Introduction
Aristolochic acid nephropathy (AAN) is a progressive
renal tubular interstitial disease caused by the ingestion of
Chinese herbal medicines or plants containing aristolochic acid
(AA) (1). In Southeast Asia and
other countries or regions where traditional Chinese medicine is
popular, herbal medicines and plants containing AA are still widely
used (2). A multicenter
retrospective cohort study in China showed that the incidence of
acute renal injury was 11.6% among nearly 660,000 hospitalized
patients, of which 40% was drug-induced and 16% may have been
caused by Chinese herbal medicine and plants containing AA
(3). A considerable proportion of
patients with AAN have impaired renal function and progress to
end-stage renal disease (ESRD) (4).
Patients with ESRD require long-term renal replacement therapy,
which necessitates a large number of medical resources. Therefore,
an in-depth understanding of the pathogenesis of AAN as well as the
development of effective prevention and treatment measures is
important.
Rapamycin, a specific inhibitor of mammalian target
of rapamycin (mTOR), is a novel and effective immunosuppressant
that has a protective effect on the kidney in the state of
transplantation (5,6). Rapamycin is reported to improve renal
function and renal tissue ultrastructure in a rat renal
ischemia-reperfusion injury model by inhibiting renal tubular
epithelial cell apoptosis (7). The
results of our previous study demonstrated that rapamycin could
effectively attenuate AAN (8).
However, the mechanism underlying the regulation of AAN by
rapamycin remains to be elucidated.
Autophagy is initiated by lysosomes and can be
defined as the process by which intracellular fragments, which are
widely present in eukaryotic cells, are degraded (9). In this process, damaged organelles and
misfolded proteins are phagocytized to form autophagosomes, which
then fuse with lysosomes to form autolysosomes. These degrade
autophagosomes and ensure dynamic cell balance (10). Autophagy is indispensable for
maintaining intracellular homeostasis and adapting to stress states
(including hunger, energy deprivation, oxidative stress and
hypoxia) and ensures cell survival (11). Similarly, AAN leads to organelle
dysfunction, including mitochondrial and endoplasmic reticulum
stress and induces specific autophagy, resulting in the clearance
of damaged organelles (12).
However, this is a compensatory mechanism; when stimulation
persists or increases, cellular homeostasis is disrupted, leading
to apoptosis (13,14). A previous study showed that enhanced
renal autophagy can inhibit the apoptosis of kidney cells and serve
a protective role in AAN (15).
mTOR is a serine-threonine protein kinase that belongs to the
phosphatidylinositol 3 kinase protein kinase family (16). It serves a key regulatory role in
many biological processes, such as cell proliferation, survival,
differentiation and autophagy (17–19).
Studies have shown that inhibition of phosphorylated (p-)mTOR can
activate autophagy and promote phagocytosis of damaged organelles
and misfolded proteins, thereby protecting cells from death
(20,21). Nevertheless, as a specific mTOR
inhibitor, rapamycin can activate autophagy to maintain
AA-stimulated renal cell homeostasis and may, therefore, inhibit
apoptosis and exert a protective effect in AAN.
The present study investigated the underlying
molecular mechanism of action of rapamycin against AAN. The
findings suggested that rapamycin attenuated AAN by blocking the
expression of p-mTOR and its downstream substrate p-S6K1, thereby
activating autophagy in a mouse model of AAN and in AA-treated
renal tubular epithelial cells in vitro, which was
associated with the inhibition of cell apoptosis and improved
AA-induced renal damage.
Materials and methods
Animal models
Animal experiments were approved by the Animal
Ethics Committee of Shenzhen University (approval no.
SUGH-A-02104). A total of 30 six-week-old C57BL/6 male mice
weighing 20–25 g were purchased from the Shanghai SLAC Laboratory
Animal Co. Ltd. All mice were housed in a room maintained at
moderate temperature (20±2°C) and humidity (40–60%) and were
subjected to a 12-h light/dark cycle. Mice were provided free
access to food and water for the duration of the present study.
They were adaptively fed for 7 days and randomly divided into three
groups as follows: A control group (Ctrl group, n=10), an
aristolochic acid-induced group (AAI group, n=10) and a
rapamycin-treated group (RAP group, n=10). Mice in the control
group were administered 0.9% normal saline at a volume equivalent
to that of the injected drug in the other two groups. The AAI and
RAP groups were intraperitoneally injected with aristolochic acid
(AA; Sigma-Aldrich; Merck KGaA) at a dose of 2.5 mg/kg/day for 6
weeks. After 2 h, mice in the RAP group were intraperitoneally
injected with rapamycin solution (Sigma-Aldrich; Merck KGaA) at a
dose of 1 mg/kg/day. After 6 weeks, all mice were anesthetized with
isoflurane (3% for induction and 2% for maintenance). Subsequently,
1 ml of arterial blood was quickly drawn from the abdominal aorta.
Mice were euthanized by the immediate removal of the heart after
exsanguination and death of mice was confirmed by the lack of
breathing and reflexes of paw withdrawal. Arterial blood and kidney
tissues were collected for subsequent studies.
Serum creatinine and blood urea
nitrogen
The supernatant obtained after centrifuging the
arterial blood (300 × g for 20 min at 4°C) of each mouse was stored
at −80°C until further analysis. Serum creatinine and urea nitrogen
levels were detected using enzyme-linked immunosorbent assay kits
(Serum creatinine, cat. no. MM-44455M1; serum urea nitrogen, cat.
no. MM-0692M1; Jiangsu Baolai Biotechnology Co., Ltd.) according to
the manufacturer's protocols.
Hematoxylin and eosin (H&E)
staining
Kidney tissues were fixed in 4% (v/v)
paraformaldehyde for 48 h at room temperature, then placed in
ethanol solution for gradient dehydration (75, 85, 95 and 100%) and
made transparent in xylene for 30 min. Finally, the transparent
kidney tissues were embedded in paraffin and sliced into 5-µm thick
sections. The slices were incubated overnight in an oven at 65°C,
then dewaxed in xylene for 30 min and rehydrated in fractionated
ethanol series for histological examination. Next, the sections
were stained with H&E (Beijing Solarbio Science &
Technology Co., Ltd.) according to the standardized H&E
staining procedure with Hematoxylin for 10 min and Eosin for 15 sec
at room temperature and observed under a bright field using a DP73
microscope (Olympus Corporation; magnification, ×400).
Periodic acid-Schiff (PAS)
staining
Sections of renal tissues (5-µm thick) were stained
using the PAS staining kit (Muto Pure Chemicals Co., Ltd.)
following the manufacturer's instructions. Briefly, sections were
treated with 1% (w/v) periodic acid for 10 min at room temperature.
Then, the sections were treated with Schiff's reagent for 30 min at
37°C after washing three times with distilled water. The staining
reaction was terminated using three treatments with sulfurous acid
solution. The samples were dried and observed under a bright field
using a DP73 microscope (Olympus Corporation; magnification,
×400).
Cell culture
HK-2 cells were purchased from Kunming Cell Bank,
Chinese Academy of Sciences and cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) in a 5% CO2 incubator at 37°C.
HK-2 cells were used for in vitro experiments and were
divided into three groups as follows: Untreated group (Control
group), HK-2 cells induced with 10 µg/ml AA for 24 h (AAI group)
and HK-2 cells pre-treated with 50 mM rapamycin for 2 h and then
stimulated with 10 µg/ml AA for 24 h (RAP group).
Western blotting
RIPA lysis buffer was used to lyse renal tissues and
HK-2 cells. After lysis, total proteins were extracted and
quantified using a protein assay kit (cat. no. P0006C; Beyotime
Institute of Biotechnology). Proteins (40 µg/lane) were separated
using 10% sodium dodecyl sulfate-polyacrylamide gels and
transferred onto polyvinylidene difluoride membranes, which were
blocked with 5% fat-free milk for 2 h at room temperature. The
membranes were incubated with primary antibodies against p-mTOR
(phospho S2448; cat. no. ab109268; 1:1,000; Abcam), mTOR (cat. no.
ab32028; 1:1,000; Abcam), p-ribosomal S6 protein kinase (p-S6K1;
phospho T252 for mouse and phospho T229 for human; cat. no.
ab59208; 1:1,000; Abcam), S6K1 (cat. no. ab32359; 1:1,000; Abcam),
p62 (cat. no. 13121; 1:1,000; Cell Signaling Technology, Inc.),
Beclin-1 (cat. no. 3738; 1:1,000; Cell Signaling Technology, Inc.),
light chain 3 (LC3; cat. no. 4108; 1:1,000; Cell Signaling
Technology, Inc.), Bcl-2 (cat. no. 3498; 1:1,000; Cell Signaling
Technology, Inc.) and Bax (cat. no. 2772; 1:1,000; Cell Signaling
Technology, Inc.) at 4°C overnight and then washed with buffer (TBS
with 0.1% Tween-20). Then, a horseradish peroxidase-conjugated
antibody (cat. no. A25012; 1:5,000; Abbkine Scientific Co.) was
added to the membranes and samples were incubated at room
temperature for 1 h. Lastly, the protein bands were visualized
using an enhanced chemiluminescence system. Western primary and
secondary antibody removal solution (cat. no. P0025; Beyotime
Institute of Biotechnology) was used for re-probing the PVDF
membranes. Protein expression levels were normalized to that of
GAPDH (cat. no. 5174; 1:10,000; Cell Signaling Technology, Inc.)
and quantified with ImageJ software (version 1.46; National
Institute of Health).
Flow cytometry
HK-2 cells apoptosis in each group were evaluated by
flow cytometry using Annexin V-FITC Apoptosis Detection kit
(Dojindo Molecular Technologies, Inc.), following the
manufacturer's protocol. Briefly, the cells were washed twice with
PBS at room temperature, and then re-suspended in 200 µl 1X
AnnexinV Binding Solution and the cell concentration was adjusted
to 1×106/ml. Then, 100 µl cell suspension was added to a
new tube, then Annexin V-FITC (5 µl) and PI (5 µl) were added and
vortexed gently. Following incubation for 15 min at room
temperature in the dark, 400 µl 1X Annexin V Binding Solution was
added and gently mixed. Finally, the cells were analyzed using a
FACScan flow cytometer (BD Biosciences) and FlowJo software (FlowJo
LLC 10.0.7) was used to analyze the data. Flow cytometry was
performed in triplicate and repeated at least three times.
Immunofluorescent staining
An HK-2 cell monolayer was fixed with 4%
paraformaldehyde at room temperature for 1 h. To detect autophagy,
cells were blocked with 10% normal goat serum with 0.2% Triton
X-100 for 1 h at room temperature. Cells were then incubated with
primary antibodies against LC3B (cat. no. NB100-2220; 1:200; Novus
Biologicals, LLC) overnight at 4°C. After washing three times with
PBS, the cells were incubated with Alexa Fluor 488 goat anti-mouse
IgG (cat. no. Ab150113; 1:500; Abcam) for 2 h at room temperature.
Subsequently, the cells were washed three times with PBS and
stained with 4′,6-diamidino-2-phenylindole (Beijing Solarbio
Science & Technology Co., Ltd.) for 30 min at 37°C in the dark.
Images were captured using an epifluorescence microscope (Olympus
Corporation; magnification, ×200). Mean area fraction of LC3B was
quantified using Image-Pro plus 6.0 (Media Cybernetics, Inc.).
Statistical analysis
All experiments were independently repeated three
times. Data are presented as the mean ± standard deviation.
GraphPad Prism 8.0 (GraphPad Software, Inc.) was used to analyze
the results using one-way analysis of variance. Multiple
comparisons between groups were analyzed using Tukey's post-hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
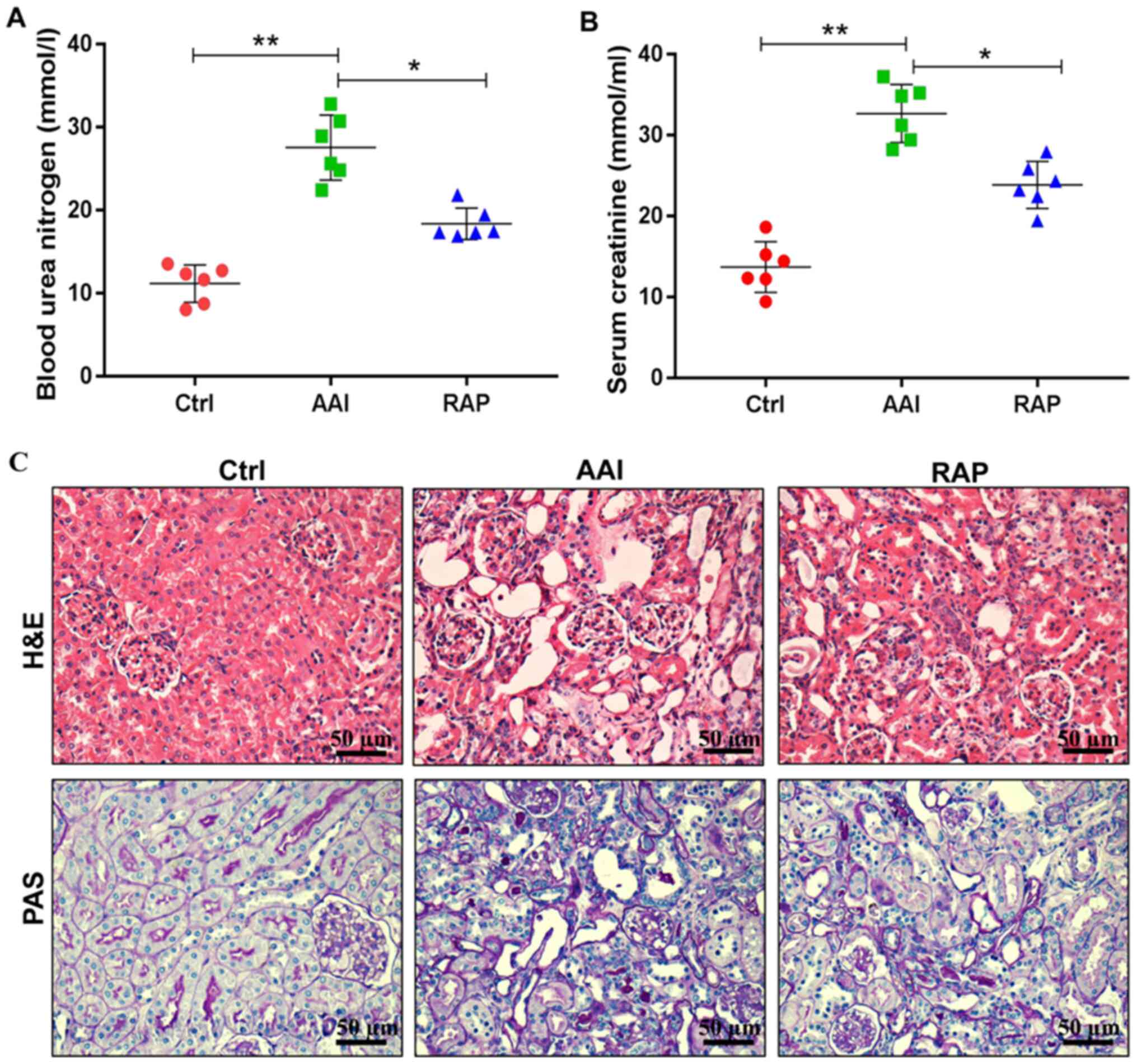
Rapamycin ameliorates AA-induced renal
dysfunction and structural abnormalities
Studies have shown that AA can lead to renal
dysfunction and structural abnormalities in rodents (22,23).
Consistent with previous reports, the present study found that AA
induced significant renal insufficiency, as evidenced by
significantly increased levels of blood urea nitrogen (Fig. 1A) and serum creatinine (Fig. 1B). In addition, H&E and PAS
staining demonstrated that, compared with the Ctrl group, AAI group
mice presented notable tubular dilatation and necrosis, increased
tubulointerstitial area, mesangial expansion and glomerular
hypertrophy (Fig. 1C). However,
these negative effects were reversed following the intraperitoneal
injection of rapamycin. Treatment with rapamycin decreased the
circulating levels of blood urea nitrogen and serum creatinine and
markedly improved renal tubular lesions, mesangial matrix expansion
and hypertrophic changes. Collectively, these results indicated
that supplementation of rapamycin protects against AA-induced renal
damage in mice.
Inhibition of mTOR by rapamycin
promotes AA-induced renal autophagy and thereby suppresses
apoptosis of kidney tissues in vivo
mTOR serves a significant role in the progression of
many diseases and is the main target for regulating autophagy
(24). Autophagy is activated in
AA-induced renal injury, but further activation of renal autophagy
has a protective effect in AAN (15). Autophagy is a method of cellular
adaptation that prevents cell death resulting from external
stimulation. During this process, damaged organelles, including the
mitochondria and endoplasmic reticulum, are removed (11,25).
Therefore, the present study determined whether rapamycin could
modulate the renal activity of mTOR in a mouse AAN model, thereby
activating renal autophagy and protecting against AA-induced renal
damage. To validate the hypothesis, western blotting was performed
to determine the renal expression of p-mTOR and its downstream
substrate p-S6K1 as well as the autophagic markers, including p62,
Beclin1 and LC3II/LCI, in all groups. P-mTOR and p-S6K1 expression
was increased in kidney tissues from AA-treated mice with increased
renal autophagy, as evidenced by upregulated renal expression of
p62, Beclin1 and LC3II/LCI. As expected, rapamycin treatment
effectively prevented the AA-induced increase in mTOR and S6K1
phosphorylation, which led to marked elevations in AA-induced renal
expression of the autophagy markers (Fig. 2). This suggested that inhibiting
mTOR activity by rapamycin further activated renal autophagy.
Autophagy and apoptosis are two connected pathological processes
involved in the development of AAN (26). To determine the effects of rapamycin
on renal apoptosis in AAN mice, the protein expression of Bcl-2 and
Bax, common markers of apoptosis, was assessed in kidney tissues
using western blotting. The results indicated that the kidney
tissue of AA-treated mice presented with decreased expression of
Bcl-2 and increased expression of Bax, which were reversed by
rapamycin treatment (Fig. 2A, G and
H), suggesting that rapamycin inhibited apoptosis in the
kidneys of AA-treated renal injury mice. Taken together, these
observations indicate that rapamycin supplementation inhibits the
renal activity of mTOR, which promotes renal autophagy, thereby
probably suppressing the apoptosis of kidney tissues in mice with
AA-induced renal injury.
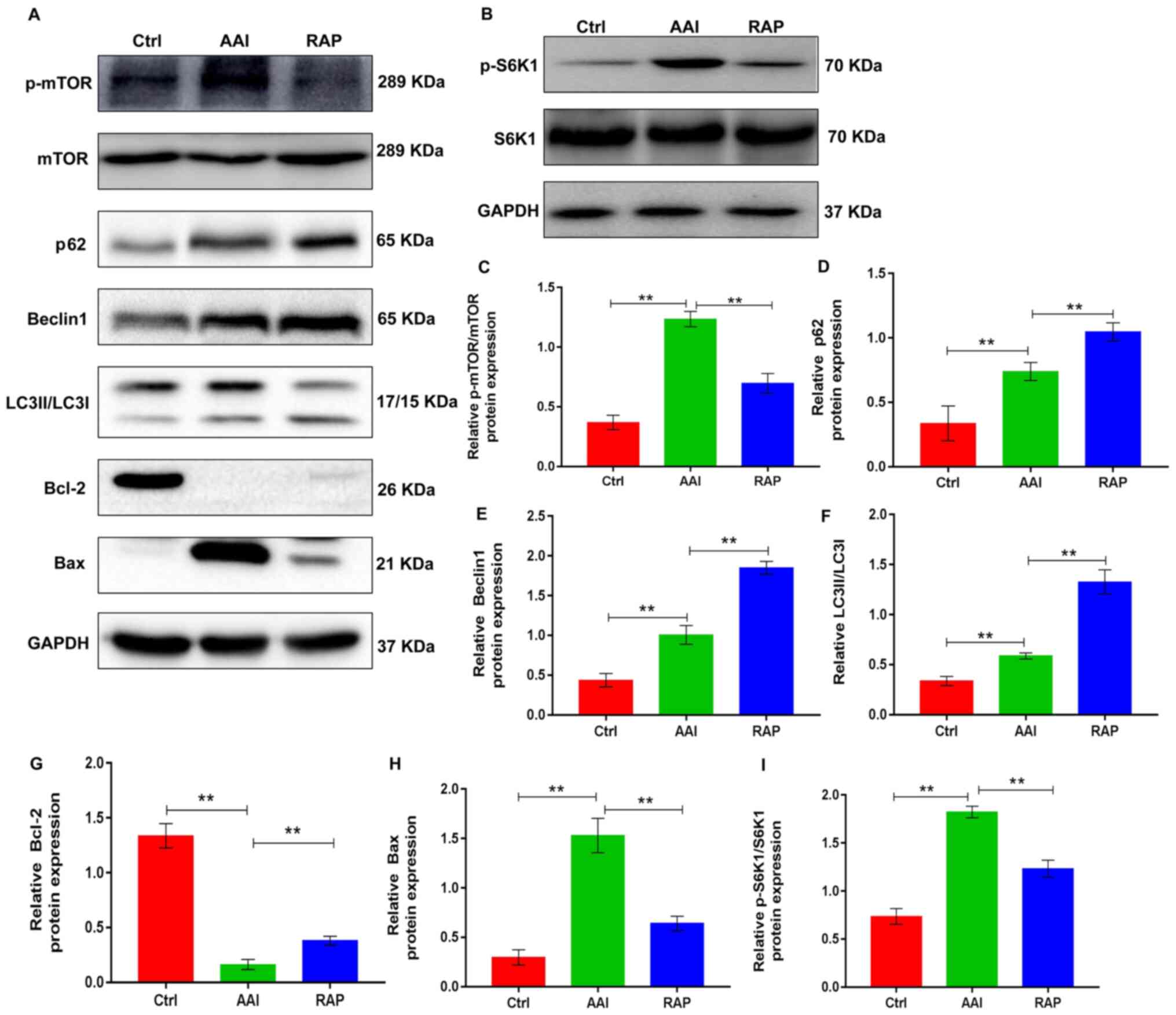 | Figure 2.Rapamycin promotes AA-induced renal
autophagy and suppresses apoptosis in the kidney tissues of mice by
inhibiting mTOR activity. (A) Representative western blots showing
the protein expression levels of p-mTOR, mTOR p62, Beclin1, LC3 and
Bcl-2 in kidney tissues; GAPDH was used as a loading and
normalization control. (B) Representative western blots showing the
protein expression levels of p-S6K1 and S6K1 in kidney tissues;
GAPDH was used as a loading and normalization control. Renal
expression of (C) p-mTOR/mTOR, (D) p62, (E) Beclin1, (F) LC3, (G)
Bcl-2, (H) Bax and (I) p-S6K1/S6K1 were quantified using ImageJ.
Results are presented as the mean ± standard deviation.
**P<0.01. All experiments were performed three times;
n=10/group. AA, aristolochic acid; p-, phosphorylated; LC3, light
chain 3; S6K1, ribosomal S6 protein kinase 1; Ctrl, control group;
AAI, AA-induced group; RAP, rapamycin treatment group. |
Rapamycin inhibits mTOR activity and
promotes AA-induced renal autophagy in HK-2 cells
Renal tubular epithelial cells are used to study AAN
(27). In the mouse model of the
present study, AA treatment induced severe damage to renal tubular
epithelial cells. Thus, the pro-autophagy activities of rapamycin
was next evaluated using an in vitro experimental model of
the AA-challenged HK-2 human renal tubular epithelial cell line.
Western blotting revealed that AA treatment markedly increased the
expression of p-mTOR and p-S6K1, whereas the treatment of HK-2
cells with rapamycin reduced the AA-induced increase in p-mTOR and
p-S6K1 (Fig. 3A-B and F-G).
Notably, AA treatment resulted in the activation of autophagy in
HK-2 cells and was accompanied by increased protein expression of
p62, Beclin-1 and LC3II/LCI. However, the inhibitory action of
rapamycin on mTOR further promoted AA-induced autophagy in HK-2
cells, as evidenced by higher protein expression of p62, Beclin-1
and LC3II/LC3I, compared to AA treatment (Fig. 3A-3E). Moreover, cellular
immunofluorescence staining with anti-LC3B antibody revealed that
rapamycin promoted AA-induced renal autophagy in HK-2 cells
(Fig. 3H and I). These data
suggested that rapamycin inhibited the activity of mTOR, which
mediates stronger autophagy in AA-treated HK-2 cells.
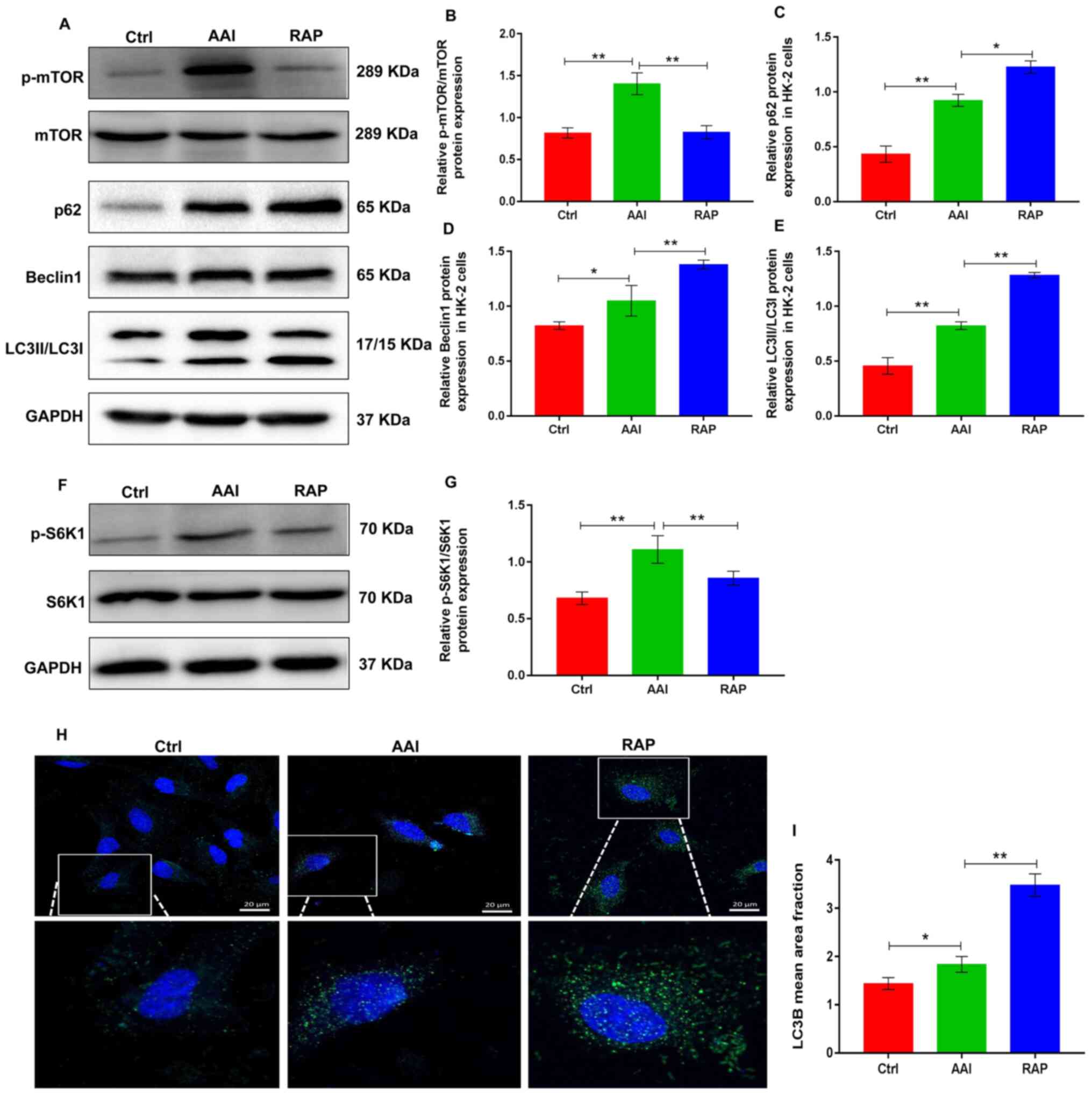 | Figure 3.Rapamycin inhibits p-mTOR expression
and promotes AA-induced renal autophagy in HK-2 cells. (A)
Representative images showing the protein expression of p-mTOR,
mTOR, p62, Beclin1 and LC3 in HK-2 cells determined using western
blotting. GAPDH was used as an internal control. Relative protein
expression of (B) p-mTOR/mTOR, (C) p62, (D) Beclin1 and (E) LC3
were quantified using ImageJ. (F) Representative images showing the
protein expression of p-S6K1 and S6K1 in HK-2 cells determined
using western blotting. GAPDH was used as an internal control. (G)
Relative protein expression of p-S6K1/S6K1 was quantified using
ImageJ. (H) Detection of autophagosomes by LC3B immunofluorescence
staining in HK-2 cells; small green dots indicate autophagosome
formation (scale bar=25 µm). (I) Mean area fraction of LC3B in each
group was quantified using Image-Pro plus 6.0. Results are
presented as the mean ± standard deviation. *P<0.05;
**P<0.01. All experiments were performed three times. p-,
phosphorylated; LC3, light chain 3; AA, aristolochic acid; S6K1,
ribosomal S6 protein kinase 1; Ctrl, control group; AAI, AA-induced
group; RAP, rapamycin treatment group. |
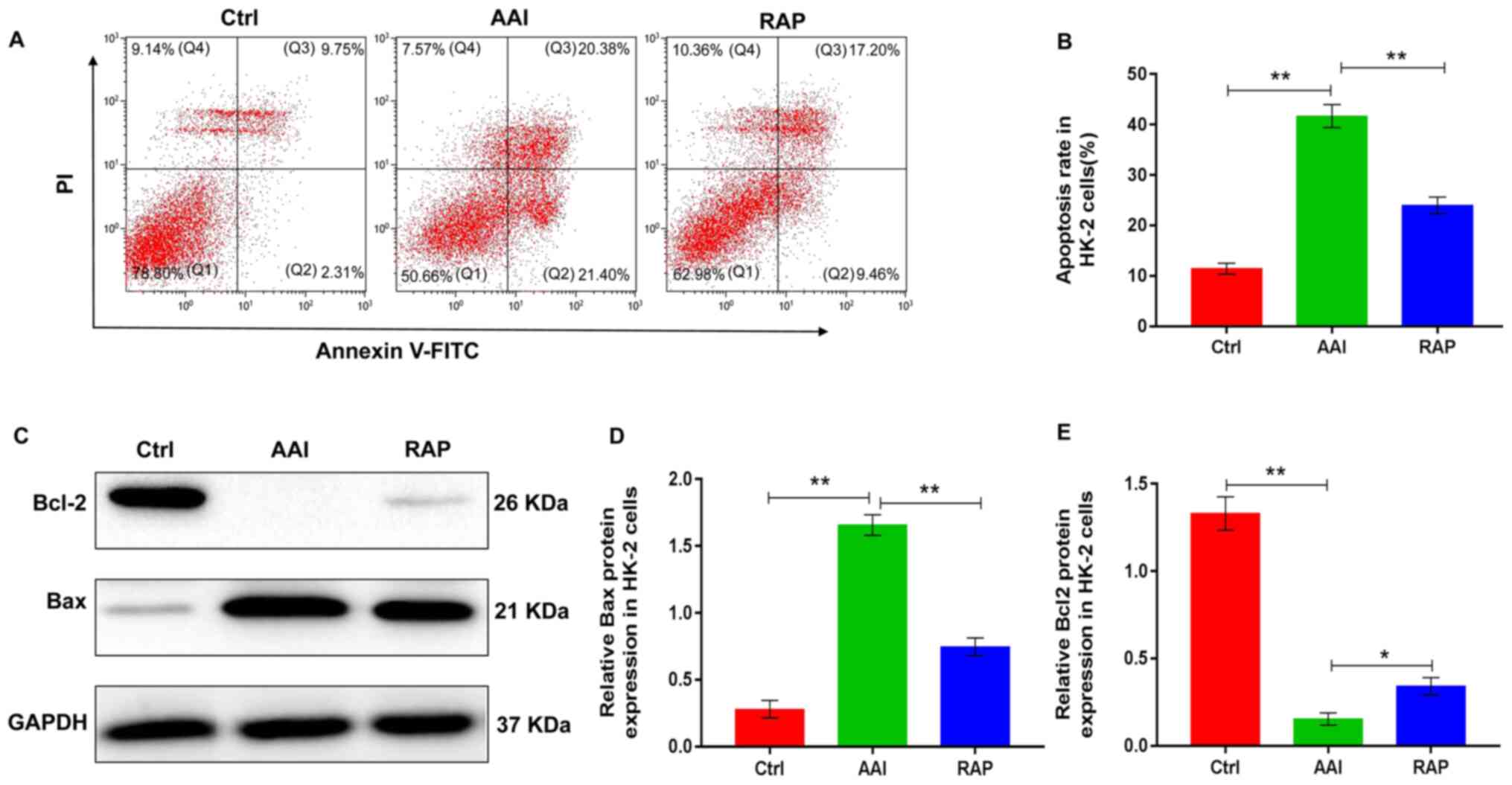
Rapamycin inhibits AA-induced
apoptosis in HK-2 cells
The effect of rapamycin on autophagy in AA-treated
HK-2 cells may affect cell survival. Therefore, the effects of
rapamycin on renal apoptosis was determined using HK-2 cells. Flow
cytometry results revealed that AA treatment significantly
increased the rate of HK-2 cell apoptosis. Conversely, rapamycin
supplementation reversed the AA-induced increase in HK-2 cell
apoptosis (Fig. 4A and B).
Additionally, to further explore the role of rapamycin in the renal
apoptosis of HK-2 cells, the protein expression of Bcl2 and Bax was
evaluated using western blotting. Following AA treatment, the
expression of Bcl2 was significantly reduced, whereas rapamycin
replenishment was associated with a marked elevation in Bcl2
expression levels compared with that in the AAI group. Treatment
with AA notably increased the expression of Bax; however, the
AA-induced increase in Bax expression was significantly reduced
following treatment with rapamycin compared to that observed in the
AAI group (Fig. 4C-4E).
Collectively, the observations indicated that rapamycin
supplementation inhibited AA-induced apoptosis in HK-2 cells likely
by activating autophagy.
Discussion
The results of the present study provided new
insights into the mechanism underlying the improvement of AAN by
rapamycin in mice. Rapamycin significantly ameliorated AA-induced
renal dysfunction and structural injury. These effects of rapamycin
were associated with the promotion of renal autophagy and
inhibition of kidney-cell apoptosis. The data demonstrated, for the
first time to the best of the authors' knowledge, that rapamycin
protects the kidney from AA-induced renal damage by potentiating
the mTOR-mediated autophagy signaling pathway.
AAN, a variant of interstitial nephritis leading to
ESRD and urothelial malignancy, was originally reported in Belgium
in a group of patients who ingested slimming pills made from
powdered root extracts of Chinese herbs containing AA (28). Unfortunately, there is no proven and
effective treatment method for AAN. The present study demonstrated
that blocking the mTOR signaling pathway by rapamycin effectively
ameliorated AAN, as evidenced by a reduction in blood urea nitrogen
and serum creatinine levels, as well as an improvement in the
structural units of the kidney tissues. The findings suggested that
rapamycin may be a promising pharmacological strategy for the
treatment of AAN. However, the renal protective mechanism of
rapamycin in AAN requires further investigation.
Autophagy is an adaptive response to intracellular
or extracellular stress, including hypoxia, nutrient or growth
factor deprivation, oxidative damage and other destructive injuries
(29). Stress-induced autophagy
supports cell survival by clearing and recovering damaged
macromolecules, protein aggregates and organelles (30). Thus, it is generally regarded as a
self-protection mechanism in cells. However, upon persistent
stimulation or beyond a certain threshold, this protective effect
is compromised and eventually leads to cell death (31). Accumulating evidence indicates that
autophagy is predominantly regulated by the mTOR-dependent
signaling pathway (32). As a
specific mTOR inhibitor, rapamycin binds to mTOR complex I through
fk506 binding protein 12 and directly inhibits the protein
expression of p-mTOR (33).
Notably, previous studies have demonstrated that renal autophagy is
activated in AAN; furthermore, enhanced renal autophagy can prevent
AA-induced renal damage in mice (14,15).
Thus, it was hypothesized that the nephroprotective role of
rapamycin in AAN is probably mediated by mTOR-induced activation of
autophagy. Consistent with the findings of the abovementioned
studies, the present study showed that AA treatment for 6 weeks
resulted in a sharp increase in p-mTOR and its downstream substrate
p-S6K1 expression and a significant elevation in p62, Beclin-1 and
LC3II/LC3I expression in the kidney tissues of mice. By contrast,
replenishment of rapamycin reversed the increased expression of
p-mTOR and p-S6K1 and induced the protein expression of p62,
Beclin-1 and LC3II/LC3I. Consistent with these results, the direct
treatment of HK-2 cells with AA, with or without rapamycin
supplementation, resulted in similar findings. The results
suggested that rapamycin inhibited the activity of mTOR, which in
turn, mediated stronger autophagic activity in AAN. The activation
of renal autophagy induced by AA may be a compensatory
stress-protective mechanism; continuous AA stimulation
decompensates this adaptive protection and does not inhibit the
progression of AAN. Rapamycin significantly inhibited the
phosphorylation of mTOR and S6K1, which resulted in further
activation of renal autophagy and exerted a cytoprotective effect
to maintain the survival of renal cells that were under AA
stimulation. These findings explained the renal protective
mechanism of rapamycin in AAN. Notably, AA induced high expression
of p-mTOR and also activated autophagy in mouse kidney tissues and
HK-2 cells. This may be due to mTOR exerting other biological
effects in AAN, which are independent of AA-induced renal
autophagy. Other physiological and pathological effects of mTOR,
including the regulation of cell differentiation, inflammatory
action and glucose and lipid metabolism in AAN, are worthy of
further research.
Autophagy and apoptosis are two important cellular
processes that involve complex and intersecting protein networks
(34). Autophagy promotes cell
survival by accelerating cellular metabolism and assisting the
removal of mitochondria following cellular injury, which can
regulate apoptosis (35).
Mitochondrial damage induces the release of metabolic hydrolase and
caspase activator into the cytoplasm from the mitochondria, which
degrades certain proteins in the cytoplasm and induces apoptosis
(36). However, activated autophagy
intercepts and degrades damaged mitochondria and also selectively
removes activated caspase-8 to prevent apoptosis (37,38).
The present study demonstrated that AA treatment promotes apoptosis
in HK-2 cells in vitro and the renal tissues of mice in
vivo. Although the inhibition of mTOR phosphorylation by
rapamycin further activated renal autophagy in a mouse model of AAN
and AA-challenged HK-2 cells, it is unclear whether the induction
of autophagy by rapamycin is responsible for apoptosis observed in
the present study. Rapamycin markedly inhibited apoptosis in
AA-treated HK-2 cells and renal tissues of mice. Taken together,
the data supported a role for rapamycin as an effector to promote
AA-induced autophagy, which probably thereby inhibited apoptosis in
AAN by decreasing the renal expression of p-mTOR.
The present study had some limitations. First,
although other studies have demonstrated that enhanced renal
autophagy can inhibit the apoptosis of kidney cells and serve a
protective role in AAN (12,15),
whether the beneficial effect of mTOR inhibition on apoptosis
disappears in the case of co-treatment with an autophagy inhibitor,
such as the chloroquine, should be investigated. In the future,
further experiments will be conducted to verify this hypothesis.
Second, the inhibitory effect of rapamycin on mTOR activity in
untreated with AA cells should be verified to ensure the
rationality of the experimental grouping, which is also a
limitation that should be noted for future experiment group
design.
To summarize, the present study demonstrated that
rapamycin was highly effective in preventing the progression of
AAN. Functionally, it was found that rapamycin effectively
inhibited the renal activity of mTOR signal pathway in AAN, which
in turn, promoted renal autophagy, thus probably enhancing the
anti-apoptosis ability of kidney cells and protecting mice from
AA-induced renal injury. Therefore, because rapamycin has been used
clinically as an effective immunosuppressant to prevent renal
transplant rejection, it shows potential as a therapy for future
medical interventions in the management of AAN.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China of Xiangdong Yang
(grant no. 81670660); the Shandong Important Research Plans Fund of
Xiangdong Yang (grant no. GG201809250293); the Natural Science
Foundation of Shenzhen University General Hospital of Fan Lin
(grant no. SUGH2018QD071); and the Natural Science Foundation of
Shenzhen University General Hospital of Yeping Ren (grant no.
SUGH2020QD011).
Availability of data and materials
All data used or analyzed during the present study
are available from the corresponding author on reasonable
request.
Authors' contributions
FL, YQL, LLT, XHX and YPR performed the experiments.
XDY and BCC conceived and designed the study. FL, YFS and XLZ
analyzed the data and drafted the manuscript. XDY and YQL reviewed
and edited the manuscript. XDY and FL assessed and confirmed the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Animal experiments were approved by the Animal
Ethics Committee of Shenzhen University (approval no.
SUGH-A-02104).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jadot I, Declèves AE, Nortier J and Caron
N: An Integrated View of Aristolochic Acid Nephropathy: Update of
the Literature. Int J Mol Sci. 18:182017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luciano RL and Perazella MA: Aristolochic
acid nephropathy: Epidemiology, clinical presentation, and
treatment. Drug Saf. 38:55–64. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu X, Nie S, Liu Z, Chen C, Xu G, Zha Y,
Qian J, Liu B, Han S, Xu A, et al: Epidemiology and Clinical
Correlates of AKI in Chinese Hospitalized Adults. Clin J Am Soc
Nephrol. 10:1510–1518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Redeker S, Oppe M, Visser M, Busschbach
JJV, Weimar W, Massey E and Ismail S; ‘Nierteam aan Huis’
consortium, : Cost-effectiveness of a home-based group educational
programme on renal replacement therapies: A study protocol. BMJ
Open. 9:e0256842019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hahn D, Hodson EM, Hamiwka LA, Lee VW,
Chapman JR, Craig JC and Webster AC: Target of rapamycin inhibitors
(TOR-I; sirolimus and everolimus) for primary immunosuppression in
kidney transplant recipients. Cochrane Database Syst Rev.
12:CD0042902019.PubMed/NCBI
|
|
6
|
Tarasewicz A, Dębska-Ślizień A, Rutkowska
B, Szurowska E and Matuszewski M: Efficacy and safety of mammalian
target of rapamycin inhibitor use-long-term follow-up of first
tuberous sclerosis complex patient treated de novo with sirolimus
after kidney transplantation: a case report. Transplant Proc.
50:1904–1909. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang X, Yan X and Yang D, Zhou J, Song J
and Yang D: Rapamycin attenuates mitochondrial injury and renal
tubular cell apoptosis in experimental contrast-induced acute
kidney injury in rats. Biosci Rep. 38:382018. View Article : Google Scholar
|
|
8
|
Wu M, Tang L, Chen B, Zheng J, Dong F, Su
Z and Lin F: Blockade of the mTOR signaling pathway with rapamycin
ameliorates aristolochic acid nephropathy. Exp Ther Med.
19:2887–2894. 2020.PubMed/NCBI
|
|
9
|
Yu L, Chen Y and Tooze SA: Autophagy
pathway: Cellular and molecular mechanisms. Autophagy. 14:207–215.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levine B and Kroemer G: Biological
Functions of Autophagy Genes: A Disease Perspective. Cell.
176:11–42. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zeng Y, Li S, Wu J, Chen W, Sun H, Peng W,
Yu X and Yang X: Autophagy inhibitors promoted aristolochic acid I
induced renal tubular epithelial cell apoptosis via mitochondrial
pathway but alleviated nonapoptotic cell death in mouse acute
aritolochic acid nephropathy model. Apoptosis. 19:1215–1224. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang CC, Wu CT, Chen LP, Hung KY, Liu SH
and Chiang CK: Autophagy induction promotes aristolochic
acid-I-induced renal injury in vivo and in vitro. Toxicology.
312:63–73. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jin C, Miao X, Zhong Y, Han J, Liu Q, Zhu
J, Xia X and Peng X: The renoprotective effect of diosgenin on
aristolochic acid I-induced renal injury in rats: Impact on
apoptosis, mitochondrial dynamics and autophagy. Food Funct.
11:7456–7467. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zeng Y, Yang X, Wang J, Fan J, Kong Q and
Yu X: Aristolochic acid I induced autophagy extenuates cell
apoptosis via ERK 1/2 pathway in renal tubular epithelial cells.
PLoS One. 7:e303122012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu Y, Li W, Hu Y, Liu Y and Sun X:
Suppression of sirtuin 1 alleviates airway inflammation through
mTOR mediated autophagy. Mol Med Rep. 22:2219–2226. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang S, Livingston MJ, Su Y and Dong Z:
Reciprocal regulation of cilia and autophagy via the MTOR and
proteasome pathways. Autophagy. 11:607–616. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li H, Zhang Y, Liu S, Li F, Wang B, Wang
J, Cao L, Xia T, Yao Q, Chen H, et al: Melatonin enhances
proliferation and modulates differentiation of neural stem cells
via autophagy in hyperglycemia. Stem Cells. 37:504–515. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He J, Ma J, Ren B and Liu A: Advances in
systemic lupus erythematosus pathogenesis via mTOR signaling
pathway. Semin Arthritis Rheum. 50:314–320. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shi B, Ma M, Zheng Y, Pan Y and Lin X:
mTOR and Beclin1: Two key autophagy-related molecules and their
roles in myocardial ischemia/reperfusion injury. J Cell Physiol.
234:12562–12568. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim JY, Leem J and Jeon EJ: Protective
effects of melatonin against aristolochic acid-induced nephropathy
in mice. Biomolecules. 10:112019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang HM, Zhao XH, Sun ZH, Li GC, Liu GC,
Sun LR, Hou JQ and Zhou W: Recognition of the toxicity of
aristolochic acid. J Clin Pharm Ther. 44:157–162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Munson MJ and Ganley IG: MTOR, PIK3C3, and
autophagy: Signaling the beginning from the end. Autophagy.
11:2375–2376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang NN, Dong J, Zhang L, Ouyang D, Cheng
Y, Chen AF, Lu AP and Cao DS: HAMdb: A database of human autophagy
modulators with specific pathway and disease information. J
Cheminform. 10:342018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Luo Z, Xu X, Sho T, Zhang J, Xu W, Yao J
and Xu J: ROS-induced autophagy regulates porcine trophectoderm
cell apoptosis, proliferation, and differentiation. Am J Physiol
Cell Physiol. 316:C198–C209. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang X, Wu J, Liu X, Wu H, Fan J and Yang
X: The protective role of Nrf2 against aristolochic acid-induced
renal tubular epithelial cell injury. Toxicol Mech Methods.
30:580–589. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Debelle FD, Vanherweghem JL and Nortier
JL: Aristolochic acid nephropathy: A worldwide problem. Kidney Int.
74:158–169. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim KH and Lee MS: Autophagy - a key
player in cellular and body metabolism. Nat Rev Endocrinol.
10:322–337. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mizushima N and Levine B: Autophagy in
human diseases. N Engl J Med. 383:1564–1576. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Papadopoli D, Boulay K, Kazak L, Pollak M,
Mallette F, Topisirovic I and Hulea L: mTOR as a central regulator
of lifespan and aging. F1000 Res. 8:F1000Faculty Rev. –998. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sato M, Seki T, Konno A, Hirai H, Kurauchi
Y, Hisatsune A and Katsuki H: Rapamycin activates mammalian
microautophagy. J Pharmacol Sci. 140:201–204. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Onishi M, Yamano K, Sato M, Matsuda N and
Okamoto K: Molecular mechanisms and physiological functions of
mitophagy. EMBO J. 40:e1047052021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abate M, Festa A, Falco M, Lombardi A,
Luce A, Grimaldi A, Zappavigna S, Sperlongano P, Irace C, Caraglia
M, et al: Mitochondria as playmakers of apoptosis, autophagy and
senescence. Semin Cell Dev Biol. 98:139–153. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang K: Autophagy and apoptosis in liver
injury. Cell Cycle. 14:1631–1642. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
D'Arcy MS: Cell death: A review of the
major forms of apoptosis, necrosis and autophagy. Cell Biol Int.
43:582–592. 2019. View Article : Google Scholar
|