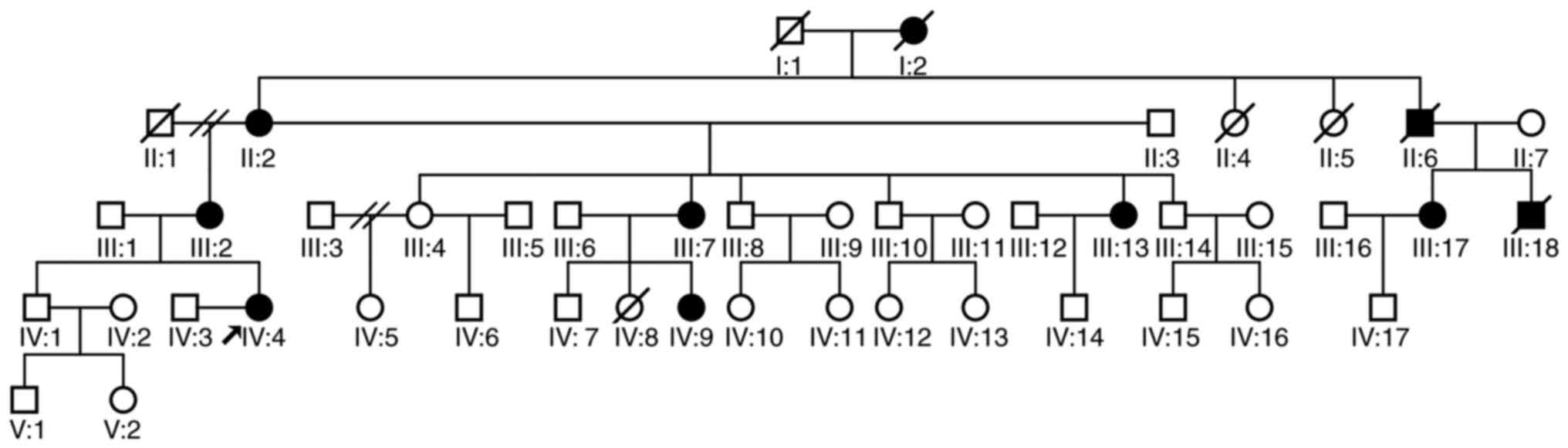
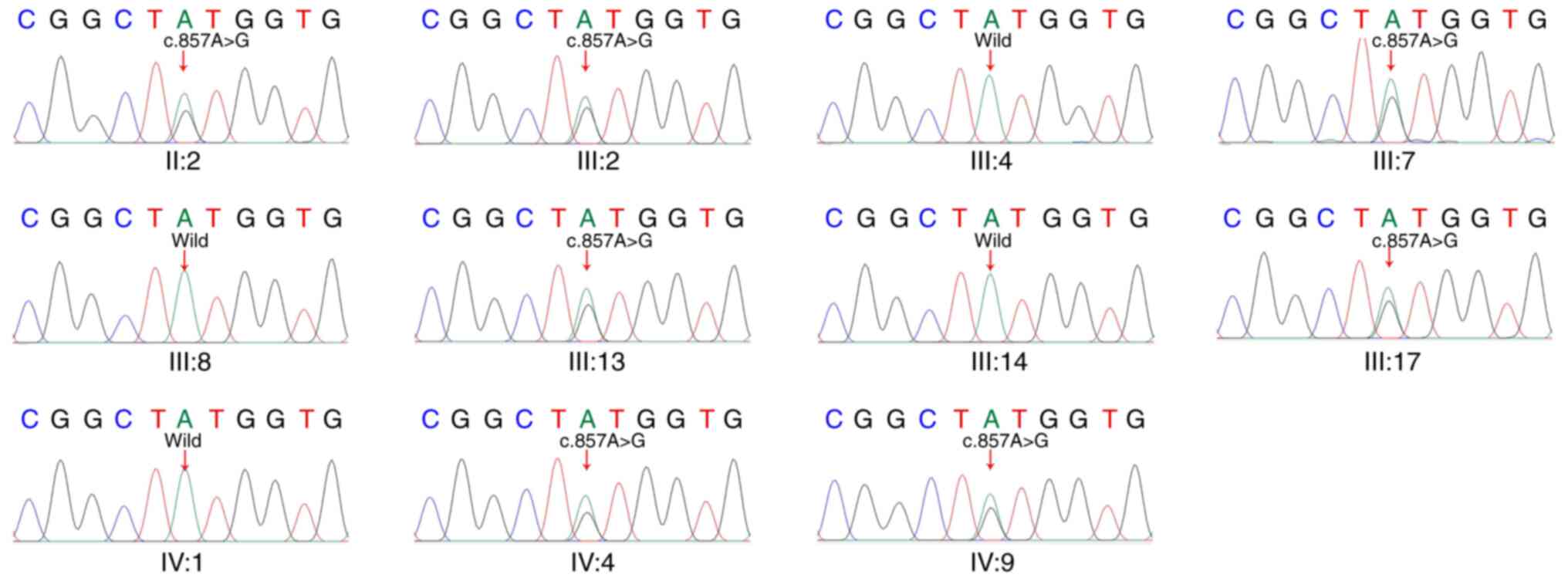
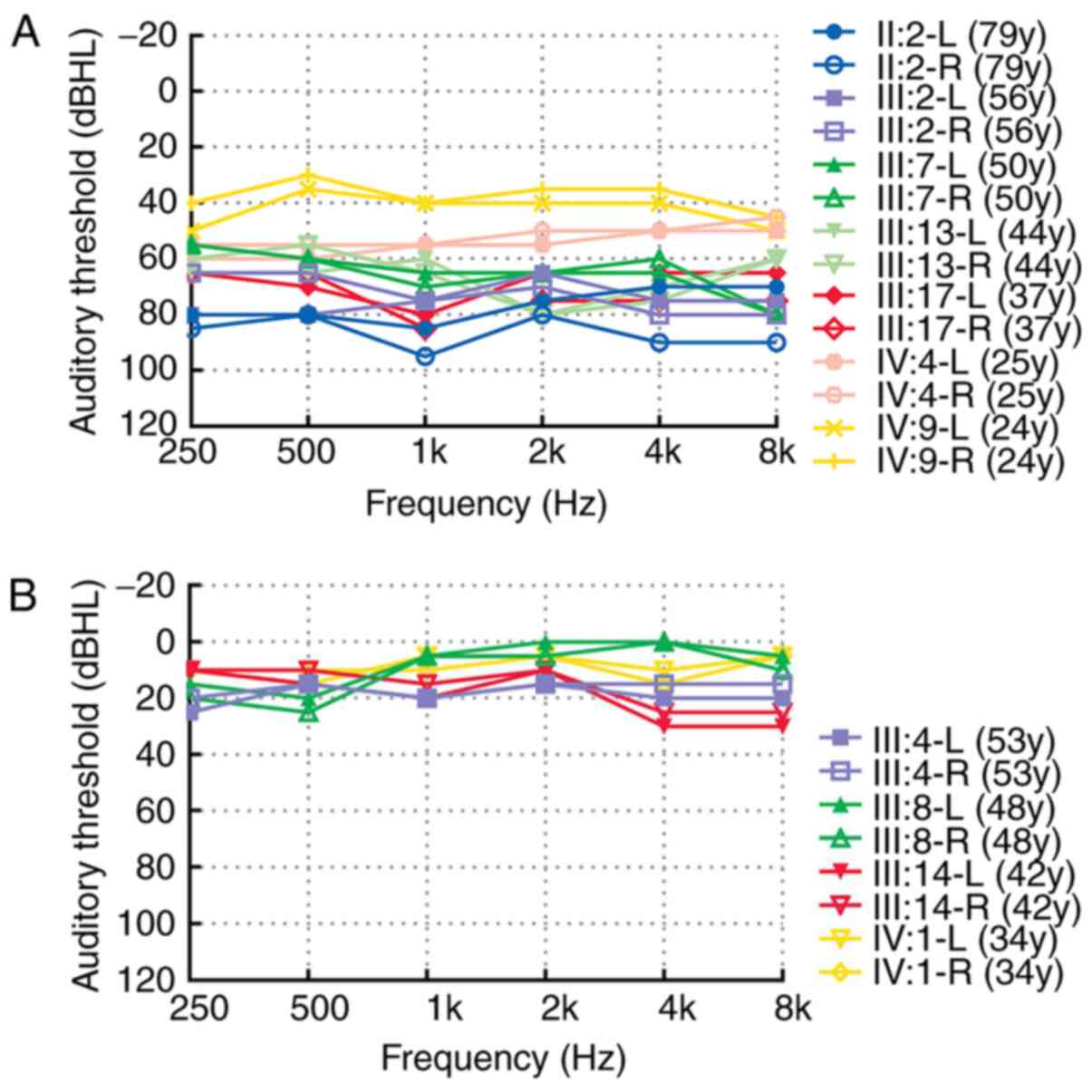
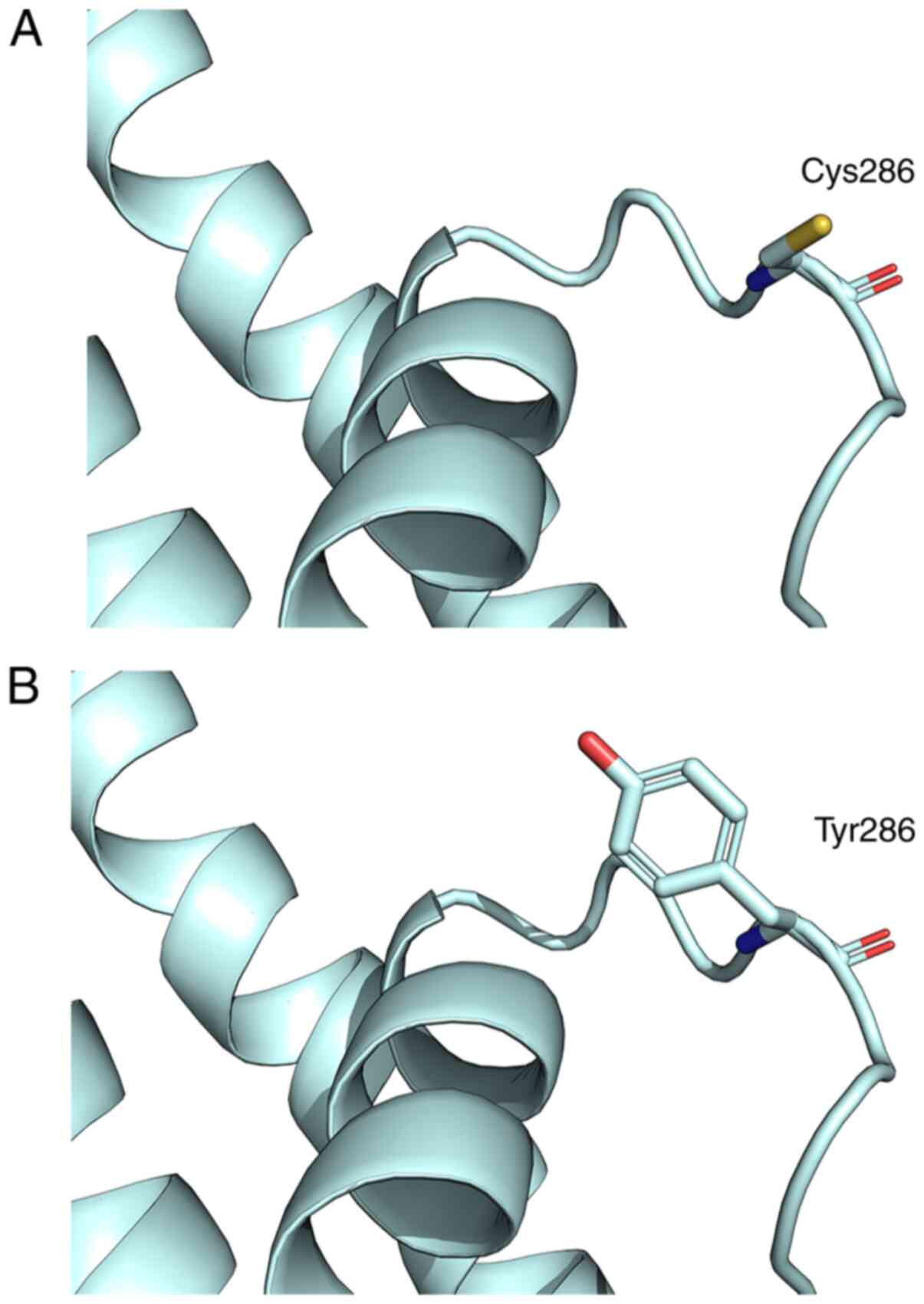
|
1
|
Liu Y, Qi J, Chen X, Tang M, Chu C, Zhu W,
Li H, Tian C, Yang G, Zhong C, et al: Critical role of spectrin in
hearing development and deafness. Sci Adv. 5:eaav78032019.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qi J, Zhang L, Tan F, Liu Y, Chu C, Zhu W,
Wang Y, Qi Z and Chai R: Espin distribution as revealed by
super-resolution microscopy of stereocilia. Am J Transl Res.
12:130–141. 2020.PubMed/NCBI
|
|
3
|
He Z, Guo L, Shu Y, Fang Q, Zhou H, Liu Y,
Liu D, Lu L, Zhang X, Ding X, et al: Autophagy protects auditory
hair cells against neomycin-induced damage. Autophagy.
13:1884–1904. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang S, Zhang Y, Dong Y, Guo L, Zhang Z,
Shao B, Qi J, Zhou H, Zhu W, Yan X, et al: Knockdown of Foxg1 in
supporting cells increases the trans-differentiation of supporting
cells into hair cells in the neonatal mouse cochlea. Cell Mol Life
Sci. 77:1401–1419. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ralli M, Balla MP, Greco A, Altissimi G,
Ricci P, Turchetta R, de Virgilio A, de Vincentiis M, Ricci S and
Cianfrone G: Work-related noise exposure in a cohort of patients
with chronic tinnitus: Analysis of demographic and audiological
characteristics. Int J Environ Res Public Health. 14:10352017.
View Article : Google Scholar
|
|
6
|
Taylor W, Pearson J, Mair A and Burns W:
Study of noise and hearing in jute weaving. J Acoust Soc Am.
38:113–120. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tao L and Segil N: Early transcriptional
response to aminoglycoside antibiotic suggests alternate pathways
leading to apoptosis in sensory hair cells in the mouse inner ear.
Front Cell Neurosci. 9:1902015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Marazita ML, Ploughman LM, Rawlings B,
Remington E, Arnos KS and Nance WE: Genetic epidemiological studies
of early-onset deafness in the U.S. school-age population. Am J Med
Genet. 46:486–491. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schrijver I: Hereditary non-syndromic
sensorineural hearing loss: Transforming silence to sound. J Mol
Diagn. 6:275–284. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qian F, Wang X, Yin Z, Xie G, Yuan H, Liu
D and Chai R: The slc4a2b gene is required for hair cell
development in zebrafish. Aging (Albany NY). 12:18804–18821. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Morton CC and Nance WE: Newborn hearing
screening-a silent revolution. N Engl J Med. 354:2151–2164. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hilgert N, Smith RJ and Van Camp G:
Function and expression pattern of nonsyndromic deafness genes.
Curr Mol Med. 9:546–564. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kubisch C, Schroeder BC, Friedrich T,
Lutjohann B, El-Amraoui A, Marlin S, Petit C and Jentsch TJ: KCNQ4,
a novel potassium channel expressed in sensory outer hair cells, is
mutated in dominant deafness. Cell. 96:437–446. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smith R and Hildebrand M: DFNA2
Nonsyndromic Hearing Loss. 1993.
|
|
15
|
Kharkovets T, Dedek K, Maier H, Schweizer
M, Khimich D, Nouvian R, Vardanyan V, Leuwer R, Moser T and Jentsch
TJ: Mice with altered KCNQ4 K+ channels implicate sensory outer
hair cells in human progressive deafness. EMBO J. 25:642–652. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kharkovets T, Hardelin JP, Safieddine S,
Schweizer M, El-Amraoui A, Petit C and Jentsch TJ: KCNQ4, a K+
channel mutated in a form of dominant deafness, is expressed in the
inner ear and the central auditory pathway. Proc Natl Acad Sci USA.
97:4333–4338. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Beisel KW, Nelson NC, Delimont DC and
Fritzsch B: Longitudinal gradients of KCNQ4 expression in spiral
ganglion and cochlear hair cells correlate with progressive hearing
loss in DFNA2. Brain Res Mol Brain Res. 82:137–149. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wollnik B, Schroeder BC, Kubisch C,
Esperer HD, Wieacker P and Jentsch TJ: Pathophysiological
mechanisms of dominant and recessive KVLQT1 K+ channel mutations
found in inherited cardiac arrhythmias. Hum Mol Genet. 6:1943–1949.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Doyle DA, Morais CJ, Pfuetzner RA, Kuo A,
Gulbis JM, Cohen SL, Chait BT and MacKinnon R: The structure of the
potassium channel: Molecular basis of K+ conduction and
selectivity. Science. 280:69–77. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gao Y, Yechikov S, Vazquez AE, Chen D and
Nie L: Impaired surface expression and conductance of the KCNQ4
channel lead to sensorineural hearing loss. J Cell Mol Med.
17:889–900. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dominguez LM and Dodson KM: Genetics of
hearing loss: Focus on DFNA2. Appl Clin Genet. 5:97–104.
2012.PubMed/NCBI
|
|
22
|
Wang H, Zhao Y, Yi Y, Gao Y, Liu Q, Wang
D, Li Q, Lan L, Li N, Guan J, et al: Targeted high-throughput
sequencing identifies pathogenic mutations in KCNQ4 in two large
Chinese families with autosomal dominant hearing loss. PLoS One.
9:e1031332014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
International Classification of
Functioning, Disability and Health. World Health Organization;
2001
|
|
24
|
Xin F, Yuan Y, Deng X, Han M, Wang G, Zhao
J, Gao X, Liu J, Yu F, Han D and Dai P: Genetic mutations in
nonsyndromic deafness patients of Chinese minority and Han
ethnicities in Yunnan, China. J Transl Med. 11:3122013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dai P, Yu F, Kang DY, Zhang X, Liu X, Mi
WZ, Cao JY, Yuan HJ, Yang WY, Wu BL and Han DY: Diagnostic methods
and clinic application for mtDNA A1555G and GJB2 and SLC26A4 genes
in deaf patients. Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi.
40:769–773. 2005.(In Chinese). PubMed/NCBI
|
|
26
|
Yuan YY, Huang DL, Yu F, Han B, Wang GJ,
Han DY and Dai P: Sequence analysis of GJB3 in Chinese deafness
population who carry one heterozygous GJB2 pathogenic mutation.
Zhonghua Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 45:287–290. 2010.(In
Chinese). PubMed/NCBI
|
|
27
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The Sequence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ
and Prins P: Sambamba: Fast processing of NGS alignment formats.
Bioinformatics. 31:2032–2034. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
1000 Genomes Project Consortium, ; Auton
A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini
JL, McCarthy S, McVean GA and Abecasis GR: A global reference for
human genetic variation. Nature. 526:68–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kent WJ: BLAT-the BLAST-like alignment
tool. Genome Res. 12:656–664. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Berman H, Henrick K and Nakamura H:
Announcing the worldwide protein data bank. Nat Struct Biol.
10:9802003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Long SB, Campbell EB and Mackinnon R:
Crystal structure of a mammalian voltage-dependent Shaker family K+
channel. Science. 309:897–903. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Landrum MJ, Lee JM, Benson M, Brown GR,
Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al:
ClinVar: Improving access to variant interpretations and supporting
evidence. Nucleic Acids Res. 46:D1062–D1067. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maljevic S, Wuttke TV and Lerche H:
Nervous system KV7 disorders: Breakdown of a subthreshold brake. J
Physiol. 586:1791–1801. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang Q, Curran ME, Splawski I, Burn TC,
Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de
Jager T, et al: Positional cloning of a novel potassium channel
gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet.
12:17–23. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Neyroud N, Tesson F, Denjoy I, Leibovici
M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, et al:
A novel mutation in the potassium channel gene KVLQT1 causes the
Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet.
15:186–189. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Brown DA: Kv7 (KCNQ) potassium channels
that are mutated in human diseases. J Physiol. 586:1781–1783. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jervell A and Lange-Nielsen F: Congenital
deaf-mutism, functional heart disease with prolongation of the Q-T
interval and sudden death. Am Heart J. 54:59–68. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hedley PL, Jorgensen P, Schlamowitz S,
Wangari R, Moolman-Smook J, Brink PA, Kanters JK, Corfield VA and
Christiansen M: The genetic basis of long QT and short QT
syndromes: A mutation update. Hum Mutat. 30:1486–1511. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Azaiez H, Booth KT, Ephraim SS, Crone B,
Black-Ziegelbein EA, Marini RJ, Shearer AE, Sloan-Heggen CM, Kolbe
D, Casavant T, et al: Genomic landscape and mutational signatures
of deafness-associated genes. Am J Hum Genet. 103:484–497. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Coucke PJ, Van Hauwe P, Kelley PM, Kunst
H, Schatteman I, Van Velzen D, Meyers J, Ensink RJ, Verstreken M,
Declau F, et al: Mutations in the KCNQ4 gene are responsible for
autosomal dominant deafness in four DFNA2 families. Hum Mol Genet.
8:1321–1328. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arnett J, Emery SB, Kim TB, Boerst AK, Lee
K, Leal SM and Lesperance MM: Autosomal dominant progressive
sensorineural hearing loss due to a novel mutation in the KCNQ4
gene. Arch Otolaryngol Head Neck Surg. 137:54–59. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sloan-Heggen CM, Bierer AO, Shearer AE,
Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT,
Campbell CA, et al: Comprehensive genetic testing in the clinical
evaluation of 1119 patients with hearing loss. Hum Genet.
135:441–450. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Borgatti R, Zucca C, Cavallini A, Ferrario
M, Panzeri C, Castaldo P, Soldovieri MV, Baschirotto C, Bresolin N,
Dalla Bernardina B, et al: A novel mutation in KCNQ2 associated
with BFNC, drug resistant epilepsy, and mental retardation.
Neurology. 63:57–65. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Weckhuysen S, Mandelstam S, Suls A,
Audenaert D, Deconinck T, Claes LR, Deprez L, Smets K, Hristova D,
Yordanova I, et al: KCNQ2 encephalopathy: Emerging phenotype of a
neonatal epileptic encephalopathy. Ann Neurol. 71:15–25. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fister P, Soltirovska-Salamon A, Debeljak
M and Paro-Panjan D: Benign familial neonatal convulsions caused by
mutation in KCNQ3, exon 6: A European case. Eur J Paediatr Neurol.
17:308–310. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lehman A, Thouta S, Mancini GM, Naidu S,
van Slegtenhorst M, McWalter K, Person R, Mwenifumbo J, Salvarinova
R; CAUSES Study, ; et al: Loss-of-function and Gain-of-function
mutations in KCNQ5 cause intellectual disability or epileptic
encephalopathy. Am J Hum Genet. 101:65–74. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jongbloed RJ, Wilde AA, Geelen JL,
Doevendans P, Schaap C, Van Langen I, van Tintelen JP, Cobben JM,
Beaufort-Krol GC, Geraedts JP and Smeets HJ: Novel KCNQ1 and HERG
missense mutations in Dutch long-QT families. Hum Mutat.
13:301–310. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Orhan G, Wuttke TV, Nies AT, Schwab M and
Lerche H: Retigabine/Ezogabine, a KCNQ/K(V)7 channel opener:
Pharmacological and clinical data. Expert Opin Pharmacother.
13:1807–1816. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Lange W, Geissendorfer J, Schenzer A,
Grotzinger J, Seebohm G, Friedrich T and Schwake M: Refinement of
the binding site and mode of action of the anticonvulsant
Retigabine on KCNQ K+ channels. Mol Pharmacol. 75:272–280. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ciliberto MA, Weisenberg JL and Wong M:
Clinical utility, safety, and tolerability of ezogabine
(retigabine) in the treatment of epilepsy. Drug Healthc Patient
Saf. 4:81–86. 2012.PubMed/NCBI
|