Introduction
Intracerebral hemorrhage (ICH) is an important
public health problem leading to high rates of death and disability
in adults and accounts for 10–15% of all cases of stroke (1). The efficacy of hemostatic therapies
for acute, spontaneous ICH is unclear (2) and no effective treatment strategies
have been clinically implemented. Recently, investigations have
focused on underlying molecular markers correlated with brain
injury after ICH onset, which have provided possible therapeutic
targets for ICH treatment.
Transforming growth factor-β1 (TGF-β1) is a
pleiotropic cytokine that has been shown to regulate a variety of
cellular processes such as proliferation, inflammation and
apoptosis (3). Among the three
isoforms of TGF-β present in mammalian cells (4), TGF-β1 was the first to be discovered
and remains the best studied. The dysregulation of the TGF-β
pathway leads to a number of human diseases including
cardiovascular disease, tissue fibrosis and cancer (5), demonstrating the essential role of the
TGF-β proteins in vivo. However, less research has been done
on TGF-β in the central nervous system (CNS).
The main sources of TGF-β1 in the injured brain are
microglia and astrocytes (6). In
1997, Wyss-Coray et al (7)
found that the local expression of TGF-β1 within the CNS parenchyma
can enhance immune-cell infiltration and intensify the CNS
impairment resulting from peripherally triggered autoimmune
responses. Conversely, more reports have been published in recent
years demonstrating the beneficial effects of TGF on nerve
function; it was reported that TGF-β1 could reactivate chronically
denervated Schwann cells and could potentially be used to prolong
regenerative responses to promote axonal regeneration (8). TGF-β1 expression was increased after
acute ischemic brain injury; it decreased infarct volume, increased
neurogenesis, and decreased apoptosis in rodent models of ischemic
stroke (9,10). Clinical epidemiological data showed
that gene polymorphisms of TGF-β1 (G800A and T869C) and their
corresponding haploids (that result in decreased TGF-β1 content)
significantly increased the risk of ICH in patients (11). The co-treatment of recombinant
tissue plasminogen activator with ginsenoside significantly
improved outcomes in patients with ICH, which could be attributed
to the ginsenoside-induced increase in TGF-β1 (12). Nevertheless, the specific mechanism
of TGF-β1 in ameliorating ICH injury has not yet been clearly
elucidated.
Nuclear factor erythroid 2-related factor 2 (Nrf2)
is a basic-leucine-zipper transcription factor that regulates the
expression of antioxidant genes by binding to the antioxidant
response element (ARE). Activation of Nrf2 has been shown to
ameliorate many systemic fibrosis-associated diseases. One study
suggested that Nrf2 inhibited TGF-β1 in hepatic stellate cells
(13). More pertinently, Nrf2 was
found to be neuroprotective after ICH; several compounds including
hemin, dimethyl fumarate and some flavonoids protected against
brain injury after ICH via mechanisms involving Nrf2 (14,15).
Specifically, microglial Nrf2 increased phagocytosis and hematoma
clearance after ICH in vitro and in vivo (16). Additionally, Nrf2 knockout mice
exhibited greater neurological deficits following ICH injury
compared with wild-type mice (17).
These findings together suggest that Nrf2 expression is closely
associated with neuroprotection and improvement following ICH
injury.
MicroRNA (miRNA) is a short-sequence non-coding RNA
that binds to complementary sequences on target messenger RNA
(mRNA) transcripts called miRNA recognition elements (MREs). MREs
are often found on the 3′ untranslated region (UTR) of the mRNA
transcript, and MRE binding by a miRNA usually prevents the mRNA
from being translated into protein (18). Following the milestone discovery of
miRNAs (19), the subsequent wave
of research established a solid association between miRNA
dysregulation and human disease. miR-93-5p was found to be
associated with inflammation, oxidative stress and cell apoptosis
(20,21). Specifically, Nrf2 has been
identified as a target of miR-93-5p that, by inhibiting Nrf2
translation, blocks its antioxidant and neuroprotective effects
(22,23). miR-93-5p has also been previously
associated with the regulation of TGF-β-mediated signaling. One
study demonstrated that miR-93-5p inhibited RUNX3, a member of the
TGF-β superfamily, which led to increased invasion and migration of
renal carcinoma cells (24).
Another study reported that miR-93-5p downregulated a TGF-β1
receptor in nasopharyngeal carcinoma (25) and a third study suggested that
miR-93-5p suppressed TGF-β1-induced fibrosis in renal tissue
(26). Despite the evidence
supporting the influence of miR-93-5p on TGF-β signaling, no one
has directly investigated the interaction between miR-93-5p and
TGF-β1, certainly not following ICH in the brain.
Competitive endogenous RNAs (ceRNAs) regulate gene
expression by competitively binding to miRNAs. For example, the
long non-coding RNA (lncRNA) ROR promoted osteogenic
differentiation of mesenchymal stem cells by functioning as a ceRNA
for miR-138 and miR-145 and could be developed into a therapeutic
strategy for bone diseases (27).
Similarly, the ceRNA regulatory pathway involving the lncRNA HCAL,
miRNAs (miR-15a, −196a, and −196b), and the LAPTM4B gene found in
hepatocellular carcinoma (HCC) suggested that ceRNA could be used
as a potential therapeutic target for HCC treatment (28). Research on the regulatory mechanisms
of ceRNA and their potential for disease treatment is still an
actively investigated research topic.
In the present study, it was hypothesized that the
3′ untranslated region (UTR) of TGF-β1 functions as a ceRNA that
sponges miR-93-5p and thereby ameliorates the effects of ICH injury
in the brain. Firstly, it was predicted that the 3′-UTR of TGF-β1
would be able to bind miR-93-5p. Secondly, it was postulated that
TGF-β1 expression would be elevated, while miR-93-5p levels would
be decreased, in a post-ICH cellular environment. Thirdly, it was
predicted that miR-93-5p-mediated inhibition of Nrf2 would be
mitigated in the presence of TGF-β1 due to competitive binding.
Finally, it was expected that TGF-β1-mediated sponging of miR-93-5p
would decrease apoptosis and increase the neuroprotective
expression of Nrf2, effectively protecting against ICH-induced
brain injury. The present findings revealed a novel
TGF-β1/miR-93-5p/Nrf2 ceRNA regulatory pathway and provided new
insight into harnessing this molecular mechanism as a potential
treatment for ICH.
Materials and methods
Bioinformatic analysis
TargetScan (http://www.targetscan.org/) and miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php)
were used to analyze the putative targets of miR-93-5p. The web
tool GeneMANIA (29) (http://genemania.org/) was used, which searches an
extensive set of functional association data, to determine the
association between Nrf2 and TGF-β1 genes.
Cell culture and treatments
As previously established, immortalized human
microglial cell line HMO6 was used in the present study (30) for the majority of the experiments,
purchased from the College of Life Science at Wuhan University
(Wuhan, China). The cells were incubated in high-glucose Dulbecco's
Modified Eagle's Medium (DMEM/high glucose; GibcoBRL/Invitrogen;
Thermo Fisher Scientific, Inc.) supplemented with 10% inactivated
fetal bovine serum (FBS; 21103-049; Life Technologies,
GibcoBRL/Invitrogen; Thermo Fisher Scientific, Inc.) and 100 U/ml
penicillin/streptomycin (GibcoBRL/Invitrogen; Thermo Fisher
Scientific, Inc.) at 37°C with 5% CO2. When cells were
80% confluent, 0.25% trypsin (Sigma-Aldrich; Merck KGaA) was used
for digestion and passaging. To investigate the mechanism of
miR-93-5p and TGF-β1 after ICH, cells were treated with thrombin
from human plasma (thrombin concentration used as in previous
literature; 20 or 40 U/ml; Sigma-Aldrich; Merck KGaA) for 48 h and
then harvested for detection (31).
To confirm the regulatory effect of miR-93-5p and
the TGF-β1 signaling pathway, HMO6 cells were exposed to thrombin
(20 U/ml) for 48 h. Then, cells were transfected for 48 h with
either a miR-93 mimic (50 nM), a miR-93 inhibitor (100 nM), a small
interfering (si) RNA specific to TGF-β1 (siTGF-β1; 50 nM), or mimic
NC, inhibitor NC, and scramble siRNA as the negative control. The
mimic, inhibitor, siRNA, and negative-control RNA were sourced from
RiboBio Co., Ltd. and used according to the manufacturers'
instructions. The transfected cells were measured for levels of
miR-93, TGF-β1, and Nrf2.
Fresh brain tissue
Fresh brain tissue was taken from 12 adult surgical
patients [six females and six males; mean age (range), 48.7 years
(12–70 years)] at the Department of Neurosurgery at The Second
Affiliated Hospital of Nanchang University. The cohort consisted of
equal numbers of patients with and without ICH; Table I describes the details regarding the
patients and the collected samples. The tissue was collected in
sterile tubes and stored in liquid nitrogen for later experiments.
Written informed consent was obtained from all tissue donors or
their parent/guardian prior for inclusion in the present study. All
the protocols used in the present study were approved by the Ethics
Committee at The Second Affiliated Hospital of Nanchang University
(Nanchang, China).
 | Table I.Data collected on patients. |
Table I.
Data collected on patients.
| Case no. | Age, years | Sex | HPI | PH | Sampling site | Type |
|---|
| 1 | 70 | M | Glioma | Hypertension | Left frontal
lobe | Cont |
| 2 | 63 | F | Glioma | None | Left thalamus | Cont |
| 3 | 43 | M | Meningioma | Hypertension | Right
cerebellopontine angle | Cont |
| 4 | 28 | M | Epilepsy | Fracture | Right frontal
lobe | Cont |
| 5 | 64 | M | DLBCL | None | Right parietal
lobe | Cont |
| 6 | 49 | F | Epilepsy | None | Left temporal
lobe | Cont |
| 7 | 43 | M | Intracerebral
hemorrhage | None | Left occipital
lobe | ICH |
| 8 | 12 | F | Moyamoya
disease | None | Left temporal
lobe | ICH |
| 9 | 49 | F | Intracerebral
hemorrhage | None | Left occipital
lobe | ICH |
| 10 | 47 | M | Moyamoya
disease | None | Left frontal
lobe | ICH |
| 11 | 54 | F | Cerebral
hemorrhage | None | Right
cerebellum | ICH |
| 12 | 62 | F | Cerebral
hemorrhage | Hypertension | Left
cerebellum | ICH |
Enzyme-linked immunosorbent assay
(ELISA)
The proinflammatory cytokines produced by activated
microglia include interleukin (IL)-1β, tumor necrosis factor-α
(TNF-α) and IL-6, which can cause cytotoxic or cytopathogenic
effects in the CNS (30). The
production of TNF-α in HMO6 cells after a 48-h incubation with
thrombin was determined in spent culture supernatants (only the
culture media collected) using ELISA kits specific for human TNF-α
(cat. no. 5YJ3MQ3F; Elabscience; kit capable of detecting TNF-α at
4.69 pg/ml). At the end of each experiment, culture supernatants
were collected, centrifuged and stored at −70°C.
Double-immunofluorescence
staining
HMO6 cells were fixed for 30 min at room temperature
in 4% paraformaldehyde in PBS (pH 7.4), and then permeabilized with
0.25% Triton X-100 for 4 min at room temperature. Slides were
incubated for 1 h in 10% goat serum/TBS, followed by incubation
with 1:100 dilution of mouse anti-Iba1 antibody (Wako Pure Chemical
Industries, Ltd.; cat. no. 012-26723) and 1:50 rabbit anti-CD68
(BIOSS, bs-20403R) at 4°C overnight. Then, the sections were
incubated with Alexa 568-conjugated donkey anti-rabbit IgG (red)
(1:200 in PTwH; Life Technologies; cat. no. A10042) and Alexa
488-conjugated donkey anti-mouse IgG (green; 1:200 in PTwH; Life
Technologies; cat. no. A32766) for 2 h at room temperature. Nuclei
were stained with DAPI (Abcam). Microgliosis was assessed by
measuring the immunoreactivity of Iba1 (expressed by all microglia)
and CD68 (expressed by activated microglia). The images were
observed under a fluorescence microscope (Olympus IX71; Olympus
Corporation; magnifications, ×40 and ×20) and Image Pro-Plus 6.0
software (1993, 2003 Media Cybernetics) was applied to calculate
the immunohistochemical optical density.
RNA/miRNA extraction and
reverse-transcription quantitative polymerase chain reaction
(RT-qPCR)
Total RNA and miRNAs were extracted from fresh brain
tissue or cultured HMO6 cells using TRIzol (Takara Bio, Inc.) and
the miRNeasy Mini kit (Qiagen GmbH), respectively. miR-93 was
reverse-transcribed using a One Step PrimeScript miRNA cDNA
synthesis kit (Takara Biotechnology Co., Ltd.) according to the
manufacturer's protocol. qPCR was performed with SYBR Green I
Master mix (Roche Diagnostics GmbH) using a LightCycler 480 (Roche
Diagnostics) as follows: 94°C for 2 min, 94°C for 30 sec, 55°C for
30 sec, 72°C for 30 sec, and 72°C for 2 min. The sequences of the
specific primers are listed in Table
II. Melting curve analyses confirmed that all the primers were
specific for their respective transcripts. Actin and U6 were used
as the reference genes. RT-qPCR was adopted to confirm the
transfection efficiency. Each reaction was performed at a 10-µl
reaction volume and in triplicate. RT-qPCR data were analyzed using
the 2−ΔΔCq method (32),
which uses the threshold or quantification cycle (Cq) to generate a
relative gene-expression value that is normalized both to the
reference gene expression and to the control experimental condition
(+/- thrombin).
| Table II.Primer sequences for RT-qPCR. |
Table II.
Primer sequences for RT-qPCR.
| Gene | Forward, 5′-3 | Reverse, 5′-3′ |
|---|
| miR-93 |
CAAAGTGCTGTTCGTGCAGGTAG |
GCTGTCAACGATACGCTACG |
| miR-181c |
AACATTCAACCTGTCGGTGAGT |
GCTGTCAACGATACGCTACG |
| U6 |
GATGACACGCAAATTCGTGAA |
GCTGTCAACGATACGCTACG |
| TGF-β1 |
AGAAGAACTGCTGCGTGCG |
TACACGATGGGCAGCGG |
| Nrf2 |
CCAGCACATCCAGTCAGAAAC |
GTCATCTACAAACGGGAAT |
| TGF-β1 3′UTR |
ATCAAGGCACAGGGGACCAG |
CTCTGGGCTTGTTTCCTCAC |
| Actin |
TGGCACCCAGCACAATGAA |
CTAAGTCATAGTCCGCCTAGAAGCA |
Western blotting
The brain samples or isolated HMO6 cells were
mechanically lysed in RIPA lysis buffer (Beyotime Institute of
Biotechnology) and an enhanced bicinchoninic protein assay kit was
used to measure protein concentrations. The protein samples were
then loaded onto a 10% SDS-PAGE gel at 50 µg per lane and the
proteins were separated. Next, the proteins were
electrophoretically transferred to a polyvinylidene difluoride
membrane (EMD Millipore). The membrane was blocked with 3% BSA
(BioSharp Life Sciences) for 1 h at room temperature, followed by
an incubation for 12 h at 4°C with primary antibodies. The primary
antibodies used were: Anti-Nrf2 (rabbit polyclonal; 1:1,000
dilution) (cat. no. GTX103322), anti-TGF-β1 (mouse polyclonal;
1:1,000 dilution) (cat. no. GTX45121; both from GeneTex, Inc.) and
anti-β-actin (Abcam; cat. no. 8226). After three washes in TBS with
0.1% Tween-20 (TBST), the membrane was probed with horseradish
peroxidase (HRP)-conjugated anti-rabbit (1:1,000; Sigma-Aldrich;
Merck KGaA; cat. no. A3812) or anti-mouse (1:1,000; Sigma-Aldrich;
Merck KGaA; cat. no. A3562) secondary antibodies for 1 h at room
temperature. The protein bands were visualized with a SuperSignal
West Pico chemiluminescence kit (Thermo Fisher Scientific, Inc.).
Finally, ImageJ 1.51j8 software (National Institutes of Health) was
used to measure the relative density of the proteins, which was
normalized to the loading control β-actin. The experiments were
performed in triplicate.
Dual luciferase reporter assay
The 3′UTR and coding sequence (CDS) of TGF-β1 were
amplified from the genomic DNA of human brain tissue and subcloned
into the pcDNA4.0 (Addgene; cat. no. MLCC1153; 5.1kb) or psiCHECK2
(Addgene; cat. no. P0197; 6.3kb) dual luciferase reporter plasmids.
Specifically, pcDNA4.0 luciferase reporters containing TGF-β1-CDS
(1,173 bp), TGF-β1-3′UTR-WT (729 bp), or TGF-β1-3′UTR-MUT (729 bp;
TGF-β1 with a mutated 3′UTR sequence) were constructed. The
psiCHECK2 reporters contained either Nrf2 3′UTR (486 bp) or TGF-β1
3′UTR (729 bp). The constructed vectors were transfected into 293T
cells and HMO6 cells with Lipofectamine® 3000
Transfection Reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
For some experiments, the luciferase reporter plasmids were
co-transfected with an miR-93 mimic (or the control; mimic NC),
miR-93 inhibitor (or the control; inhibitor NC), siTGF-β1
(5′-AAGGGCTACCATGCCAACTTC-3′) (33), or the scramble control siTGF-β1-NC,
purchased from RiboBio (Guangzhou RiboBio Co., Ltd.). Luciferase
activity was analyzed as previously described (34). Each assay was performed in
triplicate.
Terminal deoxynucleotidyl transferase
dUTP nick end labeling (TUNEL) assay
TUNEL assays were performed with the one step TUNEL
kit (Tiangen Biotech Co., Ltd.) according to the manufacturer's
instructions. Briefly, the transfected HMO6 cells were fixed at
room temperature in 4% paraformaldehyde in PBS on
poly-(L-lysine)-coated slides, rinsed with PBS, and permeabilized
with 0.1% Triton X-100. Then, the slides were washed with PBS, and
the cells were incubated with 50 µl TUNEL reaction mixture for 1 h
at 37°C in the dark. The TUNEL-stained cells were observed under a
confocal laser scanning microscope (Olympus IX71; Olympus
Corporation; magnification, ×10) using 405-nm excitation and 568-nm
emission, and cells with red fluorescence were defined as apoptotic
cells.
Statistical analysis
All the data are expressed as the means ± SD.
Statistical analyses were conducted with GraphPad Prism 6.01
software (GraphPad Software, Inc.). The independent Student's
t-test was used to compare differences between two groups. More
than three groups were analyzed by one-way analysis of variance
followed by multiple comparisons using Tukey's test. Statistical
significance for intergroup differences was assessed by the
χ2 test or Fisher's exact test for categorical
variables. Correlation analysis was performed using the Pearson
linear correlation analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-93-5p targets both TGF-β1 and Nrf2. A previous
study showed that activated Nrf2 could trigger the expression of
ARE-regulated heme oxygenase-1 (HO-1), and that Nrf2 played
anti-oxidative and anti-inflammatory roles that were
neuroprotective following ICH (35). Based on literature reviews and
gene-target prediction databases, such as TargetScan and
miRTarBase, it was found that miR-93-5p had a binding site on Nrf2
mRNA (context++ score percentile, 85) (22,36).
Since TGF-β1 has been heavily investigated as a potential mediator
of ICH, its association with miR-93-5p was also analyzed.
Bioinformatics analysis using TargetScan and miRTarBase indicated
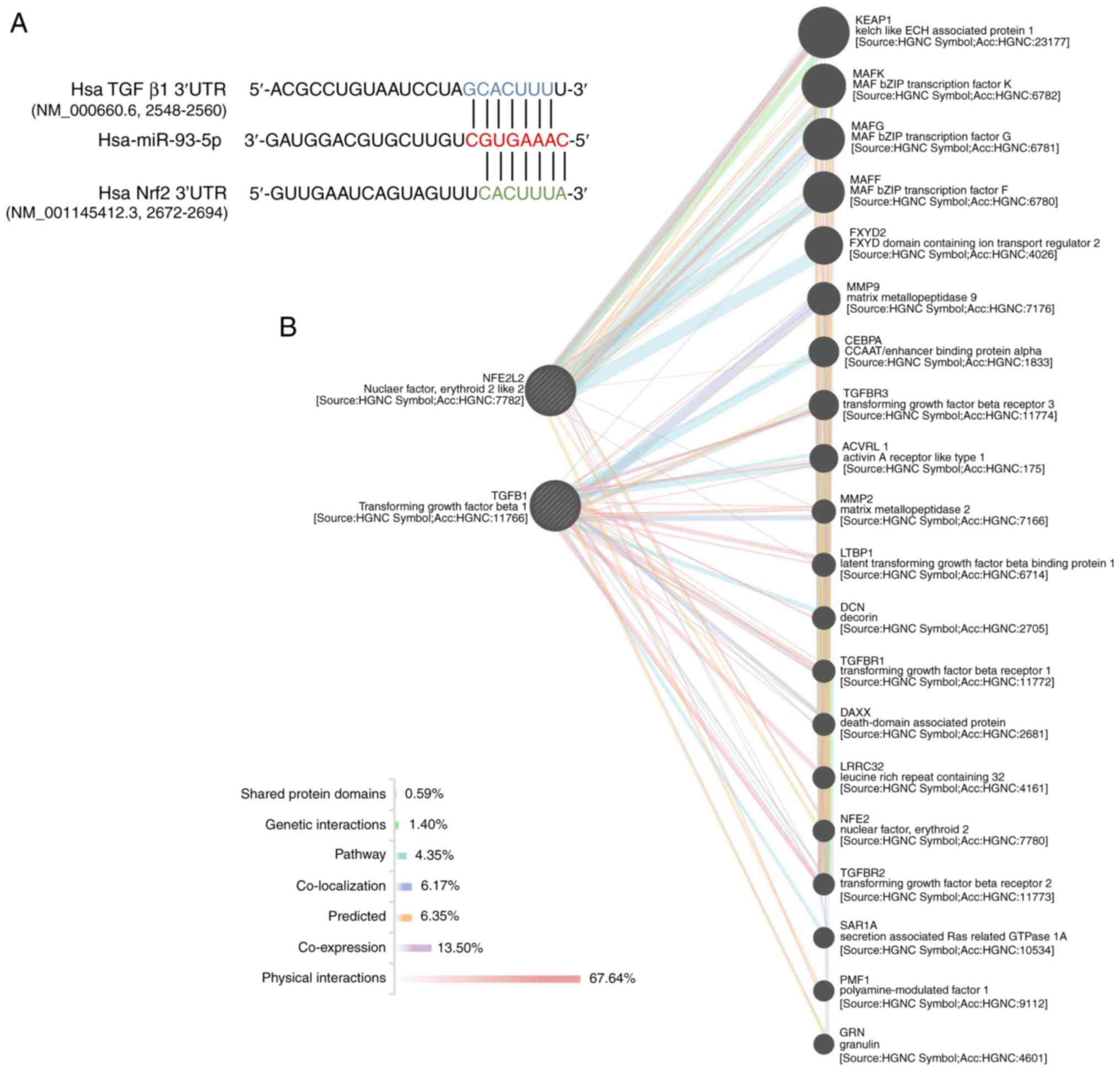
that miR-93-5p bound to the 3′UTR of TGF-β1 mRNA (Fig. 1A). In order to examine the potential
connection between TGF-β1, miR-93-5p and Nrf2, GeneMANIA was used
for protein-protein interaction network and gene co-expression
analyses. No direct association was found between Nrf2 and TGF-β1
(Fig. 1B), suggesting that
miR-93-5p was the association to regulate them both (hereafter
miR-93 represents miR-93-5p).
Relative expression levels of miR-93, TGF-β1 and
Nrf2 in HMO6 cells. Microglia are the resident immune cells of the
CNS. After ICH, microglia become activated, obtain an ameboid
morphology, and release proinflammatory cytokines (37). A previous study revealed that
excessive activation or lack of microglial regulation can cause
neurotoxicity (30); microglia are
important sources of pro-inflammatory and oxidative-stress factors,
such as tumor necrosis factor (TNF), nitric oxide, interleukin and
other neurotoxic substances. Therefore, a human microglial cell
line (HMO6) was used in the present study as an in vitro
host and simulated the ICH environment by treatment with thrombin
from human plasma. HMO6 cells were treated with thrombin at
concentrations of 20 and 40 U/ml and their morphology was
visualized under a bright-field microscope (Fig. 2B). In addition, inflammatory factors
like TNF-α were detected in the cell supernatants using ELISA kits
(Fig. 2A). The expression of TNF-α
in cells treated with 40 U/ml thrombin was higher compared with
that of cells treated with 20 U/ml thrombin (all P<0.05). This
result corresponded with the greater impairment in cellular
morphology of the 40 U/ml thrombin-treated cells compared with the
20 U/ml thrombin-treated cells; more cells formed clusters after 40
U/ml thrombin treatment. Despite the higher TNF-α levels implying a
more robust ICH-like cellular environment, the highly rounded and
clustered microglial cells caused by 40 U/ml thrombin were
unsuitable for experiments. Based on the aforementioned findings,
HMO6 cells were treat with 20 U/ml thrombin in the subsequent
experiments. Furthermore, immunofluorescence staining of Iba1 and
CD68 observed in these HMO6 cells revealed that the thrombin
treatment successfully activated human microglia (Fig. 2C). By verifying the presence of
activated, ameboid-shaped microglia releasing inflammatory
cytokines, an effective in vitro model of ICH was
established.
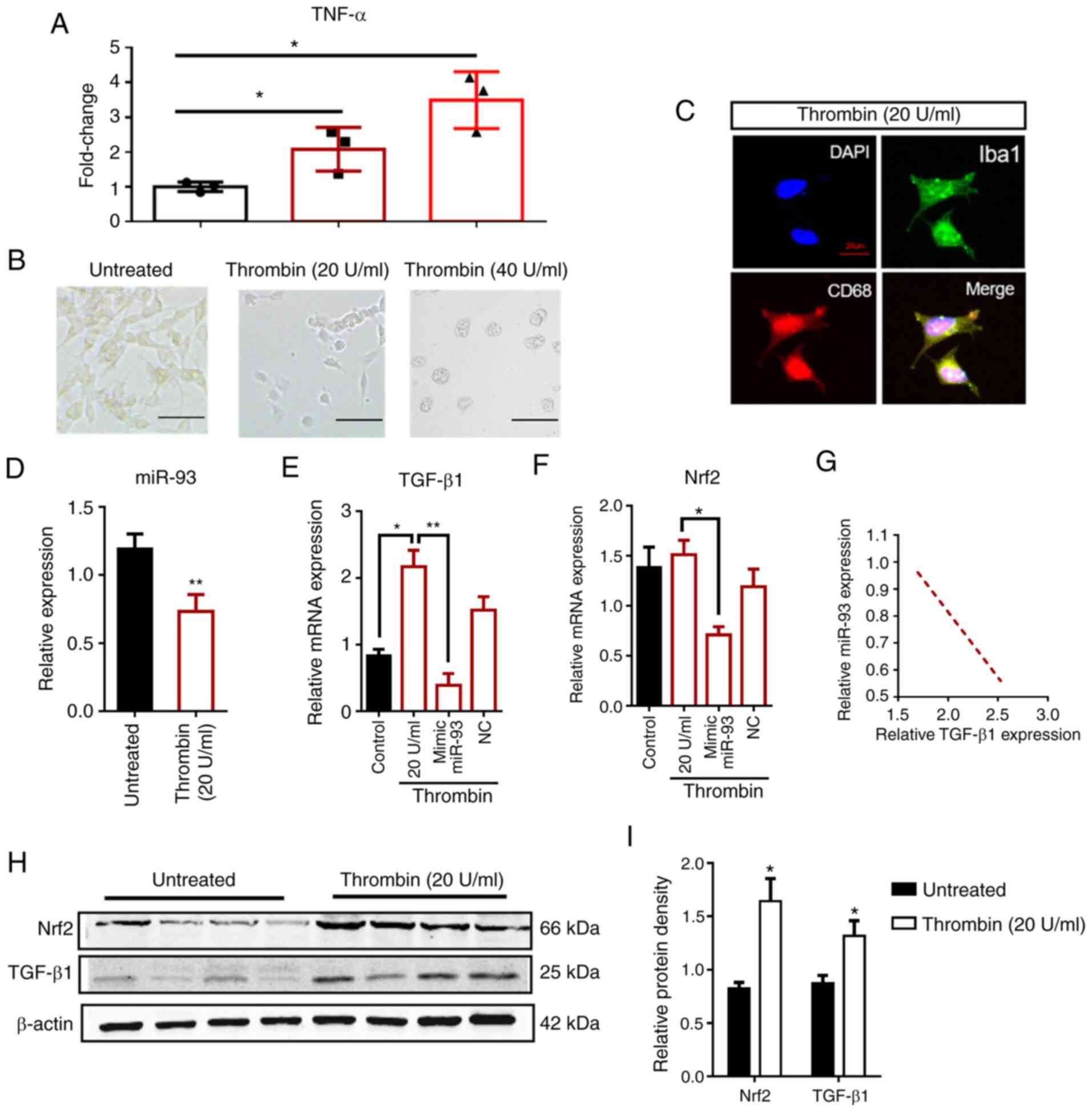 | Figure 2.Morphology and expression of HMO6
microglial cells after thrombin treatment. (A) TNF-α mRNA levels in
thrombin-treated HMO6 cells are elevated significantly after 48 h,
in a dose-dependent manner, compared with control cells
(untreated). (B) Phase contrast microscopy showing the morphology
of HMO6 cells that are untreated or treated with thrombin at
concentrations of 20 or 40 U/ml. Scale bar, 50 µm. (C) Images of
Iba1 (green) and CD68 (red) immunoreactivity in HMO6 cells, treated
with 20 U/ml thrombin. DAPI (blue) labels the cell nuclei. Scale
bar, 20 µm. (D) miR-93 expression in HMO6 cells with or without
thrombin treatment. (E and F) The modulation of Nrf2 or TGF-β1 mRNA
levels in thrombin-treated HMO6 cells after transfection with
miR-93 mimic or the corresponding control (mimic NC) was assessed
by reverse transcription-quantitative PCR. The red border
represents the thrombin-treated groups. (G) TGF-β1 and miR-93 RNA
expression levels in HMO6 cells were negatively correlated as
assessed by Pearson correlation analysis (r=−0.967; P <0.05).
(H) Western blotting images showing the protein levels of TGF-β1
and Nrf2 in HMO6 cells after thrombin (20 U/ml) treatment. β-actin
was the loading control. (I) Quantification of the TGF-β1 and Nrf2
protein levels in untreated or thrombin-treated HMO6 cells. n=6 for
each group. Data are presented as the means ± SD. *P<0.05,
**P<0. 01. TNF, tumor necrosis factor; miR, microRNA; TGF,
transforming growth factor; Nrf2, nuclear factor erythroid
2-related factor 2. |
Western blotting and RT-qPCR were used to detect
changes in miR-93, TGF-β1, and Nrf2 expression in
thrombin-treated HMO6 cells. The RT-qPCR results revealed that the
expression of miR-93 was decreased (P<0.05), Nrf2 mRNA
levels were not significantly changed, and TGF-β1 mRNA was
significantly elevated (P<0.01) in thrombin-treated cells
(Fig. 2D-F). Nrf2 mRNA levels were
not significantly changed, whereas Nrf2 protein levels were
increased, indicating that the increase in Nrf2 protein levels was
regulated by microRNA during translation. After transfection with
an miR-93 mimic, the mRNA expression levels of Nrf2 and
TGF-β1 were both decreased (P<0.05), indicating that
miR-93 could negatively regulate both these mRNAs. Pearson's
correlation analysis showed that TGF-β1 (r=−0.967;
P<0.05) expression was negatively correlated with miR-93 after
ICH (Fig. 2G). The protein levels
of Nrf2 and TGF-β1 were increased in thrombin-treated cells
(P<0.05; Fig. 2H and I), which
further confirmed that both TGF-β1 and Nrf2 were associated with
the injury following ICH. The effect of thrombin treatment on Nrf2
protein levels but not mRNA levels suggested that there may be
post-transcriptional regulation of Nrf2 after ICH. The significant
correlation between miR-93 expression and TGF-β1 and
Nrf2 expression of thrombin-treated HMO6 cells supports the
hypothesis that TGF-β1 functions as a ceRNA by sponging miR-93.
Expression patterns of miR-93, TGF-β1 and Nrf2 in
human brain tissue are consistent with thrombin-treated HMO6 cells.
The demographic characteristics of the participants are shown in
Table III. A total of 12
participants with new-onset ICH were recruited into the present
study (2 males and 4 females; median age, 44.50±17.21 years)
together with 15 healthy volunteers (4 males and 2 females; median
age, 52.83±15.82 years). There was no significant difference in the
age or sex distribution between the patients with ICH and healthy
volunteers. In order to verify the credibility of the collected
human brain tissue, the level of miR-181c was additionally tested,
whose decreased expression patterns after ICH have previously been
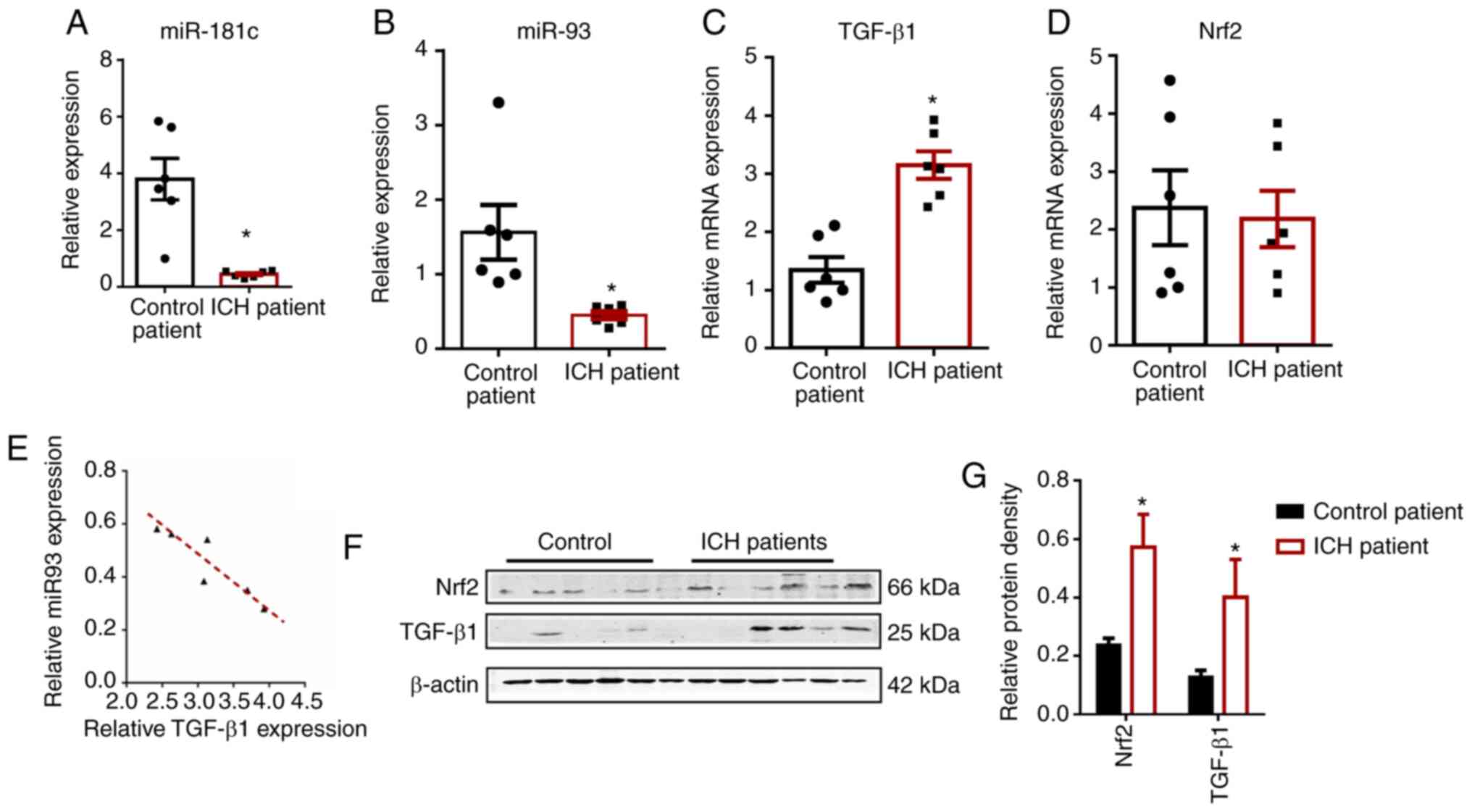
established (Fig. 3A). The results
for miR-181c were consistent with previous reports (P<0.05)
(38), confirming that the human
brain samples were reliable. Detection of mRNA and miRNA expression
in the brain tissue of patients with ICH and control (n=6 per
group; Table I) revealed that
miR-93, TGF-β1 and Nrf2 levels were consistent with
the expression levels in HMO6 cells. The RT-qPCR results
demonstrated that the expression of miR-93 was markedly lower,
Nrf2 did not change, and TGF-β1 expression was
significantly higher (P<0.05) in ICH brain tissue compared with
the corresponding non-ICH control tissue (Fig. 3B-D); this pattern was consistent
with the trend of expression in the thrombin-exposed HMO6 cells. In
addition, Pearson's correlation analysis revealed a negative
correlation between TGF-β1 expression and miR-93 after ICH
(r, −0.908; P<0.05; Fig.
3E).
| Table III.Clinical features of patients with
ICH and normal controls. |
Table III.
Clinical features of patients with
ICH and normal controls.
| Baseline
characteristics | Controls (n=6) | ICH (n=6) | P-values |
|---|
| Age, mean ± SD
years | 52.83±15.82 | 44.50±17.21 | 0.798 |
| Male sex, n% | 66.70 | 33.30 | 0.567 |
| Hypertension,
n% | 33.30 | 16.70 | 0.545 |
| Fracture, n% | 16.70 | – | NA |
| Smoking history,
n% | 50.00 | 16.70 | 0.545 |
Western blotting (Fig.
3F and G) data showed that the protein expression of TGF-β1 and
Nrf2 in the tissue from patients with ICH was significantly higher
compared with that in control tissue (P<0.05); this
post-translational expression pattern was also consistent with that
of the ICH model in HMO6 cells.
TGF-β1 3′UTR functions as a ceRNA by sponging
miR-93. After transfection, the relative expression level of miR-93
was measured via RT-qPCR, to verify transfection efficiency. The
results demonstrated that the expression of miR-93 in HMO6 cells of
the miR-93 mimics group was markedly elevated when compared with
the control groups (P<0.05), and declined in miR-93 inhibitor
group (P<0.05). However, the expression of TGF-β1
decreased significantly in the siTGF-β1 group in comparison with
the control groups (P<0.05; Fig. 4A
and B). Considering that TGF-β1 and Nrf2 mRNA were predicted
targets of miR-93, and that miR-93 showed low expression in brain
tissue from patients with ICH, it was hypothesized that TGF-β1 mRNA
may act as a ceRNA for miR-93. In order to examine the potential
ceRNA function of TGF-β1, a dual luciferase reporter assay was used
to directly test whether Nrf2 and TGF-β1 are targets of miR-93. The
results showed that luciferase activity was decreased in 293T cells
co-transfected with miR-93 and Nrf2-3′UTR or TGF-β1-3′UTR
(P<0.01; Fig. 4C-E), indicating
that miR-93 could negatively regulate both; this result
corroborated what was predicted by the bioinformatics analysis:
miR-93 targets both Nrf2 and TGF-β1 mRNA. To determine whether
miR-93 acts as a bridge between TGF-β1 and Nrf2 and to support the
hypothesized ceRNA function of TGF-β1, a miR-93 mimic or inhibitor
was used to simulate in 293T cells an environment with or without
miR-93 and measured luciferase activity. As shown in Fig. 4F, the luciferase activity of
Nrf2-3′UTR increased when co-transfected with TGF-β1-3′UTR-WT,
which offset the negative regulation of miR-93 mimic on Nrf2
(Fig. 4D). However, in the
TGF-β1-3′UTR-MUT and in the context of inhibitor 93, this effect
disappeared (Fig. 4F and G).
Co-transfection of the miR-93 mimic and the coding sequence of
TGF-β1 (TGF-β1-CDS) in 293T cells also failed to increase the
luciferase activity of Nrf2-3′UTR (Fig.
4F), indicating that the coding region of TGF-β1 has no direct
effect on the expression of Nrf2. These results indicate that the
comprehensive mechanism by which TGF-β1 3′UTR increased Nrf2
expression was the sponging or competitive binding of miR-93;
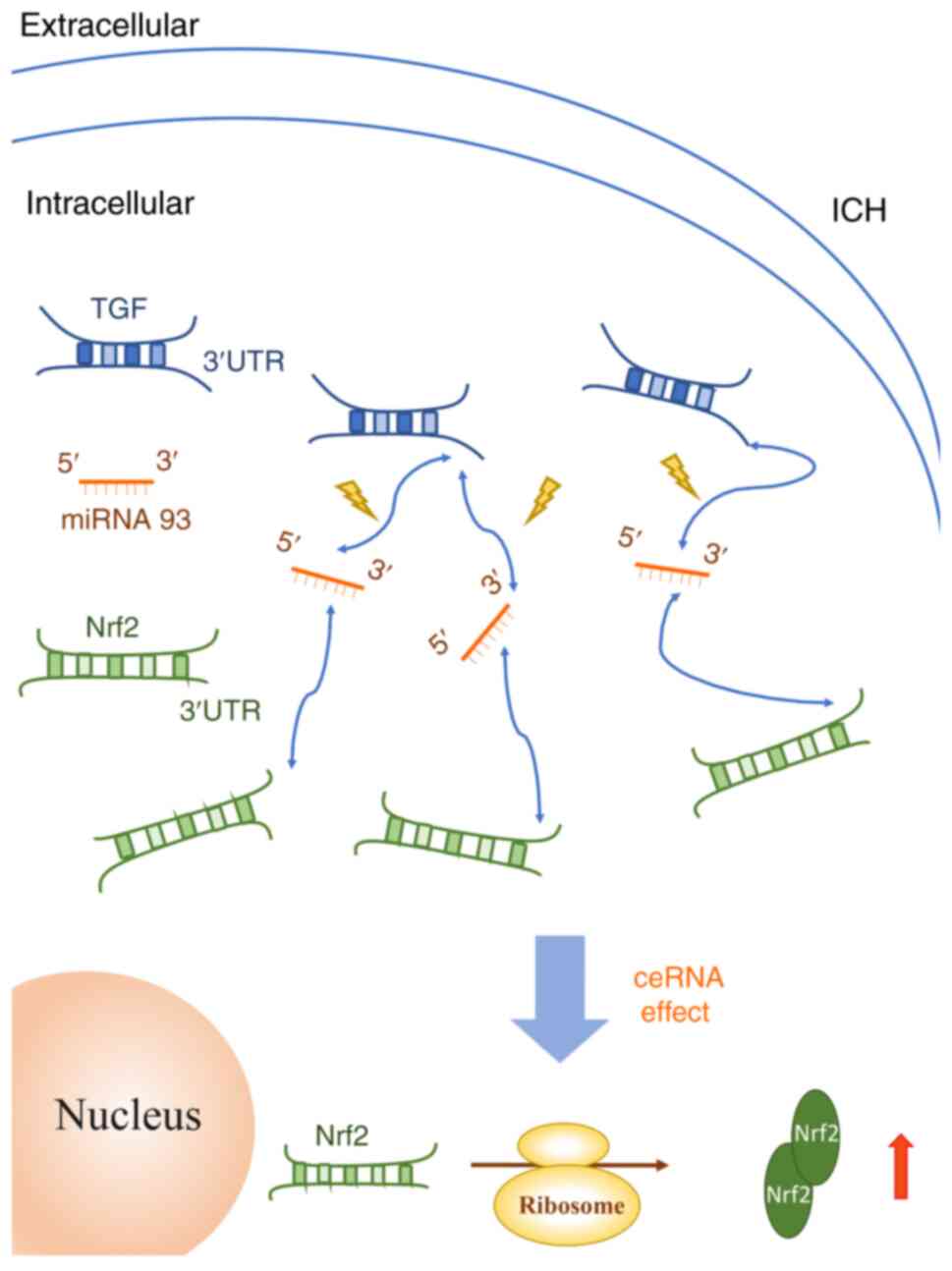
TGF-β1 competitively binds to miR-93-5p with Nrf2. When
TGF-β1 competitively binds to miR-93-5p, the activity of
miR-93 decreases, thereby upregulating the expression of Nrf2
(Fig. 5).
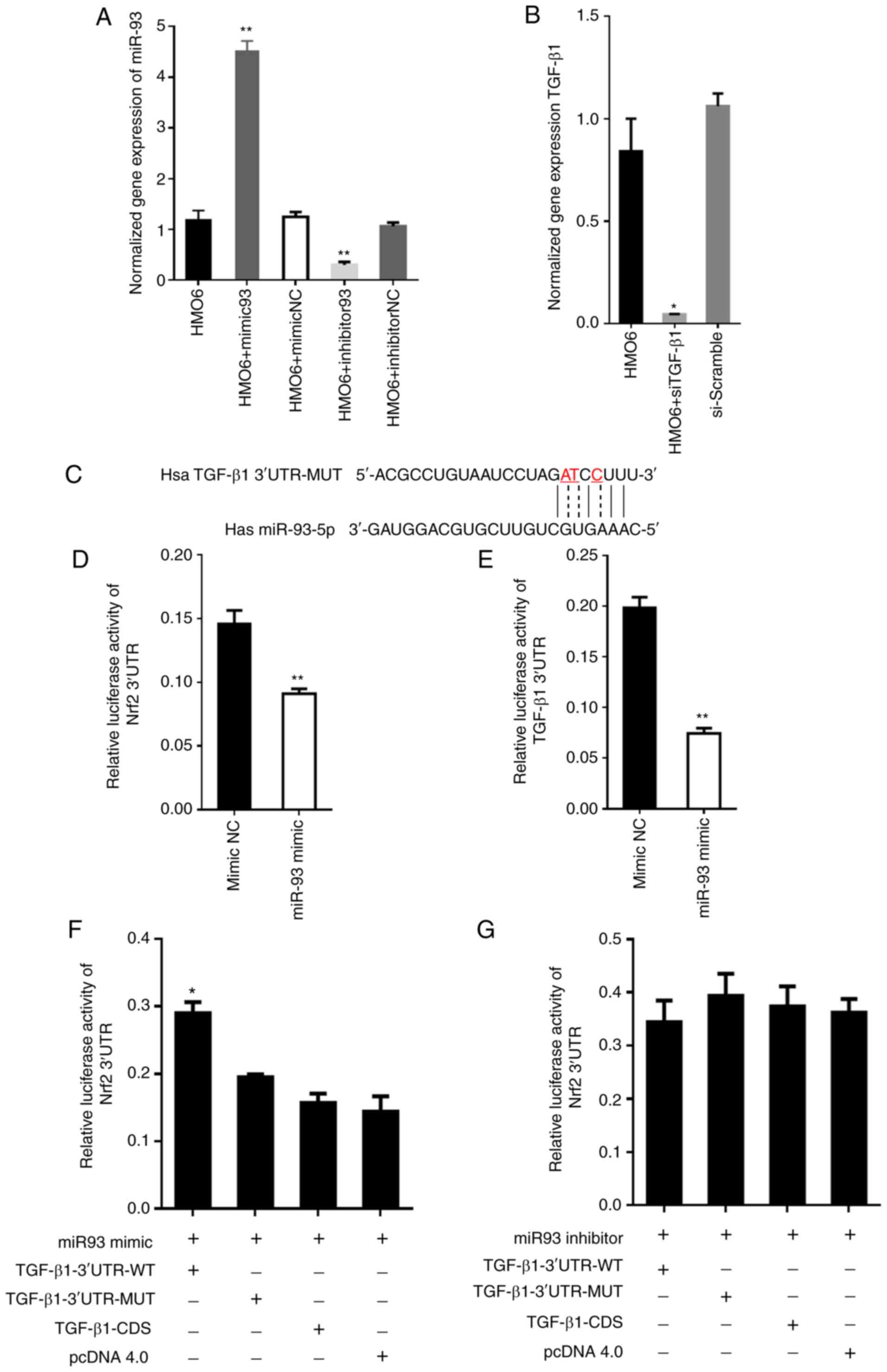 | Figure 4.TGF-β1 competes with Nrf2 mRNA for
binding to miR-93. (A) Expression of miR-93 in HMO6 cells of each
group after transfection. The expression of miR-93 was
significantly elevated in miR-93 mimics group and declined in
miR-93 inhibitor group, **P<0.05 vs. control group. (B)
Expression of TGF-β1 in HMO6 cells of each group after
transfection. The expression of TGF-β1 was significantly
declined in the siTGF-β1 group. *P<0.05 vs. control group. (C)
Mutated nucleotides in the TGF-β1 3′-UTR sequence (indicated
by the dotted line and red font) were generated in the seed region
of miR-93 to abolish binding. The mutated sequence is labeled
TGF-β1−3′-UTR-MUT. (D and E) A dual luciferase reporter
assay was used to test whether TGF-β1 and Nrf2 were
targets of miR-93. 293T cells were co-transfected with (d)
TGF-β1−3′-UTR or (e) Nrf2−3′-UTR and miR-93 mimic or
mimic control. **P<0.01. (F) 293T cells were co-transfected with
miR-93 mimic + TGF-β1−3′UTR-WT, miR-93 mimic +
TGF-β1−3′-UTR-MUT, miR-93 mimic + TGF-β1-CDS, or
miR-93 mimic + empty vector pcDNA 4.0. Under these different
miR-93/TGF-β1 conditions, the expression of Nrf2 was
assessed by measuring relative luciferase activity. Data are
presented as the means ± SD of three independent experiments.
*P<0.05 by Student's t-test. 3′UTR WT is the endogenous 3′-UTR
sequence and 3′UTR MUT is the mutated 3′-UTR sequence. (G) The same
experimental setup, measures and analyses were conducted as in (F),
except with the miR-93 inhibitor. miR, microRNA; RT-qPCR, reverse
transcription-quantitative PCR; TGF, transforming growth factor;
Nrf2, nuclear factor erythroid 2-related factor 2; si, small
interfering RNA; UTR, untranslated region; WT, wild type; MUT,
mutant; NC, negative control; CDS, coding DNA sequence. |
Effects of miR-93 and TGF-β1 on the apoptosis of
HMO6 cells. Next, the functional role of miR-93 after ICH was
investigated by artificially overexpressing miR-93 (via miR-93
mimic) or knocking it down (with the miR-93 inhibitor) in
thrombin-treated HMO6 cells. The results of western blot analysis
showed that both Nrf2 and TGF-β1 protein levels were decreased in
cells transfected with the miR-93 mimic (P<0.05; Fig. 6A), which is consistent with the
luciferase assay results (Fig. 4D and
E). Nrf2 and TGF-β1, which are neuroprotective against ICH,
were elevated in cells transfected with an inhibitor of miR-93
(P<0.05). This result suggested that miR-93 inhibited Nrf2 and
TGF-β1 and can thereby aggravate brain damage after ICH (Fig. 6B). The loss of TGF-β1 was also
simulated using siRNA technology and found that Nrf2 expression was
decreased in siTGF-β1-transfected cells compared with those
transfected with the scramble control (P<0.05; Fig. 6C). The experiment confirmed that
Nrf2 expression is inhibited by miR-93 when TGF-β1 is unavailable
to competitively bind or sponge miR-93.
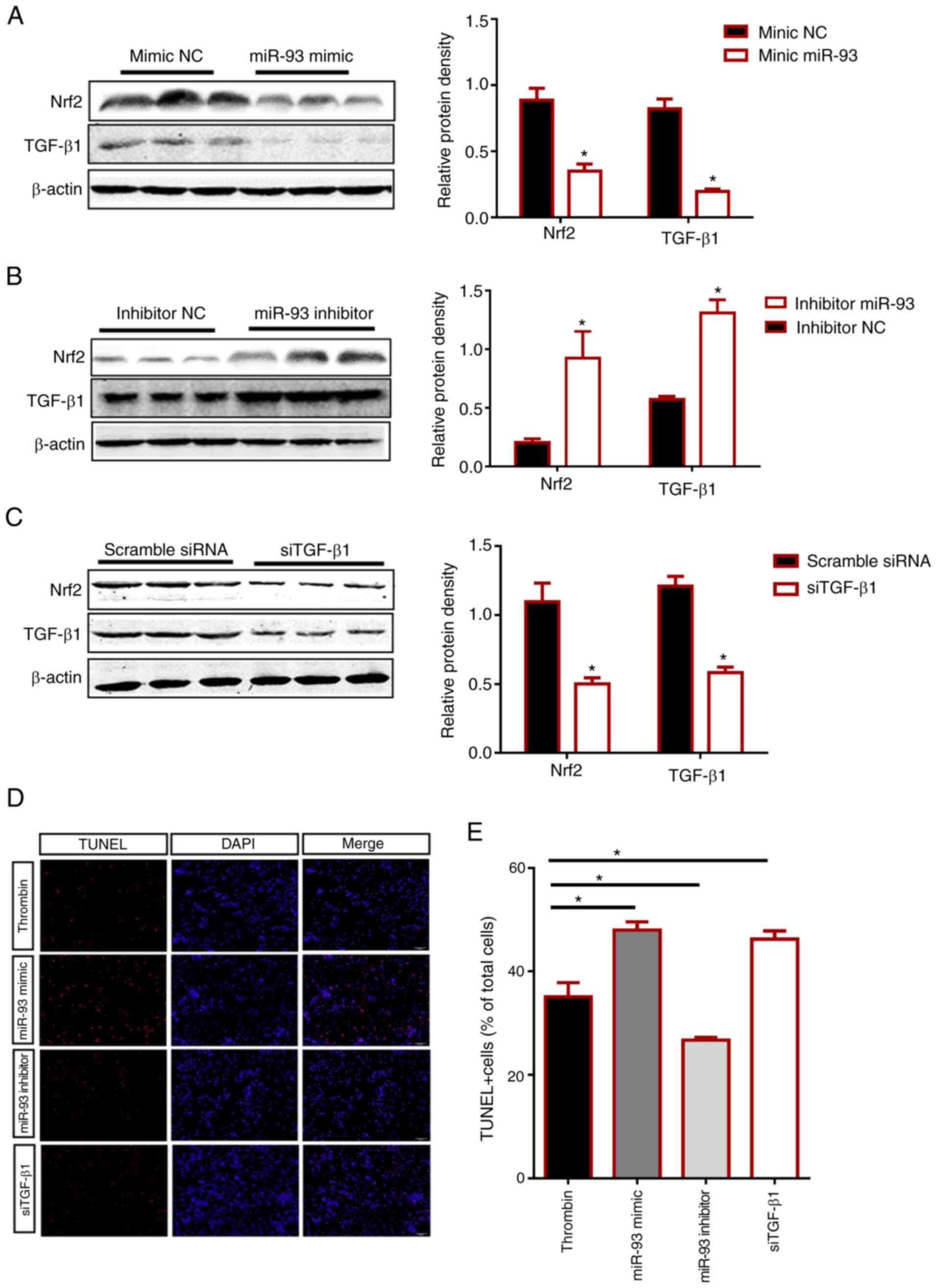 | Figure 6.Effects of manipulating miR-93 and
TGF-β1 levels on Nrf2 expression and apoptosis. (A-C) Protein
expression levels of TGF-β1 and Nrf2 in HMO6 cells transfected for
48 h with (A) miR-93 mimic, (B) miR-93 inhibitor or (C) siTGF-β1
were determined by western blotting. The negative controls in (A),
(B), and (C) are mimic NC, inhibitor NC and scramble siRNA,
respectively. Representative western blotting images and
quantification of the group data are shown. (D) HMO6 cells were
treated with 48 h of thrombin treatment (20 U/ml). Then, apoptotic
cell death was measured by counting TUNEL-stained cells (red) after
transfection with miR-93 mimic, miR-93 inhibitor or siTGF-β1. DAPI
(blue) labels cell nuclei. Scale bar, 100 µm. (E) The bar graph
quantifies the number of TUNEL-positive (TUNEL+) cells in the
different groups as a percentage of total cells. The group data
represent three separate experiments and are expressed as the means
± SD. *P<0.05 by Student's t-test. miR, microRNA; TGF,
transforming growth factor; Nrf2, nuclear factor erythroid
2-related factor 2; si, small interfering RNA; NC, negative
control. |
TUNEL staining showed that thrombin control compared
with siTGF-β1 and miR-93 mimic-transfected HMO6 cells exhibited
enhanced apoptosis (P<0.05). However, the number of
TUNEL-positive cells in the miR-93 inhibitor-transfected group was
significantly lower than the thrombin group (20 U/ml) and even
further decreased compared with the miR-93 mimic and
siTGF-β1-transfected groups (P<0.05; Fig. 6D). Collectively, the findings
revealed that miR-93 inhibited TGF-β1 and Nrf2 expression and
aggravated cellular apoptosis after ICH.
Discussion
The ceRNA mechanism was first proposed by PierPaolo
Pandolfi (39) of Harvard Medical
School in a study published in Cell in 2011. This newly discovered
gene-expression regulator competed with target mRNA for competitive
binding to the same miRNA molecule, resulting in a relative
decrease in the activity of the miRNA. The decreased miRNA activity
upregulated the expression level of the miRNA's target genes and
played a regulatory role in post-transcriptional gene expression.
Since the discovery of ceRNAs, these regulatory molecules have been
proposed as therapeutic targets for human diseases. Most research
on ceRNA regulatory mechanisms has focused on tumors and A previous
study showed that ceRNA may provide novel therapeutic options for
ischemic stroke (40)
Various types of RNA molecules, such as pseudo-gene
transcripts, long-chain non-coding RNA (lncRNA) and mRNA, can act
as ceRNA to shield the inhibition or degradation of another mRNA by
miRNA (39). Those mRNAs that
contain complementary binding regions to the miRNA seed sequence
[often at the 3′-UTR since it retains the miRNA binding site
(18)] are highly likely to be
involved in gene regulation by acting as ceRNAs. Considering that
the amount of mRNA in the cell exceeds most other types of cellular
RNA, and the target sequences of miRNA are mostly located on mRNA,
it is reasonable to propose that mRNAs are critical competitors for
miRNA binding. In addition, it was found that 3′UTRs on mRNAs not
only act as cis regulatory elements that alter the stability of
their own mRNA transcripts but may also act in trans to modulate
gene expression through miRNA binding (41,42).
This is particularly relevant given the recent identification of
3′-UTRs that are expressed independently from the protein-coding
sequences to which they are normally linked (43). Nevertheless, most studies
investigated lncRNA that function as ceRNA and few studies have
focused on 3′UTRs as ceRNA (44,45)
Based on the expression patterns of TGF-β1, Nrf2 and
miR-93 that were observed in the brain tissue of patients with ICH,
it was hypothesized that the 3′-UTR of TGF-β1 acted as a ceRNA
sponge for miR-93, impairing its inhibitory effect on Nrf2 and
promoting Nrf2 expression. miR-93 was markedly downregulated under
ICH conditions in human brain tissue and in a human microglial cell
line. The consequent increase in Nrf2 expression ameliorated ICH
injury, as indicated by the decrease in apoptosis that was observed
in the in vitro model of ICH. Importantly, it was revealed
that two neuroprotective factors, TGF-β1 and Nrf2, are associated
with miR-93 and are mutually regulated following ICH via a
ceRNA-mediated mechanism.
As a multifunctional cytokine, TGF-β1 is expressed
by neurons, astrocytes and microglia in the CNS, where it regulates
astrocytic regeneration and differentiation and neuronal survival.
Clinical studies found that the plasma TGF-β1 level was closely
associated with the prognosis of patients with ICH, suggesting that
TGF-β1 could promote the recovery of neurological function. The
literature indicates that TGF-β1 is closely associated with
ICH-induced inflammatory injury, but it remains unclear whether
TGF-β1 could directly protect neurons (46). The present study presents the first
report of a ceRNA regulatory mechanism in ICH, based on the
competitive sponging activity of TGF-β1.
Regarding the association between TGF-β1 and miR-93,
there have been reports that miR-93 promoted the TGF-β-induced
epithelial-to-mesenchymal transition via the downregulation of
NEDD4L in lung cancer cells (47).
However, there were no prior studies indicating the correlation
between miR-93 and TGF-β1 expression in the brain. In the present
study, a mechanism of action for miR-93 in ICH was established and
a more comprehensive understanding of how miR-93 aggravated brain
damage during ICH was established. Specifically, miR-93 was shown
to directly bind the 3′UTR of Nrf2 (predicted by bioinformatics and
confirmed by luciferase assay). The downregulation of miR-93
increased the expression of Nrf2, consistent with previous reports
(22).
Previous research confirmed that HO-1 inhibited the
nuclear translocation of the NF-κB p65 subunit to block the cascade
of many downstream inflammatory factors, thereby increasing the
nuclear translocation of Nrf2 and resulting in a neuroprotective
effect on early brain damage caused by ICH (48). Other studies indicated that Nrf2 in
microglia augmented antioxidative capacity, phagocytosis and
hematoma clearance after ICH (16).
In corroboration, the present data showed that increasing Nrf2
expression by inhibition of miR-93 decreased apoptosis in human
microglial cells.
The present study has some limitations. Firstly,
only the existence of the TGF-β1/miR-93/Nrf2 pathway in humans was
confirmed. The present study did not harness the power of animal
models to examine the effects of miR-93 on neurological behaviors,
hematoma size, or prognosis after ICH. However, it is not often
that a promising point of therapeutic intervention is verified in
humans; our results are not restricted by the ever-present question
of translation/replication from animals to humans. Future attempts
to manipulate the ceRNA function of TGF-β1 as a potential ICH
treatment would be strengthened by the knowledge that the mechanism
exists in humans and can therefore be clinically applied. Secondly,
some studies have used a treated HMO6 cell line to reflect the
human brain injury (49,50); but there are still others, such as
SH-SY5Y cell line or primary neurons, that can perform similar
experiments. The reason why HMO6 cells were used is that most of
the evidence suggests that microglia cells are activated shortly
after ICH and this activation contributes to secondary ICH-induced
brain injury. The HMO6 cell line as human microglia are closer to
the present study's experimental principle. The in vitro
thrombin-toxicity model of ICH in primary human microglia cultured
from resected brain tissue may not fully reflect the post-ICH
changes in human microglia or other brain cell types in
vivo. Furthermore, since the patient samples are limited, the
present study failed to assess apoptosis in the patient samples.
Finally, it is important to note that the control samples of human
brain tissue did not represent a healthy brain environment. The
patients who provided the control brain tissue did not have ICH but
did exhibit other serious diseases, including epilepsy and glioma.
Therefore, it is possible that these neurological diseases
influenced the expression levels of Nrf2, TGF-β1 and miR-93, making
the measurements an inaccurate baseline for comparison with the ICH
condition. Nevertheless, the relatively even distribution of
diseases and sampling locations strengthened the justification for
a “non-ICH” control group.
In summary, the present study elucidated a novel
function of TGF-β1, miR-93 and Nrf2 in ICH. The ceRNA mechanism
activated following ICH involved the 3′UTR of TGF-β1 serving as a
ceRNA to sponge miR-93, thereby increasing the expression of Nrf2
and decreasing apoptosis. The TGF-β1/miR-93/Nrf2 pathway could
provide novel opportunities for ICH treatment; for instance,
neuroprotection could be achieved by artificially increasing TGF-β1
expression in a localized manner or decreasing miR-93 expression
after ICH to increase the expression of Nrf2. The next step towards
a potential drug target is the verification of the TGF-β1, Nrf2 and
miR-93 binding sites. Future challenges will be to understand why
such ceRNA regulatory networks exist, how they may have evolved
(perhaps via pseudogenes), and the consequences of perturbing
them.
Acknowledgements
The authors would like to thank Dr Dai Baowen and Dr
Pei Pang from the Department of Neurology at Huazhong University of
Science (Wuhan, China) for their assistance with this work.
Funding
This work was supported by National Natural Science
Foundation of China (grant nos. 81660209, 81760221 and 81960221)
and the National Science & Technology Foundational Resource
Investigation Program of China (grant no. 2018FY100900).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW, XC, XW and ZC performed the experiments. DL, YO,
BB, YZ, ZQ and MY analyzed the data. ZC and XY designed the study.
HW and ZC confirm the authenticity of all the raw data. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was performed in accordance with
the recommendations outlined in the Guide for the Care and Use of
Laboratory Animals and in accordance with the National Institutes
of Health, and was approved by the Committee on the Ethics of
Affiliated Hospital of Jiujiang University (permit no. 2017001).
Written informed consent was obtained from all tissue donors or
their parent/guardian prior for inclusion in the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ICH
|
intracerebral hemorrhage
|
|
TGF-β1
|
transforming growth factor-β1
|
|
ceRNAs
|
competitive endogenous RNAs
|
|
Nrf2
|
nuclear factor erythroid 2-related
factor 2
|
|
siRNA
|
small interfering RNA
|
|
CNS
|
central nervous system
|
|
ARE
|
antioxidant response element
|
|
MREs
|
miRNA recognition elements
|
|
lncRNA
|
long non-coding RNA
|
|
HCC
|
hepatocellular carcinoma
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IL-1β
|
interleukin 1β
|
|
HO-1
|
heme oxygenase-1
|
|
NC
|
non-specific control
|
|
WT
|
wild type
|
|
MUT
|
mutant
|
|
CDS
|
coding DNA sequence
|
References
|
1
|
Qureshi AI, Tuhrim S, Broderick JP, Batjer
HH, Hondo H and Hanley DF: Spontaneous intracerebral hemorrhage. N
Engl J Med. 344:1450–1460. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Al-Shahi Salman R, Law ZK, Bath PM,
Steiner T and Sprigg N: Haemostatic therapies for acute spontaneous
intracerebral haemorrhage. Cochrane Database Syst Rev.
4:CD0059512018.PubMed/NCBI
|
|
3
|
Moses HL, Roberts AB and Derynck R: The
discovery and early days of TGF-β: A historical perspective. Cold
Spring Harb Perspect Biol. 8:a0218652016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Esebanmen GE and Langridge WHR: The role
of TGF-beta signaling in dendritic cell tolerance. Immunol Res.
65:987–994. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gordon KJ and Blobe GC: Role of
transforming growth factor-beta superfamily signaling pathways in
human disease. Biochim Biophys Acta. 1782:197–228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Brionne TC, Tesseur I, Masliah E and
Wyss-Coray T: Loss of TGF-beta 1 leads to increased neuronal cell
death and microgliosis in mouse brain. Neuron. 40:1133–1145. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wyss-Coray T, Borrow P, Brooker MJ and
Mucke L: Astroglial overproduction of TGF-beta 1 enhances
inflammatory central nervous system disease in transgenic mice. J
Neuroimmunol. 77:45–50. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sulaiman W and Nguyen DH: Transforming
growth factor beta 1, a cytokine with regenerative functions.
Neural Regen Res. 11:1549–1552. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma M, Ma Y, Yi X, Guo R, Zhu W, Fan X, Xu
G, Frey WH II and Liu X: Intranasal delivery of transforming growth
factor-beta1 in mice after stroke reduces infarct volume and
increases neurogenesis in the subventricular zone. BMC Neurosci.
9:1172008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buisson A, Lesne S, Docagne F, Ali C,
Nicole O, MacKenzie ET and Vivien D: Transforming growth
factor-beta and ischemic brain injury. Cell Mol Neurobiol.
23:539–550. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kumar P, Kumar A, Misra S, Sagar R, Farooq
M, Kumari R, Vivekanandhan S, Srivastava AK and Prasad K:
Association of transforming growth factor-β1 gene C509T, G800A and
T869C polymorphisms with intracerebral hemorrhage in North Indian
Population: A case-control study. Neurol Sci. 37:353–359. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen J, Bai Q, Zhao Z, Sui H and Xie X:
Ginsenoside represses symptomatic intracerebral hemorrhage after
recombinant tissue plasminogen activator therapy by promoting
transforming growth factor-beta1. J Stroke Cerebrovasc Dis.
25:549–555. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zeng J, Chen Y, Ding R, Feng L, Fu Z, Yang
S, Deng X, Xie Z and Zheng S: Isoliquiritigenin alleviates early
brain injury after experimental intracerebral hemorrhage via
suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome
activation by promoting Nrf2 antioxidant pathway. J
Neuroinflammation. 14:1192017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhao X, Sun G, Zhang J, Ting SM, Gonzales
N and Aronowski J: Dimethyl fumarate protects brain from damage
produced by intracerebral hemorrhage by mechanism involving nrf2.
Stroke. 46:1923–1928. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang CF, Cho S and Wang J:
(−)-Epicatechin protects hemorrhagic brain via synergistic Nrf2
pathways. Ann Clin Transl Neurol. 1:258–271. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao X, Sun G, Ting SM, Song S, Zhang J,
Edwards NJ and Aronowski J: Cleaning up after ICH: The role of Nrf2
in modulating microglia function and hematoma clearance. J
Neurochem. 133:144–152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao X, Sun G, Zhang J, Strong R, Dash PK,
Kan YW, Grotta JC and Aronowski J: Transcription factor Nrf2
protects the brain from damage produced by intracerebral
hemorrhage. Stroke. 38:3280–3286. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee RC, Feinbaum RL and Ambros V: The C.
elegans heterochronic gene lin-4 encodes small RNAs with antisense
complementarity to lin-14. Cell. 75:843–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Singh B, Ronghe AM, Chatterjee A, Bhat NK
and Bhat HK: MicroRNA-93 regulates NRF2 expression and is
associated with breast carcinogenesis. Carcinogenesis.
34:1165–1172. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fabbri E, Borgatti M, Montagner G, Bianchi
N, Finotti A, Lampronti I, Bezzerri V, Dechecchi MC, Cabrini G and
Gambari R: Expression of microRNA-93 and Interleukin-8 during
Pseudomonas aeruginosa-mediated induction of proinflammatory
responses. Am J Respir Cell Mol Biol. 50:1144–1155. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang P, Liang X, Lu Y, Zhao X and Liang J:
MicroRNA-93 downregulation ameliorates cerebral ischemic injury
through the Nrf2/HO-1 defense pathway. Neurochem Res. 41:2627–2635.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Larki P, Ahadi A, Zare A, Tarighi S,
Zaheri M, Souri M, Zali MR, Ghaedi H and Omrani MD: Up-regulation
of miR-21, miR-25, miR-93, and miR-106b in gastric cancer. Iran
Biomed J. 22:367–373. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Parodi AS, De Guerrero LB, Astarloa L,
Cintora A, Gonzalez CC, Maglio F, Magnoni C, Milani H, Ruggiero H
and Squassi G: Immunization against Argentinian hemorrhagic fever
using an attenuated strain of Junin virus. IV. Evaluation of
clinical and immunological results. Medicina (B Aires). 30:1–3.
1970.(In Spanish).
|
|
25
|
Lyu X, Fang W, Cai L, Zheng H, Ye Y, Zhang
L, Li J, Peng H, Cho WC, Wang E, et al: TGFβR2 is a major target of
miR-93 in nasopharyngeal carcinoma aggressiveness. Mol Cancer.
13:512014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ma J, Zhang L, Hao J, Li N, Tang J and Hao
L: Up-regulation of microRNA-93 inhibits TGF-β1-induced EMT and
renal fibrogenesis by down-regulation of Orai1. J Pharmacol Sci.
136:218–227. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Feng L, Shi L, Lu YF, Wang B, Tang T, Fu
WM, He W, Li G and Zhang JF: Linc-ROR promotes osteogenic
differentiation of mesenchymal stem cells by functioning as a
competing endogenous RNA for miR-138 and miR-145. Mol Ther Nucleic
Acids. 11:345–353. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie CR, Wang F, Zhang S, Wang FQ, Zheng S,
Li Z, Lv J, Qi HQ, Fang QL, Wang XM, et al: Long noncoding RNA HCAL
facilitates the growth and metastasis of hepatocellular carcinoma
by acting as a ceRNA of LAPTM4B. Mol Ther Nucleic Acids. 9:440–451.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Montojo J, Zuberi K, Rodriguez H, Bader GD
and Morris Q: GeneMANIA: Fast gene network construction and
function prediction for Cytoscape. F1000 Res. 3:1532014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nagai A, Nakagawa E, Hatori K, Choi HB,
McLarnon JG, Lee MA and Kim SU: Generation and characterization of
immortalized human microglial cell lines: Expression of cytokines
and chemokines. Neurobiol Dis. 8:1057–1068. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang MD, Wang Y, Xia YP, Dai JW, Gao L,
Wang SQ, Wang HJ, Mao L, Li M, Yu SM, et al: High serum MiR-130a
levels are associated with severe perihematomal edema and predict
adverse outcome in acute ICH. Mol Neurobiol. 53:1310–1321. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Qi W, Chen X, Holian J, Mreich E, Twigg S,
Gilbert RE and Pollock CA: Transforming growth factor-beta1
differentially mediates fibronectin and inflammatory cytokine
expression in kidney tubular cells. Am J Physiol Renal Physiol.
291:F1070–F1077. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Liu D, Huang HZ, Wang ZH, Hou TY,
Yang X, Pang P, Wei N, Zhou YF, Dupras MJ, et al: A novel
microRNA-124/PTPN1 signal pathway mediates synaptic and memory
deficits in alzheimer's disease. Biol Psychiatry. 83:395–405. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yin XP, Wu D, Zhou J, Chen ZY, Bao B and
Xie L: Heme oxygenase 1 plays role of neuron-protection by
regulating Nrf2-ARE signaling post intracerebral hemorrhage. Int J
Clin Exp Pathol. 8:10156–10163. 2015.PubMed/NCBI
|
|
36
|
Liu Z, Zhang F, Zhao L, Zhang X, Li Y and
Liu L: Protective effect of pravastatin on myocardial ischemia
reperfusion injury by regulation of the miR-93/Nrf2/ARE signal
pathway. Drug Des Devel Ther. 14:3853–3864. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Taylor RA and Sansing LH: Microglial
responses after ischemic stroke and intracerebral hemorrhage. Clin
Dev Immunol. 2013:7460682013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yin M, Chen Z, Ouyang Y, Zhang H, Wan Z,
Wang H, Wu W and Yin X: Thrombin-induced, TNFR-dependent miR-181c
downregulation promotes MLL1 and NF-κB target gene expression in
human microglia. J Neuroinflammation. 14:1322017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Salmena L, Poliseno L, Tay Y, Kats L and
Pandolfi PP: A ceRNA hypothesis: The Rosetta Stone of a hidden RNA
language? Cell. 146:353–358. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen F, Zhang L, Wang E, Zhang C and Li X:
LncRNA GAS5 regulates ischemic stroke as a competing endogenous RNA
for miR-137 to regulate the Notch1 signaling pathway. Biochem
Biophys Res Commun. 496:184–190. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rastinejad F and Blau HM: Genetic
complementation reveals a novel regulatory role for 3′ untranslated
regions in growth and differentiation. Cell. 72:903–917. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rastinejad F, Conboy MJ, Rando TA and Blau
HM: Tumor suppression by RNA from the 3′ untranslated region of
alpha-tropomyosin. Cell. 75:1107–1117. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mercer TR, Wilhelm D, Dinger ME, Soldà G,
Korbie DJ, Glazov EA, Truong V, Schwenke M, Simons C, Matthaei KI,
et al: Expression of distinct RNAs from 3′ untranslated regions.
Nucleic Acids Res. 39:2393–2403. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Fang L, Du WW, Yang X, Chen K, Ghanekar A,
Levy G, Yang W, Yee AJ, Lu WY, Xuan JW, et al: Versican
3′-untranslated region (3′-UTR) functions as a ceRNA in inducing
the development of hepatocellular carcinoma by regulating miRNA
activity. FASEB J. 27:907–919. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fan Z, Kim S, Bai Y, Diergaarde B and Park
HJ: 3′-UTR shortening contributes to Subtype-Specific cancer growth
by breaking stable ceRNA crosstalk of housekeeping genes. Front
Bioeng Biotechnol. 8:3342020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Taylor RA, Chang CF, Goods BA, Hammond MD,
Mac Grory B, Ai Y, Steinschneider AF, Renfroe SC, Askenase MH,
McCullough LD, et al: TGF-β1 modulates microglial phenotype and
promotes recovery after intracerebral hemorrhage. J Clin Invest.
127:280–292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Qu MH, Han C, Srivastava AK, Cui T, Zou N,
Gao ZQ and Wang QE: miR-93 promotes TGF-β-induced
epithelial-to-mesenchymal transition through downregulation of
NEDD4L in lung cancer cells. Tumour Biol. 37:5645–5651. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yin XP, Chen ZY, Zhou J, Wu D and Bao B:
Mechanisms underlying the perifocal neuroprotective effect of the
Nrf2-ARE signaling pathway after intracranial hemorrhage. Drug Des
Devel Ther. 9:5973–5986. 2015.PubMed/NCBI
|
|
49
|
Sheikh AM, Yano S, Mitaki S, Haque MA,
Yamaguchi S and Nagai A: A Mesenchymal stem cell line (B10)
increases angiogenesis in a rat MCAO model. Exp Neurol.
311:182–193. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Narantuya D, Nagai A, Sheikh AM, Masuda J,
Kobayashi S, Yamaguchi S and Kim SU: Human microglia transplanted
in rat focal ischemia brain induce neuroprotection and behavioral
improvement. PLoS One. 5:e117462010. View Article : Google Scholar : PubMed/NCBI
|