Introduction
Pulmonary fibrosis (PF) is a common, chronic and
incurable disease, which is characterized by lung tissue scarring
and the destruction of lung tissue structure (1). Despite the development of various
detection methods and clinical treatments, PF remains a major
public health problem that inevitably leads to a decline in lung
function, progressive respiratory failure and high mortality
(2,3). Increasing evidence suggests that a
possible mechanism for the pathogenesis of PF involves
epithelial-mesenchymal transition (EMT) (4,5), a
transdifferentiation process in which epithelial cells are
ultimately converted into motility mesenchymal cells with increased
collagen deposition and other extracellular matrix components
characteristic of alveolar epithelial cells (AECs). The main
mediator of EMT is TGFβ-1, which can upregulate the expression of
pro-fibrotic genes, such as vimentin, α-smooth muscle actin
(α-SMA; ACTA2) and collagen 1 and downregulate the
expression of epithelial cell-related genes, such as CDH1.
TGFβ-1-induced EMT serves a crucial role in fibrosis in various
organs (6) and AECs are among its
important target cells, which can convert into a mesenchymal
phenotype through TGFβ-1-induced EMT, thereby promoting PF.
MicroRNAs (miRNAs/miRs) are small, noncoding RNAs
that inhibit gene expression by binding to the 3′-untranslated
regions (3′-UTR) of the mRNA of their target genes. There is
increasing evidence that miRNAs serve critical roles in various
biological processes, including cell proliferation,
differentiation, apoptosis and regulation of the EMT process
(7–9). The miRNA miR-320a-3p is known to be
involved in the regulation of EMT and studies have shown that it
serves important roles in the regulation of the EMT process in
various tumors (10,11). However, whether miR-320a-3p can
regulate the EMT process in PF has yet to be investigated.
The present study used the Gene Expression Omnibus
(GEO) and ArrayExpress databases as relevant data sources to
analyze the differential expression of miR-320a-3p in PF and normal
lung tissues and then verified the relative expression levels of
miR-320a-3p in clinical samples. In addition, the regulatory
mechanism of miR-320a-3p mediating the attenuation of PF was
explored through experimental cellular studies.
Materials and methods
Data acquired from the GEO and
ArrayExpress databases
The relevant expression level data of miR-320a-3p in
PF tissues and normal tissue specimens were obtained from the GEO
(http://www.ncbi.nlm.nih.gov/gds/) and
the ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) databases. The
following key words were used as search terms: ‘lung fibrosis’,
‘idiopathic pulmonary fibrosis (IPF)’, ‘microRNA (miRNA)’,
‘miR320a’. The following criteria were strictly followed: i) Study
type: Expression profiling by microarray; ii) datasets were derived
from Homo sapiens; iii) datasets included interstitial lung
fibrosis tissues and normal tissues; iv) the datasets included
>5 samples in each group; and v) the expression data of miR320a
from the patients and control subjects were clearly defined and
provided. Potentially relevant datasets were further screened by
reviewing titles, summaries and overall design. Relevant full
articles and samples information were evaluated in order to
identify studies that met the eligibility criteria. The data of
eligible datasets and series were current to June 2020. Basic
information about enrolled datasets are listed in Table SI.
Collection of clinical samples
The PF tissue samples (n=12; sex ratio between male
and female was 1:2; age range, 39–78 years) were collected by
bronchoscopic lung cryobiopsy performed in the Department of
Respiratory Medicine of the First Affiliated Hospital of Chongqing
Medical University. Normal control samples (n=17; sex ratio between
male and female was 1:1.43; age range, 47–77 years) were obtained
from normal tissues adjacent to cancer tissues collected in the
Department of Cardiothoracic Surgery of the First Affiliated
Hospital of Chongqing Medical University. The recruitment dates for
the patients were between June and December 2020. The two sets of
samples were collected and frozen in liquid nitrogen for subsequent
RNA extraction and reverse transcription-quantitative (RT-q) PCR.
The collection and analysis procedures of all samples were approved
by the Ethics Research Institute of the First Affiliated Hospital
of Chongqing Medical University (approval no. 2020-147-2) and
written informed consent was obtained from all the participants.
Information on patients is listed in Table SII.
Cell culture and transfection
The adenocarcinomic human alveolar basal epithelial
cell line, A549, was provided by the Stem Cell Bank, Chinese
Academy of Science (Shanghai, China). Cells were cultured in
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.),
supplemented with 10% fetal bovine serum Premium (PAN-Biotech GmbH)
and 100 IU/ml penicillin and 100 µg/ml streptomycin (Gibco; Thermo
Fisher Scientific, Inc.) in a humidified incubator at 37°C with a
5% CO2 atmosphere. A549 cells were transfected with
miR-320a-3p mimic, signal transducer and STAT3 short interfering
(si)RNA or corresponding negative control (NC; Shanghai GenePharma
Co., Ltd.) using Lipofectamine® 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Following transfection for 24 h, the cells were
treated with or without TGF-β1 (PeproTech, Inc.) for 48 h and then
harvested for subsequent experiments. The si-NC and mimic-NC were
non-targeting sequences. The HBLV-h-STAT3-3×flag-mcherry-PURO
(HBLV-h-STAT3) and HBLV-mcherry-PURO (HBLV-NC) were constructed by
Hanbio Biotechnology Co., Ltd. A549 cells were transfected with
HBLV-h-STAT3 or HBLV-NC and puromycin was used to select cells with
functional transfection status (DIYIBio Inc.). The transfection
sequences are listed in Table
I.
 | Table I.Transfection sequences used in the
present study. |
Table I.
Transfection sequences used in the
present study.
| Name | Sequence
(5′-3′) |
|---|
|
hsa-miR-320a-3pmimics-F |
5′-AAAAGCUGGGUUGAGAGGGCGA-3′ |
|
hsa-miR-320a-3pmimics-R |
5′-GCCCUCUCAACCCAGCUUUUUU-3′ |
| Negative
control-F |
5′-UUGUACUACACAAAAGUACUG-3′ |
| Negative
control-R |
5′-GUACUUUUGUGUAGUACAAUU-3′ |
| si-STAT3-F |
5′-GCAACAGAUUGCCUGCAUUTT-3′ |
| si-STAT3-R |
5′-AAUGCAGGCAAUCUGUUGCTT-3′ |
| si-Negative
control-F |
5′-UUCUCCGAACGUGUCACGUTT-3′ |
| si-Negative
control-R |
5′-ACGUGACACGUUCGGAGAATT-3′ |
RT-qPCR
A549 cells (1×105 per well) were seeded
in the 6-well plate for total RNA extraction. Total RNA was
isolated from clinical samples and cultured cells using RNAiso Plus
(Takara Bio, Inc.) according to the manufacturer's instructions.
RNA was reverse transcribed using the PrimeScript RT Reagent kit
with genomic DNA (gDNA) Eraser (Takara Bio, Inc.). The gDNA
elimination reaction was performed for 2 min at 42°C. Reverse
transcription was conducted for 15 min at 37°C and 5 sec at 85°C.
The qPCR analysis was performed using the CFX96 real-time PCR
system (Bio-Rad Laboratories, Inc.) and the TB Green Premix EX Taq
II PCR kit (Takara Bio, Inc.). Amplification consisted of an
initial 30 sec incubation at 95°C, followed by 40 cycles of 5 sec
at 95°C and 30 sec at 60°C. Data were expressed as the relative
differences between the control and treated cells after
normalization to GAPDH expression. To determine the relative
expression levels of miRNAs, the U6 small nuclear RNA was used for
normalization. The relative expression levels of the mRNA and miRNA
was calculated using the 2−ΔΔCq method (12). Primers for miR-320a-3p and U6 were
purchased from GenePharma Co. Ltd., while those for GAPDH,
ACTA2, vimentin, CDH1 were purchased from Takara Bio, Inc. The
sequence of the primers used are listed in Table II. All experiments were repeated
three times.
| Table II.Primers used in the present
study. |
Table II.
Primers used in the present
study.
| Name | Sequence
(5′-3′) |
|---|
| GAPDH-F |
5′-CTTTGGTATCGTGGAAGGACTC-3′ |
| GAPDH-R |
5′-GTAGAGGCAGGGATGATGTTCT-3′ |
| ACTA2-F |
5′-CCCTTGAGAAGAGTTACGAGTTG-3′ |
| ACTA2-R |
5′-CATGATGCTGTTGTAGGTGGTT-3′ |
| CDH1-F |
5′-CGATTCAAAGTGGGCACAGATG-3′ |
| CDH1-R |
5′-GTAGGTGGAGTCCCAGGCGTAG-3′ |
|
vimentin-F |
5′-TCTGGATTCACTCCCTCTGGTT-3′ |
|
vimentin-R |
5′-ATCGTGATGCTGAGAAGTTTCGT-3′ |
|
hsa-miR-320a-3p-F |
5′-ATGAGAAAAAGCTGGGTTGAGA-3′ |
|
hsa-miR-320a-3p-R |
5′-TATGGTTTTGACGACTGTGTGAT-3′ |
| U6-F |
5′-CAGCACATATACTAAAATTGGAACG-3′ |
| U6-R |
5′-ACGAATTTGCGTGTCATCC-3′ |
Western blot analysis
The total protein content of the cells was extracted
by using radio immunoprecipitation assay (RIPA) lysis buffer
(Beyotime Institute of Biotechnology) which contained 1%
phosphatase inhibitor (Wuhan Boster Biological Technology, Ltd.)
and 1% phenylmethanesulfonyl fluoride (Beyotime Institute of
Biotechnology). Total protein concentration was measured using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). For western blotting, 20 µg of protein was loaded
into each lane of the 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis gels, followed by electrophoresis and protein
transfer to polyvinylidene fluoride membranes (EMD Millipore).
Following transfer, the membranes were blocked with QuickBlock™
blocking buffer for western blotting (Beyotime Institute of
Biotechnology) for 15 min at room temperature. The primary
antibodies rabbit anti-E-cadherin (1:10,000; cat. no. ab40772),
rabbit anti-vimentin (1:3,000; cat. no. ab92547), rabbit anti-α-SMA
(1:3,000; cat. no. ab32575) and rabbit anti-GAPDH (1:10,000; cat.
no. ab181602) were purchased from Abcam. The rabbit
anti-phosphorylated (p-)STAT3 (Tyr705, 1:2,000; cat. no. 9145),
mouse anti-STAT3 (1:1,000; cat. no. 9139), rabbit anti-p-SMAD3
(Ser423/425, 1:1,000; cat. no. 9520) and rabbit anti-SMAD3
(1:1,000; cat. no. 9523) were purchased from Cell Signaling
Technology, Inc. Immunoblots were probed with the corresponding
primary antibody at 4°C overnight and subsequently with the
appropriate secondary antibody (1:8,000; cat. no. ZB-2301; OriGene
Technologies, Inc.) for 1 h at room temperature. The protein bands
on the membranes were visualized with enhanced chemiluminescence
reagent (Vazyme Biotech Co., Ltd.) and images were captured using
an automatic Fusion FX Edge chemiluminescence image analysis system
(Vilber Lourmat Sa). Densitometric analysis of the blot images was
performed using the Fusion Capt software v.18.06 (Vilber Lourmat
Sa). GAPDH was used as an internal normalizer.
Immunofluorescence staining and
fluorescence microscopy
Cells (5×104 per well) were seeded and
grown in 6-well plates with cell culture silicon slides. Following
treatment, the cells were fixed in 4% paraformaldehyde for 20 min
at room temperature, then permeabilized with 0.2% Triton X-100 in
PBS for 20 min at room temperature (rinsed three times with PBS
between each step). Non-specific binding sites were blocked with 3%
BSA (Wuhan Servicebio Technology Co., Ltd.) for 30 min at room
temperature. The cells were then incubated overnight at 4°C with
the corresponding primary antibody at a 1:200 dilution. Afterwards,
the cells were incubated with the appropriate
fluorescein-conjugated secondary antibody for 1 h in the dark at
37°C. Primary antibodies and secondary antibodies were purchased
from Wuhan Servicebio Technology Co., Ltd. The fluorescent stain
4′,6-diamidino-2-phenylindole (DAPI) was used to stain cell nuclei
for 10 min in the dark at room temperature before image
acquisition. The images were acquired using a fluorescence
microscope (Nikon Corporation) with ×40 magnification. The red
fluorescence indicated positive antibody expression and the blue
fluorescence indicated nuclear DAPI staining. Mean gray value
analysis was performed using the ImageJ software v1.53e (National
Institutes of Health).
Firefly and Renilla dual-luciferase
assay
The target genes of miR-320a-3p were identified
using the miRanda (http://www.microrna.org/microrna/home.do), miRWalk
(http://mirwalk.umm.uni-heidelberg.de/) and PITA
(http://genie.weizmann.ac.il/pubs/mir07/mir07_data.html)
algorithms. The dual-luciferase reporter system was used to
validate the target relationship between miR-320a-3p and
STAT3. Human STAT3 3′-UTR containing a 3′-UTR with
mutated (mu) sequences complementary to the seed sequence of
miR-320a-3p was inserted into the pSI-Check2 vector (Promega
Corporation). The wild type (wt) STAT3 3′-UTR sequence was also
inserted into the pSI-Check2 vector. Luciferase reporter plasmid
was constructed and provided by Hanbio Biotechnology Co., Ltd. The
A549 cells were grown in 96-well plates and then co-transfected
with miR-320a-3p mimic or NC and pSI-Check2-STAT3-mu-3′-UTR or wt
pSI-Check2-STAT3-3′-UTR using transfection reagent LipoFiter
(Hanbio Biotechnology Co., Ltd.). Then, 6 h after transfection, the
medium was replaced with fresh medium and after 48 h, the
dual-luciferase reporter system (Promega Corporation) was used to
measure luciferase activity. The normalized luciferase activity was
expressed as the ratio of Renilla luciferase activity to
firefly luciferase activity (Rluc/fluc).
Statistical analysis
Data were expressed as the mean ± standard deviation
and SPSS v25 software (IBM Corp.) was used for data analysis.
Independent-samples t-test was used for the analysis of data
between two groups. Differences among three or more groups were
compared using one-way analysis of variance followed by Tukey's
post hoc test. GraphPad Prism v8.0 (GraphPad Software, Inc.) was
used to draw the box plot, symbols and lines plot and column bar
graph. P<0.05 was considered to indicate a statistically
significant difference.
Results
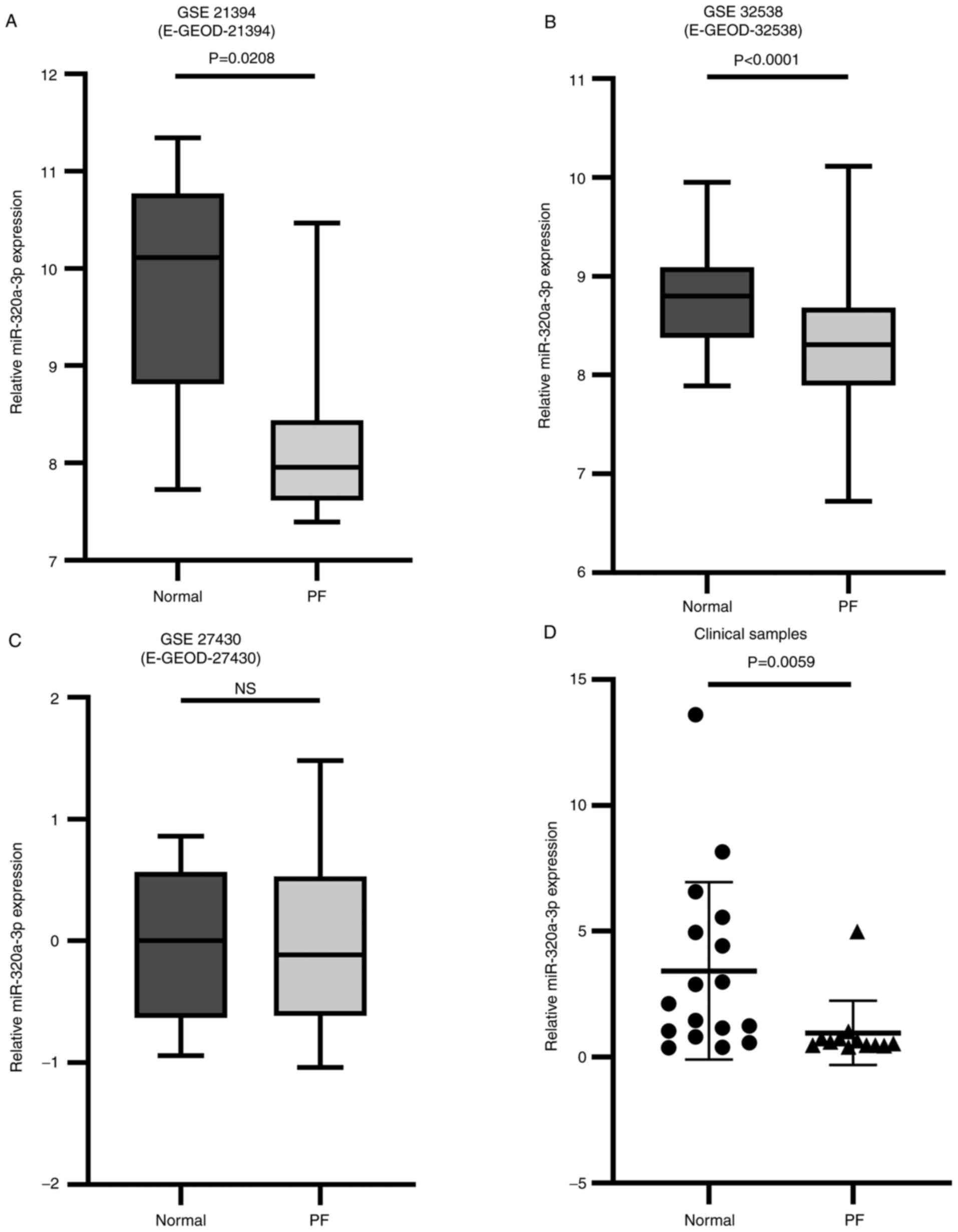
Expression of miR-320a-3p in PF
A total of 3 datasets were selected for the present
study, according to the inclusion criteria (GSE21394/E-GEOD-21394,
GSE32538/E-GEOD-32538 and GSE27430/E-GEOD-27430). With the
combination of datasets from the GEO and ArrayExpress database, a
total of 174 PF samples were included. Sample sizes ranged from
13–142 subjects and the pathological types include the usual
interstitial pneumonia/idiopathic pulmonary fibrosis (UIP/IPF),
cryptogenic organic pneumonia (COP), desquamative interstitial
pneumonia (DIP), nonspecific interstitial pneumonia (NSIP),
connective tissue disease-associated interstitial lung disease
(CTD-ILD) and respiratory bronchiolitis-associated interstitial
lung disease (RB-ILD). It was found that two results in the
analysis of three datasets showed that the relative expression
levels of miR-320a-3p were significantly lower in PF tissue samples
compared with normal tissue samples (P<0.05, P<0.0001;
Fig. 1A and B). The remaining
analyses showed that the relative expression levels of miR-320a-3p
decreased in the PF group, but the difference was not statistically
significant (Fig. 1C). Based on the
results of the analysis of the datasets, clinical tissue samples of
PF patients (n=12) and normal control subjects (n=17) were
collected. The pathological types of samples of PF included
UIP/IPF, NSIP, COP and CTD-ILD. The tissue of the control group was
confirmed to be normal tissue by pathology. The data showed that
the relative expression levels of miR-320a-3p were markedly
decreased in PF tissue samples compared with normal tissue samples
(Fig. 1D).
miR-320a-3p is decreased in
TGF-β1-stimulated A549 cells and elevates expression levels of
miR-320a-3p inhibited EMT
The A549 cell lines is widely used as a model system
to study the mechanisms of the EMT process in PF. In the present
study, qPCR analysis was first used to determine the relative
expression levels of miR-320a-3p in A549 cells stimulated for 48 h
with TGF-β1 at different concentrations (0, 1, 3, 5 and 10 ng/ml).
It was found that TGF-β1 significantly reduced the miR-320a-3p
expression levels compared with the control group and the optimal
inhibitory effect was obtained at a concentration of 10 ng/ml
(Fig. 2A). Subsequently, the levels
of miR-320a-3p in A549 cells stimulated with 10 ng/ml of TGF-β1 was
determined at various time points (0, 24 and 48 h) and it was found
that the optimal inhibition was achieved at 48 h (Fig. 2B). After transfecting A549 cells
with miR-320a-3p mimic and NC, followed by treatment with TGF-β1
(10 ng/ml) for 48 h, qPCR analysis was used to determine the
expression levels of mRNA of CDH1, vimentin and
ACTA2. The results showed that compared with the TGF-β1
treatment group, miR-320a-3p mimic treatment increased the
expression of mRNA of CDH1 and significantly decreased the
expression of mRNA of vimentin and ACTA2. Western
blot analysis and immunofluorescence staining was also used to
examine the expression of the epithelial phenotype marker
E-cadherin and the mesenchymal phenotype markers vimentin and
α-SMA. The results showed that compared with the corresponding
control group, TGF-β1-induced EMT increased the expression of
vimentin and α-SMA and significantly reduced the expression of
E-cadherin. miR-320a-3p mimic treatment attenuated the above
effects, that is, it restored the expression of E-cadherin and
reduced the expression of vimentin and α-SMA (Fig. 2C-E). These findings indicated that
TGF-β1 can induce EMT changes in A549 cells and miR-320a-3p mimic
can inhibit those EMT changes. The transfection efficiency of
miR-320a-3p mimic is shown in Fig.
2F.
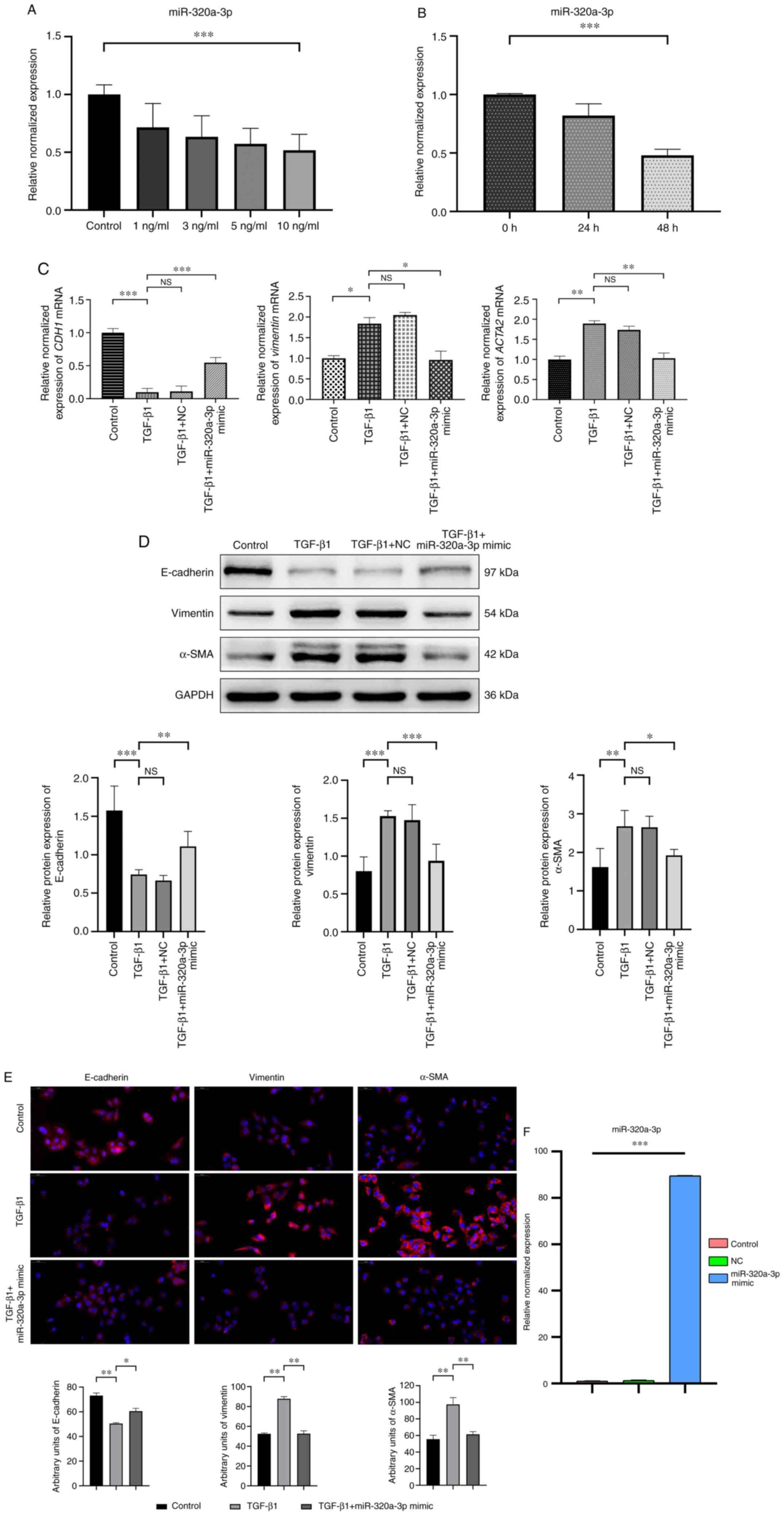 | Figure 2.Expression levels of miR-320a-3p in
A549 cells and the effects of miR-320a-3p on EMT in
TGF-β1-stimulated A549 cells. (A) A549 cells were treated with
different concentrations (0, 1, 3, 5 and 10 ng/ml) of TGF-β1 for 48
h. (B) A549 cells were treated with 10 ng/ml TGF-β1 for 0, 24 and
48 h. The relative expression levels of miR-320a-3p in A549 cells
measured by RT-qPCR. (C) A549 cells were treated with or without
TGF-β1 (10 ng/ml) for 48 h after transfection with miR-320a-3p
mimic or NC. The relative expression levels of CDH1,
vimentin and ACTA2 were determined by RT-qPCR. (D) A549
cells were treated with or without TGF-β1 (10 ng/ml) for 48 h
following transfection with miR-320a-3p mimic or NC. To confirm the
occurrence of EMT, the expression of E-cadherin, vimentin and α-SMA
were determined by western blot analysis. (E) Immunofluorescence
staining showed that the levels of vimentin and α-SMA were reduced
in miR-320a-3p mimic treated cells compared with those in cells
treated with TGF-β1, while levels of E-cadherin were increased.
E-cadherin, vimentin and α-SMA (red); DAPI-counterstained nuclei
(blue). Magnification, ×40, scale bar=50 µm. (F) The transfection
efficiency of miR-320a-3p mimic was determined by RT-qPCR. Error
bars represent the standard deviation of three experiments.
*P<0.05, **P<0.01, ***P<0.001. miR, microRNA; EMT,
epithelial-mesenchymal transition; NC, negative controls; α-SMA,
α-smooth muscle actin; NS, no significant difference; RT-qPCR,
reverse transcription-quantitative PCR. |
STAT3 is essential for the modulation
of TGF-β1-induced EMT in A549 cells by miR-320a-3p
To further investigate the mechanism, several
bioinformatics analyses were conducted and the results revealed
that STAT3 is a potential target of miR-320a-3p. A dual-luciferase
reporter assay was then conducted and revealed that the miR-320a-3p
mimic significantly inhibited the luciferase activity in the wt
pSI-Check2-STAT3-3′-UTR group compared with the mimic-NC group
(P<0.01; Fig. 3A and B).
However, the miR-320a-3p mimic did not influence the luciferase
activity of pSI-Check2-STAT3-mut-3′-UTR. Western blot analysis also
showed that miR-320a-3P inhibited the expression of both STAT3 and
p-STAT3 in TGF-β1-stimulated A549 cells, but not the ratio of
p-STAT3 and STAT3 (Fig. 3C),
indicating that miR-320a-3p directly regulated STAT3 expression.
STAT3 is a member of a family of cytokine-responsive transcription
factors and has been identified as being hyperphosphorylated during
EMT (13). Accordingly, it was
investigated whether the expression of STAT3 is involved in
TGF-β1-induced EMT in A549 cells. Therefore, A549 cells were
transfected with STAT3 siRNA and NC siRNA and then treated them
with TGF-β1 (10 ng/ml) for 48 h. The results showed that, compared
with the control group, STAT3 siRNA reduced the expression of
vimentin and α-SMA, while the expression of E-cadherin was
significantly increased (Fig. 3D).
To further confirm that miR-320a-3p mimic inhibited TGF-β1-induced
EMT in A549 cells through a STAT3-dependent mechanism, miR-320a-3p
mimic was co-transfected with HBLV-h-STAT3 or HBLV-NC into A549
cells. Compared with mock-transfected cells, HBLV-h-STAT3
significantly increased the expression of STAT3. In addition, the
upregulation of STAT3 inhibited the effect of the miR-320a-3p mimic
on the expression of E-cadherin and significantly increased the
expression of vimentin, α-SMA and p-STAT3 (Fig. 3E). In summary, the above data
indicated that miR-320a-3p mimic can attenuate TGF-β1-induced EMT
in A549 cells by targeting STAT3 and p-STAT3. The transfection
efficiency of si-STAT3 and HBLV-h-STAT3 are shown in Fig. 3F and G.
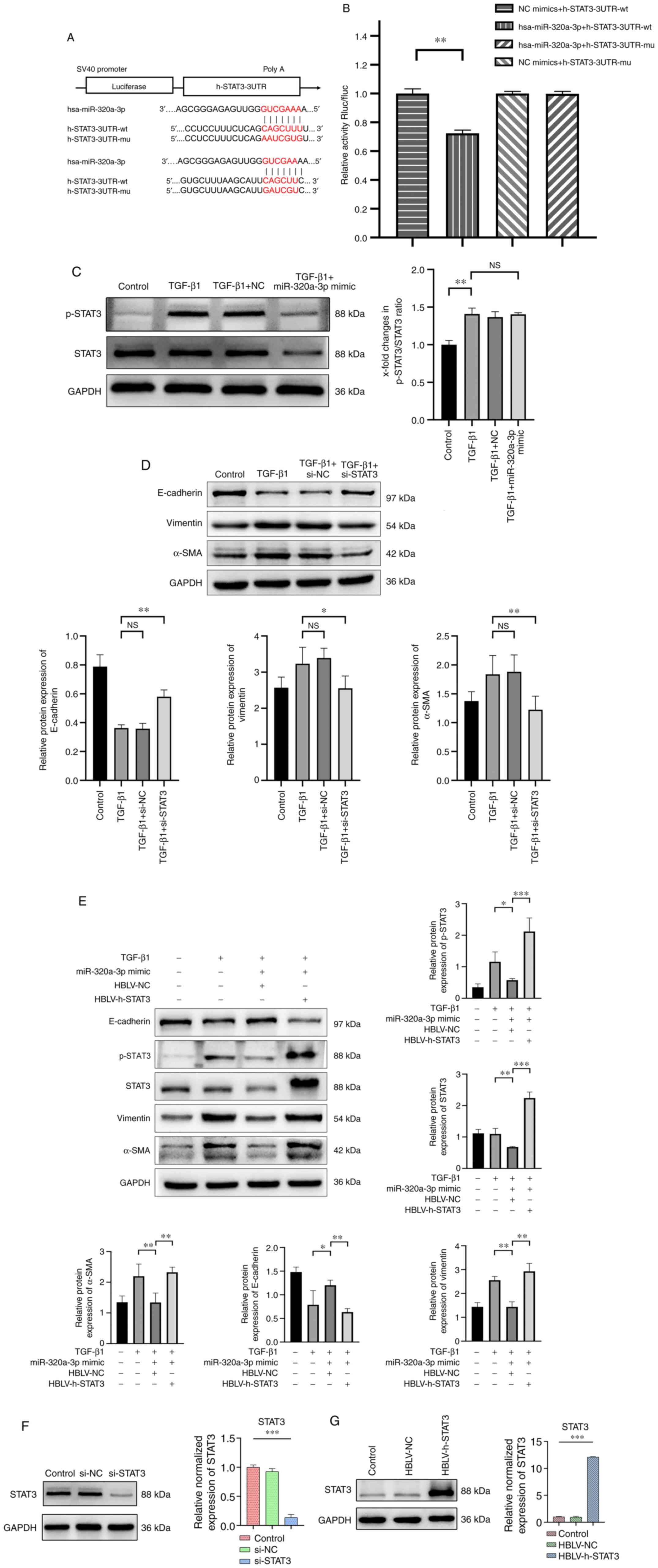 | Figure 3.STAT3 is a target gene of
miR-320a-3p and effects of STAT3 on EMT in A549 cells induced by
TGF-β1. Dual-luciferase reporter assay. (A) The STAT3 3′-UTR region
containing the wild-type (wt) or mutant (mu) binding site for
miR-320a-3p. (B) Relative Rluc/fluc activity of cells
co-transfected with either wt pSI-Check2-STAT3-3′-UTR or
pSI-Check2-STAT3-mut-3′-UTR and miR-320a-3p mimic or corresponding
NC. (C) A549 cells were treated with or without TGF-β1 (10 ng/ml)
for 48 h after transfection with miR-320a-3p mimic or NC. The
expression of p-STAT3, STAT3 were determined by western blot
analysis and their ratio were analyzed. (D) A549 cells were treated
with or without TGF-β1 (10 ng/ml) for 48 h after transfection with
STAT3 siRNA or NC siRNA. To confirm the effects of STAT3 on the EMT
in A549 cells, the expression of E-cadherin, vimentin and α-SMA
were determined by western blot analysis. (E) A549 cells were
treated with or without TGF-β1 (10 ng/ml) for 48 h after
co-transfection with miR-320a-3p mimic and HBLV-h-STAT3 or HBLV-NC.
The expression of E-cadherin, vimentin, α-SMA, p-STAT3 and STAT3
were determined by western blot analysis. The transfection
efficiency of (F) si-STAT3 and (G) HBLV-h-STAT3 were determined by
western blot analysis. Error bars represent the standard deviation
of three experiments. *P<0.05, **P<0.01, ***P<0.001. miR,
microRNA; EMT, epithelial-mesenchymal transition; UTR; wt,
wild-type; mu, mutant; Rluc/fluc, Renilla luciferase/firefly
luciferase; NC, negative controls; p-, phosphorylated; si, short
interfering; α-SMA, α-smooth muscle actin; NS, no significant
difference. |
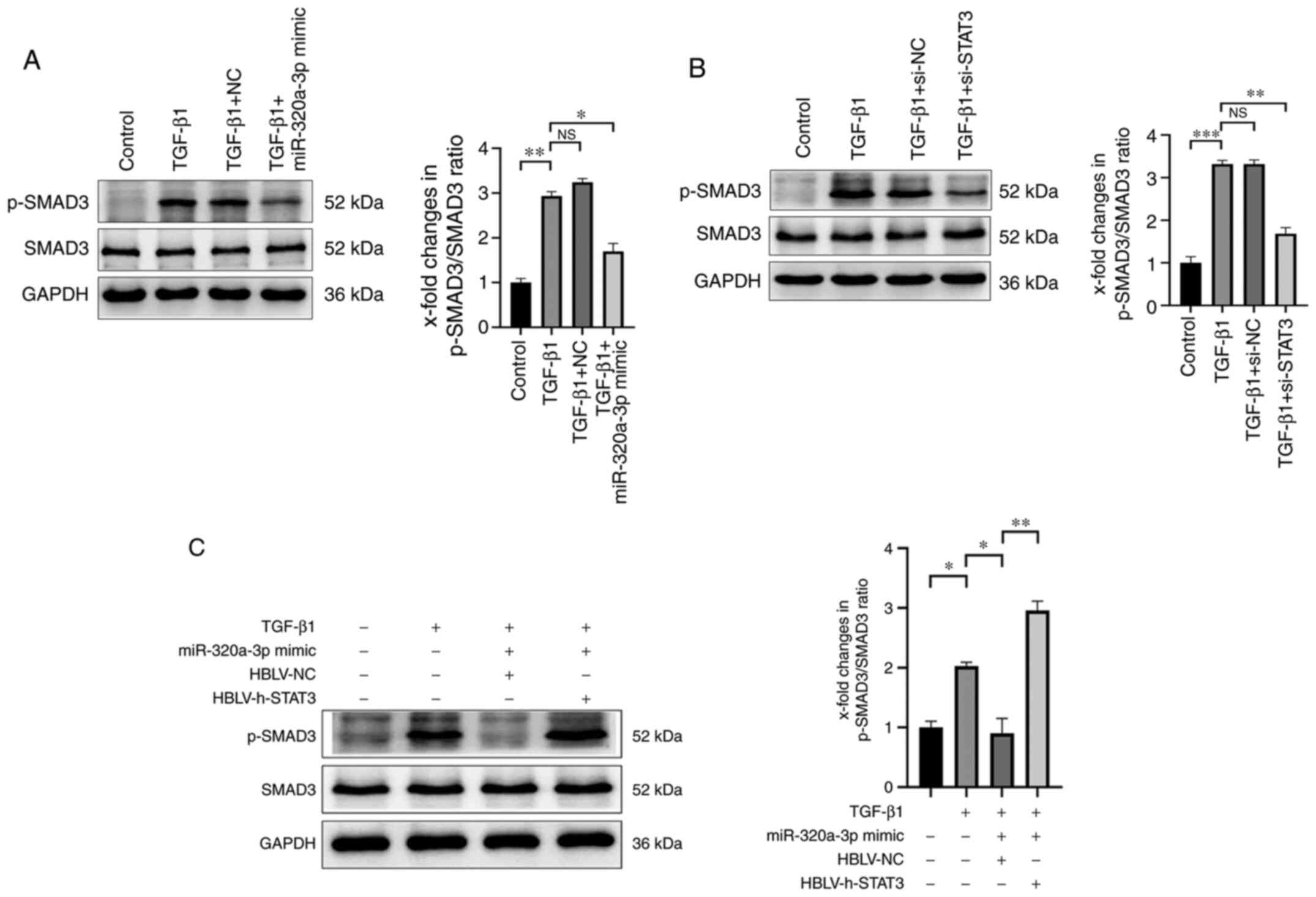
miR-320a-3p negatively regulates the
phosphorylation of SMAD3
Following transfection of A549 cells with
miR-320a-3p mimic, the expression of p-SMAD3 induced by TGF-β1 was
significantly reduced, while total SMAD3 was unaffected (Fig. 4A). Therefore, it was investigated
whether the inhibitory effect of miR-320a-3p on the phosphorylation
of SMAD3 was mediated by STAT3. To this end, the A549 cells were
first transfected with STAT3 siRNA and it was confirmed that
knocking down STAT3 led to reduced expression of p-SMAD3 induced by
TGF-β1 (Fig. 4B). Then,
transfecting A549 cells with HBLV-h-STAT3 revealed that
overexpression of STAT3 can significantly increase the expression
of p-SMAD3 induced by TGF-β1 (Fig.
4C). Taken together, these findings suggested that miR-320a-3p
inhibited TGF-β1-induced EMT in A549 cells partly by modulating the
expression of STAT3 and p-SMAD3.
Discussion
EMT of epithelial cells serves a functional role in
the development and progression of PF. Increasing evidence has
shown that miRNAs can regulate PF (14–16).
The present study first undertook to mine the expression data of
miR-320a-3p in PF tissue samples by analyzing the datasets in the
GEO and ArrayExpress databases. Although there was a considerable
amount of high-throughput data in these databases, only three
datasets that met the inclusion criteria were found. As it was
implausible that the three datasets could produce an effective
meta-analysis, only the statistical results of each data set
analysis are listed. Of these, two results revealed that the
relative expression levels of miR-320a-3p in PF tissue samples were
significantly reduced compared with those in normal tissue samples.
The results from the remaining analysis showed that the relative
expression levels of miR-320a-3p were not statistically different
between PF and normal tissue samples. Accordingly, the present
study undertook to determine the relative expression levels of
miR-320a-3p in clinical PF tissue samples. The pathological types
of the clinical samples in the present study included UIP/IPF,
NSIP, COP and CTD-ILD and the results confirmed that the relative
expression levels of miR-320a-3p were markedly decreased in PF
tissues sample compared with those in normal tissue samples,
suggesting that miR-320a-3p might be have a regulatory role in EMT
changes during PF.
A549 cells are adenocarcinomic human type II
alveolar epithelial cells. After 3 days of in vitro culture,
primary type II alveolar epithelial cells will be transformed into
type I alveolar epithelial cells. For this reason, A549 cells have
become a standard cell model to study the process of EMT in PF
in vitro (17,18). The role served by miR-320a-3p in the
EMT process in PF was confirmed by qPCR analysis of the expression
of miR-320a-3p in A549 cells treated with or without TGF-β1, which
revealed that treatment with TGF-β1 decreased the relative
expression levels of miR-320a-3p in a time- and
concentration-dependent manner. Based on the aforementioned
experimental results, 10 ng/ml for 48 h was chosen as the optimal
TGF-β1 treatment dose for subsequent experiments. Western blot
analysis and immunofluorescence staining of miR-320a-3p
mimic-transfected A549 cells stimulated with TGF-β1 showed that
overexpression of miR-320a-3p restored the epithelial phenotype of
A549 cells. suggesting a suppressive role for miR-320a-3p in PF
progression.
STAT3, a member of the STAT family of protein, is a
cytoplasmic transcription factor that can be activated by multiple
cytokines. The activation of STAT3 regulates a variety of
biological functions, including apoptosis, migration, proliferation
and differentiation (19,20). The role of STAT3 in fibrosis remains
to be elucidated. According to different experimental subjects,
related studies have produced conflicting results (21–23).
Regarding PF, the majority of studies reveal elevated levels of
p-STAT3 in patients with PF (24–26).
Evidence supports a role for STAT3 activation in PF. Zehender et
al (25) reports that STAT3
contributes to induces the transformation of fibroblasts into
myofibroblasts, leading to abnormal accumulation of extracellular
matrix (ECM). Pedroza et al (20) demonstrate that the STAT3 signaling
pathway is aberrantly activated in lung biopsies from patients with
IPF and in lung tissue from PF animal models. Considering the
importance of STAT3 in the development of PF, STAT3 would be a
potential target for intervention of the EMT process in PF. The
present study used bioinformatics tools and dual-luciferase assay
to show that miR-320a-3p directly regulated STAT3. The expression
of p-STAT-3 was shown to be elevated in TGF-β1-treated A549 cells
and subsequent experiments confirmed that overexpression of
miR-320a-3p can significantly inhibit such high expression of STAT3
and p-STAT3 levels. The present study found that the ratio of
p-STAT3 and STAT3 did not decrease in the TGF-β1 + miR-320a-3p
mimic group compared with the TGF-β1 treatment group. This may be
because the reduction of p-STAT3 follows the total STAT3 and
miR-320a-3p does not affect the phosphorylation process of STAT3.
Furthermore, western blot analysis demonstrated that knocking down
STAT3 could suppress the expression of multiple EMT markers in
TGF-β1-treated A549 cells, whereas overexpression of STAT3 could
aggravate the above-mentioned EMT performance. These results
indicated that miR-320a-3p reduced the expression of p-STAT3 by
binding to STAT3, thereby inhibiting the EMT process in PF.
It also illustrated the role served by STAT3 in the regulation of
the expression of EMT-related genes in A549 cells stimulated by
TGF-β1.
TGF-β is a multifunctional cytokine key mediator in
the initiation of tissue repair, especially in the kidney, liver
and lung, belonging to the transforming growth factor superfamily
that includes three isoforms (TGF-β1-3) and other cytokines
(27). Among the TGF-β isoforms,
TGF-β1 is the one most associated with PF. Elevated levels of
TGF-β1 are reported in the bleomycin (BLM)-induced PF rat model as
well as in patients with IPF (28,29).
Differentiation of fibroblasts and synthesis of ECM proteins caused
by overproduction of TGF-β1 contribute to the pathogenesis of PF
(30). The signal transduction of
the TGF-β family is mediated by TGFβRII, which recruits TGFβRI and
activates various signaling pathways, including pathways mediated
by Smad signaling proteins (31).
Smads act as intracellular effectors in response to TGF-β and once
inside the nucleus, Smad complexes bind regulatory elements and
induce the transcription of key genes associated with EMT (31). In addition, activation by Smads
promotes fibroblast proliferation, differentiation and ECM
remodeling (32). Crosstalk between
the Smad and STAT3 pathways has been proposed. A cooperative role
of STAT3 and TGF-β1/SMAD3 has been reported in hepatic fibrosis
(33), while in HaCaT and HepG2
cells a physical direct interaction between STAT3 and SMAD3
attenuating the TGF-β1-induced response has been suggested
(34). These results varied
depending on the cell type and disease model studied and, although
conflicting, indicate crosstalk between TGF-β1 and STAT3 signaling
pathways. The present study observed that miR-320a-3p mimic and
STAT3 siRNA could reduce the phosphorylation of SMAD3 induced by
TGF-β1 in A549 cells. By contrast, overexpression of STAT3 could
dramatically increase the levels of p-SMAD3, implying that STAT3
might induce SMAD3 activation in the EMT process of type II
alveolar epithelial cells.
The present study demonstrated that the expression
levels of miR-320a-3p were decreased in PF tissue samples compared
with normal tissue samples. Via stimulation by TGF-β1, A549 cells
exhibited the EMT phenotype and the levels of miR-320a-3p in A549
cells decreased in a time- and concentration-dependent manner.
TGF-β1 can induce EMT in A549 cells, as manifested by a decrease in
E-cadherin and an increase in vimentin and α-SMA. STAT3 was
activated in TGF-β1-treated A549 cells and the phosphorylation of
STAT3 served an important role in the development of the EMT
process. The present study also investigated the underlying
molecular mechanisms by which miR-320a-3p attenuated EMT in PF. The
phosphorylation of STAT3 and SMAD3 proteins was generally high in
A549 cells treated with TGF-β1 and their co-expression was
positively associated with TGF-β1. Higher p-STAT3 levels were
related to more prominent EMT process. Overexpression of
miR-320a-3p could attenuate the development of the EMT process in
PF by binding STAT3. The mechanism may be partly mediated through
crosstalk between the SMAD3 and STAT3 signaling. These results not
only have important implications for our understanding of STAT3
signaling in the development of PF but also for the potential role
of miR-320a-3p as a therapeutic target in PF. Overall, miR-320a-3p
had an effect that reduced the expression levels of phosphorylation
of STAT3, decreases p-SMAD3 protein levels and inhibited the
TGF-β1-induced EMT process in type II AECs. The design and main
results of the present study are shown in a schematic diagram
(Fig. 5).
Finally, the following limitations should be
considered in the present study: i) Due to various factors, the
in vivo experiment was not completed. This part of the
content served a very important role in supporting the whole
theory. Therefore, in the future, we will further explore the
effects of miR-320a-3p in PF animal models; ii) the present study
was limited to the effect of miR-320a-3p on EMT in PF and its
corresponding effect in the STAT3/SMAD3 signaling pathway. The
present study failed to cover other related factors, such as long
non-coding RNA, other genetic data or other signaling pathways.
This will also be the focus of future research.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by National Major Science
and Technology Projects of China (grant no. 2018ZX10302302003) and
National Natural Science Foundation of China (grant nos. 81601856
and 81570010).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The datasets generated and/or analyzed during the current
study are available in the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/gds) and
ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) repository.
Authors' contributions
XW, GH, YL and SG conceived and designed the
project. XW and JW acquired the data and wrote the paper. XW, JW
and GH analyzed interpreted the data. XW, JW and GH were
responsible for confirming the authenticity of the data in the
present study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The collection and analysis of all samples were
approved by the Ethics Research Institute of the First Affiliated
Hospital of Chongqing Medical University (approval no. 2020-147-2)
and written informed consent was obtained from all the
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kim MH, Jung SY, Song KH, Park JI, Ahn J,
Kim EH, Park JK, Hwang SG, Woo HJ and Song JY: A new FGFR inhibitor
disrupts the TGF-β1-induced fibrotic process. J Cell Mol Med.
24:830–840. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barratt SL, Creamer A, Hayton C and
Chaudhuri N: Idiopathic pulmonary fibrosis (IPF): An overview. J
Clin Med. 7:2012018. View Article : Google Scholar
|
|
3
|
Selman M, King TE and Pardo A: Idiopathic
pulmonary fibrosis: Prevailing and evolving hypotheses about its
pathogenesis and implications for therapy. Ann Internal Med.
134:136–151. 2001. View Article : Google Scholar
|
|
4
|
Kim KK, Kugler MC, Wolters PJ, Robillard
L, Galvez MG, Brumwell AN, Sheppard D and Chapman HA: Alveolar
epithelial cell mesenchymal transition develops in vivo during
pulmonary fibrosis and is regulated by the extracellular matrix.
Proc Natl Acad Sci USA. 103:13180–13185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Willis BC, Liebler JM, Luby-Phelps K,
Nicholson AG, Crandall ED, du Bois RM and Borok Z: Induction of
epithelial-mesenchymal transition in alveolar epithelial cells by
transforming growth factor-beta1: Potential role in idiopathic
pulmonary fibrosis. Am J Pathol. 166:1321–1332. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pardali E, Sanchez-Duffhues G,
Gomez-Puerto MC and Ten Dijke P: TGF-β-induced
endothelial-mesenchymal transition in fibrotic diseases. Int J Mol
Sci. 18:21572017. View Article : Google Scholar
|
|
7
|
Aquino-Jarquin G: Emerging role of
CRISPR/Cas9 technology for MicroRNAs editing in cancer research.
Cancer Res. 77:6812–6817. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gai YP, Zhao HN, Zhao YN, Zhu BS, Yuan SS,
Li S, Guo FY and Ji XL: MiRNA-seq-based profiles of miRNAs in
mulberry phloem sap provide insight into the pathogenic mechanisms
of mulberry yellow dwarf disease. Sci Rep. 8:8122018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mittal V: Epithelial mesenchymal
transition in tumor metastasis. Ann Rev Pathol. 13:395–412. 2018.
View Article : Google Scholar
|
|
10
|
Aljagthmi AA, Hill NT, Cooke M, Kazanietz
MG, Abba MC, Long W and Kadakia MP: ΔNp63α suppresses cells
invasion by downregulating PKCγ/Rac1 signaling through miR-320a.
Cell Death Dis. 10:6802019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang HH, Li R, Li YJ, Yu XX, Sun QN, Li
AY and Kong Y: eIF4E-related miR-320a and miR-340-5p inhibit
endometrial carcinoma cell metastatic capability by preventing
TGF-β1-induced epithelial-mesenchymal transition. Oncol Rep.
43:447–460. 2020.PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Waters DW, Blokland KEC, Pathinayake PS,
Burgess JK, Mutsaers SE, Prele CM, Schuliga M, Grainge CL and
Knight DA: Fibroblast senescence in the pathology of idiopathic
pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol.
315:L162–l172. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Huang C, Xiao X, Yang Y, Mishra A, Liang
Y, Zeng X, Yang X, Xu D, Blackburn MR, Henke CA and Liu L:
MicroRNA-101 attenuates pulmonary fibrosis by inhibiting fibroblast
proliferation and activation. J Biol Chem. 292:16420–16439. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Liang Y, Luo J, Nie J, Yin H, Chen
Q, Dong J, Zhu J, Xia J and Shuai W: XIST/miR-139 axis regulates
bleomycin (BLM)-induced extracellular matrix (ECM) and pulmonary
fibrosis through β-catenin. Oncotarget. 8:65359–65369. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang ZC, Qu ZH, Yi MJ, Shan YC, Ran N, Xu
L and Liu XJ: MiR-448-5p inhibits TGF-β1-induced
epithelial-mesenchymal transition and pulmonary fibrosis by
targeting Six1 in asthma. J Cell Physiol. 234:8804–8814. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Foster KA, Oster CG, Mayer MM, Avery ML
and Audus KL: Characterization of the A549 cell line as a type II
pulmonary epithelial cell model for drug metabolism. Exp Cell Res.
243:359–366. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lieber M, Smith B, Szakal A, Nelson-Rees W
and Todaro G: A continuous tumor-cell line from a human lung
carcinoma with properties of type II alveolar epithelial cells. Int
J Cancer. 17:62–70. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Levy DE and Darnell JE Jr: Stats:
Transcriptional control and biological impact. Nat Rev Mol Cell
Biol. 3:651–662. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pedroza M, Le TT, Lewis K,
Karmouty-Quintana H, To S, George AT, Blackburn MR, Tweardy DJ and
Agarwal SK: STAT-3 contributes to pulmonary fibrosis through
epithelial injury and fibroblast-myofibroblast differentiation.
FASEB J. 30:129–140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dai B, Cui M, Zhu M, Su WL, Qiu MC and
Zhang H: STAT1/3 and ERK1/2 synergistically regulate cardiac
fibrosis induced by high glucose. Cell Physiol Biochem. 32:960–971.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ogata H, Chinen T, Yoshida T, Kinjyo I,
Takaesu G, Shiraishi H, Iida M, Kobayashi T and Yoshimura A: Loss
of SOCS3 in the liver promotes fibrosis by enhancing STAT3-mediated
TGF-beta1 production. Oncogene. 25:2520–2530. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shigekawa M, Takehara T, Kodama T, Hikita
H, Shimizu S, Li W, Miyagi T, Hosui A, Tatsumi T, Ishida H, et al:
Involvement of STAT3-regulated hepatic soluble factors in
attenuation of stellate cell activity and liver fibrogenesis in
mice. Biochem Biophys Res Commun. 406:614–620. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Le TT, Karmouty-Quintana H, Melicoff E, Le
TT, Weng T, Chen NY, Pedroza M, Zhou Y, Davies J, Philip K, et al:
Blockade of IL-6 Trans signaling attenuates pulmonary fibrosis. J
Immunol. 193:3755–3768. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zehender A, Huang J, Györfi AH, Matei AE,
Trinh-Minh T, Xu X, Li YN, Chen CW, Lin J, Dees C, et al: The
tyrosine phosphatase SHP2 controls TGFβ-induced STAT3 signaling to
regulate fibroblast activation and fibrosis. Nat Commun.
9:32592018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu Q, Liu Y, Pan H, Xu T, Li Y, Yuan J, Li
P, Yao W, Yan W and Ni C: Aberrant expression of miR-125a-3p
promotes fibroblast activation via Fyn/STAT3 pathway during
silica-induced pulmonary fibrosis. Toxicology. 414:57–67. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morikawa M, Derynck R and Miyazono K:
TGF-β and the TGF-β family: Context-dependent roles in cell and
tissue physiology. Cold Spring Harbor Perspect Biol. 8:a0218732016.
View Article : Google Scholar
|
|
28
|
Ask K, Bonniaud P, Maass K, Eickelberg O,
Margetts PJ, Warburton D, Groffen J, Gauldie J and Kolb M:
Progressive pulmonary fibrosis is mediated by TGF-beta isoform 1
but not TGF-beta3. Int J Biochem Cell Biol. 40:484–495. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Border WA and Noble NA: Transforming
growth factor beta in tissue fibrosis. N Engl J Med. 331:1286–1292.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wynn TA and Ramalingam TR: Mechanisms of
fibrosis: Therapeutic translation for fibrotic disease. Nat Med.
18:1028–1040. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng XH and Derynck R: Specificity and
versatility in tgf-beta signaling through Smads. Annu Rev Cell Dev
Biol. 21:659–693. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Walton KL, Johnson KE and Harrison CA:
Targeting TGF-β mediated SMAD signaling for the prevention of
fibrosis. Front Pharmacol. 8:4612017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tang LY, Heller M, Meng Z, Yu LR, Tang Y,
Zhou M and Zhang YE: Transforming Growth Factor-β (TGF-β) directly
activates the JAK1-STAT3 axis to induce hepatic fibrosis in
coordination with the SMAD pathway. J Biol Chem. 292:4302–4312.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang G, Yu Y, Sun C, Liu T, Liang T, Zhan
L, Lin X and Feng XH: STAT3 selectively interacts with Smad3 to
antagonize TGF-β signalling. Oncogene. 35:4388–4398. 2016.
View Article : Google Scholar : PubMed/NCBI
|