Introduction
The cardiac toxicity that occurs during long-term
use of antitumor drugs has attracted increasing attention,
particularly antharcycline antitumor drugs. Doxorubicin (Dox) is a
highly potent antharcycline antitumor drug and is an important
component of classical chemotherapy regimens for lymphoma, leukemia
and a variety of other solid tumors, including breast cancer and
osteosarcoma. Despite the antitumor effect of Dox, it has been
reported to cause noticeable cardiac toxicity, which imposes severe
restrictions on the application of Dox and other antharcycline
antitumor drugs in clinical practice, for example, in patients with
underlying cardiac diseases. Chronic cardiac toxicity caused by Dox
is one of the most important side effects of the drug and manifests
in congestive heart failure and/or cardiomyopathy (1–2).
Previous studies have reported that the mechanism involved in the
occurrence of cardiac toxicity and dilated cardiomyopathy (DCM)
induced by Dox is associated with the damage caused by reactive
oxygen series, calcium overload, mitochondrial dysfunction,
apoptosis and autophagy (3–5). However, this remains unclear. At
present, there are a limited number of drugs that can be used for
alleviating Dox-induced cardiac toxicity. At present, dexrazoxane
is a rare drug that can alleviate myocardial toxicity caused by
anthracycline antitumor drugs, however, it displays adverse
reactions, including neutropenia, and its underlying mechanism of
action is not completely understood (6). The development of a safer and more
effective drug or method to antagonize the cardiac toxicity
resulting from Dox and other antharcycline antitumor drugs poses a
significant challenge for modern medicine.
The pathological changes in the heart that are
caused by Dox in adult patients are predominantly DCM. Therefore,
Dox-induced cardiomyopathy is considered as the primary method to
construct animal models of DCM (1).
It has been confirmed in rabbit and rat animal models of DCM that
Dox causes clinical symptoms, such as cardiac dilatation,
arrhythmia, cardiac insufficiency and even heart failure in some
cases, which is highly similar to the occurrence and development of
DCM in patients (7–8). Myocardial fibrosis plays a crucial
role in the occurrence and development of Dox-induced cardiac
damage and DCM (9); however, the
underlying mechanisms are complex and are not completely
understood. Some studies have demonstrated that autophagy,
endoplasmic reticulum stress (ERS) and cell apoptosis are
potentially involved in the occurrence and development of
myocardial fibrosis (10–12). Despite this, the specific regulatory
mechanism of Dox-induced myocardial fibrosis remains unclear.
Therefore, a further exploration of this mechanism is essential for
understanding the pathogenesis of Dox-induced myocardial fibrosis
and DCM.
Myocardial fibrosis is frequently caused by
increased extracellular matrix (ECM) synthesis and its delayed
degradation for various reasons, thus resulting in extensive ECM
deposition in myocardial interstitium. The imbalance between MMPs
and TIMPs is associated with ECM deposition, as well as the
occurrence and development of myocardial fibrosis. TGF-β is
hypothesized to be involved in the mechanism of occurrence of
myocardial fibrosis by regulating cell growth, proliferation and
differentiation, activating cardiac fibroblasts and enhancing the
expression of MMPs (13–16).
H2S, a newly discovered gaseous signal
molecule performs a variety of anti-inflammatory, -oxidative and
-apoptotic biological functions. Previous studies have reported the
anti-myocardial fibrosis effect exerted by H2S in animal
models of diabetic cardiomyopathy, uremia cardiomyopathy and
numerous other diseases (17–19);
however, whether H2S can be effective in improving
Dox-induced myocardial fibrosis has yet to be ascertained.
The PI3K/AKT/mTOR pathway is a common cell signaling
pathway involved in various pathological and physiological
processes in mammals and participates in the regulatory mechanism
of the cell cycle and cell fate (20). It is hypothesized that this pathway
is activated by ERS and negatively regulates autophagy (21). The activation of PI3K is
hypothesized to activate and phosphorylate the downstream kinase,
AKT. mTOR, an essential downstream kinase in the PI3K/AKT pathway,
is also phosphorylated and activated by PI3K, and is a significant
factor in the initialization phase of autophagy, to negatively
regulate autophagy. Some studies have demonstrated that anoxia,
oxidative stress, excessive activation of ERS and other detrimental
factors suppress the activity of PI3K, thus inhibiting the
activation of PI3K/AKT/mTOR and inducing cell autophagy. LC3 and
Beclin-1 are autophagy markers and are commonly used for the
evaluation of the degree of autophagy of cells. LC3 is a mammalian
homologue of the yeast autophagy-related gene, Atg8. There are two
forms that can be converted to each other, namely LC3II and LC3I,
while LC3II is considered as a marker of autophagosome formation
(22–24). LC3 has a close relationship with the
formation of autophagosome and a positive association with the
number of autophagosomes (22–24).
Previous studies have demonstrated that the PI3K/AKT/mTOR pathway
plays a role in the occurrence and development of fibrosis of
liver, kidney and other tissues (25–26).
The present study aimed to construct a rat model of
Dox-induced myocardial damage and to determine whether Dox-induced
myocardial fibrosis is associated with excessive ERS, autophagy and
inhibits the PI3K/AKT/mTOR pathway signaling pathway. In addition,
to determine if H2S is able to improve Dox-induced
myocardial fibrosis by regulating the PI3K/AKT/mTOR pathway to
inhibit excessive autophagy.
Materials and methods
Experimental animals
A total of 40 adult male SD rats, weighing 200±20 g,
were purchased from Changsha Lake Animal Experimental Central
(Changsha, China). Prior to the construction of the model used, the
rats were housed individually with sufficient ventilation under a
12-h light-dark cycle at 24±1°C, and all the rats were allowed free
access to food and water. The rats were fed in accordance with
institutional policies and all the experiments were conducted with
the approval granted by the University Committee on the Use and
Care of Animals of University of South China (Hengyang, China).
Chemicals and reagents
NaHS was purchased from Sigma-Aldrich (Merck KGaA).
Dox hydrochloride was supplied by Dalian Meilun Biology Technology
Co., Ltd. PI3K p55, AKT1, mTOR, TGF-β1, MMP2, TIMP2, TIMP3,
cystathionine γ-lyase (CTH) and GAPDH were sourced from Wuhan
Boster Biological Technology, Ltd., and used at a dilution of
1:500. Protein kinase RNA-like ER kinase (PERK), protein disulphide
isomerase (PDI), DNA damage-inducible transcript 3 protein (DDIT-3)
and ER chaperone BiP (BiP) were purchased from Cell Signaling
Technology Inc., and used at a dilution of 1:1,000.
Microtubule-associated proteins 1A/1B-light chain 3 (LC3),
beclin-1, cysteine protease ATG4 (ATG4) and sequestosome-1 (P62)
were purchased from ProteinTech Group Inc., and the dilution rate
of these antibodies was 1:1,000. Anti-rat secondary antibody was
purchased from KPL, Inc., and used at a dilution of 1:8,000. Cell
lysis buffer for western blot analysis, BCA protein assay kit,
Enhanced chemiluminescence reagent kit and SDS-PAGE gel preparation
kit were sourced from Beyotime Institute of Biotechnology. The
H2S ELISA kit was supplied by Shanghai Enzyme-linked
Biotechnology Co., Ltd.
Model establishment and grouping of
rats
A total of 40 rats were allowed adaptive feeding for
one week, and the general condition of all the rats, including food
intake, mental state and weight change, was observed. All the rats
were randomly divided into either the control group, Dox group,
H2S+Dox group or H2S group, with each group
comprising of 10 rats. Dox (3.0 mg/kg) was intraperitoneally
injected 3 times/weekly for a total of 6 times in 2 weeks, to
facilitate modeling in the Dox group, while in the
H2S+Dox group Dox was diluted to 0.5 mg/ml with 0.9%
NaCl before injection for immediate use. The same dose of normal
saline was administered to the control and H2S groups by
intraperitoneal injection. Once the model had been successfully
created, NaHS (56 µmol/kg/day) was administered by intraperitoneal
injection to the rats in the H2S+Dox and H2S
group while the same dose of normal saline was administered
intraperitoneally to the rats in the control and DOX groups.
Specimen collection and
processing
After 8 weeks, the rats were weighed and
echocardiography was completed. All rats were anesthetized with an
intraperitoneal injection of 10% chloral hydrate (300 mg/kg) and
subsequently sacrificed by cervical dislocation and the hearts were
weighed after lavage with cold normal saline. The left ventricle of
the heart was fixed in 10% neutral formalin at room temperature for
24 h and preserved at 4°C until further analysis. Ventricular
muscle tissues (1 mm2) were sheared from the tissue,
fixed in 2.5% glutaraldehyde solution at room temperature and
preserved at 4°C until further analysis. Ventricular muscle tissue
(3 mm2) was added to an adequate TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.), then placed in a
sterile Eppendorf® tube without enzyme and stored at
−20°C for 1 month. The remaining tissue was stored at −80°C for
western blotting and ELISA for up to 1 year.
Echocardiography analysis
After weighing of the rats, transthoracic
echocardiography was performed by a professional echocardiologist
(Department of Ultrasound Imaging), who was blinded to the study.
An evaluation of the left ventricular function was performed by
recording and calculating the left ventricular end diastolic
diameter (LVEDD), the left ventricular end systolic diameter
(LVESD), the left ventricular ejection fraction (LVEF) and the left
ventricular short-axis shortening rate (LVFS).
Masson staining
Myocardial tissue that had been previously fixed in
10% neutral formaldehyde solution was embedded in paraffin and cut
into ~4-µm sections for MASSON staining. The tissue samples were
dewaxed for hydration, then stained with hematoxylin for 5–10 min
at room temperature, and rinsed with double-distilled water.
Following which, the samples were washed with 1% hydrochloric
acid-alcohol then double-distilled water, before being stained with
Lichun red acid magenta for 10 min at room temperature, followed by
an additional wash with double-distilled water. Subsequently 1%
phosphomolybdic acid aqueous solution was added for
differentiation, followed by staining with aniline blue for 5 min
at room temperature. The samples were then incubated with 1%
glacial acetic acid, and dehydrated in a gradient alcohol series
(70 and 90%), followed by washing with xylene and sealing with
neutral gum. The samples were then viewed in multi-fields of view
under a light microscope (magnification, ×400) for comparison of
the degree of myocardial fibrosis, which was reflected by the area
of blue staining, in each of the 4 experimental groups of rats. The
collagen fibers were stained dark blue.
Transmission electron microscopy
(TEM)
Myocardial tissues (1-mm thick) pre-fixed in 2.5%
glutaraldehyde were dehydrated in an acetone series (50% for 15
min, 70% for 15 min, 90% for 15 min, 100% for 10 min and 100% for
10 min). After dehydration, pure acetone and the embedding solution
were mixed at a ratio of 1:1. Myocardial tissue was incubated in
the acetone-embedding solution mixture at 45°C for 12 h, followed
by incubation in embedding solution for 12 h and incubation at 45°C
overnight. Subsequently, tissue sections were incubated at 60°C for
12–24 h. Tissues were sliced and stained with 3% uranyl acetate and
lead nitrate for 10–20 min at room temperature. Following washing
with double distilled water, stained sections were observed using a
transmission electron microscope.
Reverse transcription-quantitative
(RT-q)PCR
Myocardial tissue frozen in TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.) was used. Total RNA
was extracted according to the manufacturer's instructions. The
purity and concentration of RNA was determined using an ultraviolet
spectrophotometer. The reverse transcription kit was purchased from
CWBio and used according to the manufacturer's instructions to
obtain cDNA. GAPDH was used as the internal reference gene. The
primer sequences for the genes were as follows: GAPDH (product
length 252 bp) forward, 5′-ACAGCAACAGGGTGGTGGAC-3′ and reverse,
5′-TTTGAGGGTGCAGCGAACTT-3′; collagen type I α-2 chain (COL1A2;
product length 179 bp) forward, 5′-TTACCCTGGCAACATTGGTC-3′ and
reverse, 5′-CCTTGTCACCTCGAATACCTTG-3′; collagen type III α-1 chain
(COL3A1; product length 141 bp) forward, 5′-AAGGGCAGGGAACAACTGAT-3′
and reverse 5′-GGTGAAGCAGGGTGAGAAGA-3′. The following was used for
qPCR: 1 µl Template (reverse transcript), 0.5 µl forward primer (10
µmol/l), 0.5 µl reverse primer (10 mol/l), 13 µl PCR
H2O, and 15 µl 2X SYBR-Green PCR mixture. The
thermocycling conditions for PCR were as follows: Initial
denaturation at 95°C for 10 min, followed by 40 cycles of
denaturation at 95°C for 5 sec and annealing at 60°C for 30 sec.
The data was analyzed by using the 2−ΔΔCq method.
ELISA
Myocardial tissue stored at −80°C was used and the
content of H2S in the myocardial tissue was measured
using H2S content ELISA kit (cat. no. ml076943;
Enzyme-linked Biotechnology Co., Shanghai, Ltd.) according to the
manufacturer's instructions.
Western blot analysis
The left ventricular tissue that had been previously
collected from each rats in the 4 experimental groups and stored at
−80°C were used. Proteins were extracted using Cell lysis buffer
for Western and IP, PMSF (Beyotime Institute of Biotechnology) and
protein was quantified using the BCA colorimetric method. The
protein samples were heated at 99°C for 10 min. Based on the
molecular weight of the desired protein, an appropriate
concentration of SDS-PAGE (8–12%) was prepared, and proteins (14
µl) were electrophoresed and transferred to PVDF membranes. The
membranes were blocked using TBS-Tween-20 and then incubated with
primary antibodies against TIMP2 (1:500), TIMP3 (1:500), MMP2
(1:500), PERK (1:1,000), DDIT-3 (1:1,000), BIP (1:1,000), PDI
(1:1,000), Beclin-1 (1:1,000), LC3II (1:1,000), ATG4 (1:1,000), P62
(1:1,000), PI3K p55 (1:500), AKT1 (1:500), mTOR (1:500), TGF-β1
(1:500) and GAPDH (1:500) at room temperature for 1 h, followed by
incubation at 4°C overnight. Following washing with TBST, the
membranes were incubated with horseradish peroxidase-labeled rabbit
secondary antibody (1:5,000) for 90 min at room temperature.
Protein bands were visualized using an ECL kit, following rinsing
with TBST. GAPDH was used as the internal reference and Grayscale
scanning in the ImageJ software (version 1.8.0.112; National
Institutes of Health) was performed to evaluate the protein
expression level.
Statistical analysis
Statistical analysis was performed using SPSS v.18.0
software (SPSS, Inc.). The obtained data are expressed as the mean
± standard deviation (SD). One-way ANOVA was used to analyze
differences between multiple groups followed by the Tukey's
post-hoc test. P<0.05 was considered to indicate a statistically
significant difference. All experiments were repeated three
times.
Results
Mortality of rats in the experimental
groups
During the duration of the experiment, a total of 7
rats died, including 4 in the Dox group and 3 in H2S+Dox
group. A large proportion of the deaths occurred in the late stage
of the experiment, which is hypothesized to be associated with the
increase of heart failure. However, the exact cause of death was
unknown.
Amount of H2S and CTH
protein expression level in rat myocardial tissues of different
experimental groups
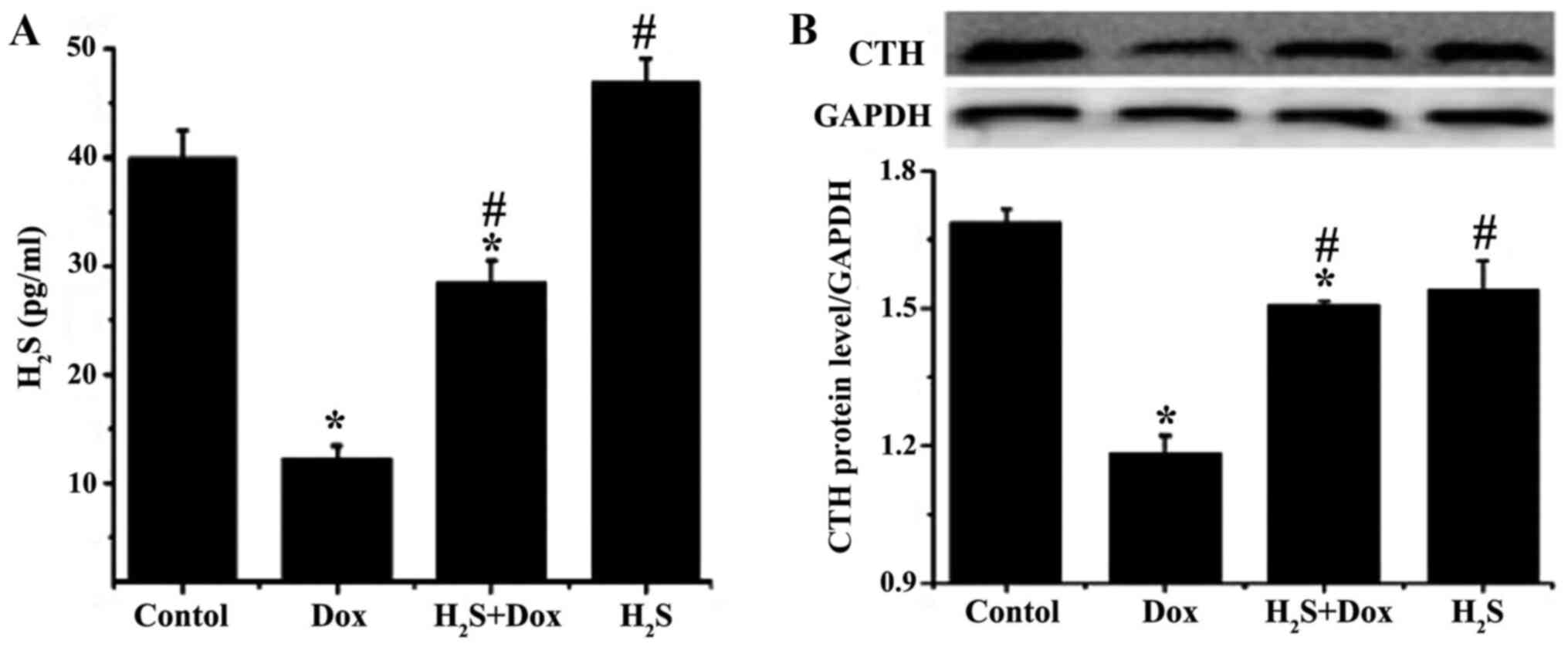
ELISA was used to determine the amount of
H2S in the myocardium and to determine if it was
associated with Dox-induced myocardial damage. The amount of
H2S in the Dox and H2S+Dox groups was
significantly reduced compared with that in the control group,
while there was no significant difference in the H2S
group (Fig. 1). The amount of
H2S was significantly higher in the H2S+Dox
and H2S groups compared with that in the Dox group. CTH
is the major catalytic enzyme, which promotes H2S
synthesis in myocardial tissue (27). CTH expression was measured using
western blot analysis and the results demonstrated that the protein
expression level of CTH was consistent with that of H2S
in each experimental group (Fig.
1).
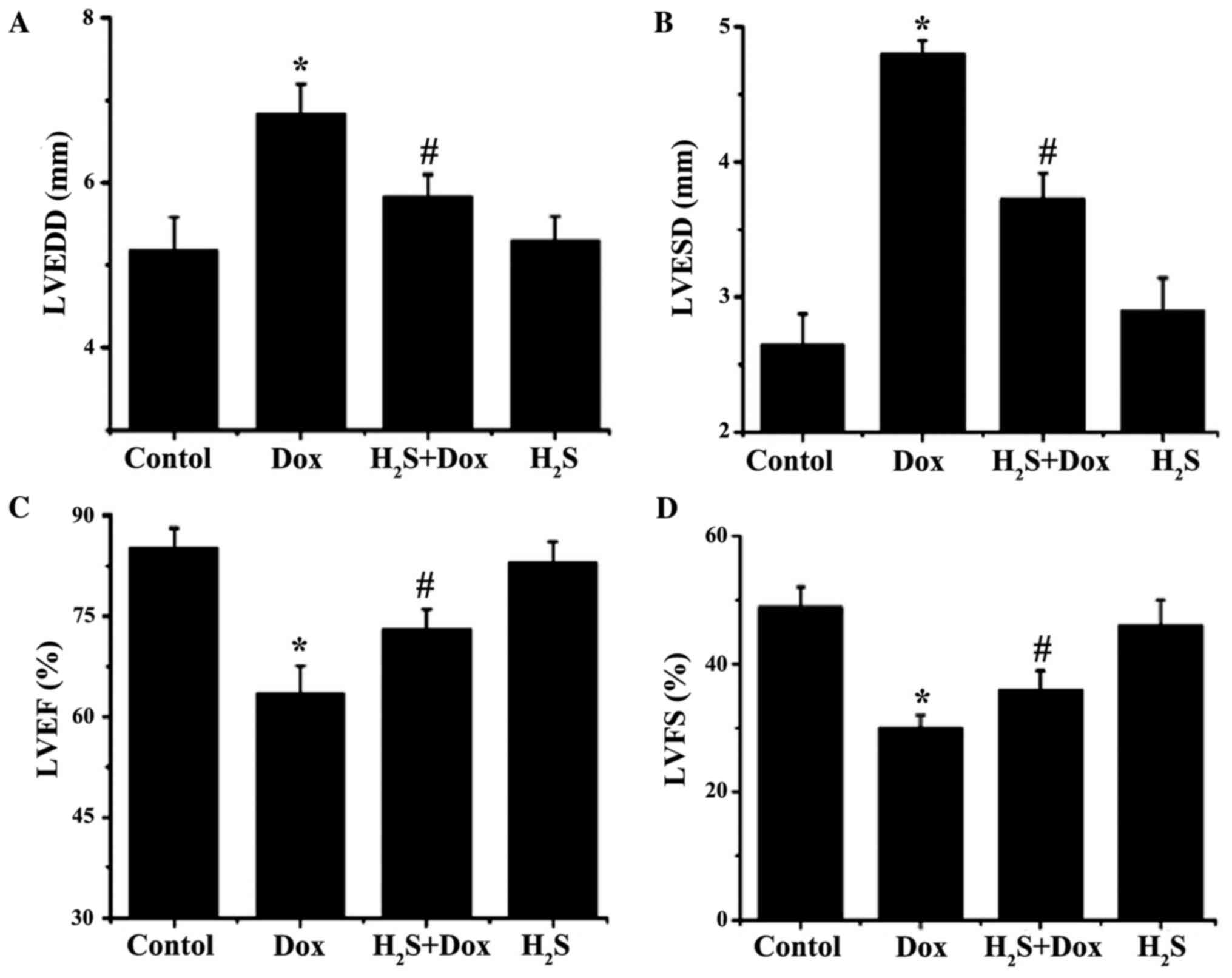
H2S improves Dox-induced
cardiac dysfunction in rats
LVEDD and LVESD were significantly increased in the
Dox group compared with that in the control group (1.65 and 2.15
mm, respectively; Fig. 2), while
LVEF and LVFS were significantly reduced (21.60 and 19.00%,
respectively; Fig. 2). LVEDD and
LVESD were significantly lower in H2S+Dox group compared
with that in the Dox group (0.99 and 1.07 mm, respectively;
Fig. 2), while LVEF and LVFS were
substantially higher (9.50 and 5.90%, respectively; Fig. 2).
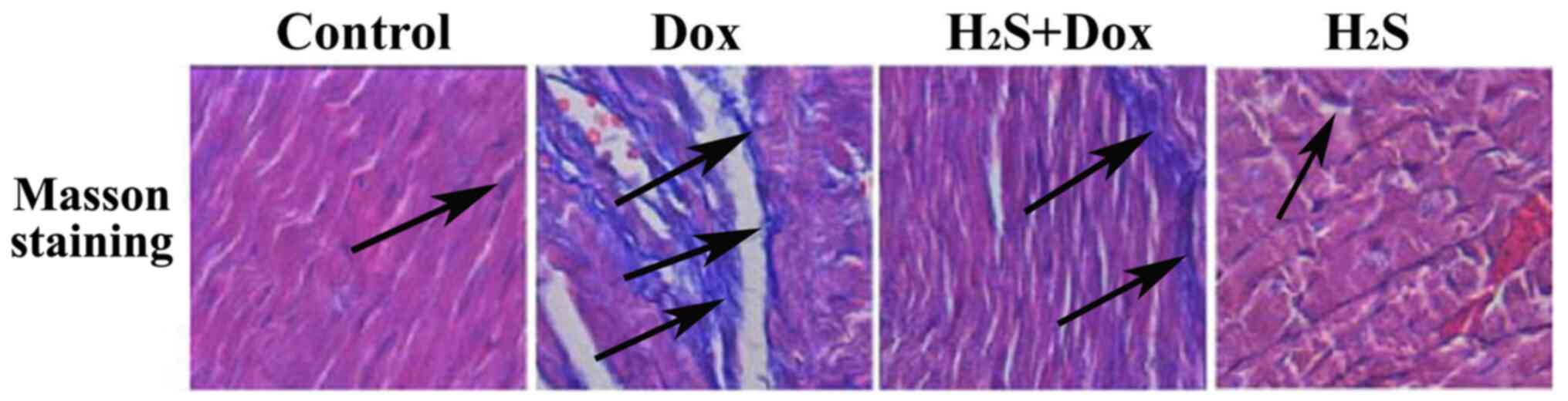
H2S improves Dox-induced
myocardial fibrosis in rats
Using Masson staining, only minimally blue-stained
fiber tissue was observed in both the control and H2S
group (Fig. 3). Myocardial cells
were disorganized and myocardial fibers were markedly increased in
the Dox group compared with the Control group, as the blue-stained
area was increased (Fig. 3).
Myocardial fibrosis improved in the H2S+Dox group
compared with that in the Dox group (Fig. 3). Additionally, the collagen volume
fraction (CVF) of myocardial tissue present in each group was
calculated using ImageJ software, which demonstrated that CVF in
the Dox group was significantly higher compared with that in the
control group, but was markedly reduced following H2S
intervention. The CVF values of the control group were found to be
similar to those of the H2S group (Table I).
 | Table I.CVF in Masson staining in the 4
experimental groups (n=3). |
Table I.
CVF in Masson staining in the 4
experimental groups (n=3).
|
| Experimental
group |
|---|
|
|
|
|---|
|
Characteristics | Control | Dox |
H2S+Dox | H2S |
|---|
| Mean CVF ± SD | 3.34±0.71 |
30.31±0.58a |
13.51±1.2a,b | 4.67±0.22 |
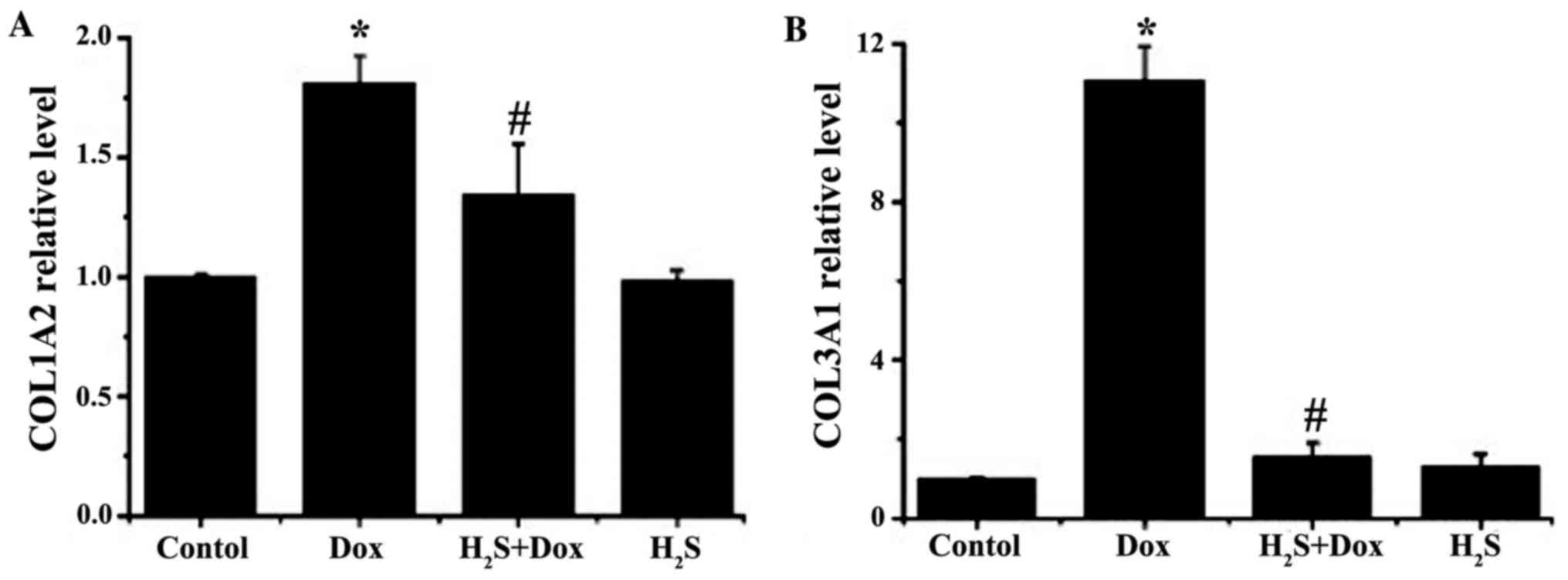
The mRNA expression levels of COL1A2 and COL3A1 in
the myocardial tissue in each experimental group were detected
using RT-qPCR and the results revealed that the expression levels
of both genes in the Dox group were significantly increased
compared with that in the control group; however, no significant
difference was found in the H2S group compared with that
in the control group. The mRNA expression levels of COL1A2 and
COL3A1 in the H2S+Dox group was significantly reduced
compared with that in the Dox group (Fig. 4).
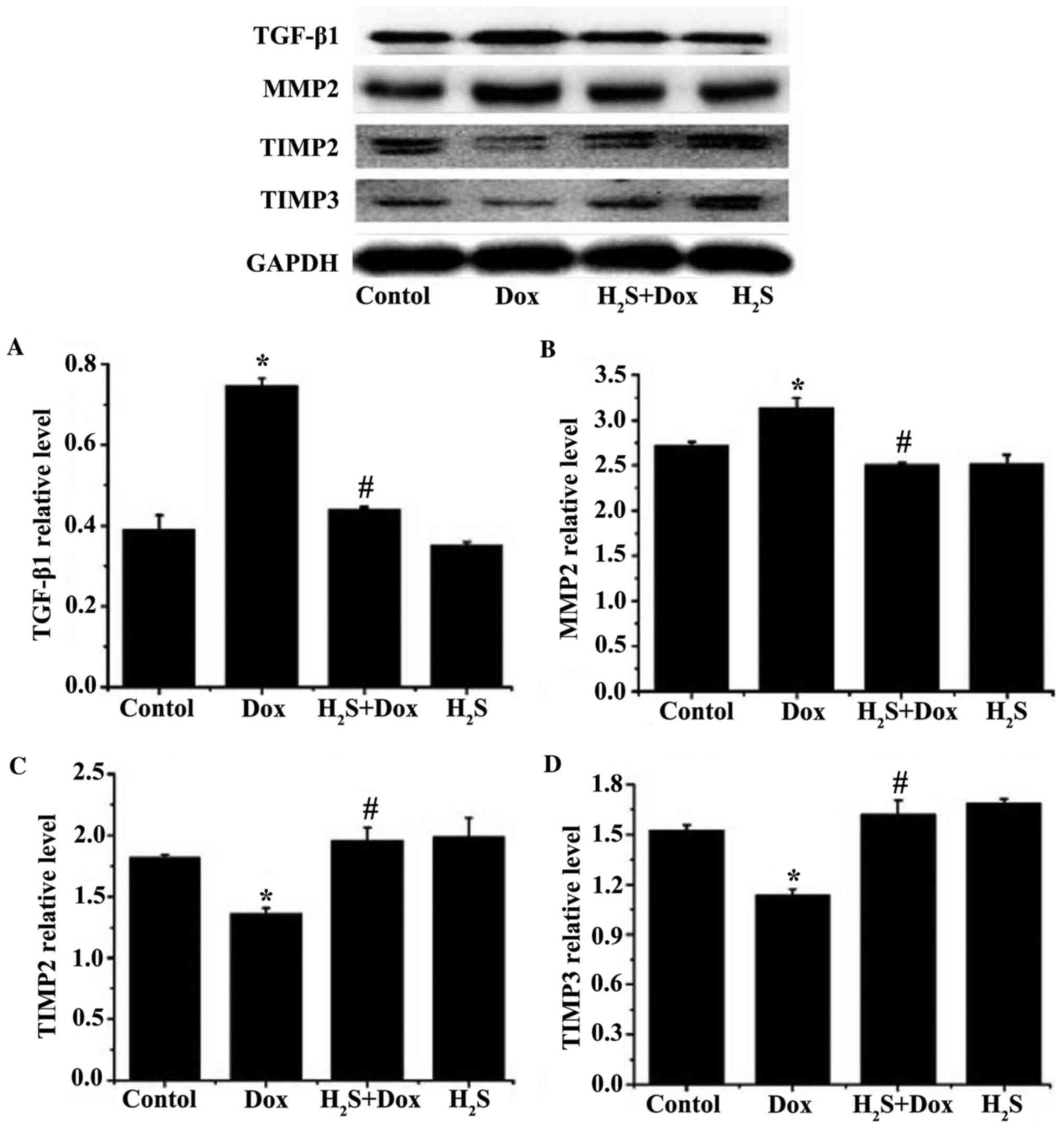
The balance of MMPs/TIMPs plays a critical role in
the synthesis and degradation of collagen, as well as the balance
of extracellular matrices. TGF-β1 is a common mediator of
myocardial fibrosis (28–29). The protein expression levels of MMP2
and TGF-β1 were significantly increased in the Dox group, while the
expression levels of TIMP2 and TIMP3 were significantly reduced
compared with that in the control group (Fig. 5). The protein expression levels of
MMP2 and TGF-β1 were significantly reduced in H2S+Dox
group, while the protein expression levels of TIMP2 and TIMP3 were
significantly increased in the H2S+Dox group compared
with that in the Dox group (Fig.
5). No significant difference was observed between the control
and H2S groups for any of the aforementioned proteins
(Fig. 5).
Effects of H2S on
myocardial ultrastructure in rats with Dox-induced
cardiomyopathy
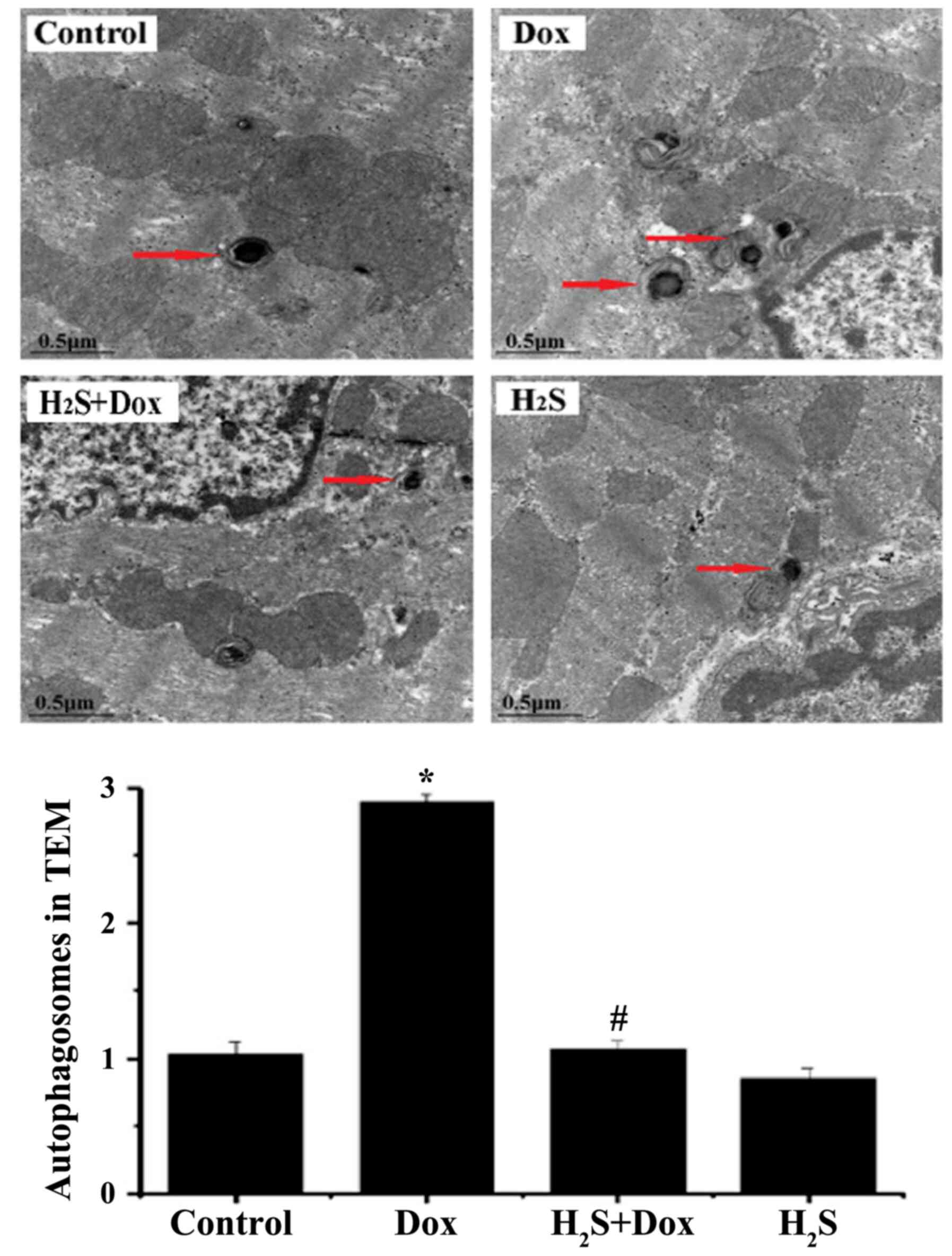
TEM was used to observe myocardial fibers and
autophagosomes in the 4 experimental groups. The myocardial tissues
of the control and H2S groups were arranged neatly with
no edema or necrosis (Fig. 6). Part
of the myocardial tissue in the Dox group displayed disorder of
myocardial tissue, edema and lytic necrosis. In addition, a higher
number of autophagosomes and autophagic vacuoles were observed in
the Dox group compared with that in the control group. In the
H2S+Dox group, myocardial fiber arrangement disorder and
edema were improved and the numbers of autophagosomes were lower
compared with that in the Dox group (Fig. 6).
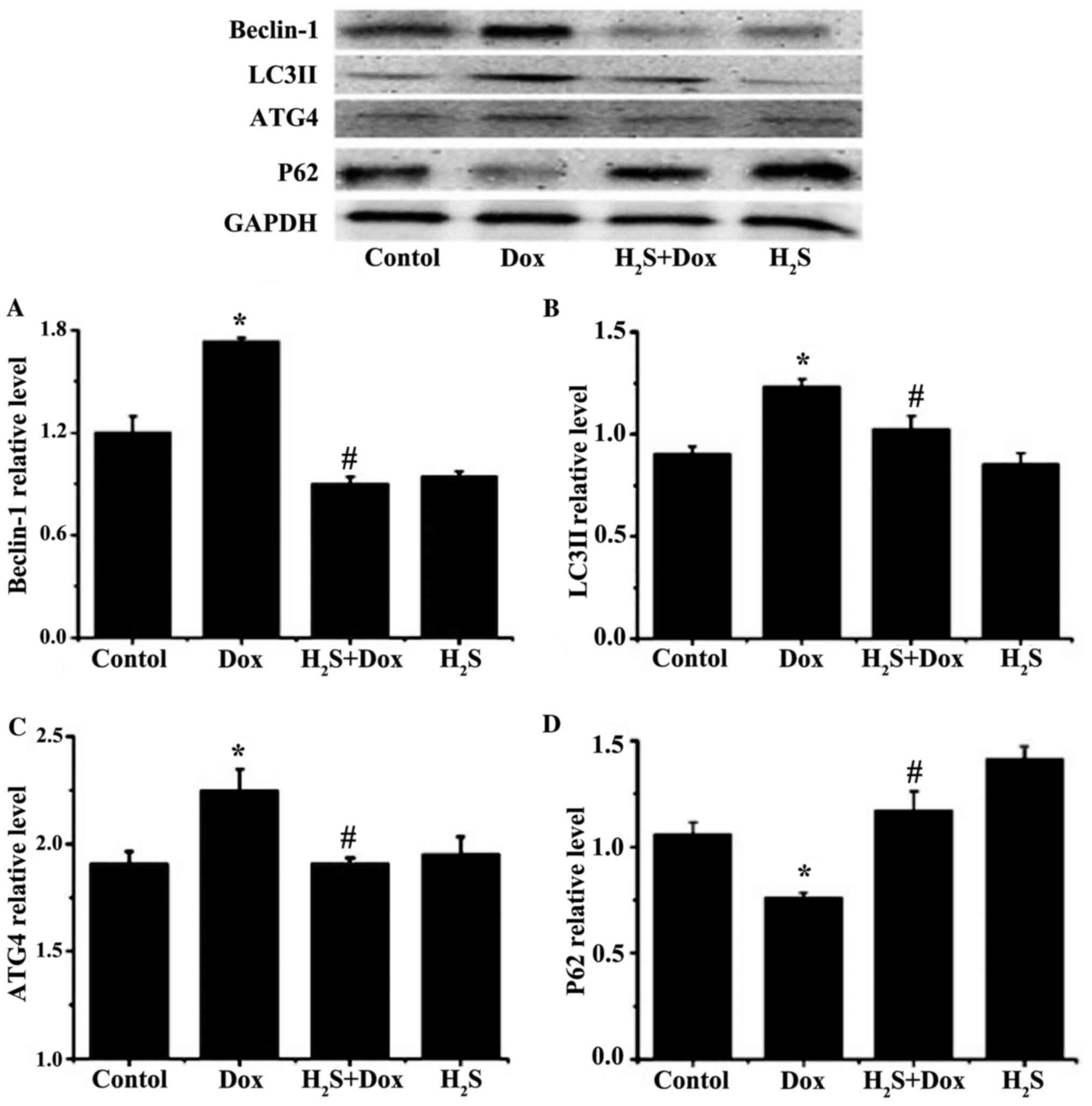
H2S inhibits autophagy in
rats with Dox-induced cardiomyopathy
The protein expression levels of beclin-1, LC3II and
ATG4 were significantly increased in the Dox group compared with
that in the control group (Fig. 7).
In contrast, the expression level of P62 was significantly
decreased in the Dox group compared with that in the control group
(Fig. 7). There was no significant
difference observed in the H2S group compared with that
in the control group for all the aforementioned proteins. The
expression levels of beclin-1, LC3II and ATG4 were significantly
reduced and the expression of P62 was significantly increased in
the H2S+Dox group compared with that in the Dox group
(Fig. 7).
H2S reduces ERS in rats
with Dox-induced cardiomyopathy
The protein expression levels of DDIT-3, PDI, BIP,
PERK in the Dox group were significantly increased compared with
that in the control group (Fig. 8),
while they were all significantly decreased in the
H2S+Dox group compared with that in the Dox group
(Fig. 8). There was no significant
difference in the aforementioned proteins between the control and
H2S groups (Fig. 8).
 | Figure 8.Protein expression levels of PDI,
BIP, DDIT-3 and PERK in the 4 experimental groups. Protein
expression levels of (A) PDI, (B) BIP, (C) DDIT-3 and (D) PERK were
determined via western blotting. Data are expressed as mean ± SD
(n=3). *P<0.05 vs. control group; #P<0.05 vs. Dox
group. H2S, hydrogen sulfide; Dox, doxorubicin; PERK,
protein kinase RNA-like ER kinase; PDI, protein disulphide
isomerase; DDIT-3, DNA damage-inducible transcript 3 protein; BiP,
ER chaperone BiP; ER, endoplasmic reticulum. |
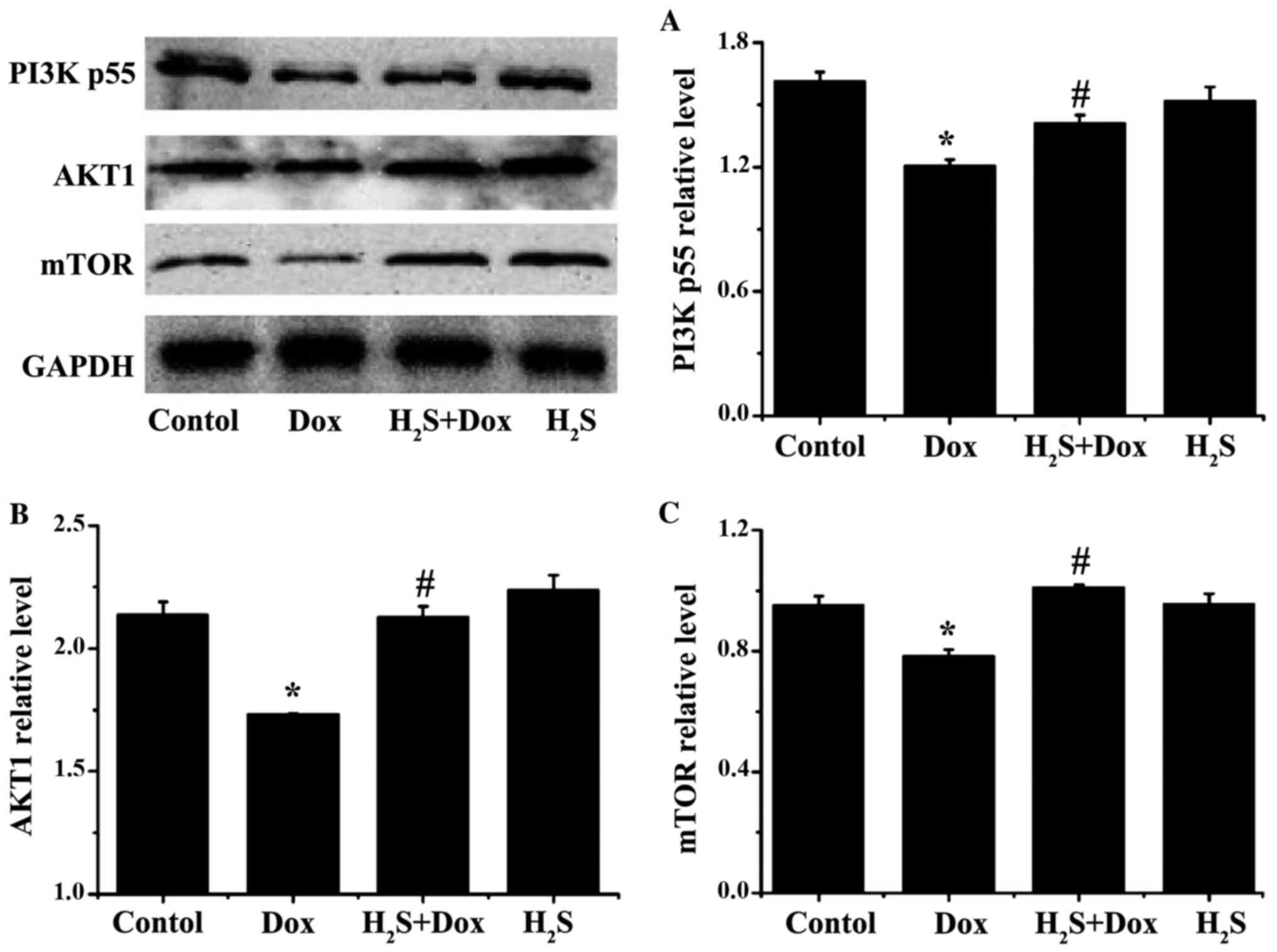
Effects of H2S on
Dox-induced change in the PI3K/AKT/mTOR signaling pathway
The protein expression levels of PI3Kp55, AKT1 and
mTOR in the Dox group were significantly reduced compared with that
in the control group (Fig. 9),
while all the proteins were significantly increased in the
H2S+Dox group compared with that in the Dox group
(Fig. 9). In addition, there was no
significant difference in the aforementioned proteins between the
control and H2S groups (Fig.
9).
Discussion
Myocardial toxicity resulting from Dox treatment is
increasingly common due to the widespread use of Dox in clinical
practice, and myocardial fibrosis could provide a significant
pathological basis for the development of Dox-induced myocardial
toxicity (30). In the present
study, a rat model of Dox-induced myocardial damage through
intraperitoneal injection of Dox was successfully constructed.
Furthermore, Masson staining demonstrated that collagenous fibers
in myocardial interstitium were disorderly arranged and collagen
deposition was significantly increased in the Dox group compared
with the control group. RT-qPCR analysis was also performed, which
demonstrated that the mRNA expression levels of COL1A2 and COL3A1
in the Dox group were significantly higher compared with that in
the control group. In addition, western blot analysis demonstrated
that the protein expression level of TGF-β1 in myocardial tissue
was higher in the Dox group compared with that in the control
group, while the expression level of MMPs/TIMPs were dysregulated.
Furthermore, ultrasonic cardiogram examination revealed the
occurrence of myocardial remodeling and cardiac insufficiency in
the rats of the Dox group. Collectively, these results demonstrated
that myocardial structural and functional remodeling occurs in the
myocardial tissues of rats following Dox induction and myocardial
fibrosis is involved in the occurrence of Dox-induced myocardial
damage. However, the mechanisms involved require further
exploration.
At present, the mechanism of Dox-induced myocardial
damage has not been adequately studied. Some studies have reported
that ERS and autophagy play a role in the internal regulatory
mechanism, Chen et al (31)
reported that isodunnianol reduced myocardial damage caused by
doxorubicin by activating protective autophagy, whereas Fu et
al (32) reported that chemical
endoplasmic reticulum chaperone molecules can alleviate Dox-induced
cardiac dysfunction. miR378 alleviates ERS and inhibits activation
of the ERS-mediated apoptosis signaling pathway in cardiomyocytes
via regulating calumenin expression, thereby reducing cardiomyocyte
apoptosis after doxorubicin-induced injury (33). Another study reported that
doxorubicin blocks autophagic flux in cardiomyocytes by impairing
lysosome acidification and lysosomal function. Moreover, reducing
autophagy initiation protects against doxorubicin cardiotoxicity
(34). Moderate activation of ERS
and autophagy is able to protect cells, while excessive activation
has an opposite effect and results in cellular damage. Some studies
have reported that ERS may induce cell autophagy through multiple
molecular mechanisms (35–36). PERK, which acts as an ERS sensor and
transmembrane protein kinase in the ER membrane is also part of a
crucial pathway to induce ERS. PERK is similar to a transmembrane
protein, as it can act as an intracellular receptor and stay on the
ER membrane of animal cells and bind to the regulatory protein,
BiP, in the ER to form a stable complex, once stress has been
detected, PERK dissociates with BiP and the activated PERK
potentially regulates ERS-induced autophagy via the PERK-DDIT-3
signaling pathway (37). In another
study, ERS was hypothesized to regulate autophagy by activating the
PI3K pathway (38). Moreover, the
findings of the present study revealed that in the Dox group, the
protein expression levels of the ERS-related proteins, such as
PERK, BiP, DDIT-3 and PDI were increased compared with the Control
group. In addition, the expression levels of the autophagy-related
proteins, such as Beclin-1, LC3II and ATG4 were also elevated,
whereas P62 was significantly decreased in the Dox group compared
with the Control group. In addition, TEM demonstrated that
autophagosomes within cells were increased in the Dox Group
compared with the control group, which indicated an upregulation of
autophagy level. The excessive ERS and autophagy observed in the
present study may play a role in the mechanism of Dox-induced
myocardial damage.
The PI3K/AKT/mTOR pathway has a widespread
distribution in cells and participates in various biological
activities, such as cell proliferation, apoptosis, transcription,
translation and the cell cycle. A previous study reported that the
PI3K/AKT/mTOR pathway is also involved in the regulation of
myocardial fibrosis (18). Some
studies have revealed that the PI3K/AKT/mTOR pathway not only plays
a role in the regulatory mechanism of differentiation of
fibroblasts and interstitial deposition, but that it is also
associated with the regulatory mechanism of autophagy (23–24).
In the present study, although myocardial fibrosis was observed in
the Dox group, the expression levels of proteins in the
PI3K/AKT/mTOR pathway were significantly decreased. The expression
levels of autophagy-related proteins, LC3B, Beclin-1 and ATG4, were
significantly elevated while the expression level of p62 was
reduced. These data suggest that the PI3K/AKT/mTOR pathway is
associated with the regulatory mechanism of autophagy of injured
myocardial cells induced by Dox.
Endogenous hydrogen sulfide is widely expressed in
the tissues of the cardiovascular system and can be produced from
CTH following the production of L-cysteine in myocardial cells.
Numerous studies have been performed to demonstrate the
physiological protective effect of CTH. In numerous pathological
models, including hypertension, myocardial infarction and diabetic
cardiomyopathy, both the changes in CTH production and the
reduction in the level of endogenous H2S have been
observed (17–19). Using different internal mechanisms,
the provision of an exogenous H2S donor, such as NaHS,
could significantly improve the level of H2S and CTH
within the human body, to produce a protective effect (17–19).
In the present study, no significant difference in the amount of
H2S was found between H2S-treated and control
rats. This could be due to the low dose of H2S that was
administered, which did not cause pathological damage, but
subsequently had little effect on the total amount of
H2S under physiological conditions. However, whether
H2S is capable of inhibiting Dox-induced myocardial
fibrosis and the internal mechanism involved remains unclear. In
the present study following H2S donor intervention,
Dox-induced myocardial fibrosis in rats was notably improved,
collagen deposition in myocardial interstitium was significantly
reduced and the mRNA expression levels of COL1A2 and COL3A1 were
reduced compared with that in the Dox group. MMPs/TIMPs imbalance
was also markedly improved and the cardiac function of rats was
restored to a certain degree, in the H2S and Dox group
compared with that in the Dox group. Additionally, following the
addition of H2S, the expression levels of the
ERS-related proteins, BIP, PERK, DDIT-3 and PDI were reduced, while
those involved in the PI3K/AKT/mTOR pathway were increased and the
autophagy-related proteins were significantly reduced. This
indicates that exogenous H2S can improve ERS and enhance
Dox-induced myocardial fibrosis in rats by inhibiting excessive
cell autophagy via the PI3K/AKT/mTOR pathway.
In the present study, it was discovered that
H2S can inhibit excessive autophagy and improve
Dox-induced myocardial fibrosis by suppressing excessive ERS and
activating the PI3K/AKT/mTOR pathway. This suggests that
H2S, an endogenous gaseous signal molecule, serves an
indispensable role in the protective mechanism of Dox-induced
myocardial damage and myocardial fibrosis by regulating ERS and
autophagy. In addition, promoting endogenous H2S
production and providing exogenous H2S can reduce the
occurrence of Dox-induced myocardial damage and myocardial
fibrosis, as well as DCM in rats. Therefore, the present study
provides a basis for a greater understanding of the pathogenesis of
Dox-induced cardiomyopathy and assists in identifying a novel
intervention target for the prevention and diagnosis of Dox-induced
cardiomyopathy. There are some limitations to the present study.
Firstly, it only explored the protective mechanism of Dox-induced
myocardial fibrosis using in vivo experiments. Thus, future
in vitro experiments would be required, which would include
using H2S inhibitors, signal pathway-specific
intervention factors and gene knockout to further clarify the
pathological mechanism involved and provide more clinical guidance
for Dox-induced cardiomyopathy.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81870230) and the
Hunan Graduate Research and Innovation Project (grant no.
CX20190765).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LN and ML performed the experiments, led the
experimental design and participated in drafting the manuscript.
JC, QW, YL and JYi performed the experiments. XZ and JZ conducted
the literature searches, and completed the verification and
revision of important knowledge content. CC and JYa helped with
designing the study, critically revising the manuscript and
approving the final version of the manuscript. LN and ML confirm
the authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
This study was granted ethical approval by the
University Committee on the Use and Care of Animals of University
of South China (Hengyang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
H2S
|
hydrogen sulfide
|
|
NaHS
|
sodium hydrosulfide
|
|
MMPs
|
matrix metalloproteinases
|
|
TIMPs
|
tissue inhibitor of
metalloproteinases
|
|
BCA
|
bicinchoninic acid
|
References
|
1
|
Jefferies JL and Towbin JA: Dilated
cardiomyopathy. Lancet. 375:752–762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Geng C, Cui C, Wang C, Lu S, Zhang M, Chen
D and Jiang P: Systematic evaluations of doxorubicin-induced
toxicity in rats based on metabolomics. ACS Omega. 6:358–366. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xia Y, Chen Z, Chen A, Fu M, Dong Z, Hu K,
Yang X, Zou Y, Sun A, Qian J and Ge J: LCZ696 improves cardiac
function via alleviating Drp1-mediated mitochondrial dysfunction in
mice with doxorubicin-induced dilated cardiomyopathy. J Mol Cell
Cardiol. 108:138–148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yao YF, Liu X, Li WJ, Shi ZW, Yan YX, Wang
LF, Chen M and Xie MY: (−)-Epigallocatechin-3-gallate alleviates
doxorubicin-induced cardiotoxicity in sarcoma 180 tumor-bearing
mice. Life Sci. 180:151–159. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang X, Li C, Wang Q, Li W, Guo D, Zhang
X, Shao M, Chen X, Ma L, Zhang Q, et al: Tanshinone IIA restores
dynamic balance of autophagosome/autolysosome in
doxorubicin-induced cardiotoxicity via targeting beclin1/LAMP1.
Cancers (Basel). 11:9102019. View Article : Google Scholar
|
|
6
|
Hasinoff BB, Patel D and Wu X: A QSAR
study that compares the ability of bisdioxopiperazine analogs of
the doxorubicin cardioprotective agent dexrazoxane (ICRF-187) to
protect myocytes with DNA topoisomerase II inhibition. Toxicol Appl
Pharmacol. 399:1150382020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gava FN, Zacché E, Ortiz EMG, Champion T,
Bandarra MB, Vasconcelos RO, Barbosa JC and Camacho AA: Doxorubicin
induced dilated cardiomyopathy in a rabbit model: An update. Res
Vet Sci. 94:115–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Q, Li Q, Na R, Li X, Liu B, Meng L,
Liutong H, Fang W, Zhu N and Zheng X: Impact of repeated
intravenous bone marrow mesenchymal stem cells infusion on
myocardial collagen network remodeling in a rat model of
doxorubicin-induced dilated cardiomyopathy. Mol Cell Biochem.
387:279–285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu X, Qi X, Lu Y, Lin C, Yuan Y, Zhu Q,
Yin Q, Li W, Li Y and Bian H: Liguzinediol protects against cardiac
fibrosis in rats in vivo and in vitro. Biomed Pharmacother.
80:260–267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu S, Chen S, Li M, Zhang B, Shen P, Liu
P, Zheng D, Chen Y and Jiang J: Autophagy activation attenuates
angiotensin II-induced cardiac fibrosis. Arch Biochem Biophys.
590:37–47. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li SJ, Liu CH, Chu HP, Mersmann HJ, Ding
ST, Chu CH, Wang CY and Chen CY: The high-fat diet induces
myocardial fibrosis in the metabolically healthy obese minipigs-The
role of ER stress and oxidative stress. Clin Nutr. 36:760–767.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeglinski MR, Davies JJL, Ghavami S,
Rattan SG, Halayko AJ and Dixon IMC: Chronic expression of Ski
induces apoptosis and represses autophagy in cardiac
myofibroblasts. Biochim Biophys Acta. 1863:1261–1268. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
González A, López B, Ravassa S, San José G
and Díez J: The complex dynamics of myocardial interstitial
fibrosis in heart failure. Focus on collagen cross-linking. Biochim
Biophys Acta Mol Cell Res. 1866:1421–1432. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang R, Jia Q, Ma SF, Wang Y, Mehmood S
and Chen Y: Exogenous H2S mitigates myocardial fibrosis in diabetic
rats through suppression of the canonical Wnt pathway. Int J Mol
Med. 44:549–558. 2019.PubMed/NCBI
|
|
15
|
Song H and Ren J: Protocatechuic acid
attenuates angiotensin II-induced cardiac fibrosis in cardiac
fibroblasts through inhibiting the NOX4/ROS/p38 signaling pathway.
Phytother Res. 33:2440–2447. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Russo I, Cavalera M, Huang S, Su Y, Hanna
A, Chen B, Shinde AV, Conway SJ, Graff J and Frangogiannis NG:
Protective effects of activated myofibroblasts in the
pressure-overloaded myocardium are mediated through smad-dependent
activation of a matrix-preserving program. Circ Res. 124:1214–1227.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xiao T, Zeng O, Luo J, Wu Z, Li F and Yang
J: Effects of hydrogen sulfide on myocardial fibrosis in diabetic
rats: Changes in matrix metalloproteinases parameters. Biomed Mater
Eng. 26 (Suppl 1):S2033–S2039. 2015.PubMed/NCBI
|
|
18
|
Liu M, Li Z, Liang B, Li L, Liu S, Tan W,
Long J, Tang F, Chu C and Yang J: Hydrogen sulfide ameliorates rat
myocardial fibrosis induced by thyroxine through PI3K/AKT signaling
pathway. Endocr J. 65:769–781. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liang B, Xiao T, Long J, Liu M, Li Z, Liu
S and Yang J: Hydrogen sulfide alleviates myocardial fibrosis in
mice with alcoholic cardiomyopathy by downregulating autophagy. Int
J Mol Med. 40:1781–1791. 2017.PubMed/NCBI
|
|
20
|
Miricescu D, Totan A, Stanescu-Spinu II,
Badoiu SC, Stefani C and Greabu M: PI3K/AKT/mTOR signaling pathway
in breast cancer: From molecular landscape to clinical aspects. Int
J Mol Sci. 22:1732020. View Article : Google Scholar
|
|
21
|
Zhang N, Zhang J, Tan YQ, Du GF, Lu R and
Zhou G: Activated Akt/mTOR-autophagy in local T cells of oral
lichen planus. Int Immunopharmacol. 48:84–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heras-Sandoval D, Pérez-Rojas JM,
Hernández-Damián J and Pedraza-Chaverri J: The role of
PI3K/AKT/mTOR pathway in the modulation of autophagy and the
clearance of protein aggregates in neurodegeneration. Cell Signal.
26:2694–2701. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thellung S, Corsaro A, Nizzari M, Barbieri
F and Florio T: Autophagy activator drugs: A new opportunity in
neuroprotection from misfolded protein toxicity. Int J Mol Sci.
20:9012019. View Article : Google Scholar
|
|
24
|
Rashid HO, Yadav RK, Kim HR and Chae HJ:
ER stress: Autophagy induction, inhibition and selection.
Autophagy. 11:1956–1977. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang TJ, Ren JJ, Zhang QQ, Kong YY, Zhang
HY, Guo XH, Fan HQ and Liu LX: IGFBPrP1 accelerates autophagy and
activation of hepatic stellate cells via mutual regulation between
H19 and PI3K/AKT/mTOR pathway. Biomed Pharmacother. 116:1090342019.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim CS, Kim IJ, Choi JS, Bae EH, Ma SK and
Kim SW: Tamoxifen ameliorates obstructive nephropathy through Src
and the PI3K/Akt/mTOR pathway. Biol Cell. 111:18–27. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Szabo C: Hydrogen sulfide, an endogenous
stimulator of mitochondrial function in cancer cells. Cells.
10:2202021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Polyakova V, Loeffler I, Hein S, Miyagawa
S, Piotrowska I, Dammer S, Risteli J, Schaper J and Kostin S:
Fibrosis in endstage human heart failure: Severe changes in
collagen metabolism and MMP/TIMP profiles. Int J Cardiol.
151:18–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
El Hajj EC, El Hajj MC, Voloshenyuk TG,
Mouton AJ, Khoutorova E, Molina PE, Gilpin NW and Gardner JD:
Alcohol modulation of cardiac matrix metalloproteinases (MMPs) and
tissue inhibitors of MMPs favors collagen accumulation. Alcohol
Clin Exp Res. 38:448–456. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu WY, Cui YK, Hong YX, Li YD, Wu Y, Li G,
Li GR and Wang Y: Doxorubicin cardiomyopathy is ameliorated by
acacetin via Sirt1-mediated activation of AMPK/Nrf2 signal
molecules. J Cell Mol Med. 24:12141–12153. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen C, Jiang L, Zhang M, Pan X, Peng C,
Huang W and Jiang Q: Isodunnianol alleviates doxorubicin-induced
myocardial injury by activating protective autophagy. Food Funct.
10:2651–2657. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Fu HY, Sanada S, Matsuzaki T, Liao Y,
Okuda K, Yamato M, Tsuchida S, Araki R, Asano Y, Asanuma H, et al:
Chemical endoplasmic reticulum chaperone alleviates
doxorubicin-induced cardiac dysfunction. Circ Res. 118:798–809.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Cui X, Wang Y, Fu Y, Guo X, Long
J, Wei C and Zhao M: Protective effect of miR378* on
doxorubicin-induced cardiomyocyte injury via calumenin. J Cell
Physiol. 233:6344–6351. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li DL, Wang ZV, Ding G, Tan W, Luo X,
Criollo A, Xie M, Jiang N, May H, Kyrychenko V, et al: Doxorubicin
blocks cardiomyocyte autophagic flux by inhibiting lysosome
acidification. Circulation. 133:1668–1687. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Song S, Tan J, Miao Y and Zhang Q:
Crosstalk of ER stress-mediated autophagy and ER-phagy: Involvement
of UPR and the core autophagy machinery. J Cell Physiol.
233:3867–3874. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Deegan S, Saveljeva S, Gorman AM and
Samali A: Stress-induced self-cannibalism: On the regulation of
autophagy by endoplasmic reticulum stress. Cell Mol Life Sci.
70:2425–2441. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu X and Long YC: Autophagy modulates
amino acid signaling network in myotubes: Differential effects on
mTORC1 pathway and the integrated stress response. FASEB J.
29:394–407. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li LJ, Chai Y, Guo XJ, Chu SL and Zhang
LS: Effects of endoplasmic reticulum stress on autophagy and
apoptosis of human leukemia cells via inhibition of the
PI3K/AKT/mTOR signaling pathway. Mol Med Rep. 17:7886–7892.
2018.PubMed/NCBI
|