Introduction
Hearing loss is one of the most common sensory
defects in humans, and is often caused by the death of sensory hair
cells (HCs) in the inner ear. HCs function in transducing sound
waves into electric signals in the inner ear (1,2).
Damage induced by a variety of intrinsic and extrinsic sources will
affect cochlear HC function. The factors involved include genetic
factors, ototoxic drugs, aging, chronic cochlear infections and
noise overexposure (3–7); notably, >60% of all cases of
deafness have been attributed to genetic factors (8–10).
Based on inheritance patterns and the presence or absence of
distinctive clinical features, hereditary deafness can be divided
into two categories: Syndromic hearing loss and non-syndromic
hearing loss. The inheritance patterns of non-syndromic hearing
loss include autosomal dominant, autosomal recessive, X-linked and
mitochondrial inheritance. Autosomal dominant non-syndromic hearing
loss (ADNSHL) accounts for ~20% of cases of hereditary hearing loss
(11,12).
In general, patients with autosomal recessive
non-syndromic hearing loss have an early age of onset and more
pronounced hearing loss. However, the principal manifestation of
ADNSHL is post-lingual progressive sensorineural hearing loss,
which begins with impairment in hearing high frequencies. Hearing
loss is genetically heterogeneous. To date, >60 loci for ADNSHL
have been mapped, and ~40 genes for ADNSHL have been identified
(Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage.
https://hereditaryhearingloss.org;
accessed March 11, 2021). ADNSHL contains a number of different
disease subtypes, DFNA2A being one of them.
The gene encoding potassium voltage-gated channel
subfamily Q member 4 (KCNQ4) (Gene MIM number 603537; Online
Mendelian Inheritance in Man. https://omim.org/entry/603537; accessed March 9,
2021).was discovered and cloned by Kubisch et al (13) in 1999. KCNQ4 is mapped to
1p34, within a region that encompasses the deafness non-syndromic
autosomal dominant 2A (DFNA2A; Phenotype MIM number 600101) locus
(Online Mendelian Inheritance in Man. https://omim.org/entry/600101; accessed March 9, 2021)
(14). The cDNA of the KCNQ4
gene, which includes 14 exons, encodes a polypeptide of 695 amino
acids with a mass of ~77 kDa. Pathogenic variants of the
KCNQ4 gene have been shown to cause progressive hearing
loss, and the gene is inherited in an autosomal dominant manner.
This gene is linked to the DFNA2A locus on chromosome 1p34,
encoding the KCNQ4 protein. It is only expressed on sensory outer
HCs, and not in the inner HCs or the stria vascularis in the inner
ear (15). The major function of
KCNQ4 protein is to contribute to potassium ion circulation in
stimulated HCs (16,17). The major process of pathogenesis
associated with KCNQ4 affects potassium recycling in the endolymph,
which subsequently changes the nature of its electrolyte milieu and
reduces the endocochlear potential (18–20).
Four subunits associate with each other to form the
voltage-gated channel. This arrangement allows potassium ions to
selectively cross the cell membrane (19). Each subunit consists of six
transmembrane domains, intracellular N- and C-termini, and a pore
region (21). Six exons (exons 2–7)
encode the six transmembrane segments (S1-S6). The S4 segment
contains the voltage sensor of the channel, whereas the S5 and S6
segments are linked through the P-loop domain to form the pore
region.
The KCNQ4 gene is one of the most commonly
mutated genes found in patients with ADNSHL, and this gene has been
shown to lead to DFNA2A. To date, ~30 pathogenic variants in this
gene have been reported as the cause of DFNA2A (The Human Gene
Mutation Database at the Institute of Medical Genetics in Cardiff;
http://www.hgmd.cf.ac.uk; accessed March 11,
2021). Variant hotspots in the KCNQ4 gene that are linked to
hearing loss have been shown to cluster around the pore region
(22); these variants result in
decreased membrane expression of the channel protein, or loss of
channel function.
The present study reported on the genetic basis of
progressive hearing loss in a Chinese family, in which a novel
missense variant c.857A>G (p.Tyr286Cys) was identified in the
pore region of the KCNQ4 channel via whole-exome sequencing (WES)
and Sanger sequencing.
Patients and methods
Family recruitment and clinical
evaluations
The proband was a 25-year-old woman. Due to hearing
loss, she visited the Department of Otolaryngology, Head and Neck
Surgery, Xijing Hospital in 2018. The patient had a family history
of hearing loss. The patient was part of a five-generation Chinese
family with 46 members. The family had autosomal dominant,
progressive, post-lingual, non-syndromic sensorineural hearing loss
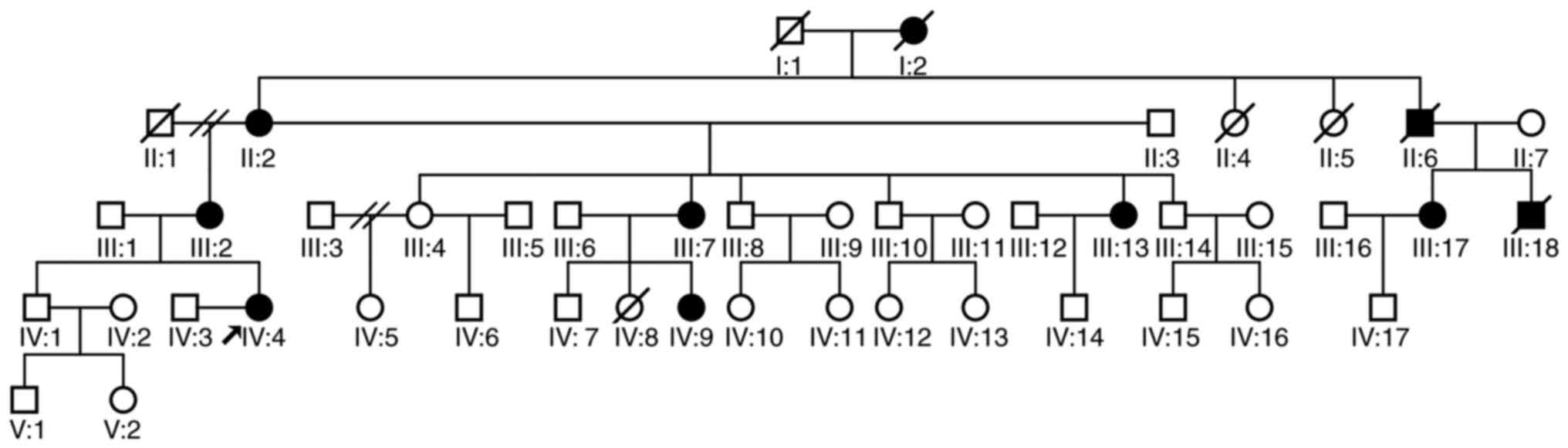
and these features are consistent with ADNSHL (Fig. 1). A total of 11 members of this
family participated in the present study, which included seven
affected and four unaffected relatives. Written informed consent
was obtained from the study participants and the study was
performed in accordance with relevant regulations of Xijing
Hospital. Other members of the family were unwilling to participate
in the present study, and since it was not possible to obtain their
consent, their information has not been used.
The following information was obtained from each
study participant, which included basic information, age of onset,
disease progression, mother's pregnancy and the study participant's
delivery, noise exposure, ototoxic drug use, head trauma,
infectious diseases, family history and other related information.
Physical examinations confirmed a family history of non-syndromic
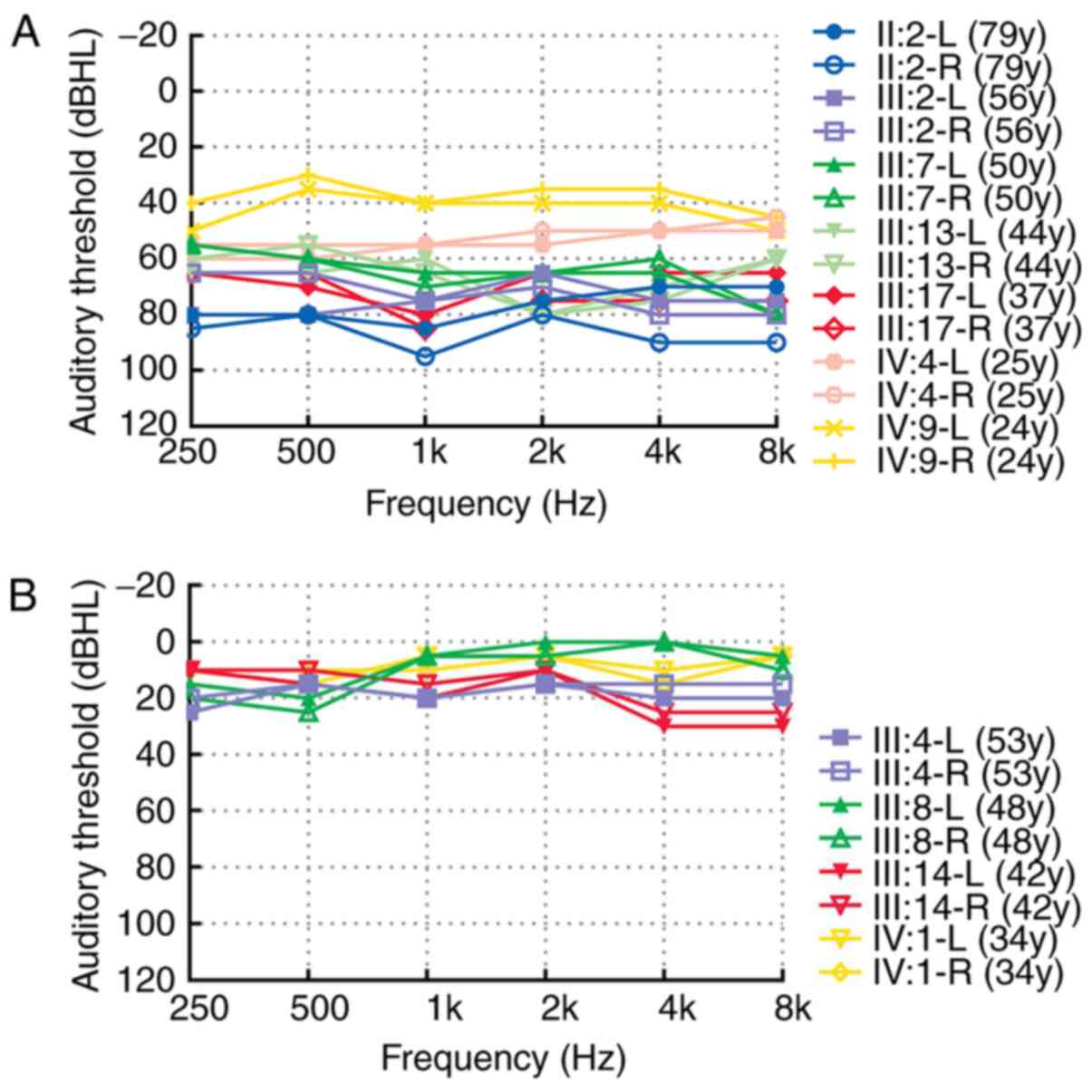
hearing loss. Audiometric evaluations and otological examinations
performed for the proband included electronic otoscope, pure tone
audiometry (PTA), acoustic impedance, distortion production
otoacoustic emission, auditory brainstem response, speech
discrimination score, electronystagmography, vestibular evoked
myogenic potential, tinnitus detection, temporal bone computed
tomography and magnetic resonance imaging. The other family members
were evaluated by PTA. Average value of the thresholds of air
conduction were determined at 500, 1,000, 2,000 and 4,000 Hz to
determine the degree of hearing loss in the family. The condition
of an individual's hearing was assessed through measuring their
hearing loss, and this was classified as mild (26–40 dB HL),
moderate (41–60 dB HL), severe (60–80 dB HL) or profound hearing
loss (≥81 dB HL) (23). Detailed
medical records were established for each family member. The
majority of the patients in this family experienced moderate
hearing loss: They were required to wear hearing aids to improve
their hearing, although they did not require treatment with drugs;
nor had they received surgery. For members of the family who were
unwilling to participate in the present study, and for whom it was
not possible to obtain their consent, their data have not been
used.
DNA extraction
Genomic DNA from the 11 study participants was
extracted from peripheral blood leukocytes using a blood DNA
extraction kit (cat. no. CW0544M; Kangwei Century Biotech Co.,
Ltd.; http://www.cwbiotech.com/goods/index/id/10199). DNA
concentration and purity were measured using an ultraviolet NanoQ
Spectrophotometer (CapitalBio Technology, Inc.).
Screening for variants in SLC26A4,
GJB2, GJB3 and mitochondrial 12S rRNA
SLC26A4, GJB2, GJB3 and mitochondrial 12S
rRNA are the most common cause of hearing loss in the Chinese
population (24). Sanger sequencing
for SLC26A4, GJB2, GJB3 and mitochondrial 12S rRNA was
performed using PCR amplification and direct sequencing of exons.
The primers referred to the studies of Dai et al (25) and Yuan et al (26). The PCR conditions and Sanger
sequencing are described in the Segregation analysis sanger
sequencing section of the text.
WES
Whole-exome capture was performed using the Agilent
SureSelect V5 enrichment capture kit (Agilent Technologies, Inc.).
The qualified genomic DNA was randomly fragmented to an average
size of 180–280 bp by Covaris S220 sonicator (Covaris Inc.). Next,
the DNA fragments were end-repaired and phosphorylated, followed by
A-tailing and ligation at the 3′-ends with paired-end adaptors
(Illumina, Inc.) with a single ‘T’ base overhang; the purification
was carried out using Agencourt SPRIselect (cat. no. B23317 Beckman
Coulter Inc.). Then, the size distribution and concentration of the
libraries (2 nM) were determined by Agilent 2100 Bioanalyzer and
quantified using PCR. Finally, the DNA library was sequenced on
Illumina HiSeq 4000 (Illumina Inc.) for paired-end 150-bp reads.
The sequence reads were aligned to the human reference genome (UCSC
hg19/hg38) (UCSC Genome Browser; https://genome.ucsc.edu; accessed March 15, 2021)
(27) using the Burrows-Wheeler
Alignment (28) to get BAM file.
SAMtools (29) and Sambamba
(30) were used to sort BAM files
and perform duplicate marking, local realignment, and base quality
recalibration for coverage and depth analysis. Duplicate reads were
removed for variants calling. ANNOVAR (31) software was used to annotate single
nucleotide polymorphism (SNP), insertion and deletion (Indel) and
copy number variation (CNV). The variants were filtered based on
the monogenic autosomal dominant trait. First, variants with minor
allele frequencies >1% in the 1000 Genomes database (32), the NHLBI Exome Sequencing Project
(ESP6500) [Exome Variant Server, NHLBI GO Exome Sequencing Project
(ESP); accessed March 11, 2021] or the Genome Aggregation Database
(gnomAD; https://gnomad.broadinstitute.org; accessed March 13,
2021) were excluded. Second, variants present in the homozygous or
hemizygous state in individuals without hearing loss were excluded.
Third, synonymous variants and intronic variants not located within
the splice site regions were excluded. Finally, variants of all
genes known to be monogenic factors for hearing loss were
systematically evaluated.
Segregation analysis sanger
sequencing
Following exome sequencing, segregation analysis of
candidate variants was completed by Sanger sequencing. Sequences of
primers for potential causative variants in the KCNQ4 gene
were designed and synthesized as follows: 5′-TTCCCTCATGATCAGGCT-3′
and 5′-ATCTTGTACCTGGATGAGGTT-3′. PCR was performed with 25 µl
reaction mixtures containing 100 ng of genomic DNA, 1 µl of the
forward and reverse primers, and 22 µl of 1.1X Golden Star T6 Super
PCR Mix (cat. no. TSE101, TsingKe Biological Technology).
Thermocycling was performed using the following program: Initial
denaturation at 98°C for 2 min, followed by 30 cycles of 98°C for
10 sec, 62°C for 10 sec, and 72°C for 10 sec and final extension at
72°C for 1 min. PCR products were purified with the Cycle Pure kit
(cat. no. D6492 OMEGA Bio-Tek) and sequenced using Applied
Biosystems 3730 DNA Analyzer (Thermo Fisher Scientific, Inc.).
Evolutionary conservation
analysis
The target sequence for alignment contained Tyr286
residue as well as its upstream and downstream amino acid residues.
Multiple sequence alignment was performed across 13 species using
BLAT (33) on the UCSC Genome
Browser (https://genome.ucsc.edu).
Three-dimensional (3D) structural
modeling
The BLAST search protocol in BIOVIA Discovery Studio
(DS) (v. 19.1.0×64; 3D EXPERIENCE Company, Dassault Systèmes)
(2019) was used to find template structures that had similar
regions to the domain of KCNQ4 protein. This was accomplished by
searching against the PDB_nr95 sequence database. The residue
sequence of the mammalian Shaker Kv1.2 potassium channel (from
Ser289 to Phe413) bore a 31% similarity to that of KCNQ4 protein
(from Ala325 to Phe449), which included the ion transport domain.
The structure was subsequently retrieved from the Protein Data Bank
(PDBID:2A79) (34,35). The Prepare Protein protocol in DS
was then used to restore missing atoms and remove water molecules.
The tertiary structure of the KCNQ4 domain was constructed based on
the template using the Build Homology Models protocol in DS.
Subsequently, the structure of the mutant (Tyr286→Cys) was
constructed using the Build Mutants Protocol, and then the model
with the highest Discrete Optimized Protein Energy score was
selected. Finally, molecular visualization and 3D images were
processed using PyMol (v2.1; Schrödinger Inc.; http://pymol.org/2/#download).
Results
Clinical description
The family selected for the study had 10 individuals
with hearing loss, three of whom (I:2, II:6, III:18) were deceased
and were unable to participate in the study. Their hearing had
begun to decline gradually at about age 20, and by the time they
are older, they had suffered severe hearing loss. The remaining
seven individuals (II:2, III:2, III:7, III:13, III:17, IV:4 and
IV:9) were all diagnosed with hearing impairment. The remainder of
the family members had good hearing, of which four members (III:4,
III:8, III:14 and IV:1), underwent PTA and their hearing was deemed
normal (Table I). The audiological
condition and clinical history of the hearing-impaired family
members showed progressive, post-lingual, symmetrical, bilateral,
non-syndromic sensorineural hearing loss. The age at onset of
hearing impairment ranged from 20–30 years, and the degree of
hearing loss was positively associated with age.
 | Table I.Phenotype and genotype of individual
family members. |
Table I.
Phenotype and genotype of individual
family members.
| Family member | Age, years | Nucleotide
change | PTA-right, dB
HL | PTA-left, dB
HL | Age of onset,
years | Noise exposure | Ototoxic drugs | Head trauma |
|---|
| II:2 | 79 | c.857A>G | 86.25 | 77.5 | 25 | No | No | No |
| III:2 | 56 | c.857A>G | 71.25 | 73.75 | 30 | No | No | No |
| III:4 | 53 | Wild-type | 17.5 | 16.25 | / | No | No | No |
| III:7 | 50 | c.857A>G | 63.75 | 63.75 | 20 | No | No | No |
| III:8 | 48 | Wild-type | 8.75 | 7.5 | / | No | No | No |
| III:13 | 44 | c.857A>G | 68.75 | 68.75 | 20 | No | No | No |
| III:14 | 42 | Wild-type | 15 | 18.75 | / | No | No | No |
| III:17 | 37 | c.857A>G | 75 | 70 | 25 | No | No | No |
| IV:1 | 34 | Wild-type | 10 | 8.75 | / | No | No | No |
| IV:4 | 25 | c.857A>G | 52.5 | 55 | 25 | No | No | No |
| IV:9 | 24 | c.857A>G | 35 | 38.75 | 30 | No | No | No |
WES identifies a causative variant in
KCNQ4
To identify the genetic cause of hearing loss, WES
was performed on this family. Variants in GJB2, GJB3,
SLC26A4 and mtDNA were excluded. Autosomal dominant inheritance
was suspected on the basis of the family pedigree, and WES was
performed on individuals III:1, III:17 and IV:4. WES detected a
novel potentially causative variant (c.857 A>G; p.Tyr286Cys) in
exon 6 of the KCNQ4 gene. This variant resulted in a
tyrosine-to-cysteine substitution at position 286 in KCNQ4 protein.
The tyrosine at position 286 is conserved throughout evolution, as
shown in Fig. 2. Subsequently,
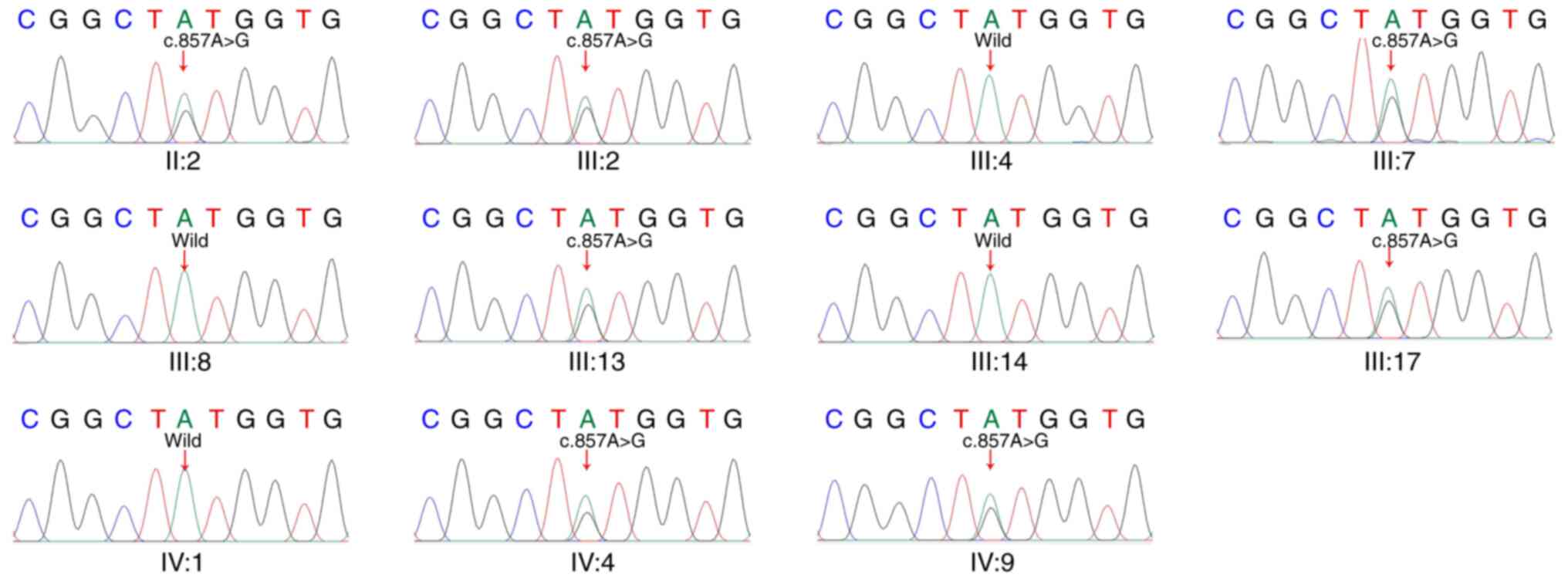
Sanger sequencing was performed to determine whether the c.857
A>G variant in the KCNQ4 gene segregated with the
affected status in this family (Fig.
3). This variant was detected in individuals II:2, III:2,
III:7, III:13, III:17, IV:4 and IV:9, and all these family members
were diagnosed as being hearing-impaired (Fig. 4A). The variant was not detected in
individuals III:4, III:8, III:14 and IV:1, who had good hearing
(Fig. 4B). The variant was found to
co-segregate with the progressive hearing loss phenotype.
Furthermore, this variant was not present in the 1000 Genomes
database or in the NHLBI Exome Sequencing Project (ESP6500). The
c.857 A>G (p.Tyr286Cys) variant in the KCNQ4 gene was
identified in ClinVar (Variation ID: 228776; (ClinVar; https://www.ncbi.nlm.nih.gov/clinvar/variation/228776;
accessed March 11, 2021) (36),
although the clinical significance was listed as ‘uncertain
significance’, and the condition was ‘not specified’ (20).
Structural modeling of p.Y286C
(p.Tyr286Cys)
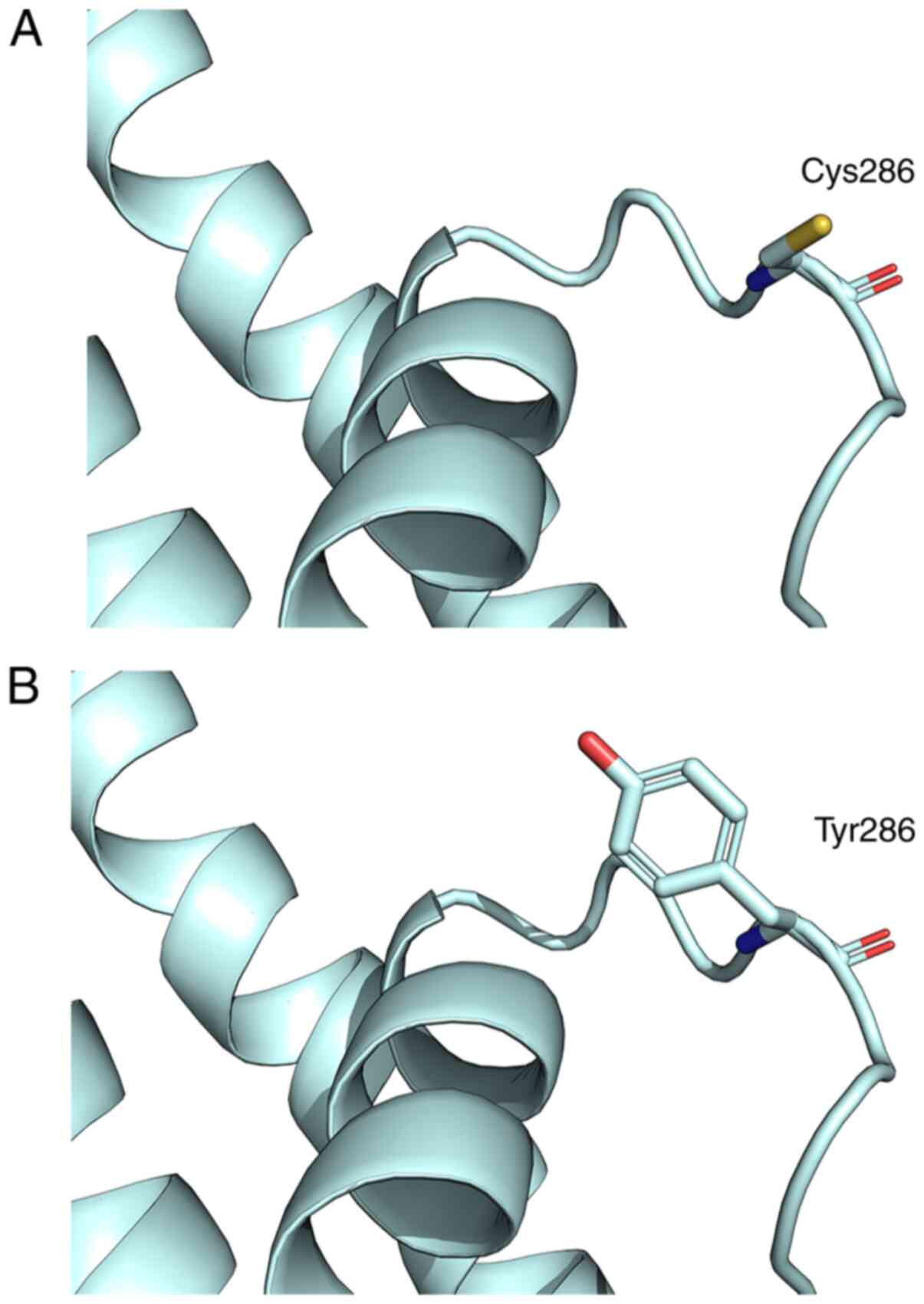
A computational tertiary protein structure
prediction of the wild-type and p.Y286C variant of KCNQ4 protein
was performed (Fig. 5). The model
covered the target sequence of KCNQ4 protein. The tyrosine residue
at position 286 of the KCNQ4 protein was identified in the pore
region of KCNQ4 protein. DS software predicted that this variant
affected the amino acid side chain due to the replacement of
tyrosine with cysteine. This variant led to the hydroxyphenyl group
in the amino acid side chain being changed to a sulfhydryl group.
This substitution led to a mismatch in the disulfide bond, which
was predicted to affect the structure of the pore region, and hence
changed the glycine-tyrosine-glycine (GYG) signature sequence of
the K+ channel pore. These three amino acids (GYG) are
found in the narrowest part of the pore (19), and variants in these three amino
acids affect its selectivity and abolish channel function in the
majority of patients.
Discussion
In the present study, a novel variant (c.857 A>G;
p.Tyr286Cys) in the KCNQ4 gene was identified in a Chinese
family with ADNSHL. The variant gene was co-segregated with
progressive hearing loss. The tyrosine at position 286 has been
shown to be well conserved across species. The replacement of
tyrosine with cysteine was suggested to affect the structure of the
pore region and abolish channel function.
The protein encoded by the KCNQ4 gene belongs
to the Kv7 channel family. The Kv7 channel family is a distinct
branch of the superfamily of voltage-gated potassium channels
encoded by KCNQ genes. There are five members in this family
(Kv7.1-Kv7.5), and the channel proteins are encoded by the
KCNQ1-KCNQ5 genes, respectively. They fulfill important
roles in the brain, heart, kidney and inner ear (37). Variants of KCNQ1 (for
example, KvLQT1) have been shown to cause heart diseases, long QT
syndrome and Jervell-Longe-Nielsen syndrome (38,39).
Variants of KCNQ2 or KCNQ3 have been causally
associated with benign familial neonatal seizures, whereas variants
in the KCNQ4 gene have been shown to cause DFNA2A (20,40).
Variants of both the KCNQ1 gene and the KCNQ4 gene
may cause hearing loss. However, deafness associated with
KCNQ1 gene is syndromic, severe, congenital and autosomal
recessive, whereas that associated with the KCNQ4 gene is
non-syndromic, progressive autosomal dominant inheritance (41,42).
To date, 40 pathogenic variants in the KCNQ4
gene have been reported to be associated with hearing loss
(Deafness Variation Database. http://deafnessvariationdatabase.org; accessed March
11, 2021) (43). All these variants
are located in exons 1, 3–8 and 14 of the KCNQ4 gene. The
majority of these variants have been linked to hearing loss, and
are clustered around the pore region. The pore region is
responsible for the ion-selectivity of the potassium channel. The
GYG signature sequence located in the narrowest region of the pore
is critical for maintaining pore structure and function (19). Missense variants at the
K+ ion selectivity filter have been shown to disrupt the
highly conserved GYG signature sequence, resulting in severely
impaired non-conducting channels to induce severe hearing loss. The
hearing loss induced by variants at the first (p.Gly285Cys;
p.Gly285Ser) and third (p.Gly287Arg) amino acids in the GYG
signature sequence has been demonstrated to be causal (13,44,45).
Furthermore, the variants in the second (p.Tyr286Cys) amino acid in
the GYG signature sequence have been documented in ClinVar by
previous investigators (20).
However, the clinical significance was denoted as ‘uncertain
significance’, and the condition as ‘not specified’. To the best of
our knowledge, the pathogenicity of the c.857 A>G (p.Tyr286Cys)
variant has not been demonstrated in previous studies (46). The novel missense variant p.Y286C in
the GYG signature sequence was co-segregated with progressive
hearing loss in the family investigated in the present study.
The reduction in Kv7 channel function induced by
KCNQ gene variants has been shown to lead to a variety of
diseases, such as epilepsy, arrhythmia and deafness (13,41,47–51).
Therefore, the use of KCNQ activators may be helpful for the
treatment of these diseases. Retigabine, a small molecule that
activates KCNQ2-5 channels, but not the KCNQ1
channel, was approved by the US Food and Drug Administration in
2011 (52,53). It was the first KCNQ
activator to be used for the treatment of epilepsy. However,
retigabine has a broad regulatory effect on almost all Kv7 channels
and other ion channels. Systemic side effects, such as dizziness,
drowsiness, memory loss, urinary retention, vertigo and slurred
speech, have been observed in patients treated with retigabine
(54). Non-selective KCNQ
activators have also been shown to cause several side effects;
therefore, the development of selective KCNQ activators is
necessary to overcome these limitations. To date, several new
small-molecule modulators (activators) of KCNQ have been
developed that may have potential benefits, while reducing adverse
effects. It may ultimately be possible to rescue Kv7 channel
functionality using KCNQ activators, thereby preventing
hearing loss in patients with DFNA2A.
Collectively, our study demonstrated that the
co-segregating heterozygous missense variant (c.857A>G;
p.Tyr286Cys) in the glycine tyrosine glycine signature sequence in
the pore region of the KCNQ4 channel was the pathogenic variants in
this ADNSHL family. This finding supports the pathogenicity of the
missense variant (c.857A>G; p.Tyr286Cys) in the KCNQ4
gene. Data presented here extend the pathogenic variant spectrum of
the KCNQ4 gene. The finding has implications in genetic
counseling for hereditary deafness and enable otolaryngologists to
select appropriate clinical interventions for patients with
ADNSHL.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural
Science Foundation of China (grant nos. 81870732 and 81800918),
National Key Research and Development Project (grant no.
2019YFB1311605), Shaanxi Provincial Science and Technology Key
Project (grant no. 2018PT-01), Xijing Boost-Free Exploration
Project (grant no. XJZT19MJ02), and Xijing Boost-Advanced
Discipline Construction Project (grant nos. XJZT14X07, XJZT18X23
and XJZT19X27).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request. The sequencing dataset has been deposited in NCBI Sequence
Read Archive, and the BioProject ID is PRJNA689907 (https://www.ncbi.nlm.nih.gov/sra/PRJNA689907).
Authors' contributions
DZ and JC conceptualized and designed the study. PL,
SW, JW and WL researched the family's history and recruited the
family members. JW performed pure tone audiometry testing, disease
diagnosis and DNA extraction. PL and SW completed PCR amplification
and Sanger sequencing. QL, WL, YY and XA were responsible for
analyzing the WES data; and QL performed the molecular genetic
studies and the sequence alignment analysis. QL drafted the
manuscript. QL and PL confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Medical Ethics
Committee of the First Affiliated Hospital of the Air Force Medical
University (approval number KY20212002-C-1), and informed consent
was obtained from all the study participants.
Patient consent for publication
Enrolled study participants and the patient provided
written informed consent for the publication of this
manuscript.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu Y, Qi J, Chen X, Tang M, Chu C, Zhu W,
Li H, Tian C, Yang G, Zhong C, et al: Critical role of spectrin in
hearing development and deafness. Sci Adv. 5:eaav78032019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qi J, Zhang L, Tan F, Liu Y, Chu C, Zhu W,
Wang Y, Qi Z and Chai R: Espin distribution as revealed by
super-resolution microscopy of stereocilia. Am J Transl Res.
12:130–141. 2020.PubMed/NCBI
|
|
3
|
He Z, Guo L, Shu Y, Fang Q, Zhou H, Liu Y,
Liu D, Lu L, Zhang X, Ding X, et al: Autophagy protects auditory
hair cells against neomycin-induced damage. Autophagy.
13:1884–1904. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang S, Zhang Y, Dong Y, Guo L, Zhang Z,
Shao B, Qi J, Zhou H, Zhu W, Yan X, et al: Knockdown of Foxg1 in
supporting cells increases the trans-differentiation of supporting
cells into hair cells in the neonatal mouse cochlea. Cell Mol Life
Sci. 77:1401–1419. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ralli M, Balla MP, Greco A, Altissimi G,
Ricci P, Turchetta R, de Virgilio A, de Vincentiis M, Ricci S and
Cianfrone G: Work-related noise exposure in a cohort of patients
with chronic tinnitus: Analysis of demographic and audiological
characteristics. Int J Environ Res Public Health. 14:10352017.
View Article : Google Scholar
|
|
6
|
Taylor W, Pearson J, Mair A and Burns W:
Study of noise and hearing in jute weaving. J Acoust Soc Am.
38:113–120. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tao L and Segil N: Early transcriptional
response to aminoglycoside antibiotic suggests alternate pathways
leading to apoptosis in sensory hair cells in the mouse inner ear.
Front Cell Neurosci. 9:1902015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marazita ML, Ploughman LM, Rawlings B,
Remington E, Arnos KS and Nance WE: Genetic epidemiological studies
of early-onset deafness in the U.S. school-age population. Am J Med
Genet. 46:486–491. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schrijver I: Hereditary non-syndromic
sensorineural hearing loss: Transforming silence to sound. J Mol
Diagn. 6:275–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qian F, Wang X, Yin Z, Xie G, Yuan H, Liu
D and Chai R: The slc4a2b gene is required for hair cell
development in zebrafish. Aging (Albany NY). 12:18804–18821. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morton CC and Nance WE: Newborn hearing
screening-a silent revolution. N Engl J Med. 354:2151–2164. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hilgert N, Smith RJ and Van Camp G:
Function and expression pattern of nonsyndromic deafness genes.
Curr Mol Med. 9:546–564. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kubisch C, Schroeder BC, Friedrich T,
Lutjohann B, El-Amraoui A, Marlin S, Petit C and Jentsch TJ: KCNQ4,
a novel potassium channel expressed in sensory outer hair cells, is
mutated in dominant deafness. Cell. 96:437–446. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smith R and Hildebrand M: DFNA2
Nonsyndromic Hearing Loss. 1993.
|
|
15
|
Kharkovets T, Dedek K, Maier H, Schweizer
M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T and Jentsch
TJ: Mice with altered KCNQ4 K+ channels implicate sensory outer
hair cells in human progressive deafness. EMBO J. 25:642–652. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kharkovets T, Hardelin JP, Safieddine S,
Schweizer M, El-Amraoui A, Petit C and Jentsch TJ: KCNQ4, a K+
channel mutated in a form of dominant deafness, is expressed in the
inner ear and the central auditory pathway. Proc Natl Acad Sci USA.
97:4333–4338. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Beisel KW, Nelson NC, Delimont DC and
Fritzsch B: Longitudinal gradients of KCNQ4 expression in spiral
ganglion and cochlear hair cells correlate with progressive hearing
loss in DFNA2. Brain Res Mol Brain Res. 82:137–149. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wollnik B, Schroeder BC, Kubisch C,
Esperer HD, Wieacker P and Jentsch TJ: Pathophysiological
mechanisms of dominant and recessive KVLQT1 K+ channel mutations
found in inherited cardiac arrhythmias. Hum Mol Genet. 6:1943–1949.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Doyle DA, Morais CJ, Pfuetzner RA, Kuo A,
Gulbis JM, Cohen SL, Chait BT and MacKinnon R: The structure of the
potassium channel: Molecular basis of K+ conduction and
selectivity. Science. 280:69–77. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao Y, Yechikov S, Vazquez AE, Chen D and
Nie L: Impaired surface expression and conductance of the KCNQ4
channel lead to sensorineural hearing loss. J Cell Mol Med.
17:889–900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dominguez LM and Dodson KM: Genetics of
hearing loss: Focus on DFNA2. Appl Clin Genet. 5:97–104.
2012.PubMed/NCBI
|
|
22
|
Wang H, Zhao Y, Yi Y, Gao Y, Liu Q, Wang
D, Li Q, Lan L, Li N, Guan J, et al: Targeted high-throughput
sequencing identifies pathogenic mutations in KCNQ4 in two large
Chinese families with autosomal dominant hearing loss. PLoS One.
9:e1031332014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
International Classification of
Functioning, Disability and Health. World Health Organization;
2001
|
|
24
|
Xin F, Yuan Y, Deng X, Han M, Wang G, Zhao
J, Gao X, Liu J, Yu F, Han D and Dai P: Genetic mutations in
nonsyndromic deafness patients of Chinese minority and Han
ethnicities in Yunnan, China. J Transl Med. 11:3122013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dai P, Yu F, Kang DY, Zhang X, Liu X, Mi
WZ, Cao JY, Yuan HJ, Yang WY, Wu BL and Han DY: Diagnostic methods
and clinic application for mtDNA A1555G and GJB2 and SLC26A4 genes
in deaf patients. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi.
40:769–773. 2005.(In Chinese). PubMed/NCBI
|
|
26
|
Yuan YY, Huang DL, Yu F, Han B, Wang GJ,
Han DY and Dai P: Sequence analysis of GJB3 in Chinese deafness
population who carry one heterozygous GJB2 pathogenic mutation.
Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 45:287–290. 2010.(In
Chinese). PubMed/NCBI
|
|
27
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The Sequence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ
and Prins P: Sambamba: Fast processing of NGS alignment formats.
Bioinformatics. 31:2032–2034. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
1000 Genomes Project Consortium, ; Auton
A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini
JL, McCarthy S, McVean GA and Abecasis GR: A global reference for
human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kent WJ: BLAT-the BLAST-like alignment
tool. Genome Res. 12:656–664. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Berman H, Henrick K and Nakamura H:
Announcing the worldwide protein data bank. Nat Struct Biol.
10:9802003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Long SB, Campbell EB and Mackinnon R:
Crystal structure of a mammalian voltage-dependent Shaker family K+
channel. Science. 309:897–903. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Landrum MJ, Lee JM, Benson M, Brown GR,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al:
ClinVar: Improving access to variant interpretations and supporting
evidence. Nucleic Acids Res. 46:D1062–D1067. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maljevic S, Wuttke TV and Lerche H:
Nervous system KV7 disorders: Breakdown of a subthreshold brake. J
Physiol. 586:1791–1801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Q, Curran ME, Splawski I, Burn TC,
Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de
Jager T, et al: Positional cloning of a novel potassium channel
gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet.
12:17–23. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Neyroud N, Tesson F, Denjoy I, Leibovici
M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, et al:
A novel mutation in the potassium channel gene KVLQT1 causes the
Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet.
15:186–189. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brown DA: Kv7 (KCNQ) potassium channels
that are mutated in human diseases. J Physiol. 586:1781–1783. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jervell A and Lange-Nielsen F: Congenital
deaf-mutism, functional heart disease with prolongation of the Q-T
interval and sudden death. Am Heart J. 54:59–68. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hedley PL, Jorgensen P, Schlamowitz S,
Wangari R, Moolman-Smook J, Brink PA, Kanters JK, Corfield VA and
Christiansen M: The genetic basis of long QT and short QT
syndromes: A mutation update. Hum Mutat. 30:1486–1511. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Azaiez H, Booth KT, Ephraim SS, Crone B,
Black-Ziegelbein EA, Marini RJ, Shearer AE, Sloan-Heggen CM, Kolbe
D, Casavant T, et al: Genomic landscape and mutational signatures
of deafness-associated genes. Am J Hum Genet. 103:484–497. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Coucke PJ, Van Hauwe P, Kelley PM, Kunst
H, Schatteman I, Van Velzen D, Meyers J, Ensink RJ, Verstreken M,
Declau F, et al: Mutations in the KCNQ4 gene are responsible for
autosomal dominant deafness in four DFNA2 families. Hum Mol Genet.
8:1321–1328. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arnett J, Emery SB, Kim TB, Boerst AK, Lee
K, Leal SM and Lesperance MM: Autosomal dominant progressive
sensorineural hearing loss due to a novel mutation in the KCNQ4
gene. Arch Otolaryngol Head Neck Surg. 137:54–59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sloan-Heggen CM, Bierer AO, Shearer AE,
Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT,
Campbell CA, et al: Comprehensive genetic testing in the clinical
evaluation of 1119 patients with hearing loss. Hum Genet.
135:441–450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Borgatti R, Zucca C, Cavallini A, Ferrario
M, Panzeri C, Castaldo P, Soldovieri MV, Baschirotto C, Bresolin N,
Dalla Bernardina B, et al: A novel mutation in KCNQ2 associated
with BFNC, drug resistant epilepsy, and mental retardation.
Neurology. 63:57–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Weckhuysen S, Mandelstam S, Suls A,
Audenaert D, Deconinck T, Claes LR, Deprez L, Smets K, Hristova D,
Yordanova I, et al: KCNQ2 encephalopathy: Emerging phenotype of a
neonatal epileptic encephalopathy. Ann Neurol. 71:15–25. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fister P, Soltirovska-Salamon A, Debeljak
M and Paro-Panjan D: Benign familial neonatal convulsions caused by
mutation in KCNQ3, exon 6: A European case. Eur J Paediatr Neurol.
17:308–310. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lehman A, Thouta S, Mancini GM, Naidu S,
van Slegtenhorst M, McWalter K, Person R, Mwenifumbo J, Salvarinova
R; CAUSES Study, ; et al: Loss-of-function and Gain-of-function
mutations in KCNQ5 cause intellectual disability or epileptic
encephalopathy. Am J Hum Genet. 101:65–74. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jongbloed RJ, Wilde AA, Geelen JL,
Doevendans P, Schaap C, Van Langen I, van Tintelen JP, Cobben JM,
Beaufort-Krol GC, Geraedts JP and Smeets HJ: Novel KCNQ1 and HERG
missense mutations in Dutch long-QT families. Hum Mutat.
13:301–310. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Orhan G, Wuttke TV, Nies AT, Schwab M and
Lerche H: Retigabine/Ezogabine, a KCNQ/K(V)7 channel opener:
Pharmacological and clinical data. Expert Opin Pharmacother.
13:1807–1816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lange W, Geissendorfer J, Schenzer A,
Grotzinger J, Seebohm G, Friedrich T and Schwake M: Refinement of
the binding site and mode of action of the anticonvulsant
Retigabine on KCNQ K+ channels. Mol Pharmacol. 75:272–280. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ciliberto MA, Weisenberg JL and Wong M:
Clinical utility, safety, and tolerability of ezogabine
(retigabine) in the treatment of epilepsy. Drug Healthc Patient
Saf. 4:81–86. 2012.PubMed/NCBI
|