Introduction
Paraquat (PQ) is a non-selective herbicide that is
widely used in developing countries (1). It is toxic to humans and animals, and
the lung is its main target organ (2). Severe lung injury often occurs after
PQ poisoning, which is characterized by alveolar epithelial cell
damage, edema, exudation and interstitial changes, as well as
inflammatory cell infiltration and progressive fibrosis that can
culminate in death due to respiratory failure. Pulmonary fibrosis
is one of the most typical manifestations of PQ poisoning (3).
The mechanism of PQ-induced pulmonary fibrosis
remains unclear. It is usually considered that inflammation and
oxidative stress may be involved in the pathogenesis of pulmonary
fibrosis (4). In the early stages
of PQ-induced acute lung injury, inflammatory cells are activated
and the influx of neutrophils and macrophages increases the level
of proinflammatory cytokines (5).
Furthermore, the production of superoxide radicals significantly
increases and directly or indirectly damages alveolar epithelial
cells (6). In addition, fibrogenic
factors such as TGF-β, collagen and α-smooth muscle actin (α-SMA)
also promote the process of pulmonary fibrosis (7).
At present, the treatment guidelines for patients
with PQ poisoning are evolving. The current treatment methods
mainly include: i) Emetic induction; ii) catharsis; iii) gastric
lavage; iv) hemodialysis; v) antioxidant treatment; vi) immune
regulation; and vii) hemoperfusion (8). Despite these treatments, the strong
toxicity of PQ poisoning and the lack of specific antidotes for it
have resulted in mortality that is still >50% (9). Therefore, the development of effective
drugs for the treatment of pulmonary fibrosis is needed.
Fluorofenidone
[1-(3-fluorophenyl)-5-methylpyridin-2(1H)-1; AKF-PD] is a novel,
small-molecule pyridone drug with clear and extensive antifibrotic
effects (10). AKF-PD inhibits
fibrosis by reducing inflammation and oxidative stress, inhibiting
cell proliferation and activation and promoting extracellular
matrix degradation (11,12). Previous studies have shown that
AKF-PD can significantly alleviate bleomycin-induced pulmonary
fibrosis in mice, inhibit the expression of TGF-β1 in
bronchoalveolar lavage fluid, reduce the expression of type I
collagen and fibronectin in lung tissue and mitigate alveolar
injury (13–15). Moreover, in human lung fibroblasts
stimulated with TGF-β1, AKF-PD significantly reduces the expression
levels of type I collagen and α-SMA (16). However, the effect of AKF-PD on
PQ-induced pulmonary fibrosis and its mechanism are still unclear
and require further study.
PI3K/Akt/mTOR is a signaling pathway that plays a
notable role in regulating cell proliferation, apoptosis, protein
synthesis, energy metabolism and autophagy (17,18).
In previous years, studies have shown that the PI3K/Akt/mTOR
signaling pathway is closely associated with the occurrence of
pulmonary fibrosis. The activation of the PI3K/Akt/mTOR signaling
pathway and insufficient autophagy promotes the occurrence and
development of pulmonary fibrosis (19–21).
The present study evaluated whether AKF-PD could
effectively alleviate pulmonary fibrosis caused by PQ poisoning,
and analyzed the potential signaling pathways associated with its
anti-fibrotic effect. First, a model of pulmonary fibrosis was
established in rats to verify the effect of AKF-PD on pulmonary
fibrosis induced by PQ poisoning. To further verify the
anti-pulmonary fibrosis mechanism of AKF-PD, HPAEpiC cells were
used to explore the regulation of AKF-PD on mTOR signaling pathway
and autophagy. The present study provided a theoretical basis for
the clinical application of AKF-PD.
Materials and methods
Reagents
PQ solution (20%) was purchased from Syngenta
Nantong Crop Protection Co., Ltd. Fluorofenidone was provided by
Xiangya School of Pharmacy, Central South University. DMEM and FBS
were purchased from Gibco (Thermo Fisher Scientific, Inc.). Cell
Counting Kit-8 (CCK-8) and trypsin were purchased from
Sigma-Aldrich (Merck KGaA). For western blot analysis, rabbit
monoclonal antibodies against TGF-β1 (1:2,000; cat. no. ab92486),
α-SMA (1:2,000; cat. no. ab32575), E-cadherin (1:1,000; cat. no.
ab231303), mTOR (1:3,000; cat. no. ab134903), phosphorylated
(p-)mTOR (1:3,000; cat. no. ab109268), PI3K (1:1,000; cat. no.
ab32089), p-PI3K (1:1,000; cat. no. ab139317), LC3 (1:4,000; cat.
no. ab232940) and p62 (1:2,000; cat. no. ab56416) were purchased
from Abcam. Mouse monoclonal antibodies against β-actin (1:2,000;
cat. no. GB11001) were purchased from Wuhan Servicebio Technology
Co., Ltd. The following ELISA kits were used: Superoxide dismutase
(cat. no. SEKH-0029; Beijing Solarbio Science & Technology Co.,
Ltd.), malondialdehyde (cat. no. SEKH-0053; Beijing Solarbio
Science & Technology Co., Ltd.), hydroxyproline (cat. no.
SEKH-0025; Beijing Solarbio Science & Technology Co., Ltd.),
lactate dehydrogenase (cat. no. SEKH-1036; Beijing Solarbio Science
& Technology Co., Ltd.), IL-1b (cat. no. SEKH-0002; Beijing
Solarbio Science & Technology Co., Ltd.), IL-10 (cat. no.
SEKH-0013; Beijing Solarbio Science & Technology Co., Ltd.),
and TNF-α (cat. no. SEKH-0047; Beijing Solarbio Science &
Technology Co., Ltd.).
Animals
Sprague-Dawley rats (weight 200–220 g; Four-week-old
male adults), were purchased from Hunan Silaike Jingda Experimental
Animal Co., Ltd. The rats were placed in a specific pathogen-free
level feeding environment for routine feeding and were provided
with food and water ad libitum. The incubator temperature
was maintained at 22±2°C with 40–60% humidity, on a 12-h light/dark
cycle. They were randomly divided into control, PQ, PQ + AKF-PD and
AKF-PD groups. The control group received 1 ml/day of normal saline
by gavage. The PQ group received 40 mg/kg of PQ by one-time
intragastric administration, then 1 ml/day of normal saline. In the
PQ + AKF-PD group, a single intragastric dose of 40 mg/kg of PQ was
administered, followed by intragastric administration of 500
mg/kg/day of AKF-PD. In the AKF-PD group, 500 mg/kg of AKF-PD was
intragastrically administered daily without PQ treatment. After 14
days of continuous administration, each rat was anesthetized with
1% pentobarbital sodium (40 mg/kg) by intraperitoneal injection
before being sacrificed by decapitation. The animal death was
confirmed by lack of responsiveness after 5 min, after which the
lung tissues were dissected. This study was approved by The
Committee of Experimental Animals of the First People's Hospital of
Hunan Province (approval no. HP2019010793).
Histopathology
The lung tissues were fixed at room temperature for
24 h in 4% paraformaldehyde, and dehydrated in ethanol of 70% (2
h), 80% (overnight), 90% (2 h) and 100% (twice in an hour) at room
temperature. Following paraffin embedding, the tissues were cut
into 4 µm sections. The sections were stained using the hematoxylin
and eosin staining kit (cat. no. C0109; Beyotime Institute of
Biotechnology) and Masson's trichrome staining kit (cat. no. C0215;
Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. After the sections were observed and
images were captured under a microscope (IX73; Olympus Corporation;
magnification, ×200), the degree of alveolitis and pulmonary
fibrosis was evaluated according to the method described by Szapiel
et al (22).
Determination of oxidative stress
parameters and collagen content
The lung tissues were collected from each group,
weighed, washed with 10% normal saline and subsequently centrifuged
at 4°C, 10,000 × g for 10 min with an automatic biochemical
analyzer. To analyze the level of oxidative stress and the degree
of fibrosis in lung tissue, the amounts of SOD, MDA, and HYP was
quantified according to the instructions of the ELISA kits.
Cell culture
Human alveolar epithelial cells (HPAEpiC cells) were
purchased from Shanghai Tong Pai Biotechnology Co., Ltd (https://www.biomart.cn/47979/index.htm).
HPAEpiC cells were cultured in DMEM containing 10% FBS, 100 U/ml
penicillin and 100 U/ml streptomycin. The cells were cultured in
37°C incubators under a 5% CO2 atmosphere and the medium
was changed every other day. When the cells grew to ~70%
confluence, HPAEpiC cells were digested with 0.25% trypsin and
subcultured at a 1:3 ratio. Cells in the logarithmic growth phase
were selected for experimental treatment. The cells were divided
into four groups: i) Control group; ii) PQ group; iii) PQ + AKF-PD
group; and iv) AKF-PD group. The control group cells were grown in
culture medium without any intervention factors. PQ group cells
were grown with the addition of 300 µmol/l PQ to stimulate cells
for 24 h. The PQ+AKF-PD group was pretreated with 1 mmol/AKF-PD for
1 h and then 300 µmol/l PQ was added for a 24-h treatment. The
AKF-PD group cells were grown with the addition of only 1 mmol/l
AKF-PD, without PQ. Cells were cultured in an incubator with 5%
CO2 at 37°C.
Cell viability examination
HPAEpiC cells in the logarithmic growth phase were
seeded in a 96-well plate and cultured in a 37°C incubator. When
the cells reached 80% confluence, the cells were incubated with PQ
at 0, 50, 100, 200, 300, 400, 500, 600, 700 and 800 µmol/l,
respectively. The cell culture supernatant was collected 24 h later
to detect the content of lactate dehydrogenase (LDH) using the
ELISA kit. Then, the adherent HPAEpiC cells were cultured in a
mixture of CCK-8 solution and culture medium at 37°C for 2 h, and
the absorbance was measured at 450 nm with a microplate reader. The
optimal concentration of PQ was selected. Next, the cells were
pretreated with increasing concentrations of AKF-PD (0.5, 1, 1.5,
2, 2.5, 3, 3.5 and 4 mmol/l) for 1 h, then stimulated with PQ for
24 h. Cell viability was detected by CCK-8 and LDH assay.
Detection of inflammatory factors in
each group of cells
The supernatant of each group was removed and
inflammatory factors, including IL-1β, IL-6 and TNF-α, were
measured using an ELISA kit. All the operation steps were strictly
in accordance with the kit instructions.
Immunofluorescence
The levels of p62 were also detected by using
immunofluorescence. Coverslips covered with HPAEpiC cells were
rinsed with PBS for 3 min at room temperature. Then, the cells were
fixed with 4% paraformaldehyde for 15 min at room temperature and
permeabilized with 0.3% Triton X-100 (Sigma-Aldrich; Merck KGaA)
for 15 min at room temperature. The coverslips were then washed
with PBS for 3 times, followed by incubating 3% BSA (Sigma-Aldrich;
Merck KGaA) for 30 min at room temperature. The cells were
incubated at 4°C overnight in rabbit anti-p62 primary antibody
(cat. no. ab51480; 1:300; Abcam) and were then incubated in
Cy3-AffiniPure Goat Anti-Rabbit IgG (cat. no. Ab45360; 1:200;
Abcam) at 37°C for 1 h. Finally, the cells were washed three times
with PBS and then incubated with DAPI (cat. no. D9435;
Sigma-Aldrich; Merck KGaA) for 5 min at room temperature. Then, the
cells were observed and the images were captured under a
fluorescence microscope. All experiments were conducted
independently at least 3 times. Stained cells were visualized using
an SZX12 fluorescent microscope (Olympus Corporation;
magnification, ×200).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from HPAEpiC cells was extracted with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), and reverse-transcribed to cDNA using the PrimeScript™ RT
Reagent kit with gDNA Eraser (Takara Biotechnology Co., Ltd.),
according to the manufacturer's protocols. The cDNA templates were
amplified using the TB Green® Fast qPCR Mix (Takara
Biotechnology Co., Ltd.). The following primers were used: α-SMA,
5′-CGGGACATCAAGGAGAAACT-3′ and reverse 5′-CCCATCAGGCAACTCGTAA-3′;
TGF-β, forward 5′-TCGACATGGAGCTGGTGAAA-3′ and reverse
5′-GAGCCTTAGTTTGGACAGATCTG-3′; β-actin, forward
5′-CAAATGTGTTCAGCTCAGCCAGCA-3′ and reverse
5′-CTGGAAGGTGGACAGCGAGG3′. Samples were denatured at 95°C for 30
sec, and then PCR amplification was achieved by 40 cycles at 95°C
for 5 sec and 60°C for 15 sec using the Applied Biosystems 7500
Real-Time PCR system (Thermo Fisher Scientific, Inc.). At least two
independent experiments were conducted, and β-actin was used for
normalization. The expression levels of mRNA were quantified using
the 2−ΔΔCq method (23).
Western blotting analysis
The lung tissues and HPAEpiC cells were homogenized
in RIPA lysis buffer (Thermo Fisher Scientific, Inc.). A protein
assay kit (BCA; Thermo Scientific, Inc.) was used to measure the
total protein in samples. After quantification using a BCA kit
(Thermo Fisher Scientific, Inc.), a total of 10 µg protein samples
were separated via 10% sodium dodecyl sulfate-poly-acrylamide gel
electrophoresis, and subsequently transferred to a PVDF membrane
and blocked with 5% milk (w/v) at room temperature for 1 h. The
membrane was incubated overnight with primary antibodies at 4°C.
The primary antibodies used were TGF-β1, E-cadherin, α-SMA, Akt,
p-Akt, mTOR, p-mTOR, PI3K, p-PI3K, LC3, p62 and β-actin (all
Abcam). β-Actin was used as a loading control to normalize the
data.
The membrane was washed 3 times with TBS containing
0.1% Tween-20 (TBST) and was incubated at room temperature for 1 h
with horseradish peroxidase-linked anti-rabbit antibodies (cat. no.
ab6822; Abcam) that were diluted 1:2,000 in TBST. An ECL kit (EMD
Millipore) was used for luminescent development, a
chemiluminescence instrument was used to scan the strip and Image
Lab software (v 4.0; Bio-Rad) was used to analyze the gray value of
the strip. Protein expression was normalized to β-actin.
Statistical analysis
GraphPad Prism (GraphPad Software, Inc.) and SPSS
22.0 (IBM Corp.) software were used for statistical analysis of the
data. All experiments were repeated at least three times. The data
are presented as the mean ± standard deviation. The comparisons
between two groups were analyzed using an unpaired Student's
t-test. The comparisons among multiple groups were analyzed using
ANOVA followed by Tukey's post hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
AKF-PD alleviates PQ-induced pulmonary
fibrosis in rats
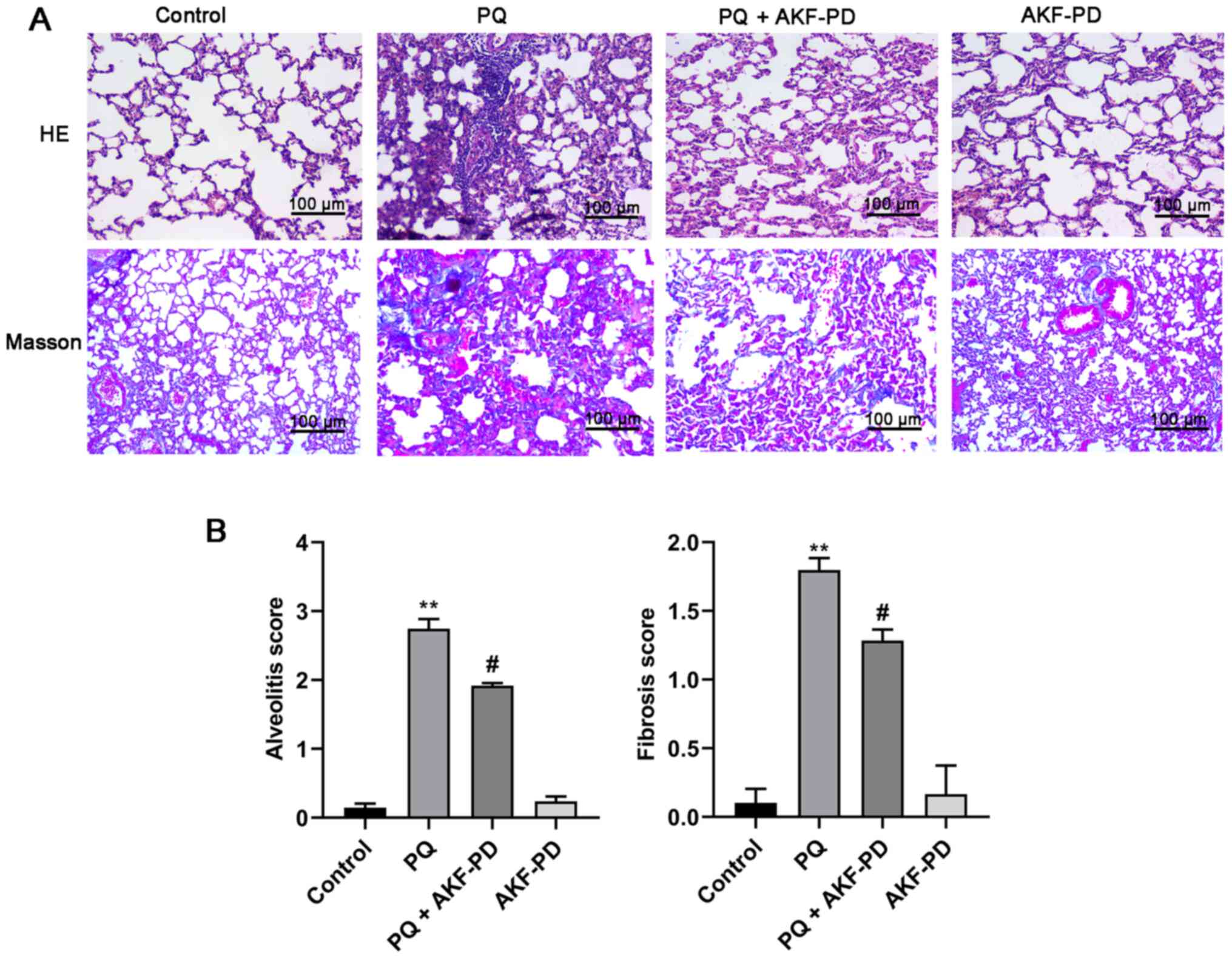
To investigate the effect of AKF-PD on PQ-induced
pulmonary fibrosis, HE and Masson staining were used to observe the
pathological changes in the lung tissue for each group. Fig. 1A shows that in the PQ group, a
significant thickening of the alveolar septa was observed with
increased deposition of collagen in lung tissues at 14 days
following PQ administration compared with the control (24). However, the degree of pulmonary
fibrosis was significantly mitigated after 14 days of AKF-PD
treatment compared with the PQ-treated rats. The degree of
alveolitis and pulmonary fibrosis was graded according to Szapiel's
semi-quantitative method (Fig. 1B).
Compared with the control group, the degree of alveolitis and
pulmonary fibrosis in the PQ group was significantly increased
(P<0.01). However, compared with the PQ group, the degree of
alveolitis and pulmonary fibrosis in the PQ + AKF-PD group was
significantly decreased (P<0.05). Additionally, there was no
significant difference between the AKF-PD group and the control
group.
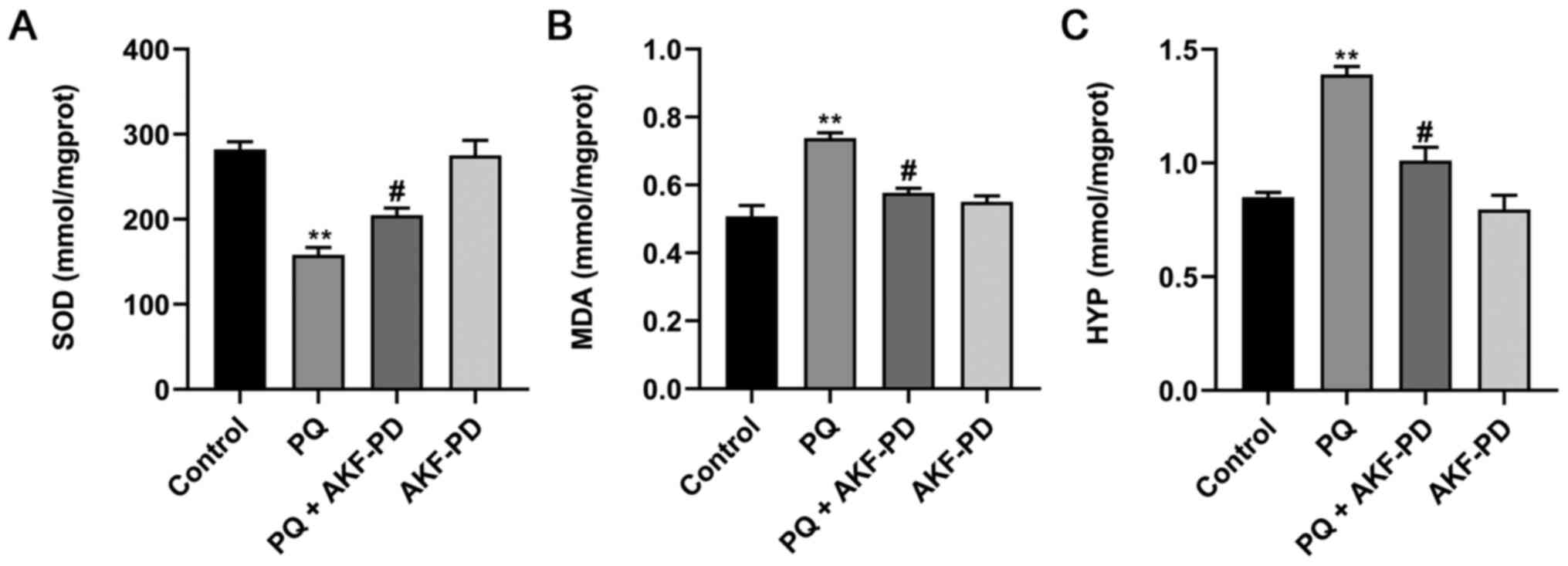
AKF-PD reduces oxidative stress
parameters and the collagen content in lung tissue
AKF-PD and PQ were administered to the rats for 14
days. The levels of SOD, MDA and HYP in lung tissues were then
evaluated to assess the impact of AKF-PD on oxidative stress and
the collagen content in PQ-poisoned rats. As shown in Fig. 2, the level of SOD significantly
decreased after 14 d of PQ treatment, but the levels of MDA and HYP
significantly increased compared with the control (all P<0.01).
However, AKF-PD treatment significantly increased the level of SOD
and decreased the levels of MDA and HYP compared with the PQ group
(all P<0.05). There was no significant difference between the
AKF-PD group and the control group.
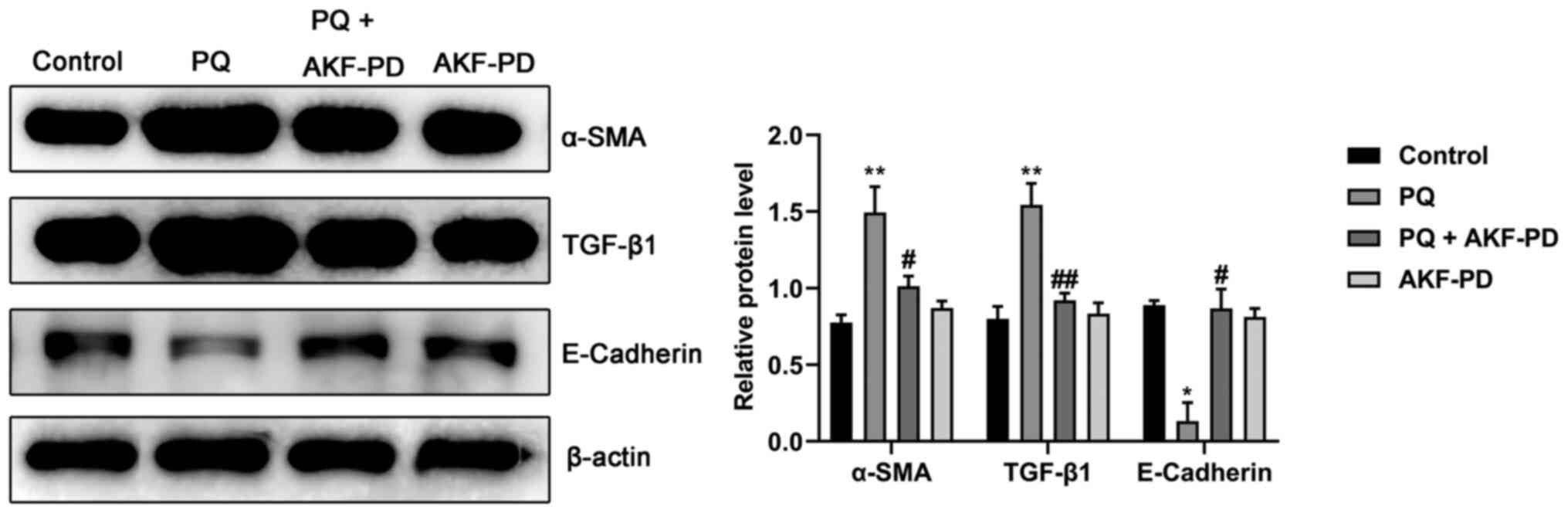
Effect of AKF-PD on the expression of
fibrosis-related proteins in the lung tissue of PQ-treated
rats
To further confirm that AKF-PD can alleviate
PQ-induced pulmonary fibrosis, the expression of fibrosis-related
proteins was measured using western blotting in the lung tissue of
rats in each group. As shown in Fig.
3, the expression levels of α-SMA and TGF-β1 protein in the
lung tissue of the PQ group significantly increased (P<0.01),
whereas the expression level of E-cadherin significantly decreased
compared with the control (P<0.05). However, AKF-PD treatment
significantly decreased the protein levels of α-SMA (P<0.05) and
TGF-β1 (P<0.01) and increased the level of E-cadherin
(P<0.05). Moreover, the expression levels of α-SMA, TGF-β1 and
E-cadherin in the AKF-PD group were not significantly different
from those in the control group.
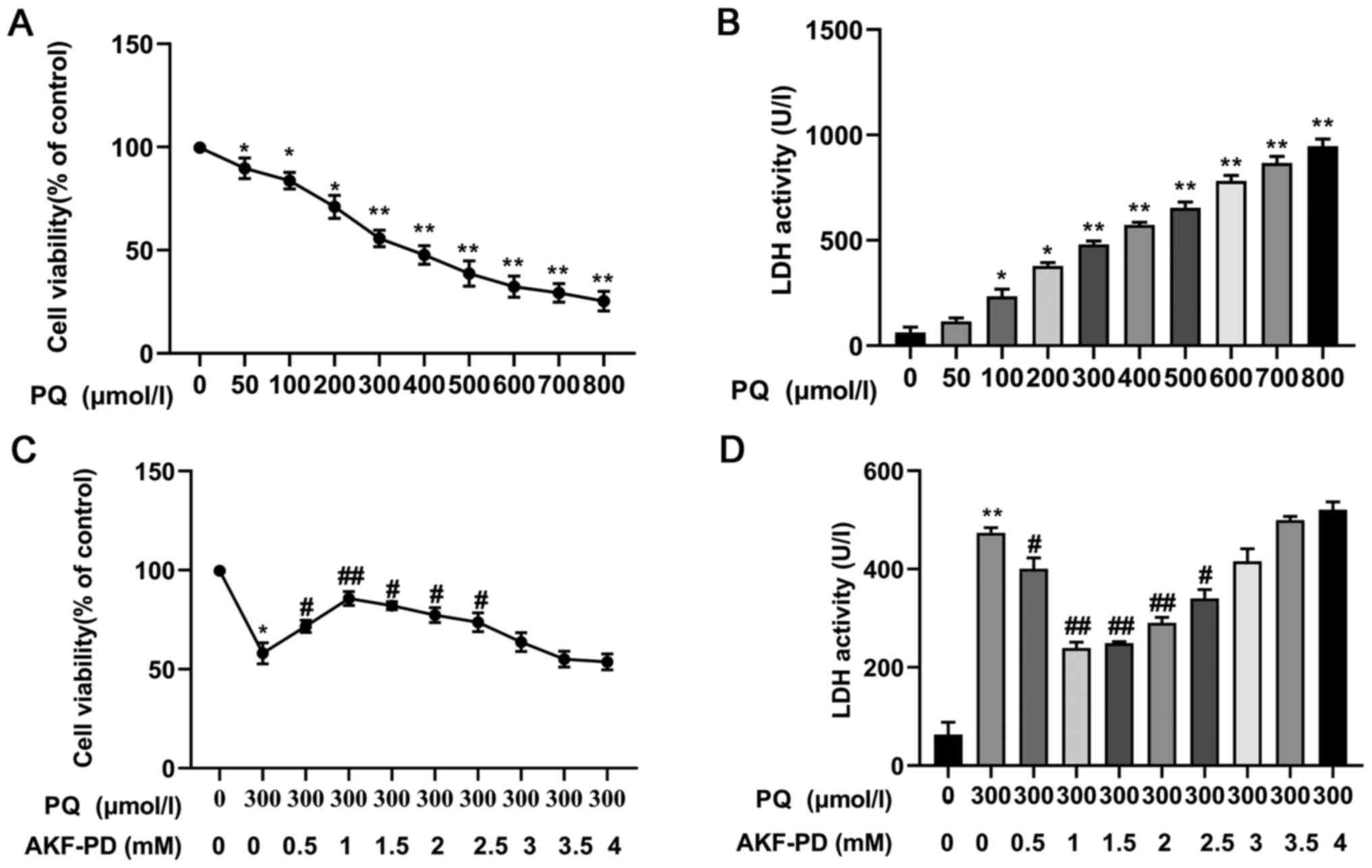
Effect of AKF-PD on cell viability
stimulated by PQ
To determine the effect of AKF-PD on the viability
of HPAEpiC cells, CCK-8 assays and LDH release assays were
performed. The cells were incubated with different concentrations
of PQ, and cell viability was measured at 24 h. As shown in
Fig. 4A, cell viability gradually
decreased significantly with the increase in the PQ concentration,
and Fig. 4B shows how LDH leakage
gradually increased significantly (all P<0.05). The median
lethal dose (IC50) of PQ was 300 µmol/l and, therefore,
the concentration of 300 µmol/l was used in subsequent experiments.
Next, the cells were treated with increasing concentrations of
AKF-PD for 1 h, then stimulated with 300 µmol/l PQ for 24 h. As
shown in Fig. 4C and D, AKF-PD
(0.5, 1, 1.5, 2 and 2.5 mmol/l) significantly ameliorated the
decrease in HPAEpiC cell viability induced by PQ, with the optimum
concentration at 1 mmol/l (P<0.05). Therefore, the concentration
of 1 mmol/l was used for subsequent experiments.
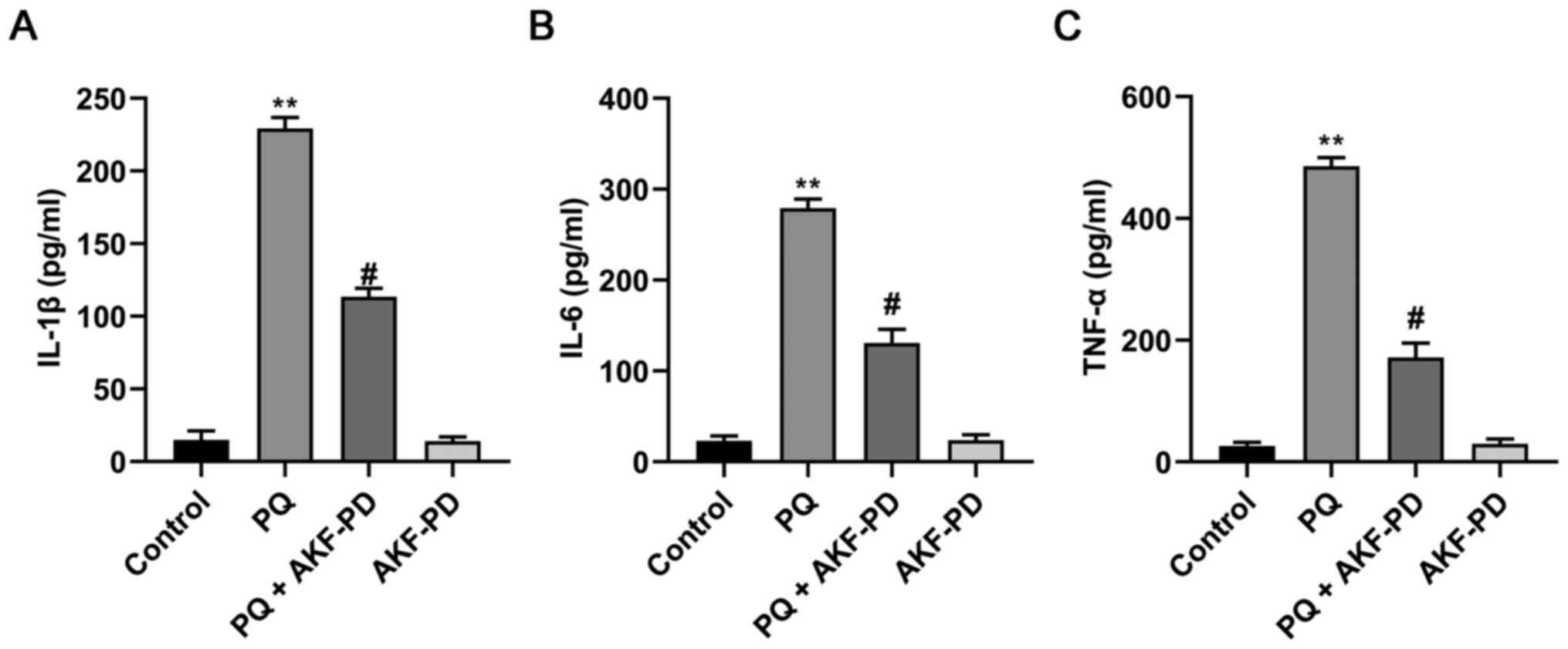
Effect of AKF-PD on the expression of
PQ-induced inflammatory factors
The concentrations of IL-1β, IL-6 and TNF-α in the
supernatant of HPAEpiC cells were quantitated using ELISA. As shown
in Fig. 5, IL-1β, IL-6 and TNF-α
levels in the PQ group significantly increased compared with the
control group (P<0.01). Compared with the PQ group, however,
IL-1β, IL-6 and TNF-α levels in the PQ + AKF-PD group significantly
decreased (P<0.05). These results suggested that AKF-PD could
inhibit PQ-induced inflammation in HPAEpiC cells.
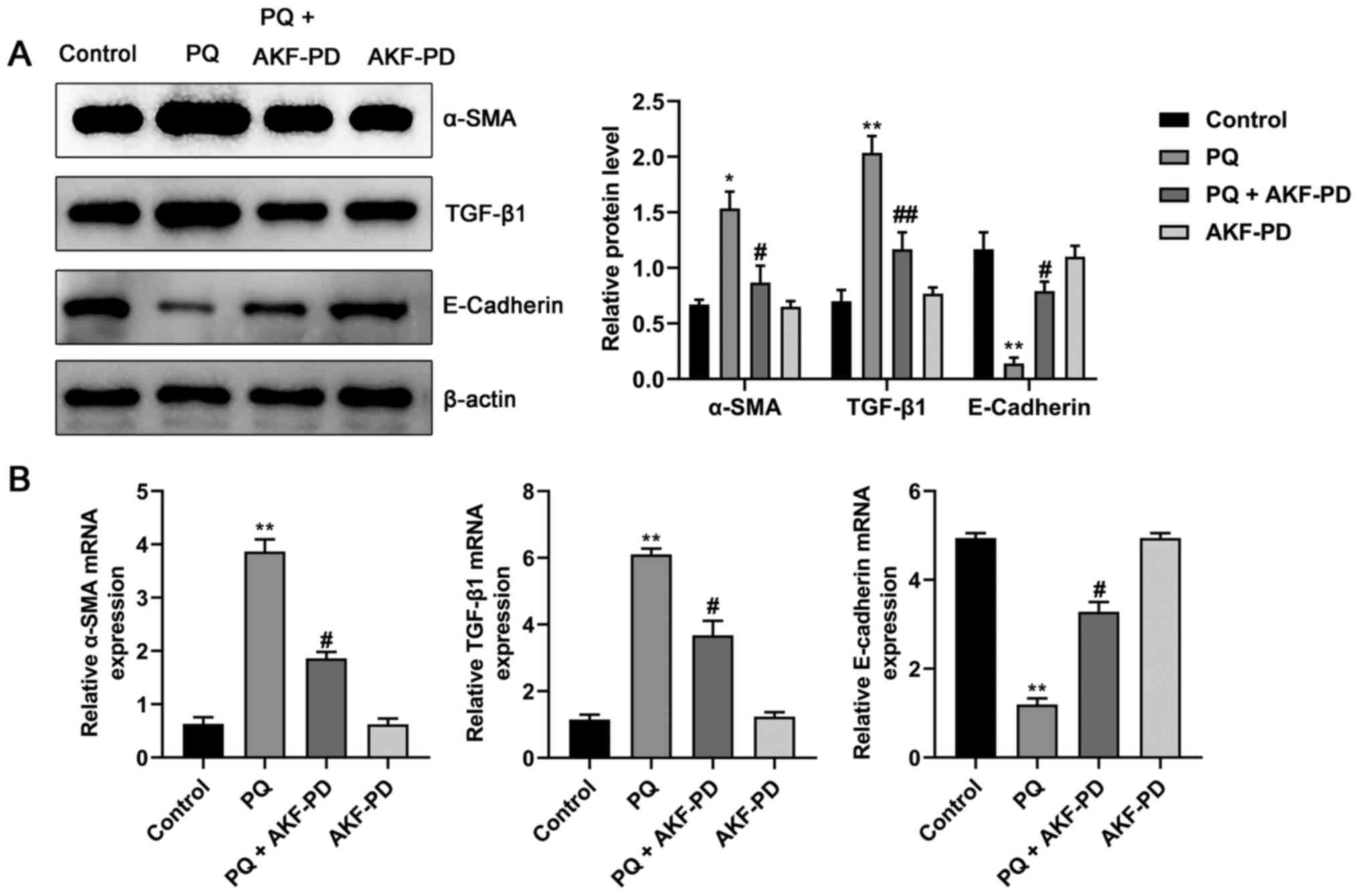
Effect of AKF-PD on the α-SMA, TGF-β1
and E-cadherin expression in HPAEpiC cells
To further verify the effect of AKF-PD on PQ-induced
pulmonary fibrosis and its potential mechanism, the protein and
mRNA levels of α-SMA, TGF-β1 and E-cadherin in HPAEpiC cells were
measured using western blotting and RT-qPCR. As shown in Fig. 6A and B, compared with the control
group, the mRNA and protein levels of α-SMA and TGF-β1 were
significantly increased, and the expression of E-cadherin protein
was significantly decreased (all P<0.05). However, compared with
the PQ group, the expression levels of α-SMA and TGF-β1 in the
PQ+AKF-PD group were significantly decreased, while the expression
of E-cadherin was significantly increased (P<0.05).
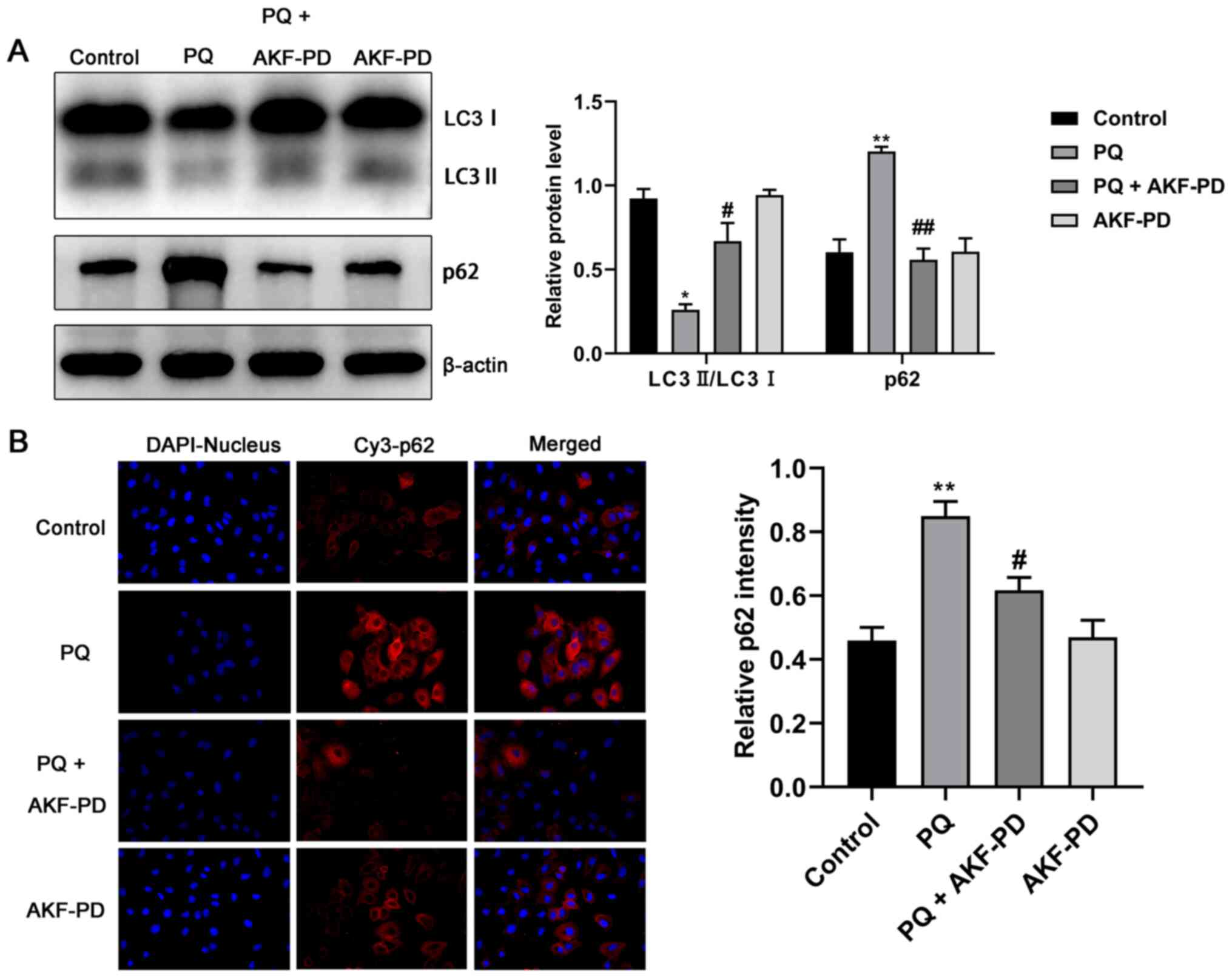
AKF-PD inhibits the development of
pulmonary fibrosis by increasing autophagy
The expression levels of autophagy-related proteins
LC3 and p62 were measured using western blotting and an
immunofluorescence test, and the results are shown in Fig. 7A. In the PQ group, LC3 II/LC3 I was
significantly decreased (P<0.05) and p62 increased (P<0.01)
compared with the control. However, in the PQ + AKF-PD group, LC3
II/LC3 I expression significantly increased (P<0.05) while p62
decreased (P<0.01) compared with the PQ group. Moreover, the
immunofluorescence images in Fig.
7B were consistent with the aforementioned results, which
indicated that AKF-PD pretreatment significantly decreased the
expression of p62 compared with PQ treated cells.
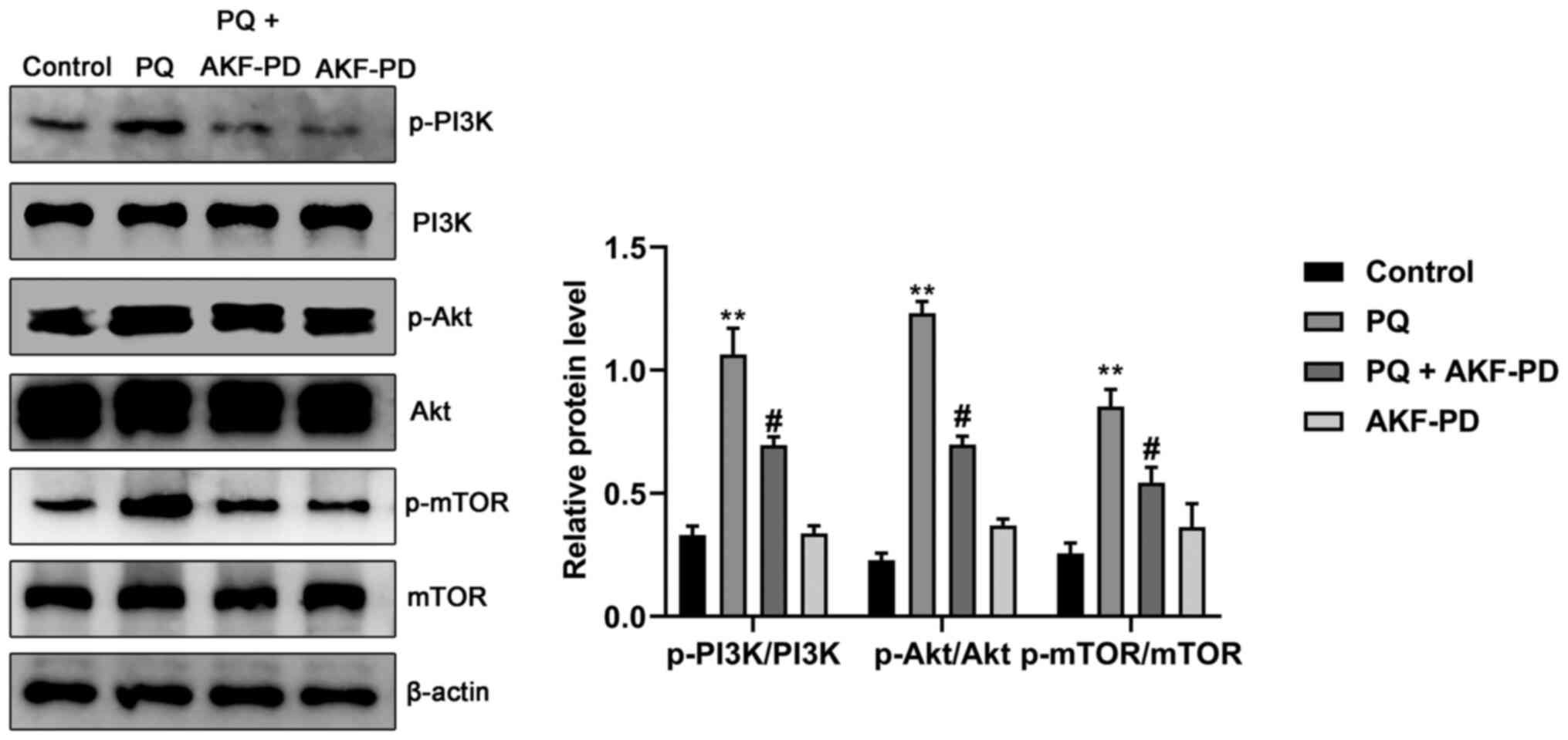
AKF-PD inhibits the development of
pulmonary fibrosis by regulating the PI3K/Akt/mTOR signaling
pathway
To investigate the potential underlying mechanisms
of AKF-PD, the expression levels of proteins associated with the
PI3K/Akt/mTOR signaling pathway were observed. As seen in Fig. 8, the p-Akt/Akt, p-mTOR/mTOR and
p-PI3K/PI3K ratios in the PQ group were significantly upregulated
compared with the control group (all P<0.01), indicating that
the PI3K/Akt/mTOR signaling pathway was activated during PQ-induced
pulmonary fibrosis. However, AKF-PD treatment significantly
reversed the PQ-induced increase in p-Akt/Akt, p-mTOR/mTOR and
p-PI3K/PI3K expression (all P<0.05). Furthermore, there was no
significant difference in the protein expression levels of
p-Akt/Akt, p-mTOR/mTOR or p-PI3K/PI3K protein in the cells treated
with only AKF-PD compared with the control group.
Discussion
PQ primarily accumulates in lung tissue, where it
causes alveolar epithelial cell rupture, edema, interstitial
changes and inflammatory cell infiltration. Moreover, increased
fibroblast proliferation and collagen deposition eventually lead to
pulmonary fibrosis (25).
Therefore, intervention and mitigation of pulmonary fibrosis are
potential strategies for treating PQ poisoning.
The results of HE and Masson staining in the present
study showed that AKF-PD treatment significantly reduced the degree
of alveolitis and fibrosis. It was preliminarily confirmed that
AKF-PD reduced the pathological damage to lung tissue in
PQ-poisoned rats. In addition, after PQ poisoning, the body
releases a large amount of oxygen free radicals, leading to a large
consumption of SOD that results in an increase in MDA (26). These two oxidative stress markers
reflect the level of damage caused by oxygen-free radicals
(27). HYP is an indicator used to
evaluate fibroblast proliferation and collagen deposition (28). Therefore, the present study
evaluated the toxicity of PQ by measuring the concentrations of
MDA, SOD and HYP in lung tissue. The results indicated that AKF-PD
reduced the MDA and HYP levels in the lung tissue of rats with PQ
poisoning and increased SOD, indicating that AKF-PD can effectively
alleviate PQ-induced oxidative stress and collagen deposition.
Epithelial-mesenchymal transition (EMT) is also an
notable part in the pathological process of pulmonary fibrosis.
Alveolar epithelial cells undergo continuous damage and gradually
transform into mesenchymal cells (29). At the same time, the expression of
E-cadherin decreases and the expression of α-SMA increases
(30). TGF-β1 is a key regulator of
EMT, and excessive activation of TGF-β1 can induce EMT in alveolar
epithelial cells, which promotes the occurrence and development of
pulmonary fibrosis (31).
In the present study, western blotting results
showed that AKF-PD reduced the expression of TGF-β1 and α-SMA in
the lung tissue of PQ-poisoned rats, and increased the expression
of E-cadherin. These results indicated that AKF-PD may reduce the
content of fibrin by inhibiting the expression of TGF-β1, thereby
alleviating pulmonary fibrosis. Furthermore, HPAEpiC cells were
used to determine the specific function of AKF-PD during the
pathogenesis of EMT. In addition, the current study demonstrated
that AKF-PD (0.5, 1, 1.5, 2 and 2.5 mmol/l) pretreatment
significantly ameliorated the decrease in HPAEpiC cell viability
induced by PQ. Notably, the protective ability of AKF-PD decreased
after 1 mmol/l, suggesting 1 mmol/l may be the maximum safety limit
of AKF-PD, and excessive drug concentration can produce toxic
effects on cells.
Autophagy is generally considered to be a
lysosomal-dependent self-degradation pathway in the development of
eukaryotes that plays a notable role in maintaining cell
homeostasis (32). A decrease in
autophagy activity is considered to be the key process in the
pathogenesis of pulmonary fibrosis (33). According to previous reports,
insufficient autophagy promotes the expression of α-SMA and
fibronectin and increases the deposition of the extracellular
matrix (34–36). LC3 and p62 are markers of autophagy.
When the expression level of LC3 II/LC3 I decreases, a large amount
of p62 accumulates in cells, which then reduces autophagy activity
and promotes lung fibrosis (37).
The present study demonstrated that AKF-PD treatment decreased the
expression of TGF-β1 and α-SMA and increased autophagy, indicating
that AKF-PD can inhibit the EMT by enhancing autophagy, thereby
alleviating pulmonary fibrosis.
The PI3K/Akt/mTOR signaling pathway plays a key
regulatory role in autophagy (38).
Under physiological conditions, growth factors and cytokines
activate tuberous sclerosis 1 proteins 1 and 2 by activating PI3K
and phosphorylating Akt, which then activates mTOR. Activated mTOR
(p-mTOR) promotes protein synthesis and inhibits autophagy by
phosphorylating downstream ribosomal protein S6 kinase 1 and
eukaryotic translation initiation factor 4E binding protein 1
(39). Continuous activation of the
PI3K/Akt/mTOR signaling pathway has been previously observed in
lung fibroblasts of patients with pulmonary fibrosis; wherein the
expression of p62 increased and the expression of LC3 II/LC3 I
decreased (40). This showed that
the activation of the PI3K/Akt/mTOR pathway in alveolar epithelial
cells decreases autophagy activity and subsequently contributes to
the pathogenesis of pulmonary fibrosis. In the present study,
AKF-PD led to a decrease in p-Akt/Akt, p-mTOR/mTOR and p-PI3K/PI3K
ratios, demonstrating that AKF-PD can inhibit the activation of the
PI3K/Akt/mTOR signaling pathway induced by PQ.
In conclusion, the current study indicated that
AKF-PD inhibited the activation of the PI3K/Akt/mTOR signaling
pathway, enhanced autophagy and suppressed the occurrence of EMT,
thereby alleviating the pulmonary fibrosis caused by PQ poisoning.
The present study explored the effect and mechanism of AKF-PD on
pulmonary fibrosis caused by PQ poisoning and laid the groundwork
for investigation into future therapeutic strategies that can be
used to treat pulmonary fibrosis.
Acknowledgements
Not applicable.
Funding
This work was supported by The Scientific Research
Project of Hunan Provincial Commission of Health and Family
Planning (grant no. B2017076), The Changsha Science and Technology
Department (grant no. kq1901051).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YJ and WL conceived the study. FJ and SL designed
the methodology, wrote the original draft manuscript, and revised
and edited the manuscript. XY, TW, and ZC analyzed the data. WL and
YJ confirm the authenticity of all the raw data. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental protocol used in this study was
approved by The Committee of Experimental Animals of the First
People's Hospital of Hunan Province.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Samsamshariat S, Vedaei A, Jahangiri S,
Gavarti MB, Sami R, Taheri A and Dorooshi G: Report of a case of
paraquat poisoning and mediastinal involvement. Adv Biomed Res.
10:52021. View Article : Google Scholar
|
|
2
|
Zhu Y, Tan J, Xie H, Wang J, Meng X and
Wang R: HIF-1α regulates EMT via the Snail and β-acatenin pathways
in paraquat poisoning-induced early pulmonary fibrosis. J Cell Mol
Med. 20:688–697. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Suntres ZE: Role of antioxidants in
paraquat toxicity. Toxicology. 180:65–77. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xiao Q, Wang W, Qi H, Gao X, Zhu B, Li J
and Wang P: Continuous hemoperfusion relieves pulmonary fibrosis in
patients with acute mild and moderate paraquat poisoning. J Toxicol
Sci. 45:611–617. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tao W, Shu Y, Miao Q and Zhu Y:
Attenuation of hyperoxia-induced lung injury in rats by
adrenomedullin. Inflammation. 35:150–157. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shen H, Wu N, Wang Y, Zhao H, Zhang L, Li
T and Zhao M: Chloroquine attenuates paraquat-induced lung injury
in mice by altering inflammation, oxidative stress and fibrosis.
Int Immunopharmacol. 46:16–22. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bai L, Li A, Gong C, Ning X and Wang Z:
Protective effect of rutin against bleomycin induced lung fibrosis:
Involvement of TGF-β1/α-SMA/Col I and III pathway. Biofactors.
46:634–644. 2020. View Article : Google Scholar
|
|
8
|
Qian J, Ye Y, Lv L, Zhu C and Ye S: FTY720
attenuates paraquat-induced lung injury in mice. Int
Immunopharmacol. 21:426–431. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu S, Hu H, Jiang Z, Tang S, Zhou Y, Sheng
J, Chen J and Cao Y: APACHE score, severity index of paraquat
poisoning, and serum lactic acid concentration in the prognosis of
paraquat poisoning of Chinese patients. Pediatr Emerg Care.
31:117–121. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang H, Zhang W, Xie T, Wang X and Ning W:
Fluorofenidone inhibits apoptosis of renal tubular epithelial cells
in rats with renal interstitial fibrosis. Braz J Med Biol Res.
52:e87722019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song C, He L, Zhang J, Ma H, Yuan X, Hu G,
Tao L, Zhang J and Meng J: Fluorofenidone attenuates pulmonary
inflammation and fibrosis via inhibiting the activation of NALP3
inflammasome and IL-1β/IL-1R1/MyD88/NF-κB pathway. J Cell Mol Med.
20:2064–2077. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma H, Peng Z, Hu G and Tao L: Effect and
mechanism of fluorofenidone on organ fibrosis. Zhong Nan Da Xue Xue
Bao Yi Xue Ban. 40:208–213. 2015.(In Chinese). PubMed/NCBI
|
|
13
|
Meng J, Zou Y, Hu C, Zhu Y, Peng Z, Hu G,
Wang Z and Tao L: Fluorofenidone attenuates bleomycin-induced
pulmonary inflammation and fibrosis in mice via restoring caveolin
1 expression and inhibiting mitogen-activated protein kinase
signaling pathway. Shock. 38:567–573. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang J, Li J, Li G, Zhang H, Wang L, Li D
and Ding J: Spermidine-mediated poly(lactic-co-glycolic acid)
nanoparticles containing fluorofenidone for the treatment of
idiopathic pulmonary fibrosis. Int J Nanomedicine. 12:6687–6704.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xu M, Li S, Wang J, Huang S, Zhang A,
Zhang Y, Gu W, Yu X and Jia Z: Cilomilast ameliorates renal
tubulointerstitial fibrosis by inhibiting the TGF-β1-Smad2/3
signaling pathway. Front Med (Lausanne). 7:6261402021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu J, Song C, Xiao Q, Hu G, Tao L and
Meng J: Fluorofenidone attenuates TGF-β1-induced lung fibroblast
activation via restoring the expression of caveolin-1. Shock.
43:201–207. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saxton RA and Sabatini DM: mTOR signaling
in growth, metabolism, and disease. Cell. 168:960–976. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alayev A and Holz MK: mTOR signaling for
biological control and cancer. J Cell Physiol. 228:1658–1664. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gui YS, Wang L, Tian X, Xue L, Ma A, Zhou
W, Zeng N, Ji Z, Cai B, Zhang H, et al: mTOR overactivation and
compromised autophagy in the pathogenesis of pulmonary fibrosis.
PLoS One. 10:e01386252015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gui X, Chen H, Cai H, Sun L and Gu L:
Leptin promotes pulmonary fibrosis development by inhibiting
autophagy via PI3K/Akt/mTOR pathway. Biochem Biophys Res Commun.
498:660–666. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rieg AD, Said S, Carolin A, Eva V, Rolf R,
Stefan U and Christian M: PDGF-BB regulates the pulmonary vascular
tone: Impact of prostaglandins, calcium, MAPK- and PI3K/AKT/mTOR
signalling and actin polymerisation in pulmonary veins of guinea
pigs. Respir Res. 19:1202018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Szapiel SV, Elson NA, Fulmer JD,
Hunninghake GW and Crystal RG: Bleomycin-induced interstitial
pulmonary disease in the nude, athymic mouse. Am Rev Respir Dis.
120:893–899. 1979.PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu MW, Su MX, Tang DY, Hao L, Xun XH and
Huang YQ: Ligustrazin increases lung cell autophagy and ameliorates
paraquat-induced pulmonary fibrosis by inhibiting PI3K/Akt/mTOR and
hedgehog signalling via increasing miR-193a expression. BMC Pulm
Med. 19:352019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Raghu G, Rochwerg B, Zhang Y, Garcia CA,
Azuma A, Behr J, Brozek JL, Collard HR, Cunningham W, Homma S, et
al: An official ATS/ERS/JRS/ALAT clinical practice guideline:
Treatment of idiopathic pulmonary fibrosis. An update of the 2011
clinical practice guideline. Am J Respir Crit Care Med. 192:e3–19.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang J, Qiao L, Li S and Yang G:
Protective effect of ginsenoside Rb1 against lung injury induced by
intestinal ischemia-reperfusion in rats. Molecules. 18:1214–1226.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu J, Chu Z, Ruan Z, Wang X, Dai T and Hu
X: Changes of intracellular porphyrin, reactive oxygen species, and
fatty acids profiles during inactivation of methicillin-resistant
staphylococcus aureus by antimicrobial blue light. Front Physiol.
9:16582018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamada M, Kuwano K, Maeyama T, Hamada N
Michihiro Yoshimi, Yoichi Nakanishi and Kasper M:
Dual-immunohistochemistry provides little evidence for
epithelial-mesenchymal transition in pulmonary fibrosis. Histochem
Cell Biol. 129:453–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Molina-Molina M, Machahua-Huamani C,
Vicens-Zygmunt V, Llatjós R, Escobar I, Sala-Llinas E,
Luburich-Hernaiz P, Dorca J and Montes-Worboys A: Anti-fibrotic
effects of pirfenidone and rapamycin in primary IPF fibroblasts and
human alveolar epithelial cells. BMC Pulm Med. 18:632018.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chitra P, Saiprasad G, Manikandan R and
Sudhandiran G: Berberine inhibits Smad and non-Smad signaling
cascades and enhances autophagy against pulmonary fibrosis. J Mol
Med (Berl). 93:1015–1031. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu Z, Li Y, Song H, He J, Li G, Zheng Y
and Li B: Collagen peptides promote photoaging skin cell repair by
activating the TGF-β/Smad pathway and depressing collagen
degradation. Food Funct. 10:6121–6134. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eskelinen E: Autophagy: Supporting
cellular and organismal homeostasis by self-eating. Int J Biochem
Cell Biol. 111:1–10. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lv X, Li K and Hu Z: Autophagy and
Pulmonary Fibrosis. Adv Exp Med Biol:. 1207:569–579. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tian K, Chen P, Liu Z, Si S, Zhang Q, Mou
Y, Han L, Wang Q and Zhou X: Sirtuin 6 inhibits epithelial to
mesenchymal transition during idiopathic pulmonary fibrosis via
inactivating TGF-β1/Smad3 signaling. Oncotarget. 8:61011–61024.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mu E, Wang J, Chen L, Lin S, Chen J and
Huang S: Ketogenic diet induces autophagy to alleviate
bleomycin-induced pulmonary fibrosis in murine models. Exp Lung
Res. 47:26–36. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Im J, Hergert P and Nho R: Reduced FoxO3a
expression causes low autophagy in idiopathic pulmonary fibrosis
fibroblasts on collagen matrices. Am J Physiol Lung Cell Mol
Physiol. 309:L552–L561. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xie T, Xu Q, Wan H, Xing S, Shang C, Gao Y
and He Z: Lipopolysaccharide promotes lung fibroblast proliferation
through autophagy inhibition via activation of the PI3K-Akt-mTOR
pathway. Lab Invest. 99:625–633. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Patel AS, Lin L, Geyer A, Haspel JA, An
CH, Cao J, Rosas IO and Morse D: Autophagy in idiopathic pulmonary
fibrosis. PLoS One. 3:e413942012. View Article : Google Scholar
|
|
39
|
Jung C, Ro S, Cao J, Otto N and Kim D:
mTOR regulation of autophagy. FEBS Lett. 584:1287–1295. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Nho R and Hergert P: IPF fibroblasts are
desensitized to type I collagen matrix-induced cell death by
suppressing low autophagy via aberrant Akt/mTOR kinases. PLoS One.
9:e946162014. View Article : Google Scholar : PubMed/NCBI
|