Introduction
Diabetic cardiomyopathy (DCM) is a complication
associated with serious microangiopathy, which is responsible for
the increased risk of morbidity and mortality in patients suffering
from diabetes (1). It has been
estimated that DCM will affect ~552 million patients globally by
2030, according to an official report (2,3).
Cardiac dysfunction and pathological structural changes, including
cardiac hypertrophy, cardiac fibrosis, cardiomyocyte apoptosis,
mitochondrial dysfunction and autophagy, occur in the development
of DCM (4–6). As previously reported, several
pathophysiological mechanisms may affect the pathogenesis of DCM,
including unbalanced energy metabolism, inflammation and oxidative
stress (7–9). Elucidating the pathogenesis of DCM
requires urgent investigation to provide an effective treatment
strategy. Inflammation and oxidative stress have been reported to
markedly affect the development and progression of diabetes, as
well as its complications (8,9).
Additionally, previous studies have indicated that uncontrolled
inflammation and oxidative stress may contribute to cardiac
dysfunction in diabetic cardiac tissues (10,11).
Extensive research has also revealed that mitochondrial injury may
serve as a central mediator in the pathology of DCM (12,13).
Therefore, identifying potential therapeutic strategies that target
chronic inflammation and oxidative stress may aid in the management
of DCM.
Sirtuins (SIRTs) are a highly conserved family of
NAD+-dependent deacetylases, which may have a broad
impact on numerous biological pathways (14). Among the SIRTs, SIRT6 is a nuclear
protein known to deacetylate histone H3 lysine 9 and H3 lysine 56
(15,16). SIRT6 has been reported to serve a
crucial role in human telomere and genome stabilization (17), glucose and lipid metabolism
(18), and inflammation and
oxidative stress (19,20). In this regard, the SIRT6 may be
potentially used to treat diabetes, immune-mediated disorders and
cardiovascular diseases. Previous studies have demonstrated that
knockout of SIRT6 can aggravate macrophage foaming and exacerbate
atherosclerosis, whereas increased expression of SIRT6 can
alleviate endothelial cell dysfunction (21,22).
According to these previous studies, SIRT6 may have a favorable
role in cardiovascular disease. However, the role and mechanism of
SIRT6 in DCM has been seldom reported.
The present study aimed to verify whether the SIRT6
inhibitor OSS-128167 aggravated myocardial injury in DCM. A mouse
model of streptozotocin (STZ)-induced type 1 diabetes and cultured
cardiomyocytes were employed in the present study. The
corresponding results supported the deleterious effect of
OSS-128167 on DCM via the aggravation of inflammation and oxidative
stress. By analyzing experimental results in vivo and in
vitro, it was confirmed that SIRT6 may be considered a
potential therapeutic target in treating DCM.
Materials and methods
Cell culture
The H9c2 cell line consists of subclonal cells
obtained from the heart tissue of embryonic rats. H9c2 cells were
provided by the Shanghai Institute of Biochemistry and Cell
Biology, Chinese Academy of Sciences. H9c2 cells were cultured in
DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 5.5
mM D-glucose, 100 U/ml penicillin, 10% FBS (Gibco; Thermo Fisher
Scientific, Inc.) and 100 mg/ml streptomycin at 37°C in a
humidified 5% CO2 incubator. Prior to experimentation,
H9c2 cells were pretreated with 20 and 50 µM OSS-128167 (Selleck
Chemicals) for 1 h, followed by stimulation with high glucose (HG;
33 mM) for various durations (both steps were conducted at 37°C).
For analysis of transforming growth factor (TGF)-β, collagen
(COL)-1, Bax, Bcl-2 and cleaved-poly ADP-ribose polymerase (PARP)
protein, cells were stimulated with HG for 36 h; for RT-qPCR
analysis of TGF-β, COL-1, Bax, Bcl-2 and cleaved-PARP mRNA, cells
were stimulated with HG for 12 h; for western blotting of tumor
necrosis factor (TNF)-α and 3-nitrotyrosine (NT), cells were
stimulated with HG for 24 h; for RT-qPCR analysis of TNF-α, cells
were stimulated with HG for 8 h; and to detect malondialdehyde
(MDA) levels, cells were stimulated with HG for 4 h.
Reagents
Selective SIRT6 antagonist OSS-128167 was dissolved
in dimethyl sulfoxide (DMSO) to obtain 50 mM stock solution, which
was diluted in DMSO for use in further experiments. The chemical
structure of OSS_128167 is shown in Fig. 1C. DMSO had a final concentration of
0.1% (v/v). All other chemical biological agents were purchased
from Sigma-Aldrich; Merck KGaA. Antibodies for SIRT6, TNF-α, COL-1,
TGF-β, PARP, 3-NT and GAPDH were purchased from Abcam.
Animal experiments
Animals were provided by the Animal Center of
Wenzhou Medical University. The present study was approved by the
Wenzhou Medical University Animal Policy and Welfare Committee
(Wenzhou, China), and experiments were conducted according to the
National Institutes of Health (NIH) guidelines regarding the care
and use of laboratory animals. A total of 40 male C57BL/6 mice
(weight, 22–26 g; age, 10 weeks; Riken BioResource Center of Japan)
were housed in an environment with room temperature maintained at
22±2.0°C and humidity at 50±5%. In addition, they were kept under a
12-h light/dark cycle, and were fed with a standard rodent diet and
water. After 1 week of acclimation, mice were intraperitoneally
injected with 50 mg/kg STZ (Sigma-Aldrich; Merck KGaA) formulated
in citrate buffer (100 mM, pH 4.5) for 5 consecutive days. A
glucometer was used to detect the levels of fasting blood-glucose
(FBG). OSS-128167 intervention was commenced once it was confirmed
that type 1 diabetes mellitus (FBG >12 mmol/l) was successfully
established. Mice with STZ-induced diabetes (STZ-DM1) were orally
administered OSS-128167 (20 or 50 mg/kg) or vehicle (1% CMC-Na)
through gavage every other day (n=10/group). As shown in Fig. S1A, the body weight and blood
glucose were recorded at certain time points. A total of 16 weeks
after administration, the mice were anesthetized with ketamine
hydrochloride (100 mg/kg body weight; Ketanest; Pfizer) and
xylazine hydrochloride (16 mg/kg body weight; Rompun 2%; Bayer).
Blood was collected from the retro-orbital vein after the mice were
anesthetized; 500 µl-1 ml blood was collected. Subsequently, the
mice were euthanized by cervical dislocation, the thoracic cavity
was opened, and the heart was removed after perfusion with normal
saline. The heart tissues were embedded in paraffin, and maintained
at 65°C for 1.5 h. Paraffin samples were subsequently stored at
4°C, while other heart tissues were placed in liquid nitrogen for
snap-freezing before further analysis. The collected blood samples
were centrifuged for 5 min (1,500 × g, 4°C) to collect the
serum.
Immunohistochemistry
Fixed heart sections (5-µm) described above were
incubated with 3% H2O2 for 30 min and blocked
for 30 min with 2% bovine serum albumin (Sigma-Aldrich; Merck
KGaA), both at room temperature in PBS. Slides were then incubated
overnight with a primary antibody against TNF-α (1:500; Santa Cruz
Biotechnology, Inc.; cat. no. sc-52746) at 4°C, followed by
incubation with a secondary antibody (1:200; Santa Cruz
Biotechnology, Inc.; cat. no. sc-516102) for 1 h and with DAB (A:B,
1:20) for 5 min at room temperature. Hematoxylin was used to stain
the cell nuclei for 3 min at room temperature and resin was used to
seal the dehydrated sections. A light microscope (magnification,
×400; Nikon Corporation) was used to capture the images.
TUNEL staining
TUNEL staining was performed using the TUNEL
Apoptosis Detection kit (R&D Systems, Inc.), according to the
manufacturer's protocol. Heart tissue sections were counterstained
with DAPI (Beyotime Institute of Biotechnology) at room temperature
for 5 min. Images were captured using the Leica A1 laser confocal
microscope (Leica Microsystems GmbH). The number of TUNEL-positive
cells was counted using ImageJ 1.52a software (NIH) in three
randomly selected fields per sample.
Hematoxylin and eosin (H&E)
staining for morphology and Masson's trichrome staining for
fibrosis
Heart tissues were fixed in 4% paraformaldehyde at
room temperature for 24 h, embedded in paraffin and sectioned into
5-µm thick samples. The tissue sections were incubated at 60–70°C
for 4–6 h, then deparaffinized in xylene at room temperature for 10
min and another subsequent 15 min to complete deparaffinization.
The sections were then rehydrated in a descending series of
ethanol, prior to being immersed in PBS trice at room temperature
for 5 min each and finally washed in distilled water at room
temperature for 20 min for subsequent staining. H&E and
Masson's trichrome staining were used to evaluate the intima-media
thickness and fibrosis content of the tissues.
For H&E staining (Beijing Solarbio Science &
Technology Co., Ltd.; cat. no. G1120), the sections were stained
with eosin for 2 min, wash in distilled water for 20 min and
stained with hematoxylin for 5 min (all at room temperature).
Subsequently, sections were washed in distilled water for 5 min
thrice, dehydrated using an ascending ethanol series and the slides
were then mounted with neutral gum.
For Masson's trichrome staining (Beijing Solarbio
Science & Technology Co., Ltd.; cat. no. G1340), the sections
were stained with ponceau staining solution dye for 5 min, washed
with distilled water for 1 min twice and incubated with 1%
phosphotungstic acid solution for 5 min. Then, the sections were
incubated with aniline blue dye solution for 5 min, treated with 1%
glacial acetic acid water for 1 min, dehydrated using an ascending
ethanol series and then stored at room temperature. The entire
staining process was performed at room temperature. A light
microscope (magnification, ×400; Nikon Corporation) was used to
observe the stained sections.
Determination of superoxide production
and the levels of cellular H2O2
Dihydroethidium (DHE) staining was used to evaluate
superoxide production. Briefly, the isolated mouse heart tissues
were embedded in OCT compound (Sakura Finetek USA, Inc.)
immediately after the heart tissues were excised from the mice,
after which they were split into frozen 5-µm sections. The sections
were then incubated in DHE in PBS (10 mmol/l) for 45 min in a dark
and moist container at 37°C. DHE was oxidized upon the reaction
with superoxide to ethidium bromide, which binds to DNA in the
nucleus and fluoresces red (23).
DHE dilutions were prepared as 1:5,000 to treat the sections, and
images were captured using the Leica A1 laser confocal microscope
(Leica Microsystems GmbH).
Determination of MDA
An MDA kit (Beyotime Institute of Biotechnology;
cat. no. S0131S) was used to determine the levels of MDA in cells
and tissues, according to the manufacturer's protocol.
Caspase 3 activity assay
PBS and 0.025% trypsin (T6325-25 g; Macklin Inc.)
were used to wash and digest H9c2 cells, respectively. The cells
then underwent centrifugation at 600 × g for 5 min at 4°C. The
supernatant was discarded, and PBS was used to wash the precipitate
twice. Lysis buffer was then added at a ratio of 100 µl lysate/2
million cells according to the kit's instructions (cat. no. C1115;
Beyotime Institute of Biotechnology). The mixture then underwent
centrifugation at 20,000 × g for 15 min at 4°C, after 15 min of
lyophilization in an ice bath. Centrifuged supernatant (50 µl) and
Ac-DEVD-pNA (10 µl) were mixed in a 96-well plate (without
generating air bubbles while mixing). Finally, the 96-well plate
was incubated for 90 min at 37°C, after which a spectrophotometer
was used to measure the optical density value at an absorbance of
405 nm.
Reverse transcription quantitative
(RT-qPCR)
According to a standard protocol, H9c2 cells were
homogenized in TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc.) to extract RNA. The two-step M-MLV Platinum SYBR
Green qPCR SuperMix- UDG kit (Thermo Fisher Scientific, Inc.) and
Eppendorf Mastercycler eprealplex detection system (Eppendorf) were
used to conduct RT and qPCR. Reverse transcription was performed at
37°C for 15 min and 85°C for 5 sec, and then maintained at 4°C. The
following thermocycling conditions were used for the qPCR: Initial
denaturation at 95°C for 3 min; followed by 50 cycles at 95°C for
15 sec and 60°C for 30 sec, and a melting curve stage at 95°C for
15 sec and 60°C for 1 min. The primers were provided by Thermo
Fisher Scientific, Inc. and primer sequences are presented in
Table SI. The Tm values were
normalized to β-actin. The 2−∆∆Cq method was used to
quantify the expression levels (24).
Immunoblotting
Cells were collected and underwent lysis using a
buffer (cat. no. AR0103-100; Boster Biological Technology). Protein
was quantified using a BCA assay and protein lysates (40 µg) were
then separated by 10% SDS-PAGE and electrotransferred to
polyvinylidene fluoride membranes. The membranes then underwent 1 h
of blocking in TBS (pH 7.6) containing 5% non-fat milk and 0.05%
Tween 20 at room temperature. The membranes were incubated with
primary antibodies overnight at 4°C and with the secondary
antibodies at room temperature for 2 h. TGF-β (1:500; cat. no.
sc-146), B-cell lymphoma-2 (Bcl-2; 1:500; cat. no. sc-7382), NF-κB
P65 (1:500; cat. no. sc-7151), cleaved (cle)-PARP (1:500; cat. no.
sc-56196) and Bcl-2-associated X (Bax; 1:500; cat. no. sc-7480)
antibodies were purchased from Santa Cruz Biotechnology, Inc. SIRT6
(1:1,000; cat. no. ab191385), COL-1 (1:1,000; cat. no. ab34710),
TNF-α (1:1,000; cat. no. ab6671), 3-NT (1:1,000; cat. no. ab61392)
and GAPDH (1:1,000; cat. no. ab8245) antibodies were provided by
Abcam. The primary and secondary antibodies (1:1,000; Cell
Signaling Technology, Inc.; cat. nos. 7076 and 7074) were diluted
in TBS-0.05% Tween-20 prior to use. The enhanced chemiluminescence
kit (Bio-Rad Laboratories, Inc.) was used to detect the specific
bands. Densitometric analysis was performed using Image Lab 5.1
software (Bio-Rad Laboratories, Inc.).
Statistical analysis
Experiments were repeated three times and data were
expressed as the mean ± SEM. GraphPad Pro Prism 6.0 (GraphPad
Software, Inc.) was used to conduct statistical analyses. Student's
t-test was used to analyze the differences between two datasets,
while a one-way ANOVA and Bonferroni correction were used to assess
multiple comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
SIRT6 inhibitor OSS-128167 aggravates
cardiac pathological abnormalities in mice with type 1 diabetes
caused by hyperglycemia
Type 1 diabetes in male C57BL/6 mice was induced by
successively injecting a low dose of STZ intraperitoneally. In the
STZ-mediated mouse model of type 1 diabetic cardiomyopathy
(STZ-DM1), the protein expression levels of SIRT6 were
significantly lower in myocardial tissues of STZ-DM1 mice compared
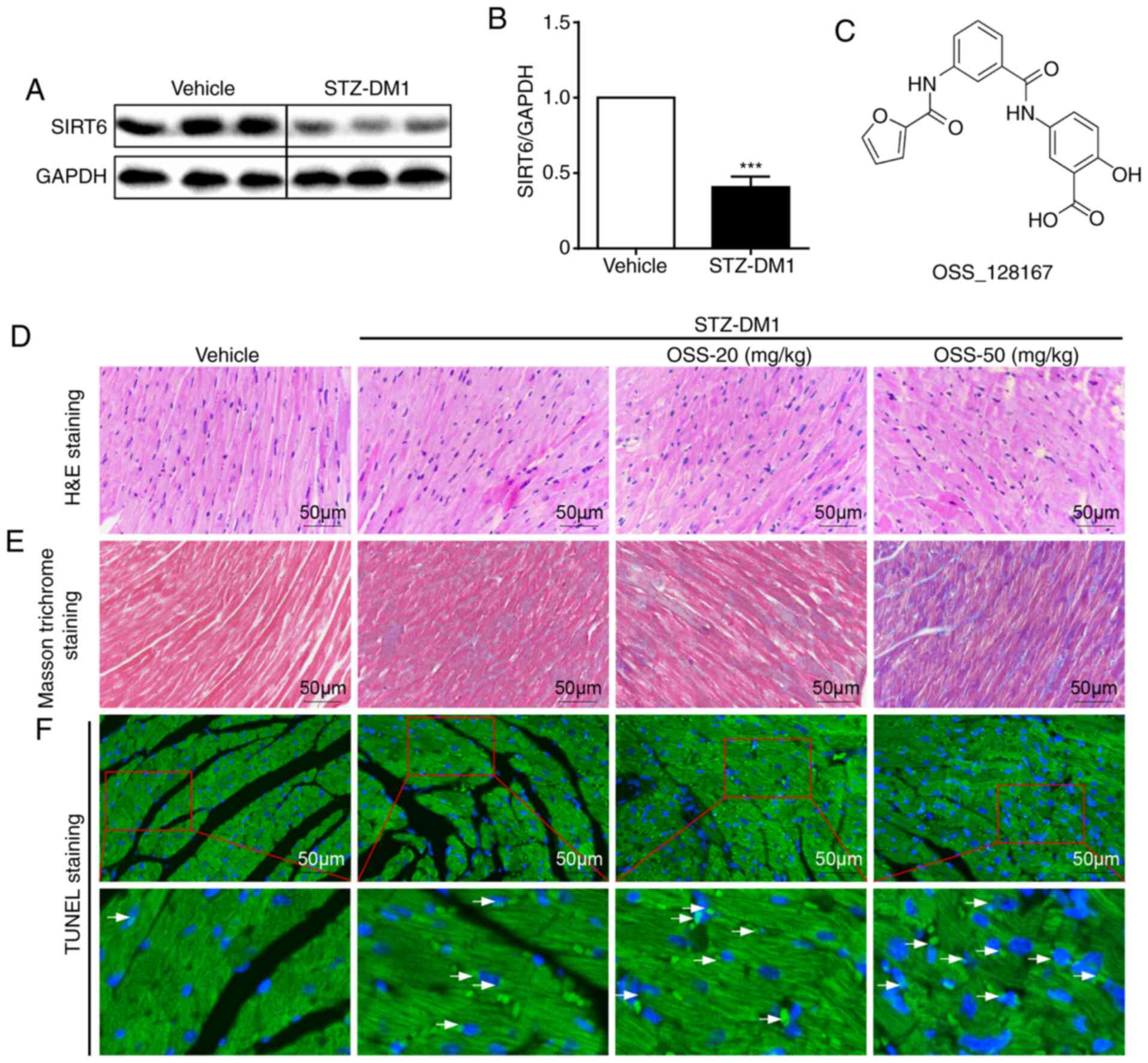
with those in the non-diabetic controls (Fig. 1A and B). Subsequently, the mouse
model of type 1 diabetic cardiomyopathy was employed to determine
whether OSS-128167 (Fig. 1C)
aggravated diabetes-induced cardiac damage. Diabetic mice were
treated with OSS-128167 (20 and 50 mg/kg) for 2 months, and the
effects of OSS-128167 on diabetic cardiac morphology were examined.
As shown using H&E and Masson's trichrome staining, diabetic
hearts exhibited an abnormal structure compared with the hearts of
normal mice. Compared with the vehicle group, H&E staining
showed myocardial fibrosis in the STZ-DM1 group was disordered,
while Masson's trichrome staining showed increased myocardial
fibrosis in the STZ-DM1 group. Notably, these structural
abnormalities were aggravated by treatment of diabetic mice with
OSS-128167 (Fig. 1D and E).
Finally, TUNEL staining was used to detect myocardial tissue
apoptosis. As shown in Fig. 1F,
OSS-128167 treatment markedly increased cell apoptosis, as
indicated by an increase in the TUNEL-positive staining area.
Notably, it was revealed that such adverse effects imposed by
OSS-128167 on cardiac morphology were not caused by metabolic
changes. OSS-128167 treatment had no impact on FBG and serum
insulin levels (Fig. S1B and C).
Moreover, compared with that in the vehicle group, heart
weight/body weight (HW/BW) was significantly increased in the
STZ-DM1 and OSS-128167 treatment groups (Fig. S1D). However, no significant
difference in HW/BW was apparent in the OSS-128167 treatment group
compared with in the STZ-DM1 group. These results indicated that
OSS-128167 exacerbated cardiac structural destruction, and
myocardial fibrosis and apoptosis in type 1 diabetic mice.
OSS-128167 aggravates diabetes-induced
myocardial inflammation and oxidative stress
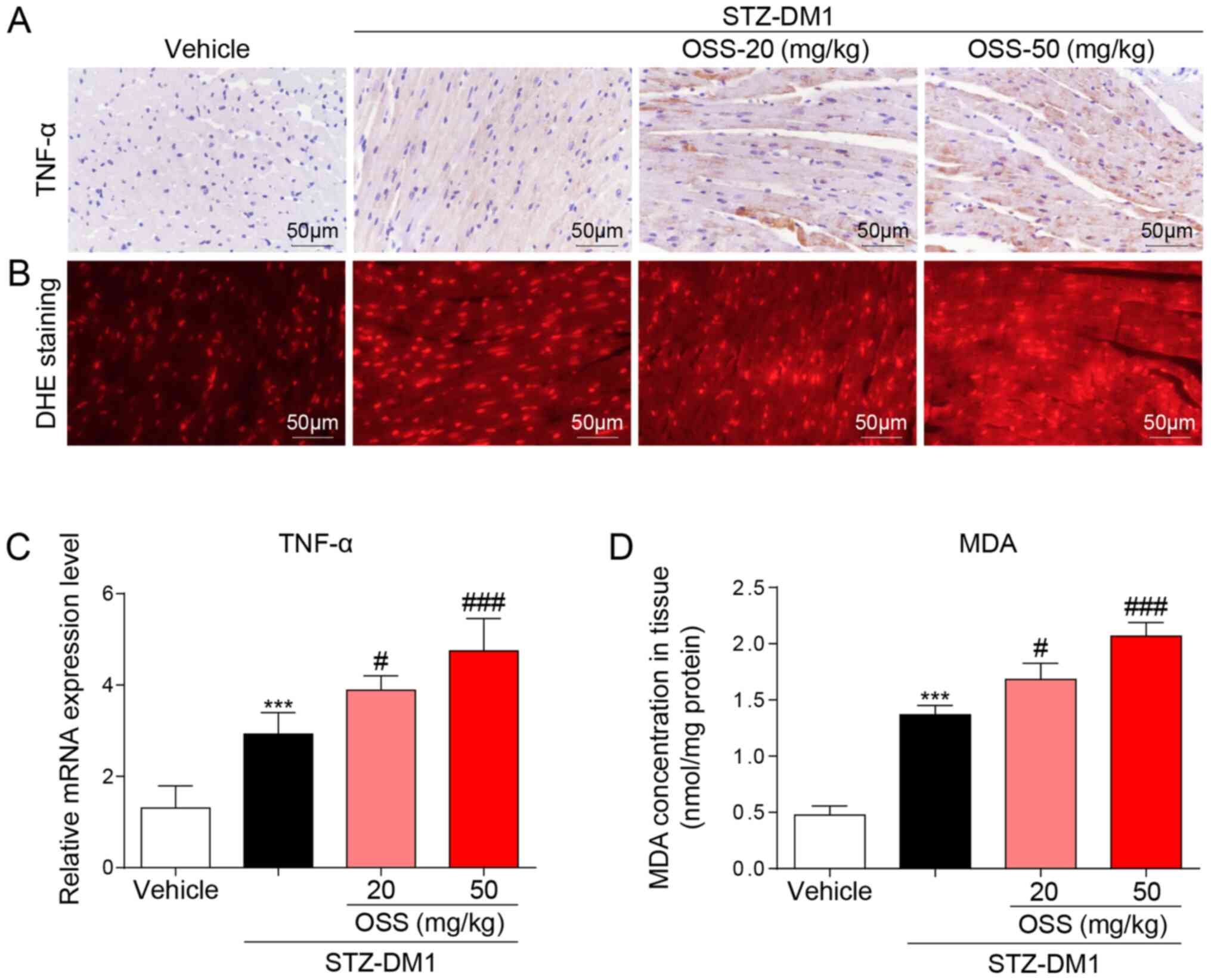
In order to determine the potential molecular
mechanisms underlying the deleterious effect exerted by OSS-128167
in vivo, the expression levels of inflammatory mediators and
the levels of oxidative stress in the myocardium were detected in
response to OSS-128167. Notably, the mRNA and protein expression
levels of the inflammatory factor TNF-α were significantly
increased in the STZ-DM1 group compared with those detected in the
vehicle group; a further increase in these levels was also detected
in the OSS-128167 treatment group (Fig.
2A and C). The biological effects of OSS-128167 on oxidative
stress were further detected in the hearts of each group. DHE and
MDA kits were used to detect oxidative stress in the myocardium;
higher levels of superoxide and MDA were detected in the hearts of
mice in the OSS-128167 group compared with those in the STZ-DM1
group (Fig. 2B and D). These
results indicated that OSS-128167 aggravated myocardial injury in
DCM by exacerbating hyperglycemia-mediated inflammation and
oxidative stress.
OSS-128167 aggravates HG-induced cell
fibrosis and apoptosis of H9c2 cells
The results of the present study were verified by
observing cultured cardiomyocytes in vitro and the potential
molecular mechanisms were explored. Briefly, H9c2 cells were
cultured and treated with HG (33 mM) to simulate hyperglycemia.
Following treatment, immunoblotting was conducted to assess the
expression levels of biomarkers for cell fibrosis (Fig. 3A). OSS-128167 significantly
increased the expression levels of pro-fibrotic markers COL-1 and
TGF-β compared with in the HG group (Fig. 3A). In addition, OSS-128167 induced a
dose-dependent increase in the mRNA expression levels of COL-1 and
TGF-β, as well as CTGF (Fig. 3B-D).
The present in vivo studies revealed that OSS-128167
increased apoptosis in the heart tissues of mice. Consequently, the
effects of OSS-128167 on HG-induced apoptosis in H9c2 cells were
examined. Under a HG challenge, the expression levels of the
apoptotic biomarker Bax were markedly upregulated by OSS-128167,
whereas the opposite was observed for Bcl-2 (Fig. 3E). Treatment with OSS-128167 also
significantly reduced the protein expression ratio of Bcl-2/Bax
compared with that in the HG group (Fig. 3F). As shown in Fig. 3G, OSS-128167 readily increased the
protein expression levels of cle-PARP, which were initially induced
by HG. Moreover, caspase-3 activity was increased in the OSS-128167
group compared with that in the HG group (Fig. 3H). These results revealed that
OSS-128167 could significantly increase myocardial fibrosis and the
expression of apoptotic proteins induced by hyperglycemia in
vivo.
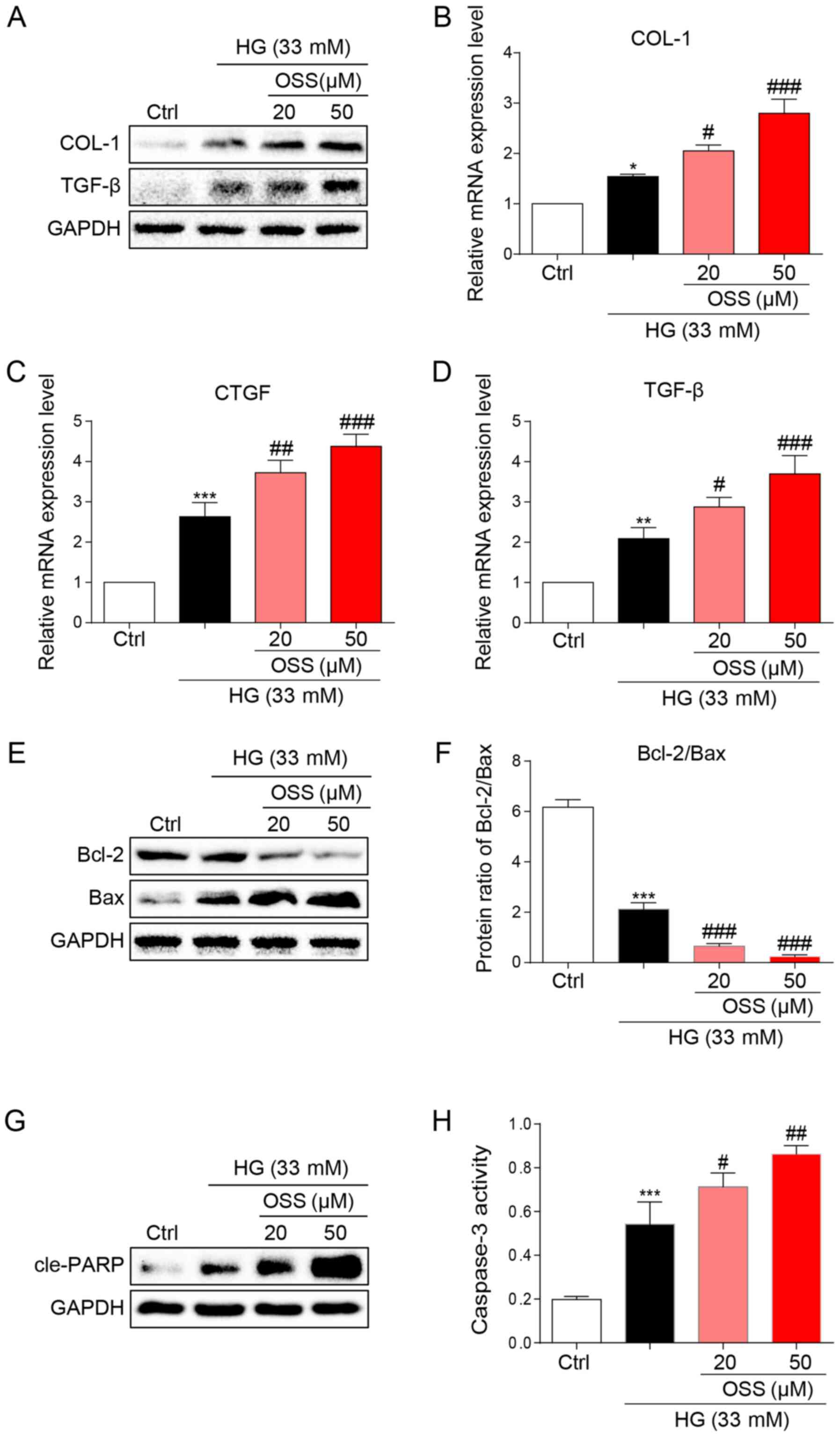 | Figure 3.OSS-128167 aggravates HG-induced
fibrosis and apoptosis of H9c2 cells. (A) Western blot analysis of
COL-1 and TGF-β in H9c2 cells. H9c2 cells underwent 1 h of
pre-incubation with a recommended dose of OSS-128167, followed by
36 h of 33 mM HG treatment. Proteins underwent immunoblotting, and
GAPDH was used as a loading control. (B-D) H9c2 cells underwent 1 h
of pre-incubation with a recommended dose of OSS-128167, followed
by 12 h of 33 mM HG incubation. Reverse transcription-quantitative
PCR was performed to determine the mRNA expression levels of COL-1,
CTGF and TGF-β. (E and F) H9c2 cells underwent 1 h of
pre-incubation with a recommended dose of OSS-128167, followed by
36 h of 33 mM HG treatment. Western blot analysis of Bcl-2 and Bax
in H9c2 cells. (G) Western blot analysis of cle-PARP in H9c2 cells.
GAPDH was used as the loading control. (H) H9c2 cells underwent 1 h
of pre-incubation with a recommended dose of OSS-128167, followed
by 12 h of 33 mM HG incubation. Caspase 3 activity assay kit was
used to determine caspase 3 activity in H9c2 cells. Data from three
independent experiments are presented. *P<0.05, **P<0.01,
***P<0.001 vs. Ctrl; #P<0.05,
##P<0.01, ###P<0.001 vs. HG. HG, high
glucose; COL-1, collagen 1; TGF-β, transforming growth factor-β;
CTGF, connective tissue growth factor; Bcl-2, B-cell lymphoma 2;
Bax, Bcl-2-associated X; cle-PARP, cleaved-poly ADP-ribose
polymerase; Ctrl, control. |
OSS-128167 aggravates HG-induced H9c2
cell inflammation and oxidative stress
The protein and mRNA expression levels of the
inflammatory cytokine, TNF-α were measured to evaluate the
pro-inflammatory biological function of OSS-128167. OSS-128167 was
revealed to significantly aggravate HG-induced increases in TNF-α
expression levels (Fig. 4A and B).
These findings were in accordance with the results of the in
vivo studies and suggested the negative impact of OSS-128167 on
cardiomyocytes through the aggravation of inflammation. Oxidative
stress also serves as an important pathological mechanism of DCM.
HG-stimulated H9c2 cells underwent immunoblotting (Fig. 4C), and the expression levels of
3-NT, a biomarker of the nitrogen free radical species, exhibited
markedly increased accumulation in H9c2 cells treated with HG which
was more prominent in the OSS-128167-treated groups. Furthermore,
MDA levels were measured in H9c2 cells. Polyunsaturated fatty acids
produce MDA via lipid peroxidation; in the present study,
OSS-128167 aggravated the HG-induced increase in MDA (Fig. 4D). These results demonstrated that
OSS-128167 aggravated HG-induced cell inflammatory responses and
oxidative stress in H9c2 cells.
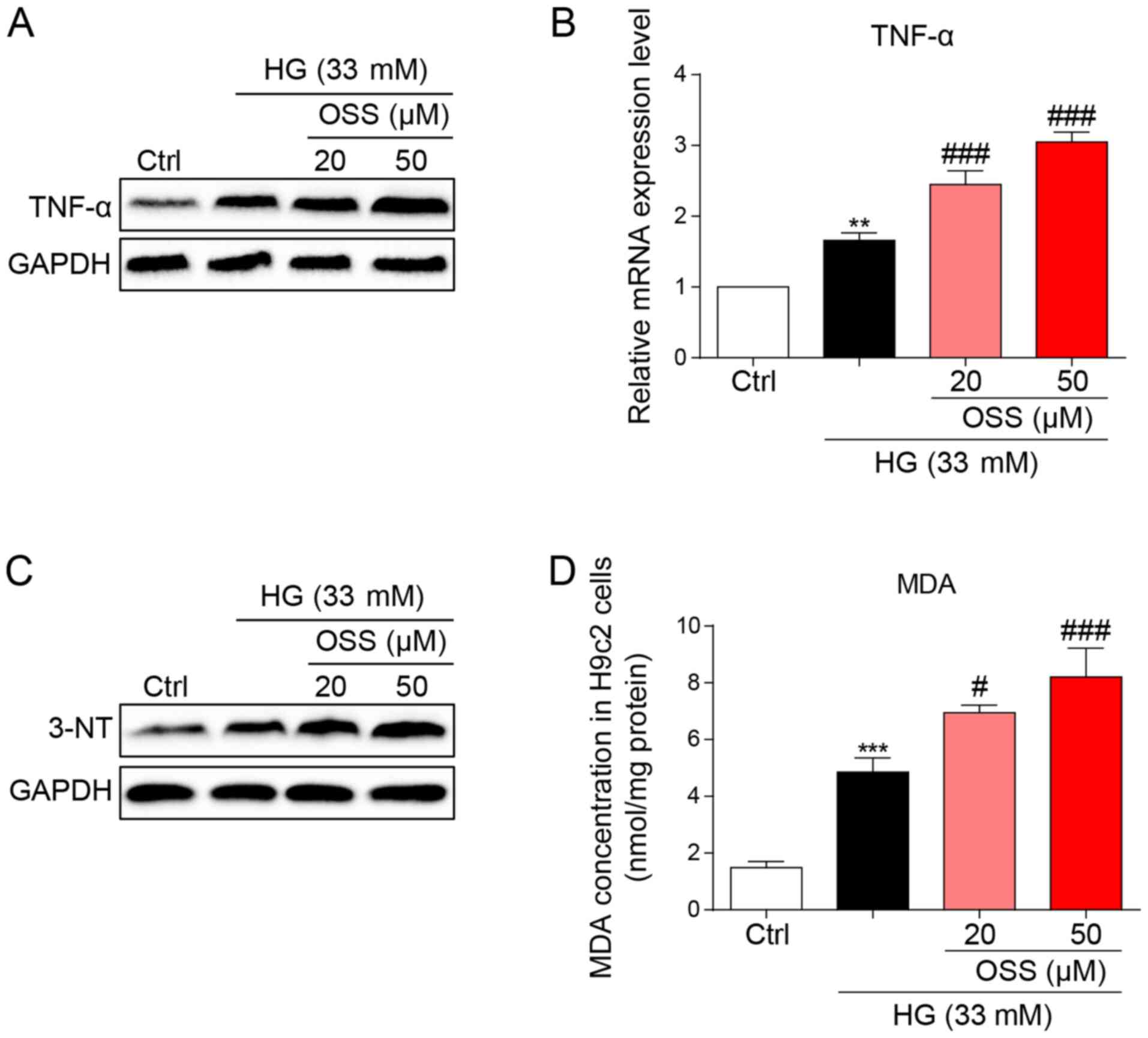 | Figure 4.OSS-128167 aggravates HG-induced
inflammation and oxidative stress in H9c2 cells. (A) H9c2 cells
underwent 1 h of pretreatment with a recommended dose of
OSS-128167, followed by 24 h of 33 mM HG treatment. Western blot
analysis of TNF-α in H9c2 cells. GAPDH was used as a loading
control. (B) H9c2 cells underwent 1 h of pretreatment with a
recommended dose of OSS-128167, followed by 8 h of 33 mM HG
incubation. Reverse transcription-quantitative PCR assay was used
to detect the mRNA expression levels of TNF-α. (C) H9c2 cells
underwent 1 h of pretreatment with a recommended dose of
OSS-128167, followed by 24 h of 33 mM HG treatment. Western blot
analysis of 3-NT in H9c2 cells. GAPDH was used as a loading
control. (D) Cells underwent 1 h of pre-treatment with a
recommended dose of OSS-128167, followed by 4 h of HG (33 mM)
treatment. MDA levels in H9C2 cells were detected. Data from three
independent experiments are shown. **P<0.01, ***P<0.001 vs.
Ctrl; #P<0.05, ###P<0.001 vs. HG. HG,
high glucose; TNF-α, tumor necrosis factor-α; 3-NT, 3-N-terminal;
MDA, malondialdehyde; Ctrl, control. |
Discussion
During the past few decades, the incidence and
prevalence of diabetes mellitus have increased worldwide; this is
primarily due to the increased incidence of diabetic complications,
such as DCM (7). Accordingly,
developing additional treatments and novel prevention strategies
for DCM is urgently required. The present study revealed that the
novel use of a SIRT6 inhibitor, OSS-128167, exacerbated cardiac
structural alterations in diabetic mice. Moreover, the present
study revealed that the cardio-pernicious effect of OSS-128167 also
led to aggravated inflammation and oxidative stress. These findings
were verified in vitro using HG-treated H9c2 cells.
Chronic and persistent inflammation, and oxidative
stress markedly affect the pathophysiology of cardiovascular
disorders induced by hyperglycemia. Persistent inflammatory factors
and ROS caused by hyperglycemia have been shown to damage normal
cellular functions and structure, causing apoptosis of
cardiomyocytes (25,26). Previous studies have demonstrated
the benefits of implementing anti-inflammatory strategies for
cardiac health (27,28). In a previous study, overexpression
of TNF-α exacerbated myocardial apoptosis by causing desmin
cleavage and modification, eventually leading to heart failure
(29). Additionally, the blockage
of TNF-α was found to significantly alleviate the inflammatory
response and myocardial fibrosis in the myocardial intima of
patients with DCM (30). In the
present study, OSS-128167 was observed to increase the expression
levels of TNF-α caused by HG or diabetes in vitro and in
vivo.
Another mechanism of oxidative stress-induced
myocardial damage also significantly affects the pathophysiological
mechanism of DCM. Oxidative stress has been shown to facilitate
myocardial fibrosis in response to HG (31). According to previous studies,
factors such as hyperglycemia, hyperlipidemia, increased free fatty
acid levels and accumulated advanced glycosylation end products can
promote the production of ROS and reactive nitrogen species in the
diabetic myocardium (32,33). In the present study, OSS-128167 was
revealed to exacerbate oxidative stress in HG-induced
cardiomyocytes, with similar results being observed in the
myocardium of type 1 diabetic mice.
Apoptosis and fibrosis of heart tissue are mediated
by inflammatory responses and oxidative stress. Therefore,
experiments were conducted to evaluate the biological effects of
OSS-128167 on hyperglycemia/HG-mediated myocardial damage in
vivo and in vitro. Hyperglycemia and HG induced the
expression of pro-fibrotic markers (COL-1, TGF-β and CTGF) and
OSS-128167 significantly increased the expression of these fibrotic
biomarkers. In addition, in STZ-induced diabetic mice, the number
of apoptotic myocardial cells detected by TUNEL staining was
markedly increased in the OSS-128167 treatment group compared with
in the STZ-DM1 group. Subsequently, in H9c2 cells under HG
challenge treatment with OSS-128167 reduced the expression levels
of the anti-apoptotic protein Bcl-2 expression, whereas it
increased the expression levels of pro-apoptotic proteins Bax and
cle-PARP. These results indicated that inhibiting the expression of
SIRT6 could exacerbate hyperglycemia-induced myocardial damage by
increasing the fibrosis and apoptosis of myocardial cells.
The present study revealed that HG induced cardiac
inflammation and oxidative stress, which may lead to the
progression of DCM. In addition, the present study provided a novel
understanding of the regulatory role of SIRT6 in cardiac injury
caused by HG. Notably, OSS-128167 was found to facilitate
inflammation, oxidative stress, fibrosis and apoptosis both in
vitro and in vivo. Although previous studies aimed to
find new therapies to prevent or treat DCM, drugs or therapeutic
targets that can completely reverse the process of DCM have not yet
been discovered. These experiments largely demonstrated that SIRT6
may serve as a novel therapeutic target for DCM. However, the
present study also has some limitations. Notably, the function of
SIRT6 in DCM was not directly assessed; a specific inhibitor of
SIRT6 was used instead. This lacks strength in explaining the role
that SIRT6 has in DCM. Future studies should investigate additional
intricate mechanisms of SIRT6 to determine if it may be used to
treat diabetes and its complications.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ was the principal investigator, designed the
study, supervised experiments and wrote the manuscript. YH, JZ, DX,
YP and YJ performed experiments. YH, JZ and DX analyzed the data.
All authors read and approved the final manuscript, and agree to be
accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved. LZ and YH confirm the authenticity of
all the raw data.
Ethics approval and consent to
participate
The animal raising and handling procedures were
performed in accordance with the Guide for the Care and Use of
Laboratory Animals. The present study was approved by the Wenzhou
Medical University Animal Policy and Welfare Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DCM
|
diabetic cardiomyopathy
|
|
SIRT6
|
sirtuin 6
|
|
STZ
|
streptozotocin
|
|
ROS
|
reactive oxygen species
|
|
TNF-α
|
tumor necrosis factor-α
|
|
DHE
|
dihydroethidium
|
|
MDA
|
malondialdehyde
|
|
HG
|
high glucose
|
|
COL
|
collagen
|
|
TGF-β
|
transforming growth factor β
|
|
CTGF
|
connective tissue growth factor
|
|
Bcl-2
|
B-cell lymphoma-2
|
|
Bax
|
Bcl-2-associated X
|
|
cle-PARP
|
cleaved poly ADP-ribose polymerase
|
|
3-NT
|
3-nitrotyrosine
|
References
|
1
|
Meagher P, Adam M, Civitarese R,
Bugyei-Twum A and Connelly KA: Heart failure with preserved
ejection fraction in diabetes: Mechanisms and management. Can J
Cardiol. 34:632–643. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Whiting DR, Guariguata L, Weil C and Shaw
J: IDF diabetes atlas: Global estimates of the prevalence of
diabetes for 2011 and 2030. Diabetes Res Clin Pract. 94:311–321.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Al-Quwaidhi AJ, Pearce MS, Sobngwi E,
Critchley JA and O'Flaherty M: Comparison of type 2 diabetes
prevalence estimates in Saudi Arabia from a validated Markov model
against the International Diabetes Federation and other modelling
studies. Diabetes Res Clin Pract. 103:496–503. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Devereux RB, Roman MJ, Paranicas M,
O'Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER and
Howard BV: Impact of diabetes on cardiac structure and function:
The strong heart study. Circulation. 101:2271–2276. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Varma U, Koutsifeli P, Benson VL, Mellor
KM and Delbridge LMD: Molecular mechanisms of cardiac pathology in
diabetes-Experimental insights. Biochim Biophys Acta Mol Basis Dis.
1864:1949–1959. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dillmann WH: Diabetic cardiomyopathy. Circ
Res. 124:1160–1162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levelt E, Gulsin G, Neubauer S and McCann
GP: MECHANISMS IN ENDOCRINOLOGY: Diabetic cardiomyopathy:
Pathophysiology and potential metabolic interventions state of the
art review. Eur J Endocrinol. 178:R127–R139. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tan Y, Zhang Z, Zheng C, Wintergerst KA,
Keller BB and Cai L: Mechanisms of diabetic cardiomyopathy and
potential therapeutic strategies: Preclinical and clinical
evidence. Nat Rev Cardiol. 17:585–607. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jia G, Hill MA and Sowers JR: Diabetic
cardiomyopathy: An update of mechanisms contributing to this
clinical entity. Circ Res. 122:624–638. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X,
Che H, Han T, Meng S, Bai Y and Wang L: LncRNA KCNQ1OT1 mediates
pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem.
50:1230–1244. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Feng W, Lei T, Wang Y, Feng R, Yuan J,
Shen X, Wu Y, Gao J, Ding W and Lu Z: GCN2 deficiency ameliorates
cardiac dysfunction in diabetic mice by reducing lipotoxicity and
oxidative stress. Free Radic Biol Med. 130:128–139. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yu LM, Dong X, Xue XD, Xu S, Zhang X, Xu
YL, Wang ZS, Wang Y, Gao H, Liang YX, et al: Melatonin attenuates
diabetic cardiomyopathy and reduces myocardial vulnerability to
ischemia-reperfusion injury by improving mitochondrial quality
control: Role of SIRT6. J Pineal Res. 70:e126982021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jubaidi FF, Zainalabidin S, Mariappan V
and Budin SB: Mitochondrial dysfunction in diabetic cardiomyopathy:
The possible therapeutic roles of phenolic acids. Int J Mol Sci.
21:60432020. View Article : Google Scholar
|
|
14
|
Finkel T, Deng CX and Mostoslavsky R:
Recent progress in the biology and physiology of sirtuins. Nature.
460:587–591. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Michishita E, McCord RA, Berber E, Kioi M,
Padilla-Nash H, Damian M, Cheung P, Kusumoto R, Kawahara TL,
Barrett JC, et al: SIRT6 is a histone H3 lysine 9 deacetylase that
modulates telomeric chromatin. Nature. 452:492–496. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang B, Zwaans BM, Eckersdorff M and
Lombard DB: The sirtuin SIRT6 deacetylates H3 K56Ac in vivo
to promote genomic stability. Cell Cycle. 8:2662–2663. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tennen RI and Chua KF: Chromatin
regulation and genome maintenance by mammalian SIRT6. Trends
Biochem Sci. 36:39–46. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kugel S and Mostoslavsky R: Chromatin and
beyond: The multitasking roles for SIRT6. Trends Biochem Sci.
39:72–81. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Khan RI, Nirzhor SSR and Akter R: A review
of the recent advances made with SIRT6 and its implications on
aging related processes, major human diseases, and possible
therapeutic targets. Biomolecules. 8:442018. View Article : Google Scholar
|
|
20
|
Singh CK, Chhabra G, Ndiaye MA,
Garcia-Peterson LM, Mack NJ and Ahmad N: The role of sirtuins in
antioxidant and redox signaling. Antioxid Redox Signal. 28:643–661.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Arsiwala T, Pahla J, van Tits LJ,
Bisceglie L, Gaul DS, Costantino S, Miranda MX, Nussbaum K, Stivala
S, Blyszczuk P, et al: Sirt6 deletion in bone marrow-derived cells
increases atherosclerosis-Central role of macrophage scavenger
receptor 1. J Mol Cell Cardiol. 139:24–32. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jin Z, Xiao Y, Yao F, Wang B, Zheng Z, Gao
H, Lv X, Chen L, He Y, Wang W and Lin R: SIRT6 inhibits cholesterol
crystal-induced vascular endothelial dysfunction via Nrf2
activation. Exp Cell Res. 387:1117442020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang L, Han J, Shan P, You S, Chen X, Jin
Y, Wang J, Huang W, Wang Y and Liang G: MD2 blockage protects
obesity-induced vascular remodeling via activating AMPK/Nrf2.
Obesity. 25:1532–1539. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fang ZY, Prins JB and Marwick TH: Diabetic
cardiomyopathy: Evidence, mechanisms, and therapeutic implications.
Endocr Rev. 25:543–567. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee TW, Kao YH, Chen YJ, Chao TF and Lee
TI: Therapeutic potential of vitamin D in AGE/RAGE-related
cardiovascular diseases. Cell Mol Life Sci. 76:4103–4115. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kolpakov MA, Sikder K, Sarkar A, Chaki S,
Shukla SK, Guo X, Qi Z, Barbery C, Sabri A and Rafiq K:
Inflammatory serine proteases play a critical role in the early
pathogenesis of diabetic cardiomyopathy. Cell Physiol Biochem.
53:982–998. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Zhang Z, Yang Y, Suo Y, Liu R,
Qiu J, Zhao Y, Jiang N, Liu C, Tse G, et al: Alogliptin prevents
diastolic dysfunction and preserves left ventricular mitochondrial
function in diabetic rabbits. Cardiovasc Diabetol. 17:1602018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Panagopoulou P, Davos CH, Milner DJ,
Varela E, Cameron J, Mann DL and Capetanaki Y: Desmin mediates
TNF-alpha-induced aggregate formation and intercalated disk
reorganization in heart failure. J Cell Biol. 181:761–775. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Westermann D, Van Linthout S, Dhayat S,
Dhayat N, Schmidt A, Noutsias M, Song XY, Spillmann F, Riad A,
Schultheiss HP and Tschöpe C: Tumor necrosis factor-alpha
antagonism protects from myocardial inflammation and fibrosis in
experimental diabetic cardiomyopathy. Basic Res Cardiol.
102:500–507. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Palomer X, Pizarro-Delgado J and
Vazquez-Carrera M: Emerging actors in diabetic cardiomyopathy:
Heartbreaker biomarkers or therapeutic targets? Trends Pharmacol
Sci. 39:452–467. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun HJ, Xiong SP, Wu ZY, Cao L, Zhu MY,
Moore PK and Bian JS: Induction of caveolin-3/eNOS complex by
nitroxyl (HNO) ameliorates diabetic cardiomyopathy. Redox Biol.
32:1014932020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barbeau PA, Holloway TM, Whitfield J,
Baechler BL, Quadrilatero J, van Loon LJC, Chabowski A and Holloway
GP: α-Linolenic acid and exercise training independently, and
additively, decrease blood pressure and prevent diastolic
dysfunction in obese Zucker rats. J Physiol. 595:4351–4364. 2017.
View Article : Google Scholar : PubMed/NCBI
|