Introduction
In clinical settings, the synthetic glucocorticoid
(GC) dexamethasone (DEX) and its derivatives are routinely used to
treat inflammatory disorders (1).
However, excessive use of DEX has been linked to negative human
health effects that include glucose metabolic dysfunction, insulin
resistance, mitochondrial dysfunction and muscle atrophy (2,3).
Muscle atrophy may be linked to long-term DEX treatment, which
leads to altered mitochondrial morphology, aggregation (4), enlargement and compromised
mitochondrial oxidative capacity (5). Due to the fact that muscle atrophy can
greatly affect the well-being of patients (6), alleviation of DEX-induced muscle
atrophy would be of great benefit to patients with inflammatory
diseases who rely on DEX treatment.
Chronic corticosteroid treatment has been shown in
numerous clinical and animal studies to cause DNA oxidative damage
resulting from disruption of mitochondrial morphology and oxidative
capacity (7). Indeed, such effects
have been reported frequently in conjunction with clinically
apparent mitochondrial dysfunction and skeletal muscle atrophy
linked to long-term and/or high-dose GC treatments (8,9).
Muscle atrophy associated with DEX treatment has been
experimentally linked to decreased protein synthesis accompanied by
increased protein degradation (10), both of which appear to be triggered
by ubiquitin-proteasome system activation involving muscle atrophy
F-box protein (atrogin-1) and muscle RING finger-1 (MuRF1) protein
(11). These two muscle-specific
proteins function as E3 ubiquitin ligases that are expressed early
during the muscle deterioration process before muscle loss becomes
apparent and thus may participate as key participants in
DEX-associated muscle atrophy.
Notably, GCs induction of muscle atrophy-associated
protein breakdown appears to involve effects on transcription
factor production, specifically of forkhead box (Fox) O1, FoxO3a
(12) and GSK3β (13), that trigger ubiquitin proteasome
system-dependent proteolysis of muscle proteins and subsequent
autophagy (14). Meanwhile, studies
of muscle atrophy induced by denervation have identified increased
mitochondrial fission and resulting fragmentation as key signals
responsible for triggering the AMP-activated kinase-forkhead box O3
(AMPK-FoxO3) signaling pathway (15,16).
Mitochondria are organelles that provide energy to cells. Matching
energy supply with energy demand is coordinated through various
processes and is critical for cellular adaptation and survival
under changing conditions (17).
Damage to mitochondrial function can lead to insufficient ATP
supply and activation of the AMPK/FoxO3 pathway to cause muscle
atrophy. After AMPK-FoxO3 pathway activation is triggered via the
metabolic sensor AMPK, FoxO3 is dephosphorylated then undergoes
nuclear translocation. Once in the nucleus, it acts as a
transcription factor that upregulates atrophy-inducing genes
expression that engage in events culminating in protein degradation
and eventual atrophy of skeletal muscle (18). In support of this mechanism,
findings of a previous in vitro study suggested that nuclear
translocation of FoxO3 led to upregulation of MuRF1 expression and
subsequent myotube atrophy (19).
Numerous studies have shed light on several putative
mechanisms that may be responsible for observed beneficial effects
of Panax ginseng compounds on muscle health. For example,
studies in rats have shown that treatment with traditional Chinese
herbal medicine Panax ginseng prevented muscle atrophy
(20), while a diabetic mouse study
demonstrated that panaxatriols extracted from ginseng exerted a
similar effect on skeletal muscle (21). In one study, treatment with the
panaxatriol Rg1, a ginsenoside, prevented degradation of C2C12
myotube muscle protein through regulation of the AKT/mTOR/FoxO
signaling pathway (22), with
prevention of atrophy attributed in another study to AKT/mTOR
pathway activation (23). Another
panaxatriol, 20(s)-ginsenoside Rg3 (S-Rg3), a major monomeric
ginsenoside also isolated from Panax ginseng, appeared to
prevent TNF-α-induced myotube atrophy (24). Our previous study demonstrated that
S-Rg3 could prevent myotube atrophy by promoting myoblast
differentiation (25). However, no
investigations of mechanisms involved in S-Rg3 prevention of
myotube atrophy have focused on S-Rg3 effects on mitochondrial
function, even though DEX is known to cause mitochondrial
dysfunction. Therefore, the present study investigated whether
S-Rg3 treatment of DEX-injured C2C12 myotubes could prevent muscle
atrophy by alleviating mitochondrial dysfunction induced by
DEX.
Materials and methods
Materials
Chemicals, reagents and kits were obtained from
commercial sources as follows: S-Rg3 (Urchem Sinopharm Chemical
Reagent Co., Ltd.), dexamethasone (DEX)and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
(Sigma-Aldrich; Merck KGaA), mouse skeletal muscle-derived C2C12
myoblasts (American Type Culture Collection), fetal bovine serum
(FBS; Clark BioScience), horse serum (Gibco; Thermo Fisher
Scientific, Inc.), antibodies against cleaved caspase-3 (cat. no.
9661), phosphorylated-AMPK (p-AMPK; cat. no. 2535), AMPK (cat. no.
2532), Bcl-2 (cat. no. 3498), Bax (cat. no. 2772), FoxO3 (cat. no.
2497), histone H3 (cat. no. 4499) and β-tubulin (cat. no. 2146)
were obtained from Cell Signaling Technology, Inc.; atrogin-1
antibody (cat. no. ab168372) and total OXPHOS Rodent WB Antibody
Cocktail (anti-complex I, II, III, IV, V; cat. no. ab110413) were
obtained from Abcam; antibody specific for muscle-specific RING
finger 1 (MuRF1; cat. no. bs-2539R) was purchased from BIOSS.
Chemiluminescence reagents was purchased from Santa Cruz
Biotechnology.
Cell culture and induction of cell
differentiation
C2C12 myoblasts were cultivated via incubation at
37°C in an atmosphere containing 5% CO2 with
humidification in medium containing Dulbecco's modified Eagle's
medium (DMEM) with 25 mM glucose, 10% FBS, 100 µg/ml streptomycin
and 100 units/ml penicillin until 70–80% confluence. Next,
myoblasts were seeded at 7.5×104 cells/well in 6-well
plates or at 5×103 cells/well in 96-well plates.
Myoblast fusion to form C2C12 myotubes was induced by culturing
cells for 5 days in DMEM containing 2% horse serum and 25 mM
glucose (26).
MTT cell viability assay
MTT assays were conducted to monitor cell viability
(27,28) as follows: C2C12 myotubes treated for
24 to 48 h with various concentrations of DEX (0, 25, 50, 100 or
200 µM) (29) or with S-Rg3 (0,
0.02, 0.2 or 2 µM) and/or DEX (200 µM) for 24 h were incubated with
MTT (0.5 mg/ml) for 4 h. All treatments were conducted at 37°C.
Next, formazan dissolved in 150 µl DMSO was added to wells then
measurements of absorbance levels of wells were taken at a
wavelength of 490 nm at room temperature. Calculations of cell
viability were performed that generated results as percentages of
viable cells relative to vehicle control.
Intracellular ATP level
measurement
After C2C12 myotubes were treated with various
concentrations of DEX (0, 25, 50, 100 or 200 µM) for 24 h, 50 µM
DEX for 0, 3, 6, 12 or 24 h or S-Rg3 (0, 0.02, 0.2 or 2 µM) and/or
DEX (50 µM) for 6 h, all at 37°C, C2C12 myotubes were lysed using
ATP lysis buffer composed of 0.5% Triton X-100, 100 mM glycine, pH
7.4 then lysates were centrifuged at 15,000 × g for 10 min at 4°C
(30). Supernatants were collected
and an ATP Bioluminescent Assay kit (Promega Corporation) was used
to measure intracellular ATP levels in supernatants on ice
according to the manufacturer's instructions.
Oxygen consumption rate (OCR)
measurement
Basal respiration rates of mitochondria were
measured via previously reported methods using a kit
(MitoXpress® Xtra Oxygen Consumption Assay; Agilent
Technologies, Inc.) (31). In
brief, C2C12 myoblasts previously seeded into 96-well microplates
(Corning, Inc.) were incubated for 5 day to allow them to
differentiate. C2C12 myotubes were then treated with various
concentrations of DEX (0, 25, 50, 100 or 200 µM) for 24 h, 50 µM
DEX for 0, 3, 6, 12 or 24 h or S-Rg3 (0, 0.02, 0.2 or 2 µM) and/or
DEX (50 µM) for 6 h, all at 37°C. OCRs were measured using a plate
reader at room temperature (Cytation 5; BioTek Instruments,
Inc.).
Transmission electron microscopy
C2C12 myotubes were treated with 50 µM DEX with or
without 0.2 µM S-Rg3 for 6 h at 37°C. After cells were fixed by 2-h
immersion in 2.5% glutaraldehyde fixative at 4°C, they were
post-fixed in 1% OsO4 at room temperature for 2 h, then
dehydrated in an ascending alcohol series and embedded in Epon
resin. Using a microtome (Leica UC7; Leica Microsystems, Inc.),
ultrathin sections (60–80 nm) were sliced from embedded cell
blocks, then sections were stained with 2% uranyl acetate and lead
citrate at room temperature for 15 min each, then examined using an
electron microscope (Tecnai G2 20 TWIN, FEI; Thermo Fisher
Scientific, Inc.). To measure DEX-induced changes in mitochondrial
number and area, images were processed using Image-Pro Plus 6.0
software (Media Cybernetics, Inc.) and data were analyzed by a
statistician who had no access to the images (blind data
analysis).
Mitochondrial membrane potential
detection
C2C12 myotubes were treated with 50 µM DEX and 0,
0.02, 0.2 or 2 µM S-Rg3 for 6 h at 37°C. A JC-1 fluorescent probe
(Beyotime Institute of Biotechnology) was used to estimate the
effect of S-Rg3 on mitochondrial membrane potential using the
manufacturer's instructions provided with the probe, but with
modifications (32). After 5-days
culture of C2C12 myoblasts seeded at 7.5×104 cells/well
in 6-well plates, cells were treated for 6 h with S-Rg3 and DEX,
each at various concentrations. Cells were next incubated in the
dark with JC-1 stain for 20 min at 37°C then stained cells were
immediately analyzed using flow cytometry (FACScan; BD
Biosciences).
RNA extraction and reverse
transcription-quantitative (RT-q) PCR
C2C12 myoblasts seeded at a density of
7.5×104 cells/well into 6-well plates were induced with
DMEM containing 2% horse serum and 25 mM glucose for 5 days, then
treated with 200 µM DEX for 24 h and collected on ice. After
extraction of total RNA using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocols, cDNA was generated from total RNA (1 µg)
via a reverse transcription kit (cat. no. KR118-02; Tiangen Biotech
Co., Ltd.) on ice, then a real-time PCR system (Eppendorf) was used
to conduct qPCR. The following thermocycling conditions were used
for the qPCR: Initial denaturation at 95°C for 5 min; followed by
40 cycles of 95°C for 15 sec, 60°C for 30 sec and 72°C for 30 sec.
Labeling of amplification products generated via qPCR was achieved
using SYBR Green Master Mix (Takara Biotechnology Co., Ltd.) and
gene-specific primers. Primer sequences were as follows: MuRF1,
5′-TGGAAACGCTATGGAGAACC-3′ (forward) and 5′-ATTCGCAGCCTGGAAGATG-3′
(reverse); Atrogin-1, 5′-CTGGCAGCAGCAGCTGAATAG−3′ (forward) and
5′-CACATGCAGGTCTGGGGCTGC−3′ (reverse); actin,
5′-AGGCCCAGAGCAAGAGAGGTA−3′ (forward) and
5′-CCATGTCGTCCCAGTTGGTAA−3′ (reverse). Amplification products
generated from housekeeping gene actin mRNA were used to normalize
the levels of the other products. Relative mRNA expression was
calculated based on the 2−∆∆Cq method (33).
Western blot analysis
C2C12 myotubes were treated with 50 or 200 µM DEX
and 0, 0.02, 0.2 or 2 µM S-Rg3 for 6 or 24 h at 37°C. After washing
cells twice with phosphate-buffered saline, they were suspended in
RIPA lysis buffer (Beyotime Institute of Biotechnology) and allowed
to lyse for 30 min at 4°C. Nuclear and cytoplasmic proteins were
isolated separately using a BestBio BB-3102 kit (BestBio). Protein
concentration was determined using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology). Lysates (30 µg total/lane)
were separated via 12% SDS-PAGE followed by electro-transfer to
polyvinylidene fluoride (PVDF) membranes. After blocking of
membranes in 5% bovine serum albumin (cat. no. 4240; BioFroxx) at
room temperature for 1 h, membranes were incubated overnight with
1:1,000 dilution of primary antibody at 4°C followed by two washes
and a 1-h incubation with 1:5,000 of horseradish
peroxidase-conjugated goat anti-rabbit IgG (cat. no.) BA1054 and
goat anti-mouse IgG (cat. no. BA1050) secondary antibodies (Boster
Biological Technology) at room temperature. Proteins were
visualized via chemiluminescence (ProteinSimple), then imaged using
a FluorChem HD2 system (ProteinSimple). Densitometry was performed
using AlphaView SA software 3.4.0.0 (ProteinSimple) (34).
Statistical analysis
All statistical analyses were performed with
GraphPad Prism 7 (GraphPad Software, Inc.) and using an unpaired
Student's t-test or one-way analysis of variance (ANOVA) followed
by Tukey's post-hoc tests. Data are expressed as the mean ±
standard deviation of three independent experimental repeats.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Induction of muscle atrophy and
mitochondrial dysfunction by dexamethasone (DEX) treatment
Results of experiments whereby C2C12 myotubes
received treatment with various doses of DEX (25, 50, 100 and 200
µM) for 24 and 48 h revealed no effect on cell viability at DEX
concentrations below 200 µM (Fig.
1A). Meanwhile, increased levels of two muscle atrophy marker
proteins, MuRF1 and atrogin-1, were observed in C2C12 myotubes
treated with 200 µM DEX (Fig. 1B).
Next, measurements of C2C12 myotube OCR and intracellular ATP level
were performed to reveal the relationship between muscle atrophy
and mitochondrial function. Subsequently it was observed that 24-h
DEX treatment (50–200 µM DEX) led to ATP deprivation (Fig. 1C) and reduced OCR (Fig. 1D). Next, C2C12 myotubes were treated
with DEX (50 µM) for various lengths of time, with the results
revealing that 6-h DEX treatment of C2C12 myotubes led to ATP
deprivation (Fig. 1E) and OCR
reduction (Fig. 1F). When
considered together, these results demonstrated that damage to
mitochondria preceded myotube atrophy, since 50 µM DEX treatment of
C2C12 myotubes for only 6 h led to mitochondrial dysfunction, while
24-h treatment with 200-µM DEX was necessary to induce myotube
atrophy. Therefore, to generate a mitochondrial damage model the
present study employed 6-h administration of 50 µM DEX and for the
myotube atrophy model administered 24-h treatment with 200 µM
DEX.
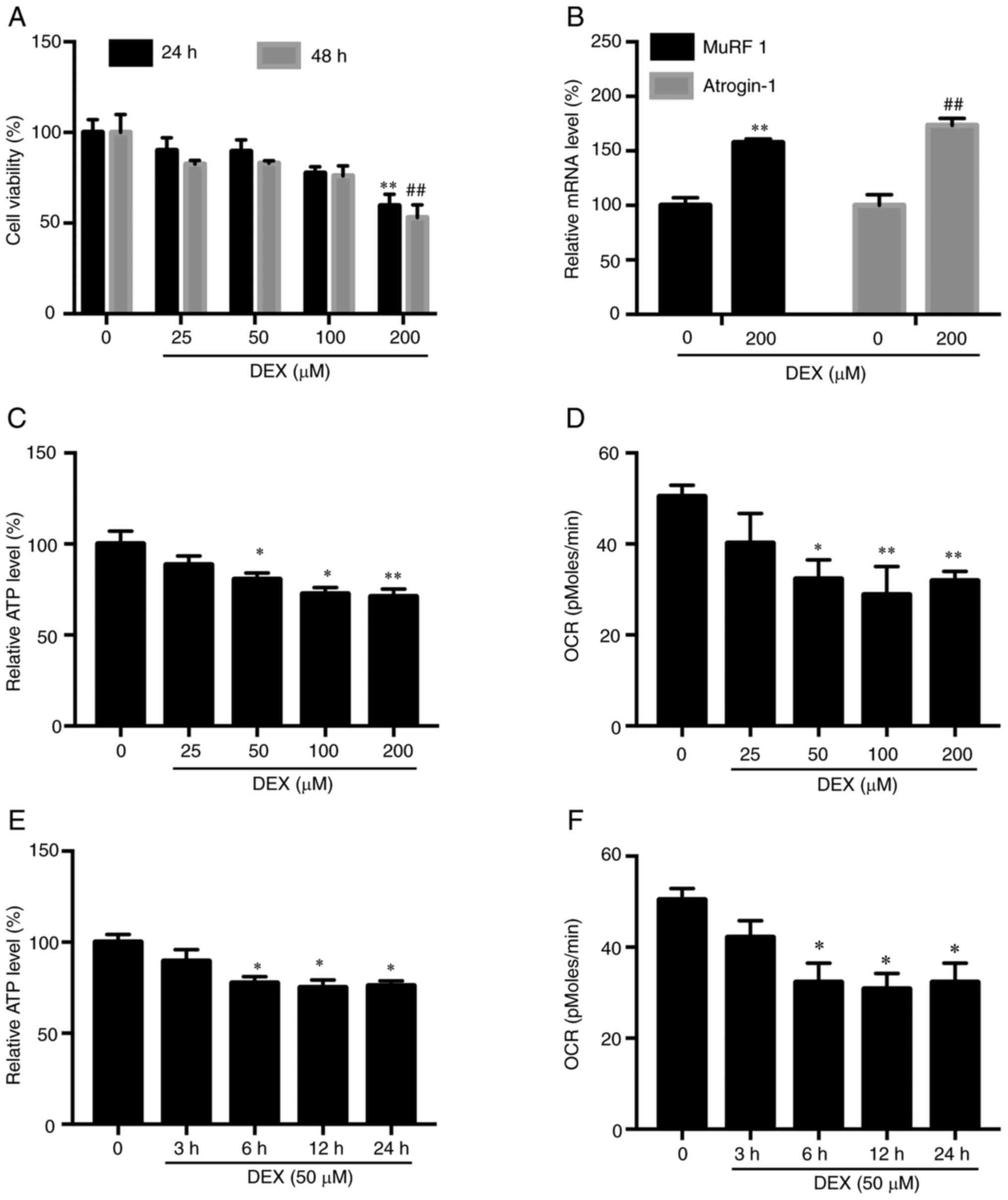 | Figure 1.Muscle atrophy and mitochondrial
dysfunction induced by DEX. (A) After 24 or 48-h treatment with
various doses of DEX (25, 50, 100 and 200 µM), no effect on C2C12
myotube viability was observed at DEX concentrations <200 µM.
**P<0.01 vs. CON (24 h); ##P<0.01 vs. CON (48 h).
Results were analyzed via one-way ANOVA with data expressed as the
mean ± standard deviation (n=3). (B) DEX-treated C2C12 myotube
atrogen-1 and MuRF1 mRNA levels were higher compared with
corresponding levels in control cells. **P<0.01 vs. CON.
(MuRF1); ##P<0.01 vs. CON (atrogin-1). Results were
analyzed via Student's t-test, with data expressed as the mean ±
standard deviation (n=3). (C) Intracellular ATP level was reduced
following 24-h treatment with various DEX doses (25, 50, 100 and
200 µM). *P<0.05 and **P<0.01 vs. CON. Results were analyzed
via one-way ANOVA, with data expressed as the mean ± standard
deviation (n=3). (D) Basal respiration (OCR) measured by
MitoXpress® Xtra Oxygen Consumption Assay was reduced at
24 h following administration of various doses of DEX (25, 50, 100
and 200 µM). *P<0.05 and **P<0.01 vs. CON. Results were
analyzed via one-way ANOVA, with data expressed as the mean ±
standard deviation (n=3). (E) Reduction of intracellular ATP level
following DEX incubation for various time points in hours.
*P<0.05 vs. CON. Results were analyzed via one-way ANOVA, with
data expressed as the mean ± standard deviation (n=3). (F)
Reduction of OCR following DEX administration for various time
points in hours. *P<0.05 vs. CON. Results were analyzed via
one-way ANOVA, with data expressed as the mean ± standard deviation
(n=3). DEX, dexamethasone; CON, 0 µM DEX; MuRF1, muscle RING
finger-1; atrogin-1, muscle atrophy F-box protein; OCR, oxygen
consumption rate. |
Treatment with S-Rg3 reverses C2C12
myotube atrophy induced by DEX
After verifying that DEX-induced myotube atrophy had
occurred as the present study's model for skeletal muscle atrophy,
effects of S-Rg3 treatment on DEX-induced injury were evaluated.
DEX treatment alone decreased C2C12 myotube cell viability to
68.68±1.21%, while S-Rg3 treatment restored C2C12 myotube cell
viability in a dose-dependent manner (Fig. 2A). The expression levels of muscle
cell-specific major muscle atrophy-associated E3 ubiquitin ligases
atrogin-1 and MuRF1 were examined to assess S-Rg3 effects on
DEX-induced skeletal muscle cell atrophy. Notably, DEX increased
expression of both atrogin-1 and MuRF1 proteins in C2C12 myotubes,
while reduced expression of the two proteins was observed following
S-Rg3 treatment (Fig. 2B-D). The
effect of S-Rg3 on apoptosis caused by DEX was also tested. The
expression levels of apoptosis-related proteins, such as cleaved
caspase-3, Bcl-2 and Bax were examined by western blot analysis.
The expression of cleaved caspase-3, which involved in the
activation cascade of caspases responsible for apoptosis execution
was decreased by S-Rg3 treatment for 24 h in DEX-induced C2C12
myotubes, and the ratio of the antiapoptotic protein Bcl-2 and
proapoptotic protein Bax was increased following treatment with
S-Rg3 for 24 h (Fig. 2E-F).
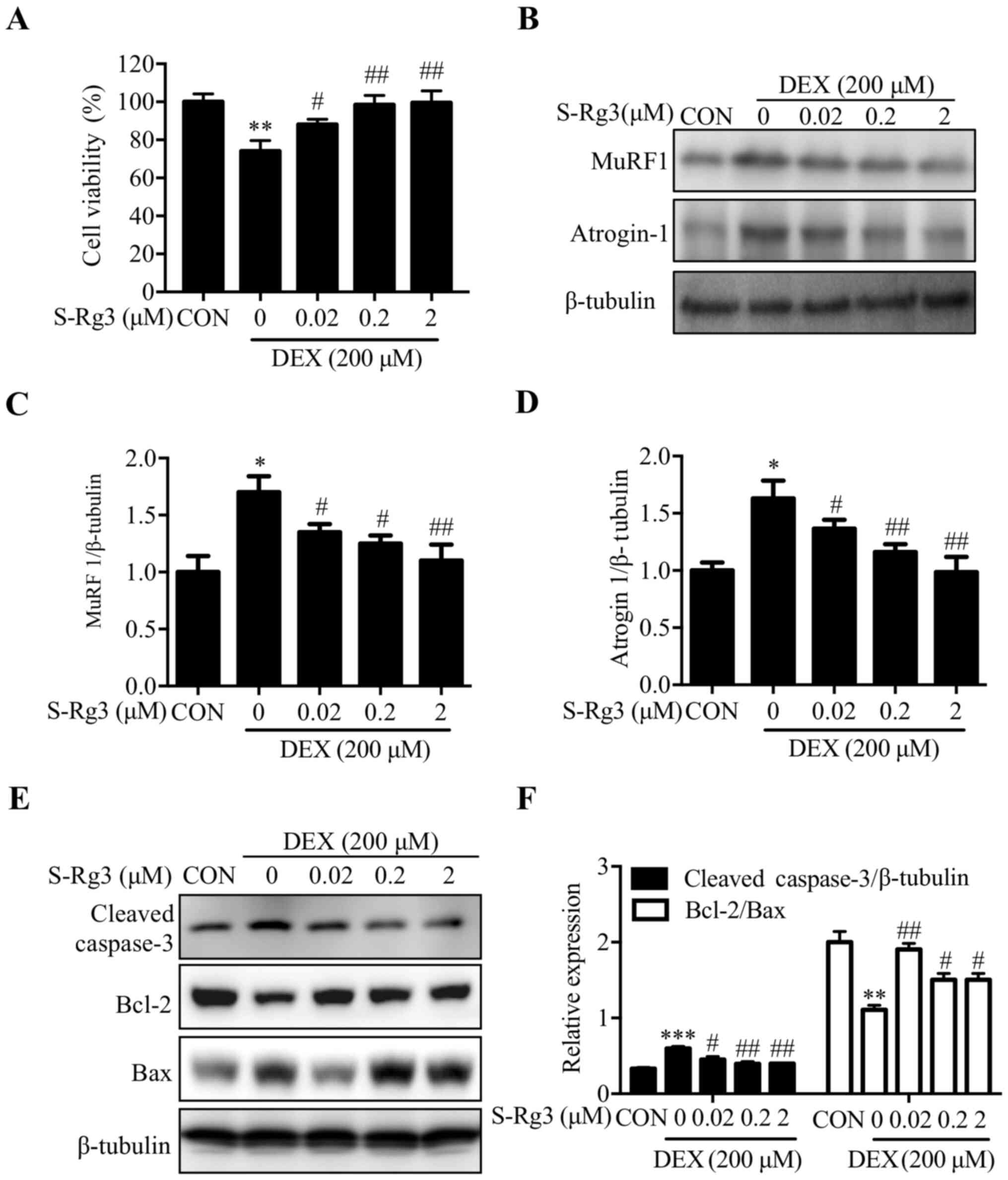 | Figure 2.S-Rg3 reversal of DEX-induced C2C12
myotube atrophy. (A) After 24-h treatment of C2C12 myotubes with
S-Rg3 (0.02, 0.2 and 2 µM) and DEX, MTT assays were conducted to
measure viability of C2C12 myotube cells. **P<0.01 vs. CON;
#P<0.01 and ##P<0.01 vs. DEX. Results
were analyzed via one-way ANOVA, with data expressed as the mean ±
standard deviation (n=3). (B) To further analyze S-Rg3 treatment
effects on skeletal muscle cell atrophy, atrogin-1 and MuRF1
expression levels were assessed via western blot analysis. (C and
D) Relative atrogin-1 and MuRF1 expression levels were quantified
via densitometric analysis, with β-tubulin serving as loading
control. *P<0.05 vs. CON; #P<0.05 and
##P<0.01 vs. DEX. Results were analyzed via one-way
ANOVA, with data expressed as the mean ± standard deviation (n=3).
(E) Following treatment of C2C12 myotubes with S-Rg3 and/or DEX for
24 h, cleaved caspase-3, Bcl-2 and Bax levels were measured via
western blot analysis. (F) Relative expression levels of cleaved
caspase-3 and Bcl-2/Bax were semi-quantified by densitometric
analyses based on β-tubulin as loading control. Results were
analyzed using a one-way ANOVA. Data are shown as the mean ±
standard deviation (n=3). **P<0.01, ***P<0.001 vs. CON;
#P<0.05, ##P<0.01 vs. DEX. S-Rg3,
20(S)-ginsenoside Rg3; DEX, dexamethasone; MuRF1, muscle RING
finger-1; atrogin-1, muscle atrophy F-box protein; CON, control
group. |
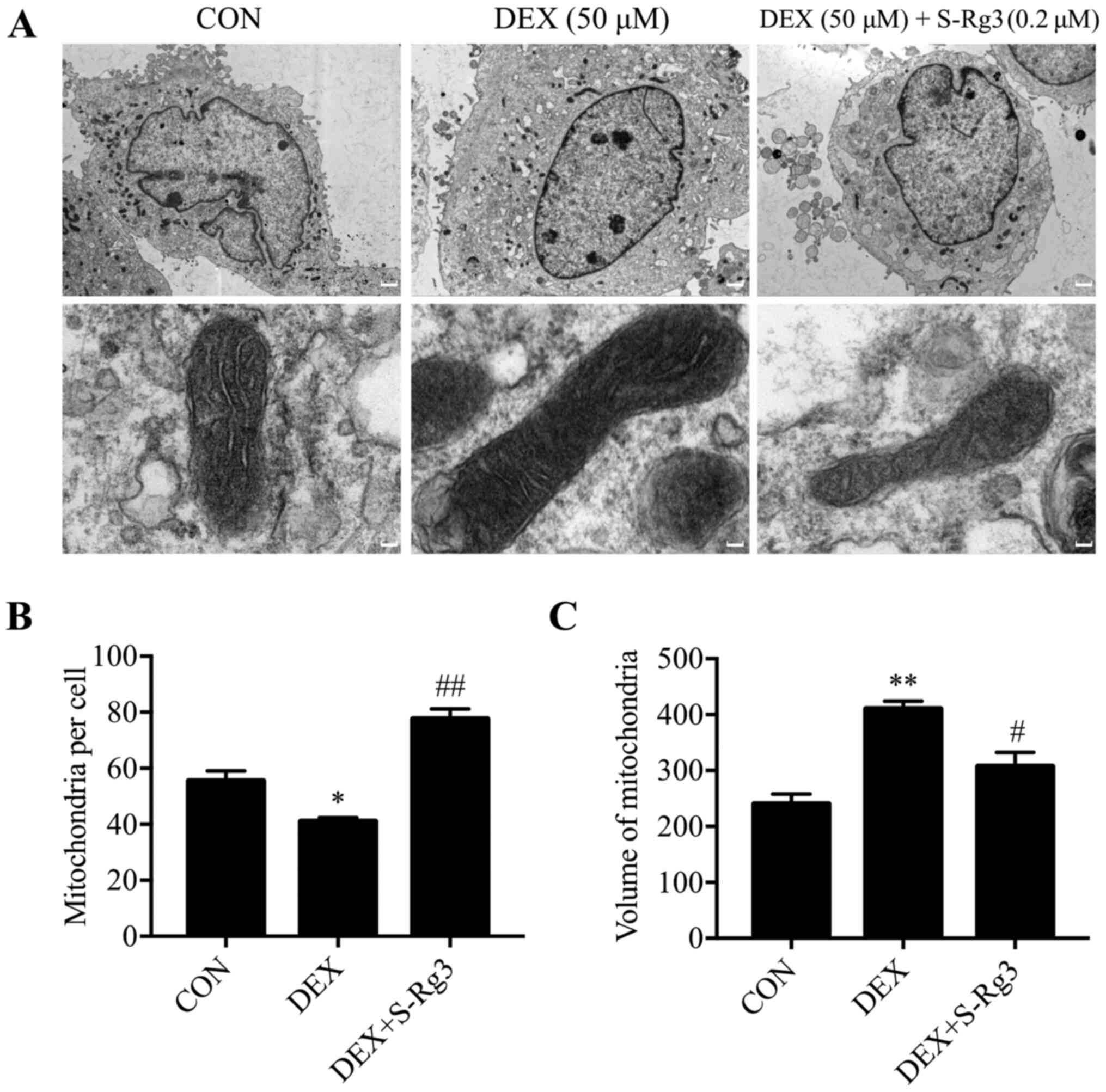
Mitochondrial morphological changes
induced by DEX are reversed by S-Rg3 treatment
Previous studies conducted using a C2C12 skeletal
muscle cell line have demonstrated that mitochondrial morphological
changes occur during mitophagy, a necessary cellular process for
removing damaged mitochondria (35,36).
Using mitochondrial volume as a morphological indicator,
DEX-induced mitochondrial injury was analyzed in C2C12 myotubes
using electron microscopy (Fig.
3A). Following DEX treatment, relatively large mitochondrial
structural changes were observed, including swollen and broken
mitochondria or the absence of mitochondria. These changes were
ameliorated by treatment with S-Rg3 as evidence that S-Rg3 restored
mitochondrial function (Fig. 3B and
C).
DEX-induced mitochondrial dysfunction
is alleviated by S-Rg3 treatment
To reveal whether an association exists between
DEX-induced muscle atrophy and mitochondrial dysfunction,
intracellular ATP levels and OCRs were measured in DEX-injured
C2C12 myotubes. Ultimately, low ATP levels were observed following
treatment with DEX that were increased following subsequent S-Rg3
treatment (Fig. 4A). In addition,
S-Rg3 treatment reversed DEX-induced mitochondrial respiratory
dysfunction (Fig. 4B), reversed
DEX-induced reduction of mitochondrial membrane potential (an early
sign of apoptosis in C2C12 myotubes) (Fig. 4C) and reversed DEX-induced
reductions of JC-1 aggregate levels from below control levels (as
revealed via staining followed by flow cytometry) to levels above
levels of untreated DEX-injured cells (Fig. 4D). Finally, as an additional
assessment of whether mitochondrial dysfunction was induced by DEX,
expression levels of key mitochondrial respiratory electron
transport chain subunit proteins of complexes II, III and V were
measured and significantly reduced expression levels of all key
subunit proteins following DEX treatment were observed; these
levels were increased following addition of S-Rg3 (Fig. 4E and F).
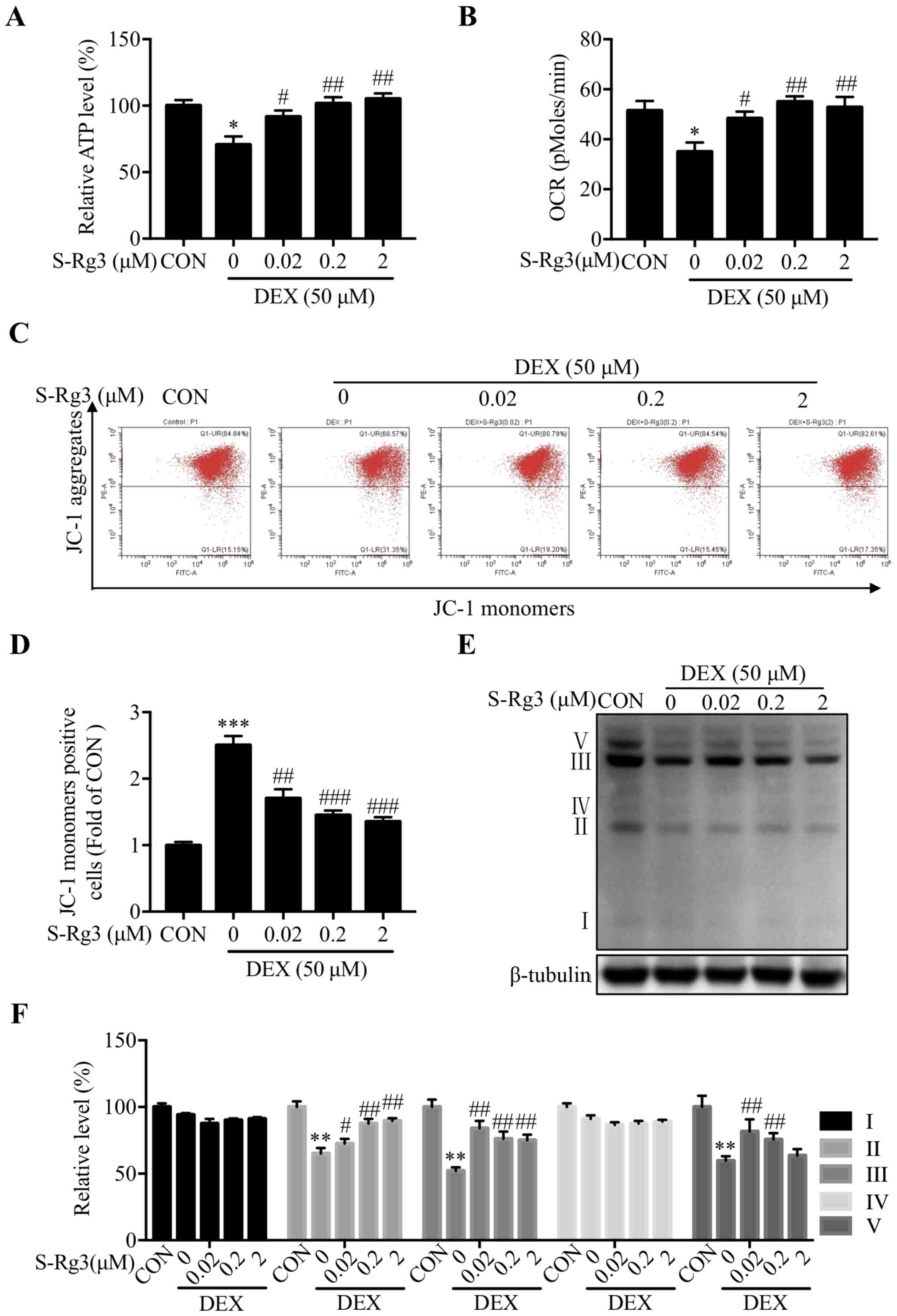 | Figure 4.S-Rg3 reversal of mitochondrial
dysfunction induced by DEX. (A) DEX-injured C2C12 myotube
intracellular ATP level was increased by S-Rg3 treatment.
*P<0.05 vs. CON; #P<0.05 and
##P<0.01 vs. DEX. Results were analyzed via one-way
ANOVA, with data expressed as the mean ± standard deviation (n=3).
(B) OCR as measured by MitoXpress® Xtra Oxygen
Consumption Assay measurement showing OCR decrease following 6-h
DEX administration that was restored following administration of
S-Rg3. *P<0.05 vs. CON; #P<0.05 and
##P<0.01 vs. DEX. Results were analyzed via one-way
ANOVA, with data expressed as the mean ± standard deviation (n=3).
(C) After 6-h DEX and/or S-Rg3 treatments, mitochondrial membrane
potential was assessed following incubation of C2C12 myotubes with
the JC-1 probe followed by flow cytometric analysis. (D) Bar graph
representing cells staining positive for JC-1 monomer then analyzed
as in (C). ***P<0.001 vs. CON; ##P<0.01 and
###P<0.001 vs. DEX. Results were analyzed via one-way
ANOVA, with data expressed as the mean ± standard deviation (n=3).
(E) Representative western blots of DEX-injured C2C12 myotube
complex I, II, III, IV and V proteins. (F) Relative expression of
complex I, II, III, IV and V proteins quantified by densitometric
analysis, with β-tubulin serving as loading control. Data are
expressed as the mean ± standard deviation (n=3). **P<0.01 vs.
CON; #P<0.05 and ##P<0.01 vs. DEX.
Results were analyzed via one-way ANOVA, with data expressed as the
mean ± standard deviation (n=3). S-Rg3, 20(S)-ginsenoside Rg3; DEX,
dexamethasone; OCR, oxygen consumption rate; CON, control
group. |
AMPK/FoxO3 signaling pathway
inhibition by S-Rg3 in the DEX-injured C2C12 myotube-based muscle
atrophy model
A critical cellular energy sensor, AMPK (37), is regulated by the intracellular AMP
to ATP ratio, with robust activation of AMPK induced by ATP
deprivation (38). In the present
study, AMPK phosphorylation increased markedly following 6-h DEX
treatment (Fig. 5A and B). In
addition, increased nuclear and decreased cytoplasmic levels of
transcription factor protein FoxO3, a known master regulator of
muscle atrophy-associated E3 ligases MuRF1 and atrogin-1 (39), were observed. These changes were
reversed by S-Rg3 treatment, which decreased AMPK phosphorylation
(Fig. 5A and B) and reduced FoxO3
nuclear translocation (Fig. 5C and
D). Therefore, these data suggested that the AMPK/FoxO3 pathway
is involved in the protective effect of S-Rg3 on mitochondrial
dysfunction.
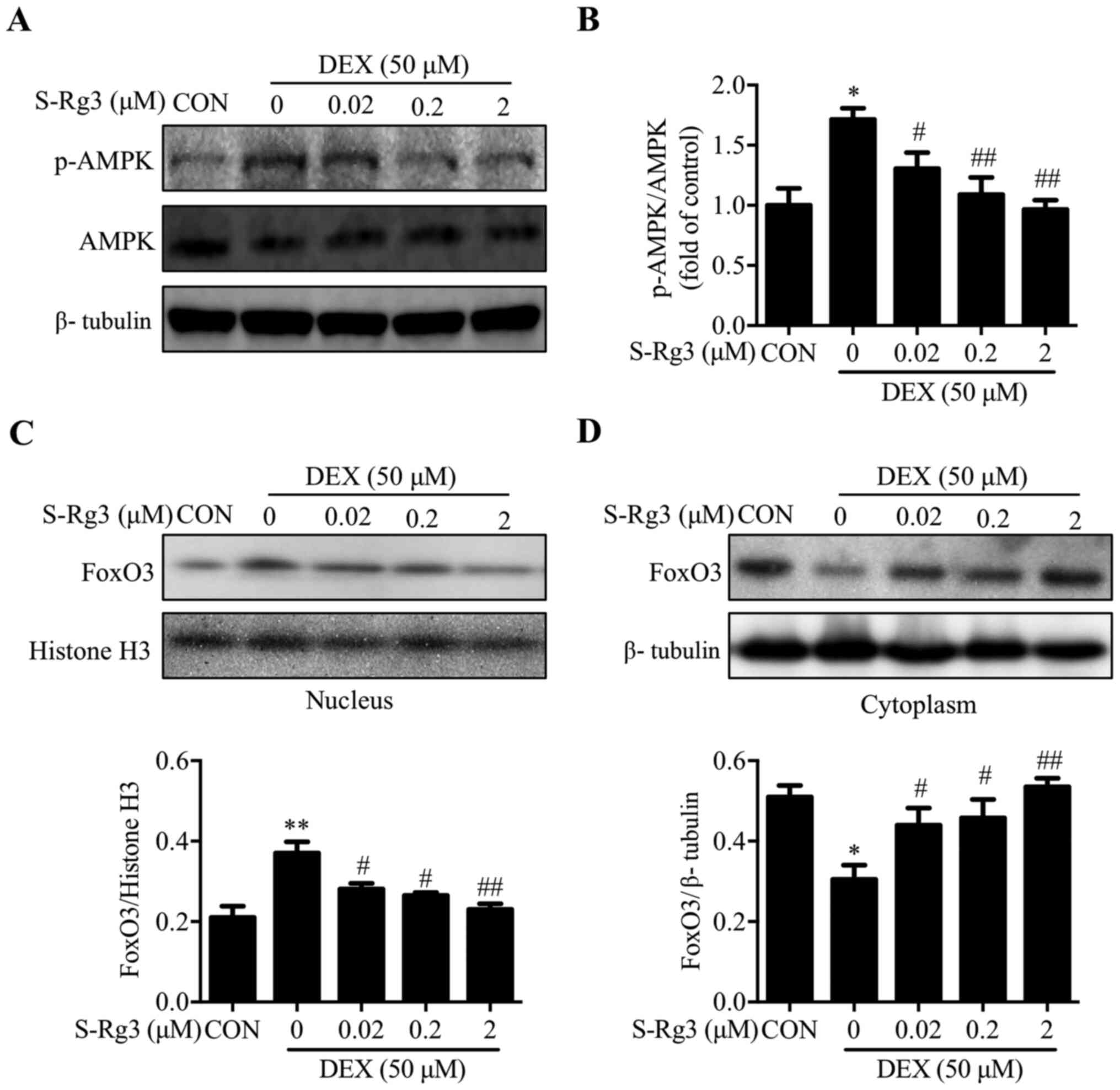 | Figure 5.S-Rg3 inhibition of the AMPK/FOXO3
signaling pathway induced in C2C12 myotubes by DEX. (A) After 6-h
S-Rg3 treatment, p-AMPK and AMPK levels in DEX-injured C2C12
myotubes were detected by western blot analysis, with β-tubulin
serving as loading control. (B) Relative expression of p-AMPK/AMPK
quantified using densitometric analysis, *P<0.05 vs. CON;
#P<0.05 and ##P<0.01 vs. DEX. Results
were analyzed via one-way ANOVA, with data expressed as the mean ±
standard deviation (n=3). (C) To further explore S-Rg3 effects,
nuclear FoxO3 expression was studied via western blot analysis,
with histone H3 serving as the nuclear protein loading control.
Relative FoxO3 expression levels were semi-quantified via
densitometric analysis. **P<0.01 vs. CON; #P<0.05,
##P<0.01 vs. DEX. Results were analyzed via one-way
ANOVA, with data expressed as the mean ± standard deviation (n=3).
(D) To further explore S-Rg3 effects, cytoplasmic FoxO3 expression
was studied via western blot analysis, with β-tubulin serving as
the cytoplasmic protein loading control. Relative FoxO3 expression
levels were semi-quantified via densitometric analysis. *P<0.05
vs. CON; #P<0.05, ##P<0.01 vs. DEX.
Results were analyzed via one-way ANOVA, with data expressed as the
mean ± standard deviation (n=3). S-Rg3, 20(S)-ginsenoside Rg3;
AMPK, AMP-activated protein kinase; FoxO3, forkhead box O3; p-,
phosphorylated; DEX, dexamethasone; CON, control group. |
Discussion
In spite of their beneficial anti-inflammatory and
other clinically useful properties, GCs are known for numerous
adverse myopathic effects (40).
For example, high-dose synthetic GC DEX treatment has been observed
to increase degradation and decrease synthesis of muscle protein
that together led to reduced muscle mass and muscle fiber thinning
(41). These myopathic effects
appear to involve triggering of catabolic signals, including
ubiquitin E3 ligases atrogin-1 and MuRF1 (42). As demonstrated in previous studies,
such effects may be counteracted by ginsenoside Rg1 (22) and ginseng protein (43), which are reported to possess
anti-atrophy effects, as well as by ginsenoside S-Rg3, which has
been shown to prevent myotube atrophy induced by TNF-α (24). The present study demonstrated that
S-Rg3 prevention of myotube atrophy was linked to decreases in
atrogin-1 and MuRF1 expression levels that subsequently
corresponded to observed increase in viability of DEX-injured C2C12
myotube cells. The present study also demonstrated that the in
vitro experimental model of myotube atrophy employed could be
used to evaluate drugs, such as S-Rg3, for effectiveness when used
for treatment of muscle atrophy.
Muscle atrophy, a clinical disorder commonly
associated with diabetes (44),
chronic obstructive pulmonary disease (45) and other chronic diseases, negatively
affects the quality of life of patients and promotes progression of
pathological disease (46). A
growing body of evidence suggests that mitochondrial dysfunction is
a key player in muscle atrophy caused by disuse and disease
(47). Meanwhile, a previous study
demonstrated that treatment with DEX can cause serious impairment
of mitochondrial function manifesting as mitochondrial loss,
dysfunctional mitochondrial respiration and disordered
mitochondrial morphology and distribution (48). To counter such adverse DEX-induced
effects, therapies targeting mitochondrial processes to increase
mitochondrial biogenesis and/or enhance mitochondrial respiration
are sought for prevention or treatment of muscle atrophy (49). In the present study, C2C12 myotube
mitochondrial dysfunction induced by 50 µM DEX administration was
associated with decreased ATP level, mitochondrial respiration
rate, mitochondrial membrane potential and mitochondrial complex
II, III and V subunit protein levels. Notably, these pathological
changes were prevented by S-Rg3 treatment.
Mitochondria function as the main energy-producing
organelles in cells, with mitochondrial functional disturbances
leading to insufficient energy supply and activation of
intracellular signaling pathways that culminates in AMPK
activation, apoptosis and/or autophagy (50,51).
Notably, intracellular ATP deprivation triggers immediate
activation of AMPK, an important energy sensor, that subsequently
causes mitochondrial biogenesis (via PGC-1α phosphorylation) or
autophagy (via ULK1 phosphorylation) (52). As demonstrated in previous studies,
AMPK participates in muscle atrophy in two ways, by phosphorylating
FoxO3 and thereby directly controlling its nuclear translocation
(53) and by participating in
AMPK/FoxO3 pathway signaling triggered by mitochondrial fission
(54). Our previous work
demonstrated that S-Rg3 protects DEX-induced muscle atrophy
probably by promoting AKT/mTOR phosphorylation and inhibiting FoxO3
nuclear transcription (25). The
present study explored the role of mitochondrial function in muscle
atrophy. The results showed that S-Rg3 treatment of DEX-injured
myotubes decreased phosphorylation of AMPK and subsequently
prevented nuclear translocation of FoxO3, effectively alleviating
mitochondrial dysfunction and preventing muscle atrophy induced by
DEX in vitro.
The present study studied GC-induced atrophy using
an in vitro muscle myotube model to gain insights into
underlying mechanisms for S-Rg3 prevention of muscle atrophy. It
was discovered that S-Rg3 treatment restored mitochondrial function
and promoted recovery from DEX-induced muscle atrophy by inhibiting
the AMPK/FoxO3 pathway activated by DEX.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. U19A2013, U20A20402
and 81973813), the National Key Research and Development Program of
China (grant no. 2017YFC1702106), the Administration of Traditional
Chinese Medicine of Jilin Province (grant no. 2020171) and the
Science and Technology Development Plan of Changchun City (grant
no. 18YJ013).
Availability of data and materials
The data used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MW analyzed data and wrote the manuscript. RJ and JL
performed the experiments and conducted the analysis of data. XX
and GS performed the statistical analysis of the data. DZ and LS
designed the study and were involved in drafting the manuscript and
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ottens TH, Nijsten MW, Hofland J, Dieleman
JM, Hoekstra M, van Dijk D and van der Maaten JM: Effect of
high-dose dexamethasone on perioperative lactate levels and glucose
control: A randomized controlled trial. Crit Care. 19:412015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gong H, Liu L, Ni CX, Zhang Y, Su WJ, Lian
YJ, Peng W, Zhang JP and Jiang CL: Dexamethasone rapidly inhibits
glucose uptake via non-genomic mechanisms in contracting myotubes.
Arch Biochem Biophys. 603:102–109. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pasieka AM and Rafacho A: Impact of
glucocorticoid excess on glucose tolerance: Clinical and
preclinical evidence. Metabolites. 6:242016. View Article : Google Scholar
|
|
4
|
Engel AG: Electron microscopic
observations in thyrotoxic and corticosteroid-induced myopathies.
Mayo Clin Proc. 41:785–796. 1966.PubMed/NCBI
|
|
5
|
Oshima Y, Kuroda Y, Kunishige M, Matsumoto
T and Mitsui T: Oxidative stress-associated mitochondrial
dysfunction in corticosteroid-treated muscle cells. Muscle Nerve.
30:49–54. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pereira RM and Freire de Carvalho J:
Glucocorticoid-induced myopathy. Joint Bone Spine. 78:41–44. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qin W, Pan J, Wu Y, Bauman WA and Cardozo
C: Protection against dexamethasone-induced muscle atrophy is
related to modulation by testosterone of FOXO1 and PGC-1α. Biochem
Biophys Res Commun. 403:473–478. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kuo T, Harris CA and Wang JC: Metabolic
functions of glucocorticoid receptor in skeletal muscle. Mol Cell
Endocrinol. 380:79–88. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ochi A, Abe T, Nakao R, Yamamoto Y,
Kitahata K, Takagi M, Hirasaka K, Ohno A, Teshima-Kondo S, Taesik
G, et al: N-myristoylated ubiquitin ligase Cbl-b inhibitor prevents
on glucocorticoid-induced atrophy in mouse skeletal muscle. Arch
Biochem Biophys. 570:23–31. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jackman RW and Kandarian SC: The molecular
basis of skeletal muscle atrophy. Am J Physiol Cell Physiol.
287:C834–C843. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gonnella P, Alamdari N, Tizio S, Aversa Z,
Petkova V and Hasselgren PO: C/EBPβ regulates dexamethasone-induced
muscle cell atrophy and expression of atrogin-1 and MuRF1. J Cell
Biochem. 112:1737–1748. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shen S, Liao Q, Liu J, Pan R, Lee SM and
Lin L: Myricanol rescues dexamethasone-induced muscle dysfunction
via a sirtuin 1-dependent mechanism. J Cachexia Sarcopenia Muscle.
10:429–444. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Weng J, Wang YH, Li M, Zhang DY and Jiang
BG: GSK3β inhibitor promotes myelination and mitigates muscle
atrophy after peripheral nerve injury. Neural Regen Res.
13:324–330. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yoshioka Y, Kubota Y, Samukawa Y,
Yamashita Y and Ashida H: Glabridin inhibits dexamethasone-induced
muscle atrophy. Arch Biochem Biophys. 664:157–166. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang SF, Zhang Y, Li B and Chen N:
Physical inactivity induces the atrophy of skeletal muscle of rats
through activating AMPK/FoxO3 signal pathway. Eur Rev Med Pharmacol
Sci. 22:199–209. 2018.PubMed/NCBI
|
|
16
|
Hu R, Wang MQ, Liu LY, You HY, Wu XH, Liu
YY, Wang YJ, Lu L, Xiao W and Wei LB: Calycosin inhibited autophagy
and oxidative stress in chronic kidney disease skeletal muscle
atrophy by regulating AMPK/SKP2/CARM1 signalling pathway. J Cell
Mol Med. 24:11084–11099. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paggio A, Checchetto V, Campo A, Menabò R,
Di Marco G, Di Lisa F, Szabo I, Rizzuto R and De Stefani D:
Identification of an ATP-sensitive potassium channel in
mitochondria. Nature. 572:609–613. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Seok YM, Yoo JM, Nam Y, Kim J, Kim JS, Son
JH and Kim HJ: Mountain ginseng inhibits skeletal muscle atrophy by
decreasing muscle RING finger protein-1 and atrogin1 through
forkhead box O3 in L6 myotubes. J Ethnopharmacol. 270:1135572020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee MK, Choi JW, Choi YH and Nam TJ:
Pyropia yezoensis protein prevents dexamethasone-induced
myotube atrophy in C2C12 myotubes. Mar Drugs. 16:4972018.
View Article : Google Scholar
|
|
20
|
Ma YL, Sun YZ and Yang HH: Protective
effect of RenShen compound and DanHuang compound on muscle atrophy
in suspended rats. Space Med Med Eng (Beijing). 12:281–283.
1999.(In Chinese). PubMed/NCBI
|
|
21
|
Takamura Y, Nomura M, Uchiyama A and
Fujita S: Effects of aerobic exercise combined with panaxatriol
derived from ginseng on insulin resistance and skeletal muscle mass
in type 2 diabetic mice. J Nutr Sci Vitaminol (Tokyo). 63:339–348.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li F, Li X, Peng X, Sun L, Jia S, Wang P,
Ma S, Zhao H, Yu Q and Huo H: Ginsenoside Rg1 prevents
starvation-induced muscle protein degradation via regulation of
AKT/mTOR/FoxO signaling in C2C12 myotubes. Exp Ther Med.
14:1241–1247. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Go GY, Lee SJ, Jo A, Lee J, Seo DW, Kang
JS, Kim SK, Kim SN, Kim YK and Bae GU: Ginsenoside Rg1 from Panax
ginseng enhances myoblast differentiation and myotube growth. J
Ginseng Res. 41:608–614. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee SJ, Bae JH, Lee H, Lee H, Park J, Kang
JS and Bae GU: Ginsenoside Rg3 upregulates myotube formation and
mitochondrial function, thereby protecting myotube atrophy induced
by tumor necrosis factor-alpha. J Ethnopharmacol. 242:1120542019.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang M, Ren J, Chen X, Liu J, Xu X, Li X,
Zhao D and Sun L: 20(S)-ginsenoside Rg3 promotes myoblast
differentiation and protects against myotube atrophy via regulation
of the Akt/mTOR/FoxO3 pathway. Biochem Pharmacol. 180:1141452020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rovetta F, Stacchiotti A, Faggi F,
Catalani S, Apostoli P, Fanzani A and Aleo MF: Cobalt triggers
necrotic cell death and atrophy in skeletal C2C12 myotubes. Toxicol
Appl Pharmacol. 271:196–205. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang M, Chen X, Jin W, Xu X, Li X and Sun
L: Ginsenoside Rb3 exerts protective properties against cigarette
smoke extract-induced cell injury by inhibiting the p38 MAPK/NF-κB
and TGF-β1/VEGF pathways in fibroblasts and epithelial cells.
Biomed Pharmacother. 108:1751–1758. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Meng F, Su X, Li W and Zheng Y:
Ginsenoside Rb3 strengthens the hypoglycemic effect through AMPK
for inhibition of hepatic gluconeogenesis. Exp Ther Med.
13:2551–2557. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gwag T, Park K, Kim E, Son C, Park J,
Nikawa T and Choi I: Inhibition of C2C12 myotube atrophy by a novel
HSP70 inducer, celastrol, via activation of Akt1 and ERK1/2
pathways. Arch Biochem Biophys. 537:21–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu J, Peng Y, Wang X, Fan Y, Qin C, Shi
L, Tang Y, Cao K, Li H, Long J and Liu J: Mitochondrial dysfunction
launches dexamethasone-induced skeletal muscle atrophy via
AMPK/FOXO3 signaling. Mol Pharm. 13:73–84. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Will Y and Dykens J: Mitochondrial
toxicity assessment in industry-a decade of technology development
and insight. Expert Opin Drug Metab Toxicol. 10:1061–1067. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen WC, Hsieh SR, Chiu CH, Hsu BD and
Liou YM: Molecular identification for
epigallocatechin-3-gallate-mediated antioxidant intervention on the
H2O2-induced oxidative stress in H9c2 rat cardiomyoblasts. J Biomed
Sci. 21:562014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang Q, Lou T, Wang M, Xue L, Lu J, Zhang
H, Zhang Z, Wang H, Jing C, Zhao D, et al: Compound K inhibits
autophagy-mediated apoptosis induced by oxygen and glucose
deprivation/reperfusion via regulating AMPK-mTOR pathway in
neurons. Life Sci. 254:1177932020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Troncoso R, Paredes F, Parra V, Gatica D,
Vásquez-Trincado C, Quiroga C, Bravo-Sagua R, López-Crisosto C,
Rodriguez AE, Oyarzún AP, et al: Dexamethasone-induced autophagy
mediates muscle atrophy through mitochondrial clearance. Cell
Cycle. 13:2281–2295. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang Y, Chen K, Ren Q, Yi L, Zhu J, Zhang
Q and Mi M: Dihydromyricetin attenuates dexamethasone-induced
muscle atrophy by improving mitochondrial function via the PGC-1α
pathway. Cell Physiol Biochem. 49:758–779. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zheng WL, Wang BJ, Wang L, Shan YP, Zou H,
Song RL, Wang T, Gu JH, Yuan Y, Liu XZ, et al: ROS-mediated cell
cycle arrest and apoptosis induced by zearalenone in mouse sertoli
cells via ER stress and the ATP/AMPK pathway. Toxins (Basel).
10:242018. View Article : Google Scholar
|
|
39
|
Sandri M, Sandri C, Gilbert A, Skurk C,
Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH and Goldberg
AL: Foxo transcription factors induce the atrophy-related ubiquitin
ligase atrogin-1 and cause skeletal muscle atrophy. Cell.
117:399–412. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
DiMauro S, Garone C and Naini A: Metabolic
myopathies. Curr Rheumatol Rep. 12:386–393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kim JW, Ku SK, Han MH, Kim KY, Kim SG, Kim
GY, Hwang HJ, Kim BW, Kim CM and Choi YH: The administration of
Fructus Schisandrae attenuates dexamethasone-induced muscle
atrophy in mice. Int J Mol Med. 36:29–42. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shimizu N, Yoshikawa N, Ito N, Maruyama T,
Suzuki Y, Takeda S, Nakae J, Tagata Y, Nishitani S, Takehana K, et
al: Crosstalk between glucocorticoid receptor and nutritional
sensor mTOR in skeletal muscle. Cell Metab. 13:170–182. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jiang R, Wang M, Shi L, Zhou J, Ma R, Feng
K, Chen X, Xu X, Li X, Li T and Sun L: Panax ginseng total protein
facilitates recovery from dexamethasone-induced muscle atrophy
through the activation of glucose consumption in C2C12 myotubes.
Biomed Res Int. 2019:37196432019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xu D, Jiang Z, Sun Z, Wang L, Zhao G,
Hassan HM, Fan S, Zhou W, Han S, Zhang L and Wang T: Mitochondrial
dysfunction and inhibition of myoblast differentiation in mice with
high-fat-diet-induced pre-diabetes. J Cell Physiol. 234:7510–7523.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gosker HR, Engelen MP, van Mameren H, van
Dijk PJ, van der Vusse GJ, Wouters EF and Schols AM: Muscle fiber
type IIX atrophy is involved in the loss of fat-free mass in
chronic obstructive pulmonary disease. Am J Clin Nutr. 76:113–119.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Atherton PJ, Greenhaff PL, Phillips SM,
Bodine SC, Adams CM and Lang CH: Control of skeletal muscle atrophy
in response to disuse: Clinical/preclinical contentions and
fallacies of evidence. Am J Physiol Endocrinol Metab.
311:E594–E604. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Romanello V and Sandri M: Mitochondrial
biogenesis and fragmentation as regulators of protein degradation
in striated muscles. J Mol Cell Cardiol. 55:64–72. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen LE, Silver WP, Seaber AV, Korompilias
AV and Urbaniak JR: Effects of dexamethasone on the contractile
function of reperfused skeletal muscle. Microsurgery. 17:313–320.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu J, Peng Y, Feng Z, Shi W, Qu L, Li Y,
Liu J and Long J: Reloading functionally ameliorates disuse-induced
muscle atrophy by reversing mitochondrial dysfunction, and similar
benefits are gained by administering a combination of mitochondrial
nutrients. Free Radic Biol Med. 69:116–128. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu J, Deng K, Pan M, Liu G, Wu J, Yang M,
Huang D, Zhang W and Mai K: Dietary carbohydrates influence muscle
texture of olive flounder Paralichthys olivaceus through
impacting mitochondria function and metabolism of glycogen and
protein. Sci Rep. 10:218112020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jager S, Handschin C, St-Pierre J and
Spiegelman BM: AMP-activated protein kinase (AMPK) action in
skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl
Acad Sci USA. 104:12017–12022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Prieto I, Alarcón CR, García-Gómez R,
Berdún R, Urgel T, Portero M, Pamplona R, Martínez-Ruiz A,
Ruiz-Sanz JI, Ruiz-Larrea MB, et al: Metabolic adaptations in
spontaneously immortalized PGC-1α knock-out mouse embryonic
fibroblasts increase their oncogenic potential. Redox Biol.
29:1013962020. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Han DS, Yang WS and Kao TW: Dexamethasone
treatment at the myoblast stage enhanced C2C12 Myocyte
differentiation. Int J Med Sci. 14:434–443. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Romanello V, Guadagnin E, Gomes L, Roder
I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz
M, et al: Mitochondrial fission and remodelling contributes to
muscle atrophy. EMBO J. 29:1774–1785. 2010. View Article : Google Scholar : PubMed/NCBI
|