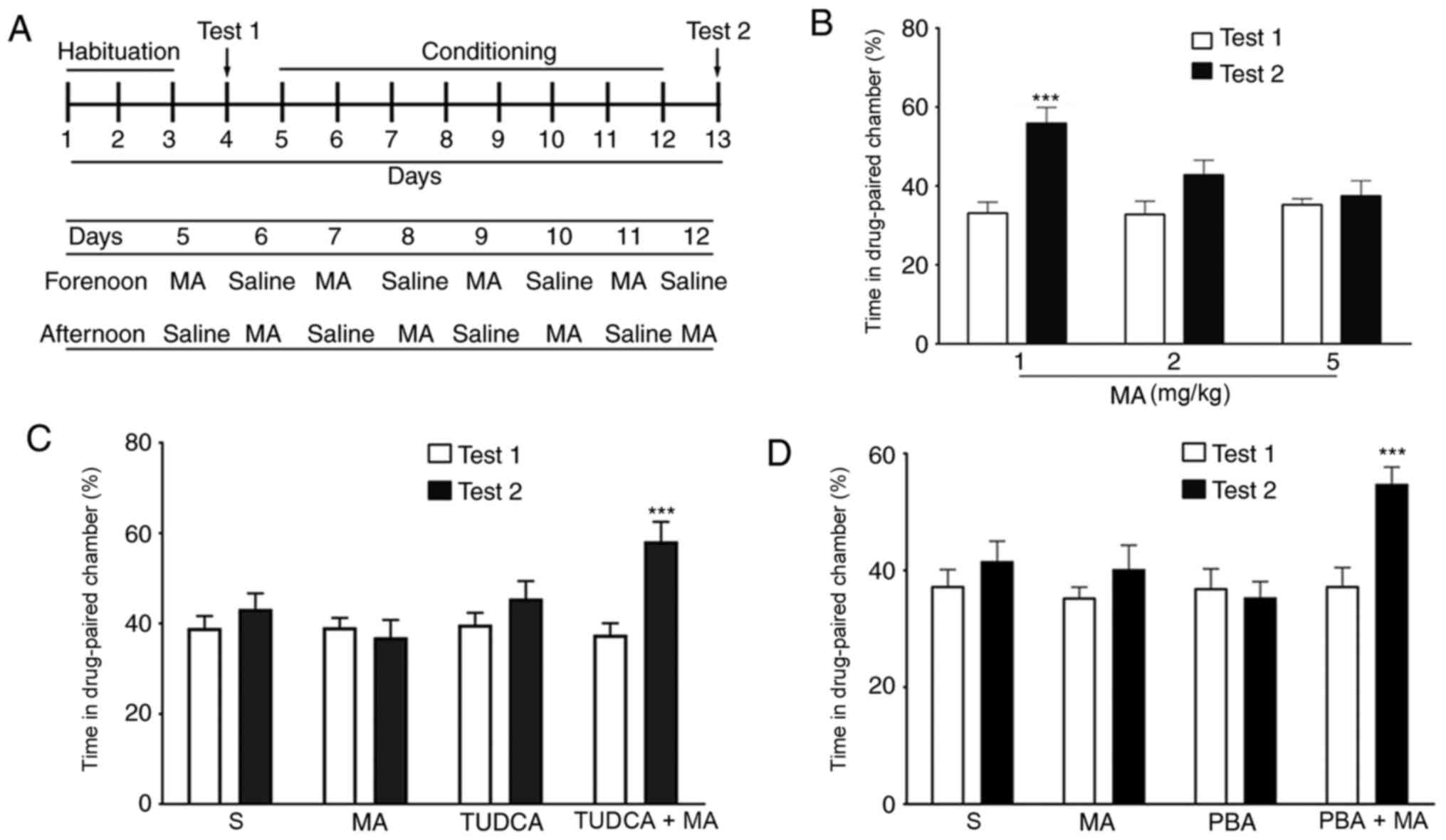
modifications (Fig. 1A). The
experiment box, which was manufactured by Anilab Software &
Instruments Co., Ltd., consisted of two distinct chambers
(17.4×13.5×15 cm3) separated by a corridor (9.8×13.5×15
cm3). Different colours (one was white, and the other
was black) and different floors (one was made of circular holes,
and the other was made of strips) in the two chambers were used so
that mice could distinguish one chamber from the other. The chamber
in which the mice stayed for a shorter duration in Test 1 was
chosen as the drug-paired chamber. The whole experiment included
four stages: Habituation, Test 1, Conditioning and Test 2. During
the 3-day habituation, mice moved freely in the chambers for 20 min
both in the morning and in the afternoon. The 8-day conditioning
was conducted with daily injections of drugs and saline,
administered alternately in the morning or the afternoon. The mice
were placed in the drug-paired chamber immediately following
administration of different does of MA (n=10/group) or TUDCA/PBA+5
mg/kg MA (n=10/group) for 30 min. When the mice were administered
saline, they were placed in the other chamber. During Test 1 and
Test 2, mice were allowed to explore the chambers freely for 15
min.
Electrophysiology
LTP of the perforant path (PP)-dentate gyrus (DG)
pathway in the hippocampus was recorded in vivo as
previously described (25), with
some modifications. Mice were first anaesthetized with urethane
(1.5 g/kg; i.p.) and then a pair of recording electrodes were
implanted into the DG of the left hemisphere at A/P: −2.0 mm, M/L:
−1.4 mm, D/V: −1.5 mm (from the dura), while a pair of stimulating
electrodes were implanted into the PP of the left hemisphere at
A/P: −3.8 mm, M/L: −3.0 mm, D/V: −1.5 mm (from the dura). A
population spike (PS) was induced using monopolar pulses (duration,
400 µsec; frequency, 1/30 Hz) using an Isolated Pulse stimulator
(A-M SYSTEMS Ltd.) and reported using a Differential AC amplifier
(A-M SYSTEMS Ltd.) and Axon Digidata 1550A Data Acquisition system
(Molecular Devices LLC). When the stabilized PS lasted for 60 min,
the stimulating current was regulated to yield a PS that was 30–50%
of the maximum amplitude, and the PS was recorded for 30 min as the
baseline. Subsequently, mice were administered with MA (1 or 5
mg/kg; i.p., n=5/group) or MA combined with pre-treatment of 200
mg/kg TUDCA or 100 mg/kg PBA (n=5/group). HFS, consisting of three
trains of 10 bursts (duration, 400 µsec; frequency, 300 Hz) with an
interval of 10 sec between each train, was given 30 min after drug
injection. The PS was recorded for 60 min using formerly single
monopolar pulses post-HFS. Data were obtained using pClamp10.0
software (Molecular Devices LLC).
Western blotting
Mice were euthanised using 5% isoflurane with an
oxygen flow rate of 1 l/min according to a previous report
(26). After the complete cessation
of the heartbeat, the hippocampal tissue (n=3/group) was dissected
on ice to extract the whole protein fraction. Western blotting was
performed as previously reported (27) to examine the expression levels of ER
stress markers, including binding immunoglobulin protein (BIP),
phosphorylated (p)-eukaryotic translation initiation factor 2α
(EIF2α), cyclic AMP-dependent transcription factor-4 (ATF-4), ATF-6
and CHOP, as well as the expression levels of
Ca2+/calmodulin-dependent protein kinase II α (CaMKIIα)
and cyclin-dependent kinase-5 (Cdk5), which are two proteins
associated with the formation of drug-evoked plasticity and drug
memory (28,29). The detailed information for
antibodies used in western blotting are presented in Table I. Values of these proteins (except
for p-EIF2α) were normalized to that of actin. The value of p-EIF2α
was normalized to that of total EIF2α. Bands were semi-quantified
using ImageJ 1.8.0.112 software (NIH).
 | Table I.Information of antibodies used for
western blotting. |
Table I.
Information of antibodies used for
western blotting.
| Antibody | Host | Supplier | Working
dilution |
|---|
| Primary
antibody |
|
|
|
|
Actin | M | Applygen
Technologies, Inc. (cat. no. C1313) | 1:5,000 |
|
BIP | R | Abcam (cat. no.
ab21685) | 1:1,000 |
|
ATF-4 | R | Abclonal Biotech
Co., Ltd. (cat. no. A18687) | 1:500 |
|
ATF-6 | R | Abclonal Biotech
Co., Ltd. (cat. no. A0202) | 1:500 |
|
p-EIF2α | R | CST (cat. no.
3398S) | 1:1,000 |
|
EIF2α | R | CST (cat. no.
5324S) | 1:500 |
|
CHOP | R | Abclonal Biotech
Co., Ltd. (cat. no. A0221) | 1:200 |
|
Cdk5 | M | Santa Cruz
Biotechnology, Inc. (cat. no. sc6247) | 1:200 |
|
CaMKIIα | M | Santa Cruz
Biotechnology, Inc. (cat. no. sc13141) | 1:500 |
| Secondary
antibody |
|
|
|
|
Anti-mouse HRP | G | Beyotime Institute
of Biotechnology (cat. no. A0216) | 1:5,000 |
|
Anti-rabbit HRP | G | Beyotime Institute
of Biotechnology (cat. no. A0208) | 1:5,000 |
Statistical analysis
In each part of the present study, three independent
experiments were performed. Data are presented as the mean ± SEM,
and statistical analysis was performed using GraphPad Prism 8.0
software (GraphPad Software, Inc.). For the CPP test, a two-way
mixed ANOVA followed by Bonferroni's multiple comparison test was
used to analyse the difference in the percentage of time spent in
the drug-paired chamber between Test 1 and Test 2. For the
electrophysiological tests, a paired t-test was used to analyse the
difference in PS amplitude between the baseline and post-HFS
measurements. For western blotting results, one-way ANOVA followed
by Bonferroni's multiple comparison test was used for analysing the
difference in protein expression levels between different groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
A dose of 5 mg/kg MA inhibits mouse
CPP behaviour by inducing ER stress
In the CPP test, 1 mg/kg MA induced a significant
increase in the percentage of time spent in the drug-paired chamber
(P<0.001), while CPP behaviour was not induced by 2 (P>0.05)
or 5 mg/kg MA (P>0.05; Fig. 1B).
To examine the role of ER stress in the inhibition of drug memory,
5 mg/kg was selected for further studies. When mice were
pre-treated with the ER stress inhibitors TUDCA (200 mg/kg, i.p.
Fig. 1C) or PBA (100 mg/kg, i.p;
Fig. 1D) 60 min before the
injection of 5 mg/kg MA, the percentage of time spent in the
drug-paired chamber in Test 2 was significantly increased compared
with that in Test 1 (P<0.001). However, when mice were
intraperitoneally injected with PBA or TUDCA alone, there was no
increase in the percentage of time spent in the drug-paired chamber
(P>0.05).
A dose of 5 mg/kg MA disturbs PP-DG
LTP in vivo via ER stress
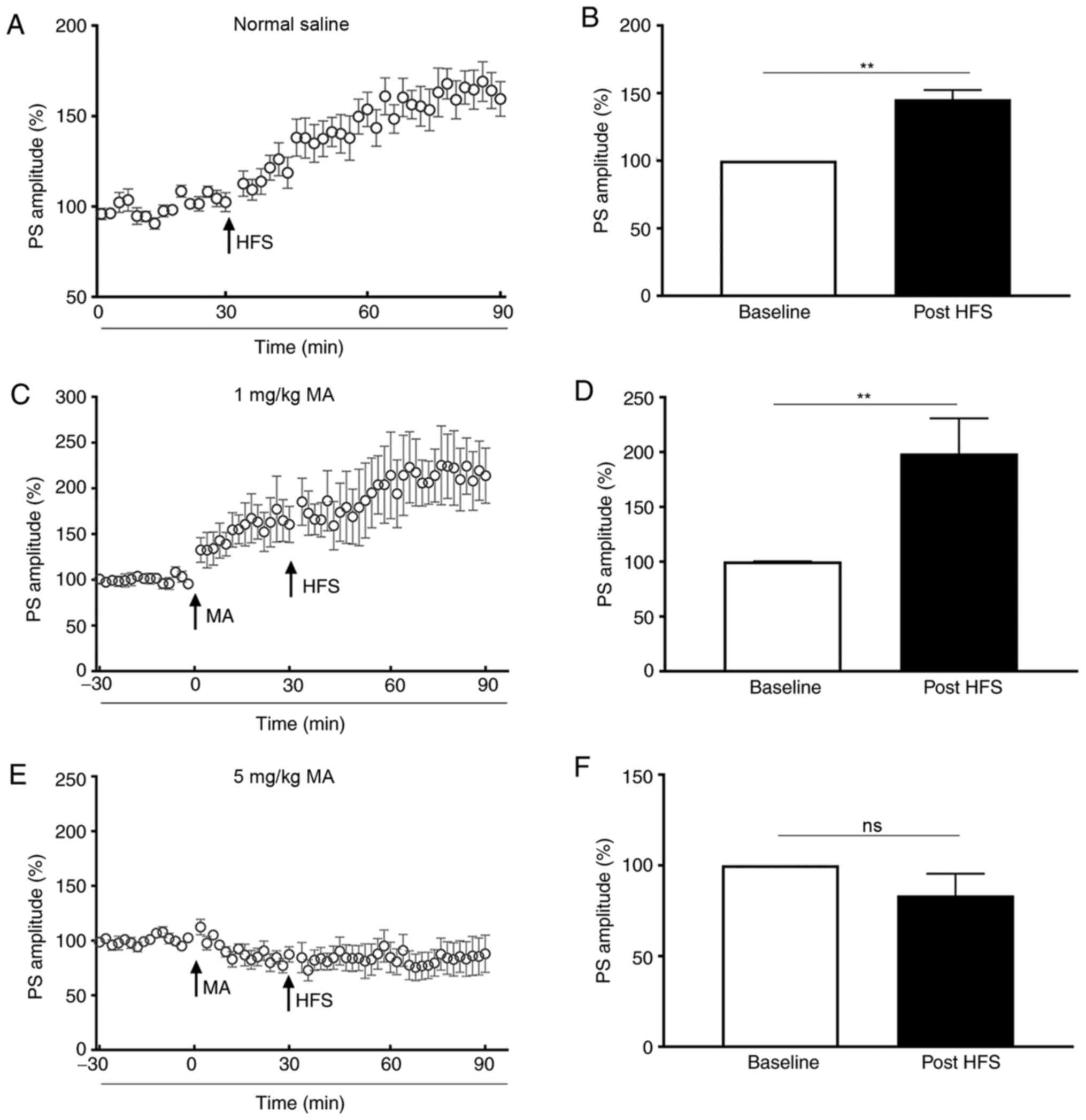
The present study demonstrated that the PS amplitude
of saline-treated mice was enhanced to 139.70±6.30% of the baseline
post-HFS (P<0.01; Fig. 2A and
B). When mice were administered an i.p. injection of 1 mg/kg
MA, the PS amplitude increased to 144.20±17.57% of the baseline
(P<0.05; bar graph was not shown), and it increased to
206.60±27.53% of the baseline (P<0.01) post-HFS (Fig. 2C and D). However, the PS amplitude
of mice treated with 5 mg/kg MA was not increased post-HFS
(P>0.05; Fig. 2E and F) in
comparison with the baseline.
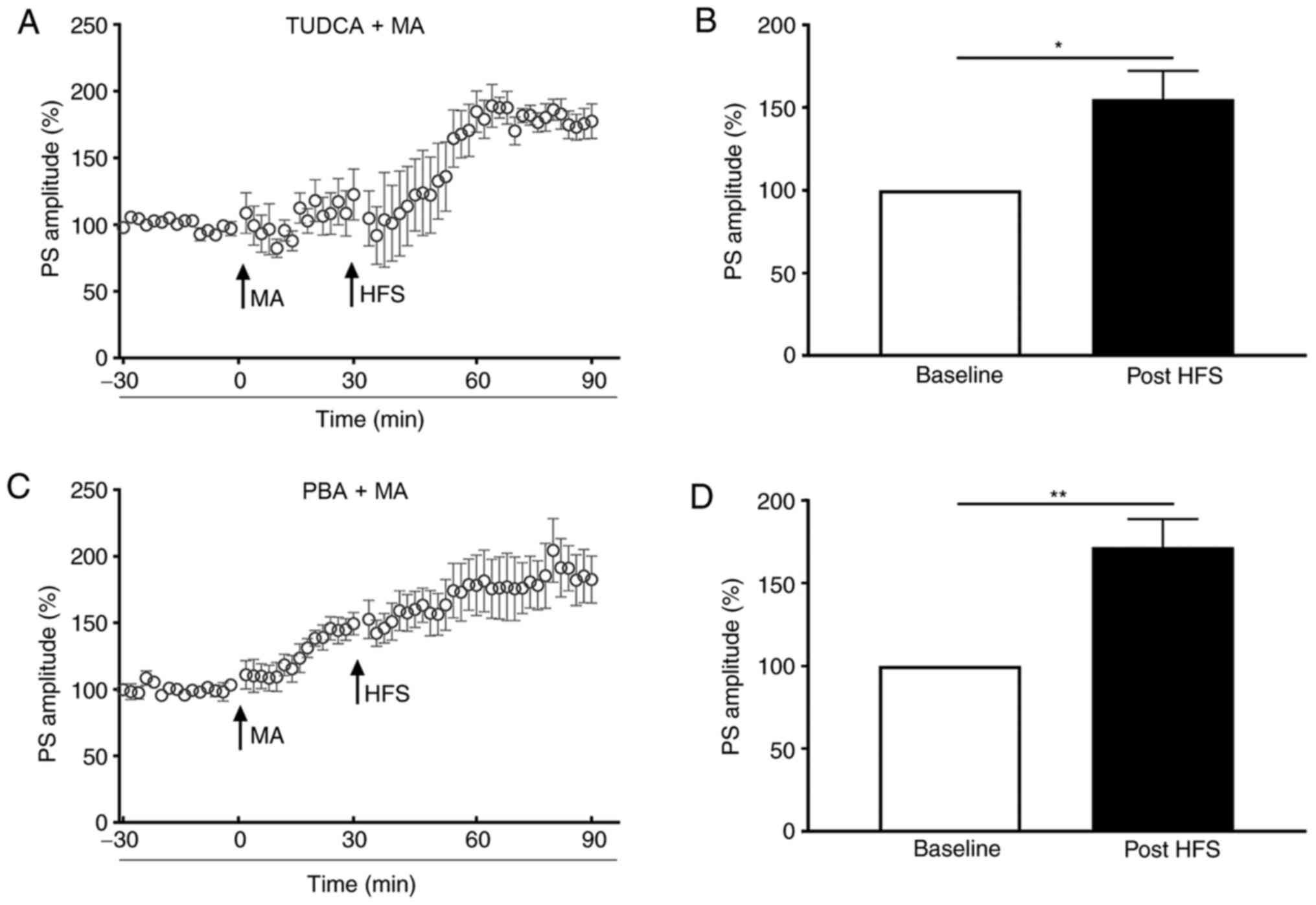
To evaluate the role of ER stress in LTP inhibition
evoked by 5 mg/kg MA, TUDCA or PBA were injected 60 min before MA
administration. In the TUDCA+MA group, the PS amplitude post-HFS
was increased to 155.1±17.09% of the baseline (P<0.05; Fig. 3A and B). For mice in the PBA+MA
group, the PS amplitude was increased to 131.90±5.05% of the
baseline (P<0.01; bar graph was not shown) when MA was injected
and it was elevated to 179.85±12.32% of the baseline post-HFS
(P<0.01; Fig. 3C and D), which
was significantly higher compared with that of the S group
(P<0.01; bar graph was not shown).
A dose of 5 mg/kg MA increases the
expression levels of ER stress markers in the hippocampus
Mice were treated with different dosages of MA (1 or
5 mg/kg) for 30 min and then whole hippocampal proteins were
extracted for western blotting. It was found that the expression
levels of the ER stress marker proteins BIP (P<0.001), ATF-4
(P<0.01), ATF-6 (P<0.001), p-EIF2α (P<0.05) and CHOP
(P<0.05) were significantly increased by 5 mg/kg MA. The
ingestion of 1mg/kg had no effect on the expression levels of BIP
(P>0.05), ATF-4 (P>0.05), ATF-6 (P>0.05) and CHOP
(P>0.05); however, it reduced the expression levels of p-EIF2α
(P<0.05; Fig. 4). When mice were
pre-treated with TUDCA, the expression levels of BIP (P<0.01),
ATF-6 (P<0.01), CHOP (P<0.05) and p-EIF2α (P<0.01) were
decreased to normal levels. Similarly, pre-treatment with PBA also
decreased the expression levels of BIP (P<0.05), ATF-6
(P<0.001), CHOP (P<0.05) and p-EIF2α (P<0.01), which were
increased by 5 mg/kg MA (Fig. 5).
Moreover, the expression of Cdk5 (P<0.001) was enhanced, while
CaMKIIα expression (P<0.05) was decreased by 5 mg/kg MA
administration. In addition, 5 mg/kg MA-induced changes in the
expression levels of Cdk5 were reversed by TUDCA (P<0.001) or
PBA (P<0.01) pre-treatment. The decreased expression level of
CaMKIIα could also be reversed by TUDCA (P<0.01) and PBA
(P<0.01) pre-treatment (Fig.
6).
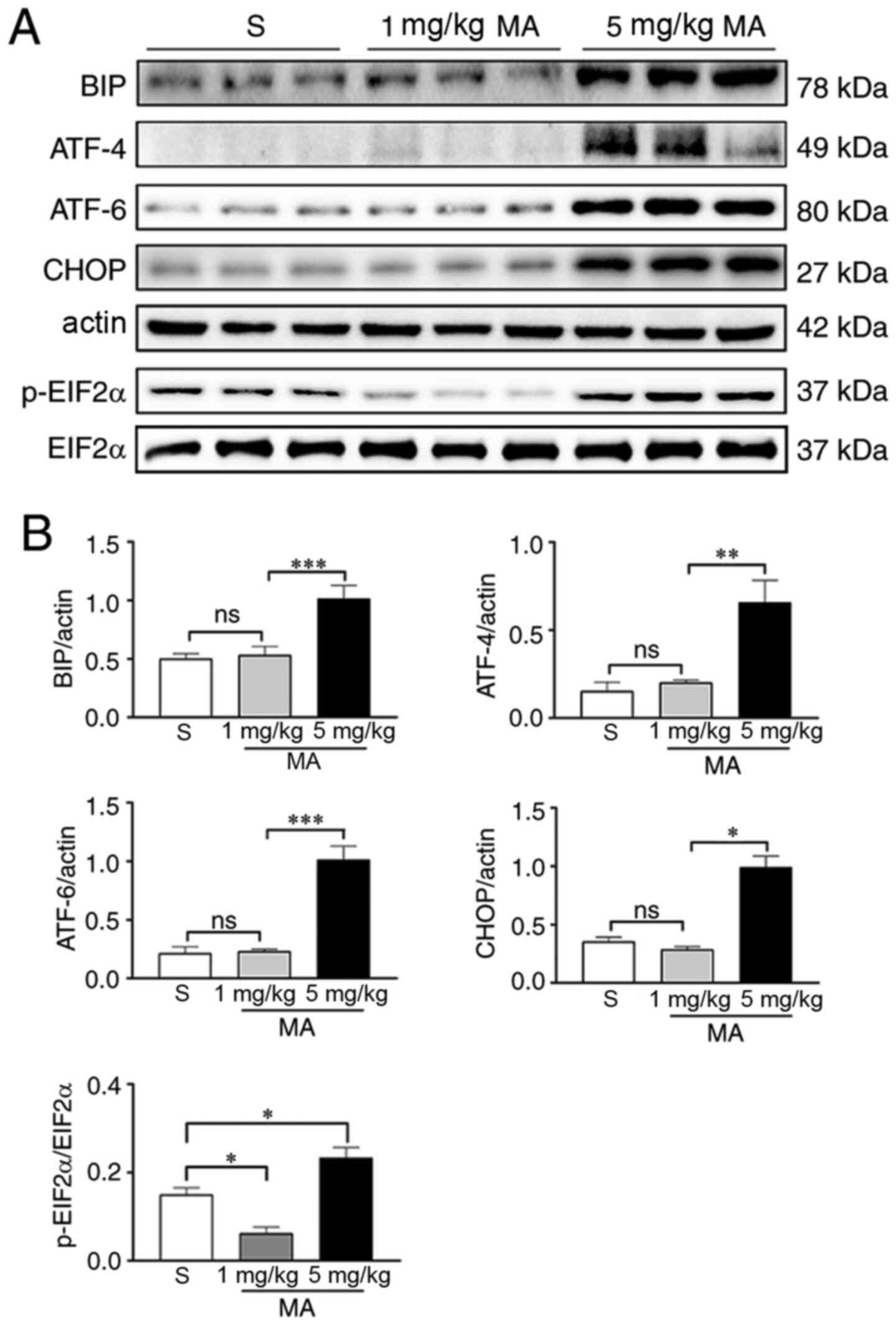 | Figure 4.Acute exposure to 5 mg/kg MA induces
ER stress in the hippocampus. (A) Western blotting results of the
ER stress markers in the hippocampus of mice treated with saline,
or 1 or 5 mg/kg MA (n=3/group). (B) Statistical analysis
demonstrated that 5 mg/kg MA, but not 1 mg/kg MA, increased the
protein expression levels of ER stress markers. Data are presented
as the mean ± SEM. *P<0.05, **P<0.01, ***P<0.001; ns
indicates P>0.05. MA, methamphetamine; ER, endoplasmic
reticulum; p-, phosphorylated; EIF2α, eukaryotic translation
initiation factor 2α; ATF, cyclic AMP-dependent transcription
factor; BIP, binding immunoglobulin protein; ns, non-significant;
S, normal saline group. |
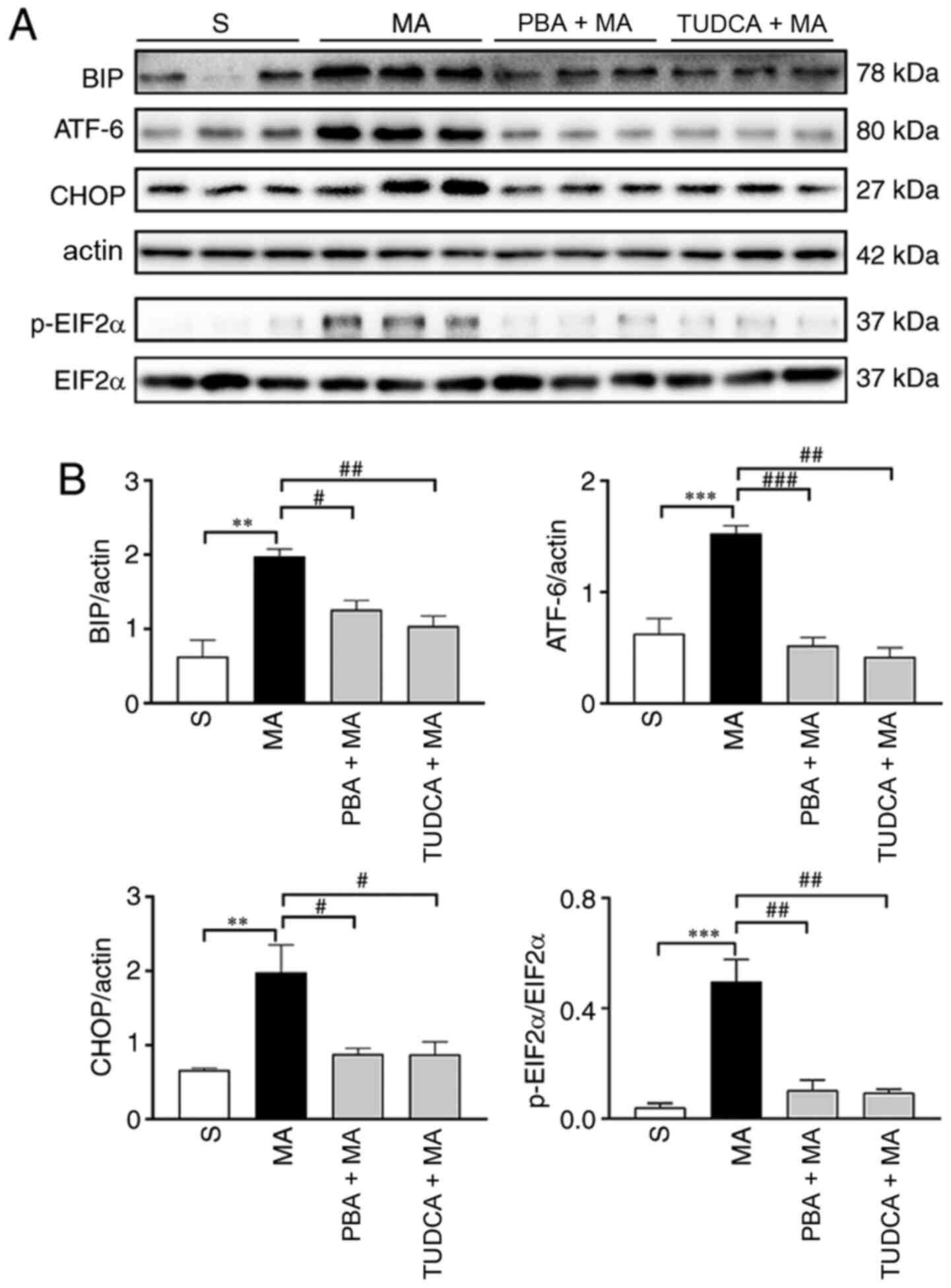 | Figure 5.MA-induced ER stress are inhibited by
the pre-treatment with PBA or TUDCA. (A) Western blotting results
of ER stress markers in the hippocampus of mice treated with
saline, 5 mg/kg MA, PBA + 5 mg/kg MA or TUDCA + 5 mg/kg MA, n=3 per
group. (B) Statistical results indicated that higher protein
expression levels of ER stress markers induced by 5 mg/kg MA were
reversed by PBA or TUDCA pre-treatment. Data are presented as the
mean ± SEM. **P<0.01, ***P<0.001; #P<0.05,
##P<0.01, ###P<0.001. MA,
methamphetamine; ER, endoplasmic reticulum; PBA, 4-phenyl butyric
acid; TUDCA, tauroursodeoxycholic acid; p-, phosphorylated; EIF2α,
eukaryotic translation initiation factor 2α; ATF, cyclic
AMP-dependent transcription factor; BIP, binding immunoglobulin
protein; S, normal saline group. |
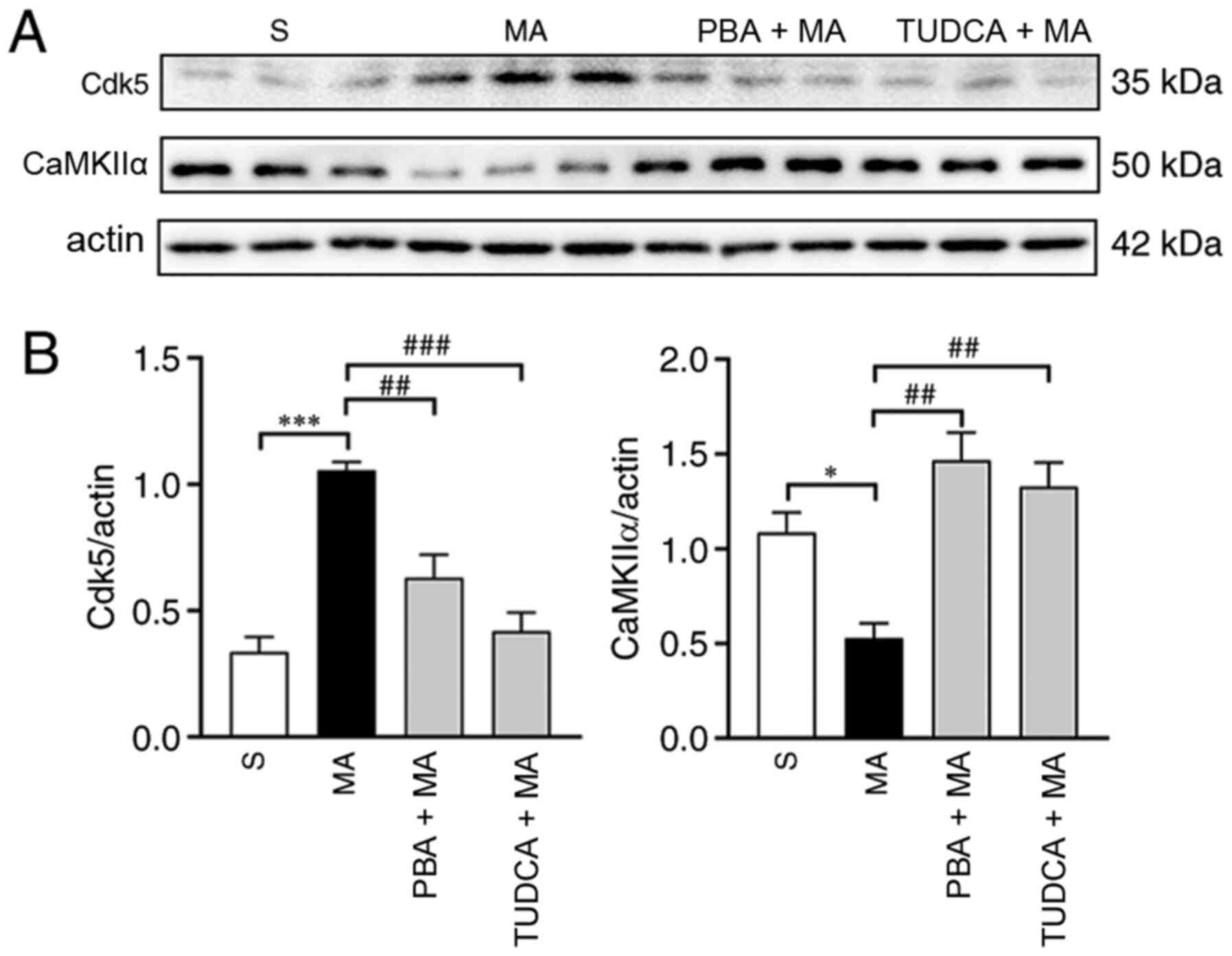 | Figure 6.Acute exposure to 5 mg/kg MA
increases the protein expression level of Cdk5 and decreases the
protein expression level of CaMKIIα, which can be reversed by TUDCA
or PBA. (A) Western blotting results of Cdk5 and CaMKIIα in the
hippocampus of mice treated with saline, 5 mg/kg MA, PBA + 5 mg/kg
MA or TUDCA + 5 mg/kg MA (n=3/group). (B) Statistical analysis
demonstrated that 5 mg/kg MA enhanced the protein expression levels
of Cdk5 and decreased the protein expression levels of CaMKIIα;
these effects were reversed by TUDCA or PBA pre-treatment. Data are
presented as the mean ± SEM. *P<0.05, ***P<0.001;
##P<0.01, ###P<0.001. MA,
methamphetamine; PBA, 4-phenyl butyric acid; TUDCA,
tauroursodeoxycholic acid; Cdk5, cyclin-dependent kinase-5;
CaMKIIα, Ca2+/calmodulin-dependent protein kinase II α;
S, normal saline group. |
Discussion
Drug addiction manifests as compulsive drug-seeking
behaviour (30). Addiction is
induced by repeated exposure to drugs, and increasing evidence has
revealed that drug-seeking is not an uncontrolled behaviour due to
a lack of willpower or a character flaw, but drug addiction is
rather a chronic disease resulting from complicated
neuroadaptations in different encephalic regions (18). Aberrant learning and memory, known
as drug memory, are involved during the formation of addiction, and
addiction is regarded as a disease of learning and memory (31). Consequently, treatment for addiction
by disrupting drug memory has been attempted (32,33).
MA, a man-made psychostimulant that is abused
worldwide, is well known for its addictive properties and the
damage it causes to multiple organs (20). MA addiction has been a concern for
researchers worldwide. Thus, the present study investigated the
mechanisms underlying MA addiction by examining it alongside the
formation of drug memory. As the ER is an important organelle for
protein assembly and folding, and as protein synthesis is of great
significance for long-term memory development (34), it was hypothesized that ER stress
may participate in the disruption of addiction memory.
In the CPP test, the present study demonstrated that
1 mg/kg, but not 2 or 5 mg/kg MA evoked CPP behaviour, which was
consistent with previously published data (35). However, when mice were pre-treated
with the ER stress inhibitors TUDCA or PBA, CPP behaviour could be
induced by 5 mg/kg MA, indicating that a high dose of MA inhibited
drug memory via ER stress.
Next, the mechanisms underlying the disturbance of
drug memory formation by MA-induced ER stress were evaluated by
studying the effects of MA administration on synaptic plasticity.
The hippocampus, a limbic structure, is important for learning to
associate specific contexts using reinforcer availability and
spatial memory storage (36).
Glutamatergic neurotransmission can be projected from the
hippocampus to multiple regions within the reward circuitry
(37) The hippocampus and the
ventral tegmental area (VTA) form a functional loop to control the
entry of information into long-term memory (37). This loop is activated when novel
information is detected in the hippocampus. Moreover, the
hippocampus-VTA loop is involved in MA-mediated place reinforcement
learning (38). Consequently, the
present study selected the PP-DG pathway in the hippocampus to
investigate the influence of MA-evoked ER stress on synaptic
plasticity, which is closely associated with the acquisition
process of MA addiction (39). As
mice underwent a 30-min conditioning period immediately after MA
injection in the CPP experiment, the present study examined the
acute effect of MA administration on LTP induction and HFS was
conducted 30 min post-MA injection. The current results suggested
that acute administration of 1 mg/kg MA induced LTP facilitation,
while 5 mg/kg MA caused a disturbance in LTP induction. However,
this disruption was attenuated when mice were pre-treated with
TUDCA or PBA. Additionally, HFS-independent enhancement of the PS
amplitude, which was conceptualized as drug-evoked synaptic
plasticity (18), was observed when
mice were injected with 1 mg/kg MA. Drug-evoked plasticity is also
reported to be induced by other addictive drugs, such as ethanol
and cocaine (40). It is also
considered to be involved in the early stages of the development of
drug addiction (41). In the
present study, 5 mg/kg MA generated drug-evoked plasticity when
mice were pre-treated with PBA, suggesting that ER stress may be
involved in the disturbance of drug-evoked synaptic plasticity.
However, drug-evoked plasticity was not observed when mice were
pre-treated with TUDCA. Nevertheless, the reasons underlying this
difference are not obvious based on the present results only.
The present study also evaluated the effects of
MA-induced ER stress on proteins involved in memory formation.
Western blotting data demonstrated that 5 mg/kg, but not 1 mg/kg,
MA induced ER stress in the hippocampus, as indicated by enhanced
expression levels of the ER stress marker proteins BIP, p-EIF2α,
ATF-4, ATF-6 and CHOP. These increases could be reversed by TUDCA
or PBA. Moreover, 5 mg/kg MA induced an increase in Cdk5
expression, as well as a decrease in CaMKIIα expression. As
previously reported, the inhibition of Cdk5 serves a key role in
facilitating cocaine-induced CPP behaviour (42). When mice were pre-treated with TUDCA
or PBA, 5 mg/kg MA-induced CPP behaviour and Cdk5 expression levels
were reversed to normal levels. CaMKIIα, an essential protein in
LTP induction, is also involved in the development and maintenance
of drug memory (43). Therefore,
the decreased expression of CaMKIIα, which was caused by 5 mg/kg MA
in the present study, was considered to serve a role in LTP
disturbance and the inhibition of drug memory. According to the
results of the behavioural, electrophysiological and molecular
studies, it was suggested that a large dosage of MA resulted in a
disturbance of drug memory formation by inhibiting hippocampal LTP
and altering the expression levels of proteins associated with
memory formation via ER stress.
To the best of our knowledge, few studies have been
reported that directly examine the relationship between addiction
and MA-induced ER stress. However, it has been revealed that the
expression of p-EIF2α, a marker protein of ER stress, is decreased
by multiple addictive drugs, such as cocaine and nicotine (44,45).
Moreover, enhanced p-EIF2α-mediated translation control could
prevent the maintenance of cocaine-induced LTP in VTA dopamine
neurons (46), which underlies
compulsive drug-seeking (18). In
accordance with this observation, the present study found that
p-EIF2α expression was enhanced by treatment with 5 mg/kg MA, which
disrupted hippocampal LTP induction and drug-seeking behaviour. It
was also observed that CPP and LTP could be induced when mice were
pre-treated with the ER stress inhibitors TUDCA or PBA, decreasing
the enhanced expression of p-EIF2α, as well as the signalling
cascades evoked by this molecule. Furthermore, the present results
indicated that the protein expression level of p-EIF2α was declined
by acute exposure to 1 mg/kg MA, which could induce both LTP and
CPP behaviour, suggesting that inhibiting the expression of p-EIF2α
might contribute to the formation of MA-induced drug memory.
Generally, researchers inhibit CPP expression to
investigate the role of certain molecules or neural circuits in the
addiction process (33,47). In the current study, however, CPP
behaviour was first found to be disturbed by 5 mg/kg MA, and it
could be induced when mice were pre-treated with ER stress
inhibitors, suggesting that MA-evoked ER stress serves a role in
drug memory inhibition caused by a high dosage of MA. Therefore, it
can be considered that the disruption of drug memory via ER stress
in the hippocampus without any adverse effects may serve an active
role in blocking the formation of drug memory. However, the
detailed mechanisms underlying the inhibition of drug-evoked
plasticity and drug-seeking behaviour caused by high-dose
MA-induced ER stress are still unclear based on the present
findings and require further investigations.
In conclusion, the present study demonstrated that a
large dose of MA could disrupt drug memory and hippocampal
drug-evoked plasticity by inducing ER stress (Fig. 7).
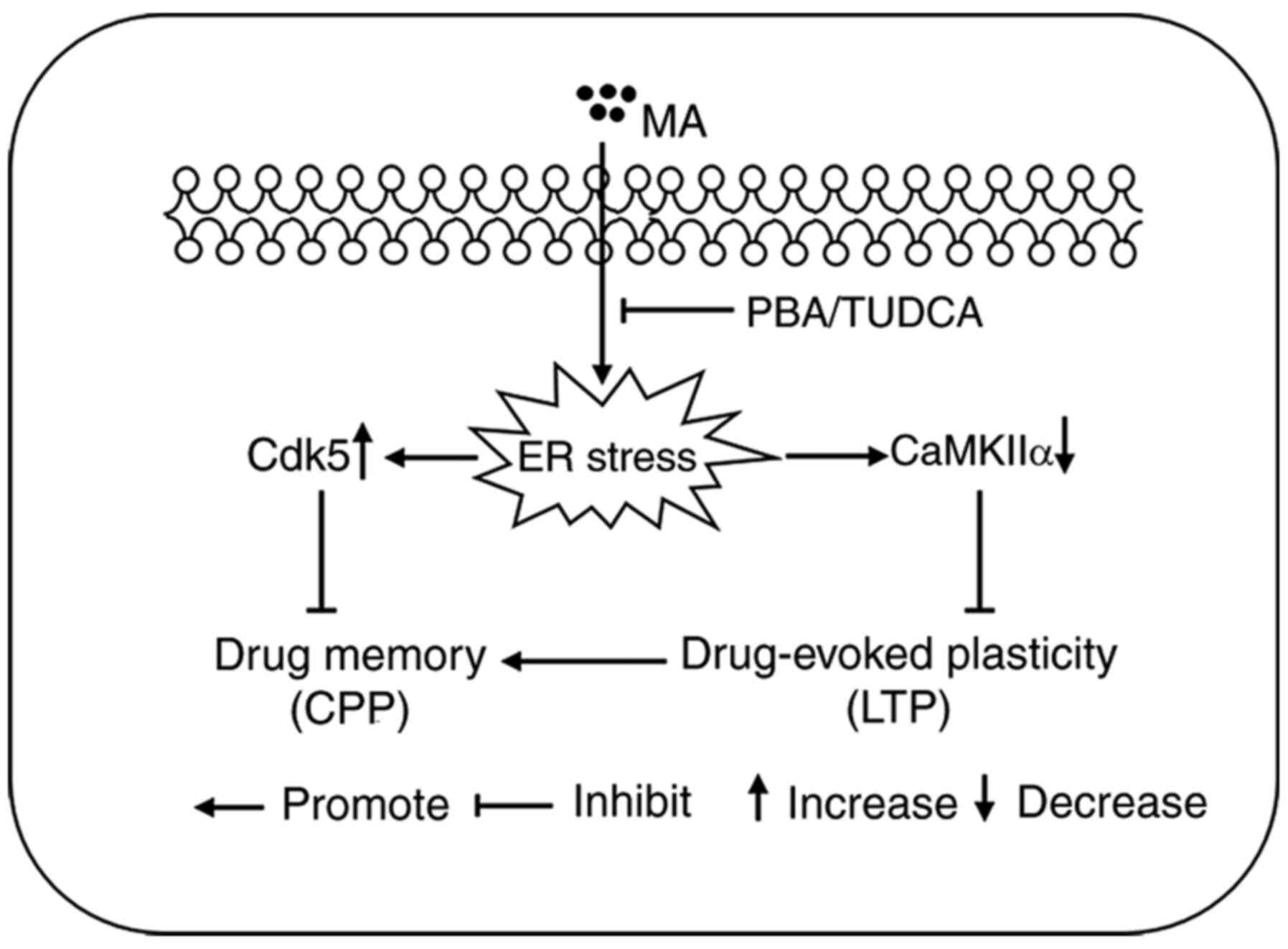 | Figure 7.Schematic diagram representing the
main findings of the present study. Acute exposure to a large dose
of MA induced a higher expression level of Cdk5 protein, and
decreased the protein expression level of CaMKIIα, both of which
are closely associated with drug-evoked plasticity and drug memory.
When mice were pre-treated with the ER stress inhibitors PBA or
TUDCA, increased protein expression of Cdk5 and decreased protein
expression of CaMKIIα were reversed. When ER stress in the
hippocampus was inhibited, 5 mg/kg MA could also induce LTP and CPP
behaviour. MA, methamphetamine; Cdk5, cyclin-dependent kinase-5;
CaMKIIα, Ca2+/calmodulin-dependent protein kinase II α;
ER, endoplasmic reticulum; PBA, 4-phenyl butyric acid; TUDCA,
tauroursodeoxycholic acid; LTP, long term potentiation; CPP,
conditioned place preference. |
Acknowledgements
Not applicable.
Funding
This work was supported by the Natural Science
Foundation of China (grant nos. 81430045 and 81871528) and
Guangzhou Sci-Tech Project (grant no. 201803010005).
Availability of data and materials
The datasets used and/or analysed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GC conducted all of the experiments with the help of
GY and HY. ZY analyzed the results. HW and RS designed the
research. ZY and RS confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experimental procedures were conducted
following the Guide for the Care and Use of Laboratory Animals of
the National Institutes of Health (NIH), and were approved by the
Animal Care and Use Committee of the Beijing Institute of
Pharmacology & Toxicology.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing
interests.
References
|
1
|
Joffe ME, Grueter CA and Grueter BA:
Biological substrates of addiction. Wiley Interdiscip Rev Cogn Sci.
5:151–171. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Koob GF and Volkow ND: Neurocircuitry of
addiction. Neuropsychopharmacology. 35:217–238. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Di Chiara G, Bassareo V, Fenu S, De Luca
MA, Spina L, Cadoni C, Acquas E, Carboni E, Valentini V and Lecca
D: Dopamine and drug addiction: The nucleus accumbens shell
connection. Neuropharmacology. 47 (Suppl 1):S227–S241. 2004.
View Article : Google Scholar
|
|
4
|
Gipson CD, Kupchik YM and Kalivas PW:
Rapid, transient synaptic plasticity in addiction.
Neuropharmacology. 76(Pt B): 276–286. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McLellan AT, Lewis DC, O'Brien CP and
Kleber HD: Drug dependence, a chronic medical illness: Implications
for treatment, insurance, and outcomes evaluation. JAMA.
284:1689–1695. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tronson N and Taylor J: Molecular
mechanisms of memory reconsolidation. Nat Rev Neurosci. 8:262–275.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Carvalho C and Takahashi R: Cannabidiol
disrupts the reconsolidation of contextual drug-associated memories
in Wistar rats. Addict Biol. 22:742–751. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hyman S: Addiction: A disease of learning
and memory. Am J Psychiatry. 162:1414–1422. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hyman S and Malenka R: Addiction and the
brain: The neurobiology of compulsion and its persistence. Nat Rev
Neurosci. 2:695–703. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Berke J and Hyman S: Addiction, dopamine,
and the molecular mechanisms of memory. Neuron. 25:515–532. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grimm JW, Hope BT, Wise RA and Shaham Y:
Neuroadaptation. Incubation of cocaine craving after withdrawal.
Nature. 412:141–142. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Y, Xue Y, Meng S, Luo Y, Liang J, Li
J, Ai S, Sun C, Shen H, Zhu W, et al: Inhibition of lactate
transport erases drug memory and prevents drug relapse. Biol
Psychiatry. 79:928–939. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hyman S, Malenka R and Nestler E: Neural
mechanisms of addiction: The role of reward-related learning and
memory. Annu Rev Neurosci. 29:565–598. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kelley AE: Memory and addiction: Shared
neural circuitry and molecular mechanisms. Neuron. 44:161–179.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Frey U and Morris RG: Synaptic tagging and
long-term potentiation. Nature. 385:533–536. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Whitlock J, Heynen A, Shuler M and Bear M:
Learning induces long-term potentiation in the hippocampus.
Science. 313:1093–1097. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pastalkova E, Serrano P, Pinkhasova D,
Wallace E, Fenton AA and Sacktor TC: Storage of spatial information
by the maintenance mechanism of LTP. Science. 313:1141–1144. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Luscher C and Malenka RC: Drug-evoked
synaptic plasticity in addiction: From molecular changes to circuit
remodeling. Neuron. 69:650–663. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grueter B, Rothwell P and Malenka R:
Integrating synaptic plasticity and striatal circuit function in
addiction. Curr Opin Neurobiol. 22:545–551. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Courtney KE and Ray LA: Methamphetamine:
An update on epidemiology, pharmacology, clinical phenomenology,
and treatment literature. Drug Alcohol Depend. 143:11–21. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
North A, Swant J, Salvatore M,
Gamble-George J, Prins P, Butler B, Mittal MK, Heltsley R, Clark JT
and Khoshbouei H: Chronic methamphetamine exposure produces a
delayed, long-lasting memory deficit. Synapse. 67:245–257. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Naidoo N: ER and aging-Protein folding and
the ER stress response. Ageing Res Rev. 8:150–159. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Z, Qin P, Deng Y, Ma Z, Guo H, Guo
H, Hou Y, Wang S, Zou W, Sun Y, et al: The novel estrogenic
receptor GPR30 alleviates ischemic injury by inhibiting
TLR4-mediated microglial inflammation. J Neuroinflammation.
15:2062018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wakida N, Kiguchi N, Saika F, Nishiue H,
Kobayashi Y and Kishioka S: CC-chemokine ligand 2 facilitates
conditioned place preference to methamphetamine through the
activation of dopamine systems. J Pharmacol Sci. 125:68–73. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gureviciene I, Ikonen S, Gurevicius K,
Sarkaki A, van Groen T, Pussinen R, Ylinen A and Tanila H: Normal
induction but accelerated decay of LTP in APP+PS1 transgenic mice.
Neurobiol Dis. 15:188–195. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Marquardt N, Feja M, Hünigen H, Plendl J,
Menken L, Fink H and Bert B: Euthanasia of laboratory mice: Are
isoflurane and sevoflurane real alternatives to carbon dioxide?
PLoS One. 13:e02037932018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cai D, Huang E, Luo B, Yang Y, Zhang F,
Liu C, Lin Z, Xie WB and Wang H: Nupr1/Chop signal axis is involved
in mitochondrion-related endothelial cell apoptosis induced by
methamphetamine. Cell Death Dis. 7:e21612016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Anderson S, Famous KR, Sadri-Vakili G,
Kumaresan V, Schmidt HD, Bass CE, Terwilliger EF, Cha JH and Pierce
RC: CaMKII: A biochemical bridge linking accumbens dopamine and
glutamate systems in cocaine seeking. Nat Neurosci. 11:344–353.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mlewski E, Krapacher F, Ferreras S and
Paglini G: Transient enhanced expression of Cdk5 activator p25
after acute and chronic d-amphetamine administration. Ann N Y Acad
Sci. 1139:89–102. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Koob G, Ahmed S, Boutrel B, Chen SA, Kenny
PJ, Markou A, O'Dell LE, Parsons LH and Sanna PP: Neurobiological
mechanisms in the transition from drug use to drug dependence.
Neurosci Biobehav Rev. 27:739–749. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
White NM: Addictive drugs as reinforcers:
Multiple partial actions on memory systems. Addiction. 91:921–949;
discussion 951–965. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li FQ, Xue YX, Wang JS, Fang Q, Li YQ, Zhu
WL, He YY, Liu JF, Xue LF, Shaham Y and Lu L: Basolateral amygdala
cdk5 activity mediates consolidation and reconsolidation of
memories for cocaine cues. J Neurosci. 30:10351–10359. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boury-Jamot B, Carrard A, Martin JL,
Halfon O, Magistretti PJ and Boutrel B: Disrupting astrocyte-neuron
lactate transfer persistently reduces conditioned responses to
cocaine. Mol Psychiatry. 21:1070–1076. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moncada D and Viola H: Induction of
long-term memory by exposure to novelty requires protein synthesis:
Evidence for a behavioral tagging. J Neurosci. 27:7476–7481. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun L, Song R, Chen Y, Yang RF, Wu N, Su
RB and Li J: A selective D3 receptor antagonist YQA14 attenuates
methamphetamine-induced behavioral sensitization and conditioned
place preference in mice. Acta Pharmacol Sin. 37:157–165. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sweatt JD: Hippocampal function in
cognition. Psychopharmacology (Berl). 174:99–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lisman J and Grace A: The hippocampal-VTA
loop: Controlling the entry of information into long-term memory.
Neuron. 46:703–713. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Keleta Y and Martinez J: Brain circuits of
methamphetamine place reinforcement learning: The role of the
hippocampus-VTA Loop. Brain Behav. 2:128–141. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ricoy U and Martinez J Jr: Local
hippocampal methamphetamine-induced reinforcement. Front Behav
Neurosci. 3:472009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lüscher C: Drug-evoked synaptic plasticity
causing addictive behavior. J Neurosci. 33:17641–17646. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ungless M, Whistler J, Malenka R and Bonci
A: Single cocaine exposure in vivo induces long-term potentiation
in dopamine neurons. Nature. 411:583–587. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Benavides DR, Quinn JJ, Zhong P, Hawasli
AH, DiLeone RJ, Kansy JW, Olausson P, Yan Z, Taylor JR and Bibb JA:
Cdk5 modulates cocaine reward, motivation, and striatal neuron
excitability. J Neurosci. 27:12967–12976. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang L, Lv Z, Hu Z, Sheng J, Hui B, Sun J
and Ma L: Chronic cocaine-induced H3 acetylation and
transcriptional activation of CaMKIIalpha in the nucleus accumbens
is critical for motivation for drug reinforcement.
Neuropsychopharmacology. 35:913–928. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Huang W, Placzek A, Viana Di Prisco G,
Khatiwada S, Sidrauski C, Krnjević K, Walter P, Dani JA and
Costa-Mattioli M: Translational control by eIF2alpha
phosphorylation regulates vulnerability to the synaptic and
behavioral effects of cocaine. Elife. 5:e120522016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Placzek A, Molfese D, Khatiwada S, Viana
Di Prisco G, Huang W, Sidrauski C, Krnjević K, Amos CL, Ray R, Dani
JA, et al: Translational control of nicotine-evoked synaptic
potentiation in mice and neuronal responses in human smokers by
eIF2α. eLife. 5:e120562016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Placzek A, Prisco G, Khatiwada S, Sgritta
M, Huang W, Krnjević K, Kaufman RJ, Dani JA, Walter P and
Costa-Mattioli M: eIF2α-mediated translational control regulates
the persistence of cocaine-induced LTP in midbrain dopamine
neurons. Elife. 5:e175172016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Fan HY, Cherng CG, Yang FY, Cheng LY, Tsai
CJ, Lin LC and Yu L: Systemic treatment with protein synthesis
inhibitors attenuates the expression of cocaine memory. Behavi
Brain Res. 208:522–527. 2010. View Article : Google Scholar
|