Introduction
Airway remodeling has been established as a key
feature of a number of respiratory diseases, including asthma
(1,2). Pathophysiologically, airway remodeling
is characterized by a series of morphological changes of airway
structures, including epithelium, basement membrane, smooth muscle
and blood vessels (3,4). Among which, changes of airway vascular
function and morphology have been observed as important components
during the process of airway remodeling (5,6). An
early study showed that vascular density and vascular area in
airway submucosa and lamina propria of children with asthma are
significantly increased (7),
suggesting a potential important role of angiogenesis in the
development of airway remodeling. A number of cellular changes have
been involved in the pathogenesis of airway vascular remodeling.
These changes include proliferation, hypertrophy, apoptosis and
migration of endothelial and vascular smooth muscle cells (VSMCs),
as well as the synthesis and degradation of extracellular matrix
(8), among which phenotypic changes
in VSMCs have been considered as an important feature (9,10).
However, the potential molecular regulator of the change in VSMC
phenotype in the pathogenesis of airway vascular remodeling remains
to be elucidated.
A disintegrin and metalloproteinase-33 (ADAM33) is a
member of the ADAM metalloproteinase family (11), which have been involved in the
pathogenesis of airway remodeling-related diseases (12). Genetic polymorphisms of ADAM33 have
been related to vulnerability to asthma (13). In addition, ADAM33 has been found to
be expressed in the airway smooth muscles and basement membranes of
almost all patients with asthma, but is absent in normal control
subjects (14). Another study
reported that the expression of ADAM33 in lung tissue is varied and
is enriched in interstitial cells (including fibroblasts and smooth
muscle cells) (15). The expression
of ADAM33 in patients with asthma is related to the severity of the
disease, the decline in respiratory function and the extent of
airway hypersensitivity, inflammation and remodeling (16,17).
Notably, our previous study in patients with asthma confirmed the
expression of ADAM33 in airway VSMCs, which could be upregulated by
IL-4 and −13 (18). Therefore, it
could be hypothesized that expression of ADAM33 in airway VSMC may
be important in regulating the proliferative phenotype of VSMCs in
airway vascular remodeling.
Previous studies have shown that the PI3K/AKT
pathways (19) and Bcl-2/Bax
proteins (20) are key regulators
for the proliferation and apoptosis of VSMCs. However, whether the
role VSMCs ADAM33 in airway vascular remodeling may involve the
regulation of PI3K/AKT and Bcl-2/Bax pathways remains to be
elucidated. Since isolation and in vitro culture of human
airway VSMCs are difficult, the present study used human aortic
smooth muscle cells (HASMCs) to evaluate the influences of ADAM33
silencing on cellular proliferation and apoptosis. The present
study aimed to provide the primary investigation into the role of
ADAM33 expressed in airway VSMCs and pathogenic airway vascular
remodeling.
Materials and methods
Patient characteristics and
samples
Lung tissue samples were collected from five
patients between February 2018 and December 2019 who received
pneumonectomy for bronchiectasis or masses at The First Affiliated
Hospital of Xinjiang Medical University (Urumqi, China). Patients
included three men and two women, three with pulmonary massive
lesions, two with bronchiectasis, aged 46–70 years old and all
without smoking history. The lung tissues with complete cross
section of bronchus >5 cm away from the pulmonary lesions were
obtained for subsequent analysis. All the procedures were approved
by the Ethics Committee of the First Affiliated Hospital of
Xinjiang Medical University (approval no. 20180130-01). All the
patients provided signed informed consent.
Immunohistochemical analysis
The lung tissues were fixed in 10% formalin for 6–8
h at 22–24°C. Briefly, after the specimens were washed with running
water, they were dehydrated with a gradient series of alcohol (75%
ethanol, 95% ethanol I, 95% ethanol II, absolute ethanol) in an
oven at 60°C for 20 min at each step. Then, the specimens were
transparentized in xylene for 20 min at each step. Finally, the
samples were immersed in a wax soaking tank for 3 h. After cooling
and solidification, they were trimmed to make wax blocks. Then
paraffin-embedded sections were then prepared at 4-µm thickness and
used for immunohistochemical analysis. After rehydration using a
gradient alcohol solution, sections of lung tissues were blocked
with 5% hydrogen peroxide for endogenous peroxidase and then
underwent antigen repairmen (6 min, twice). Then, the sections were
blocked with 5% BSA (Sangon Biotech Co., Ltd.) at room temperature
for 20 min. After incubation with primary antibody for ADAM33
(1:100; cat. no. DF9166; Affinity Biosciences) at 4°C overnight,
the slides were incubated with horseradish peroxidase-conjugated
goat anti-rabbit IgG secondary antibody (1:200; cat. no. SP-9001;
OriGene Technologies, Inc.) at 37°C for 30 min. Finally, the slides
were stained with diaminobenzidine (DAB; OriGene Technologies,
Inc.) for 5–30 sec and observed under a microscope (Olympus BX41TF;
Olympus Corporation). Pulmonary tissues with expression of ADAM33
were stained as brown or brownish-yellow granules.
Cell culture
HASMCs were purchased from Procell Life Science
& Technology Co., Ltd., and cultured with complete medium for
HASMCs (cat. no. CM-H081; Procell Life Science & Technology
Co., Ltd.). Briefly, HASMCs were taken out from liquid nitrogen and
the thawed cells were cultured in a cell incubator at 37°C,
saturated humidity and 5% CO2.
Immunofluorescence
After adjusting the cell concentration to
1×105 cells/ml, the cells were moved to slides and
cultured for 24 h until the cell adhered. The cells were fixed with
4% paraformaldehyde for 20 min, incubated with 0.5% Txition-100 for
20 min and blocked with 1% BSA (Sangon Biotech Co., Ltd.) for 30
min at room temperature. Subsequently, cells were incubated with 80
µl anti-α-actin (smooth muscle) antibody (1:100; cat. no. ab124964;
Abcam) for 2 h at 37°C, followed by incubation with 80 µl goat
anti-rabbit IgG antibody (H&L, Alexa Fluor® 488;
1:500; cat. no. ab150081; Abcam) at 37°C in the dark for 1 h, and
80 µl DAPI (1 µg/ml; cat. no. D1306; Thermo Fisher Scientific,
Inc.) at room temperature in the dark for 5 min. After washing with
PBS three times, the slides were sealed with 50% glycerin and
observed using laser confocal microscopy (magnification, ×400;
Leica Microsystems GmbH).
Construction of lentiviral vector for
ADAM33-shRNA and viral transfection
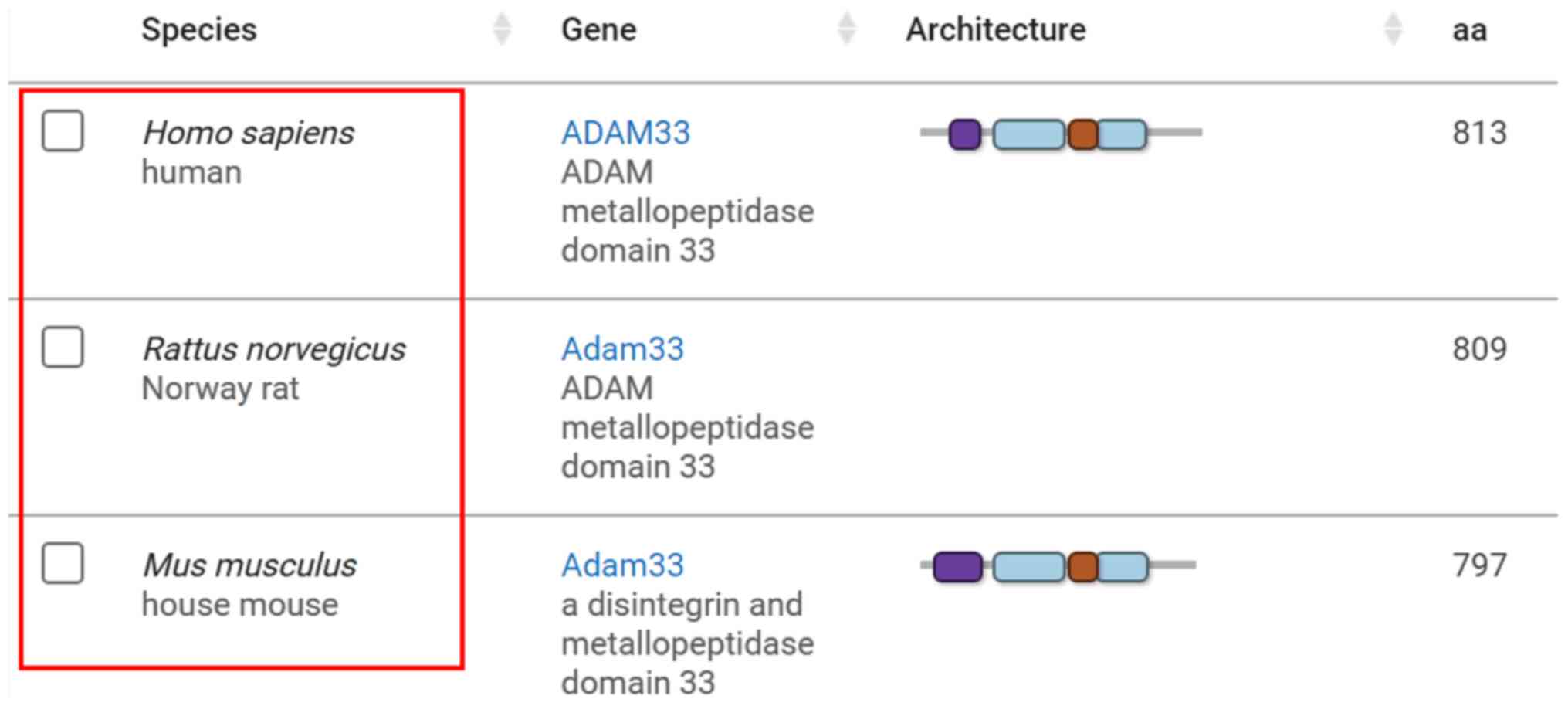
Previous studies have shown that ADAM33 is expressed
in rats and mice (21,22) and its mRNA sequence has homology
with human, as evidenced by information on the ADAM33 gene in the
NCBI database (Fig. 1; http://www.ncbi.nlm.nih.gov/gene/80332/ortholog/?scope=32524).
Lentiviral vectors carrying the short-hairpin (sh)RNA for silencing
of ADAM33 targeting three different sequences of ADAM33 [lentiviral
vector (LV)-ADAM33-shRNA1, LV-ADAM33-shRNA2 and LV-ADAM33-shRNA3])
and the negative control (NC) vectors with blanking sequence
(LV-NC) were purchased from Hanbio Biotechnology Co., Ltd. The
optimal transfection condition was confirmed with LV-NC when the
transfection was performed with a Multiplicity of Infection (MOI)
of 1×107 TU/ml, administration of HitransG P solution
(Shanghai GeneChem Co., Ltd.) and maintained for 72 h. The mRNA
sequences for ADAM33 used in the present study were: shRNA1,
GCCACTACCAAGGGCGAGTAA; shRNA2, CAGCAGGAATGCCAGCTATTA; and shRNA3,
GACTCTACCGTTCACCTAGAT.
Cell proliferation test
The HASMCs were assigned to the following
treatments: i) Blank control with no further treatment (control
group); ii) negative control treated with LV-NC; and iii) ADAM33
silencing groups treated with LV-ADAM33-shRNA1. After digesting
cells with trypsin, HASMCs with a confluence rate of 90% were
prepared into a single cell suspension of 5×104 cells/ml
in complete medium. The cells were inoculated into 96 well plates
(100 µl/ well, i.e. 5×103 cells/well) and cultured in 5%
CO2 at 37°C for 24 h. Cell Counting Kit-8 (CCK-8)
solution (100 µl of 10%; Beijing Transgen Biotech Co., Ltd.) was
added into each well. After incubation for 1 h, the optical density
(OD) values at 450 nm of the adherent cell layer from each group
were determined by enzyme-labeled instrument.
Cell apoptosis examination
The culture medium of cells from each group was
moved into a centrifuge tube, washed with PBS twice, digested with
trypsin and centrifuged at 173 × g for 5 min at room temperature.
After washing with pre-cooled PBS, the supernatant was discarded
and made into a single cell suspension. Then, 1X Binding Buffer
(100 µl) was added to suspend the cells. According to the
manufacturer's instructions of the Annexin V-PE/7-AAD Apoptosis Kit
(cat. no. AP104; MultiSciences Biotech Co., Ltd.), 5 µl Annexin
V-PE and 5 µl 7-AAD was added into each tube, and incubated in the
dark at 4°C for 15 min. Following which, 100 µl 1X Binding Buffer
was added to resuspend the cells, and then cells were passed
through a 200-mesh sieve. Flow cytometry (BD LSRFortessa™; BD
Biosciences) was then performed to determine the cellular apoptotic
status. BD FACSDiva™ Software (version 8.0.2; BD Biosciences) was
used to analyze the data. Both early apoptotic cells and late
apoptotic cells were counted. The apoptosis rate was calculated
using the following formula: (Q2 + Q4) cells/total cells.
Cell cycle analysis
The cells of each experimental group were collected
and resuspended with 500 µl pre-cooled PBS. The cell suspension was
added into 3.5 ml pre-cooled 80% ethanol and fixed at 4°C
overnight. After centrifugation at 692 × g for 5 min, the cells
were precipitated and the supernatant was discarded. After washing
with pre-cooled PBS, 500 µl PI/RNase Stabilizing Buffer (cat. no.
550825; BD Pharmingen; BD Biosciences) was added to resuspend the
cells, and then the single cell suspension was made by passing the
sample through a 200-mesh nylon screen. Samples were incubated at
4°C for 30 min. Flow cytometry (BD LSRFortessa™; BD Biosciences)
was used to detect red fluorescence and light scattering at 488 nm.
The cell DNA content and light scattering analysis were carried out
using Modfit LT3.1 analysis software (Verity Software House,
Inc.).
Reverse transcription-quantitative
(RT-q) PCR
Total RNA from HASMCs in each group was extracted
using TRIzol® (Thermo Fisher Scientific, Inc.). Then,
cDNA was synthesized from total RNA using 5X All-In-One RT
MasterMix with AccuRT Genomic DNA Removal Kit (cat. no. G492;
Applied Biological Materials, Inc.), according to the
manufacturer's instructions. The mRNA expression levels were
detected via qPCR. qPCR was performed with the SYBR premix Ex Taq
(Takara Bio, Inc.) using a Bio-Rad CFX96 detection system (Bio-Rad
Laboratories, Inc.) according to the manufacturer's instructions.
The thermocycling conditions were as follows: Predenaturation at
95°C for 2 min, denaturation at 95°C for 30 sec for a total of 40
cycles, and annealing/extension at 60°C for 30 sec for a total of
40 cycles. The sequences of primers used for RT-qPCR to determine
the expression levels of ADAM33, PI3K, AKT, ERK, Bcl-2, Bax and
β-actin were are in Table I. The
quantitative results were evaluated using the 2−∆∆Cq
method (23) and the mRNA
expression levels of the above genes were normalized against the
mRNA expression level of β-actin.
 | Table I.Primers for reverse
transcription-quantitative PCR. |
Table I.
Primers for reverse
transcription-quantitative PCR.
|
| Primer sequences
(5′→3′) |
|
|
|---|
|
|
|
|
|
|---|
| Gene | Forward | Reverse | Product size
(bp) | Tm (°C) |
|---|
| ADAM33 |
ATAGGCGTGGTGGCTCAT |
TGCGGTGTCTTGCTGTG | 112 bp | 60 |
| PI3K |
AAGAAGTTGAACGAGTGGTTGG |
GCCCTGTTTACTGCTCTCCC | 192 bp | 60 |
| AKT |
TCCTCCTCAAGAATGATGGCA |
GTGCGTTCGATGACAGTGGT | 181 bp | 60 |
| ERK |
TCTGGAGCAGTATTACGACCC |
CTGGCTGGAATCTAGCAGTCT | 134 bp | 60 |
| Bcl-2 |
GGTGGGGTCATGTGTGTGG |
CGGTTCAGGTACTCAGTCATCC | 89 bp | 60 |
| Bax |
CCCGAGAGGTCTTTTTCCGAG |
CCAGCCCATGATGGTTCTGAT | 155 bp | 60 |
| β-actin |
CATGTACGTTGCTATCCAGGC |
CTCCTTAATGTCACGCACGAT | 250 bp | 60 |
Western blotting
The experimental cells were collected and 100 µl
RIPA lysis buffer (cat. no. AR0105; Wuhan Boster Biological
Technology, Ltd.) was added. Then, the supernatant was collected by
centrifugation at 14,000 × g at 4°C for 60 min. The protein
concentration was determined using the BCA method (cat. no.
DQ111-01; Beijing Transgen Biotech Co., Ltd.) according to the
manufacturer's instructions. Then, 30 µg protein sample was loaded
per lane and resolved via 10% SDS-PAGE. Subsequently, the separated
protein was transferred to a PVDF membrane, which was then blocked
using 5% skimmed milk powder at room temperature for 1 h. The
membrane was then rinsed with TBS with 0.1% Tween-20 (TBST; three
times, 5 min/time). TBST was used to dilute primary antibodies
against β-actin (1:3,000; cat. no. D110001; Sangon Biotech Co.,
Ltd.), PI3K (1:1,000; cat. no. ab191606; Abcam), Akt (1:1,000; cat.
no. 4691T; Cell Signaling Technology, Inc.), phosphorylated (p)-Akt
(Ser473; 1:1,000; cat. no. 9271T; Cell Signaling Technology, Inc.),
ERK1/2 (1:1,000; cat. no. 9102S; Cell Signaling Technology, Inc.)
and p-ERK (Thr202/Tyr204; 1:1,000; cat. no. 9101S; Cell Signaling
Technology, Inc.). The membranes were incubated at 4°C overnight
and rinsed with TBST (three times, 5 min/time). Then, after
incubation with diluted goat anti-rabbit IgG H&L
(HRP-conjugated; 1:5,000; cat. no. ab205718; Abcam) at room
temperature for 2 h, membranes were washed with TBST again (three
times, 5 min/time). The samples were detected and imaged by a
ChemiScope 3000 Mini chemiluminescence instrument (Shanghai
Qinxiang Scientific Instrument Co., Ltd.). Densitometry was
performed by the supporting software (ChemiAnalysis; Shanghai
Qinxiang Scientific Instrument Co., Ltd.).
Statistical analysis
Continuous data are expressed as means ± standard
deviation from three independent repeats. Normally distributed data
were compared with one-way ANOVA among multiple groups followed by
LSD post hoc test. If the data did not conform to normality, they
were converted to normal distribution between subsequent analyses.
If the variance was not uniform, the Tamhane method (24) was used for comparison among multiple
groups. SPSS 19.0 software (IBM Corp.) was used for statistical
analysis. P<0.05 was considered to indicate a statistically
significant difference.
Results
Expression of ADAM33 in human airway
VSMCs
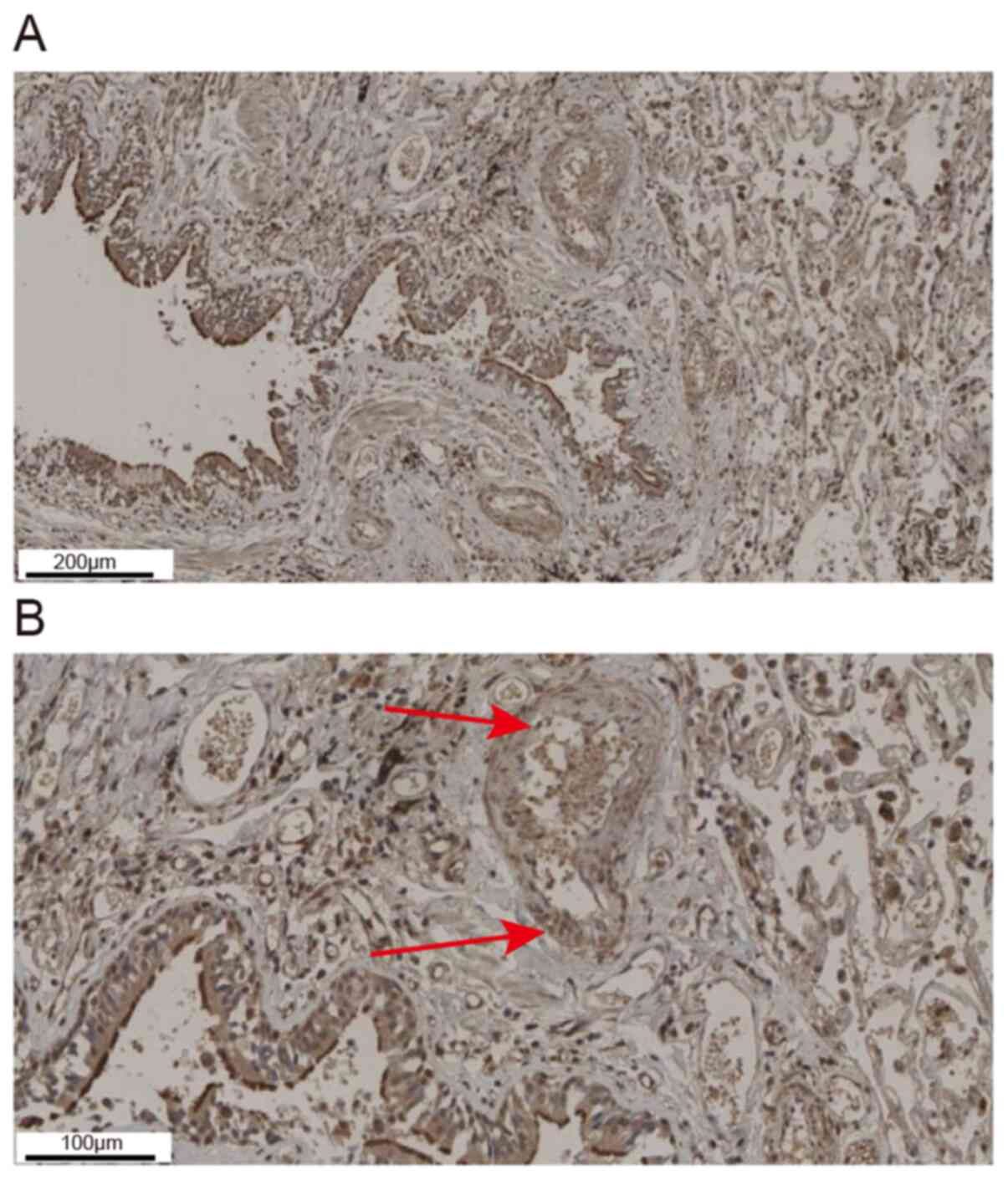
Results of immunohistochemical analysis showed that
ADAM33 was mainly expressed in epithelial cells, smooth muscle
cells, endothelial cells and interstitial inflammatory cells in the
lung tissue of asthmatic patients (Fig.
2A). As indicated by the arrows, ADAM33 positive staining could
also be seen in airway VSMCs (Fig.
2B).
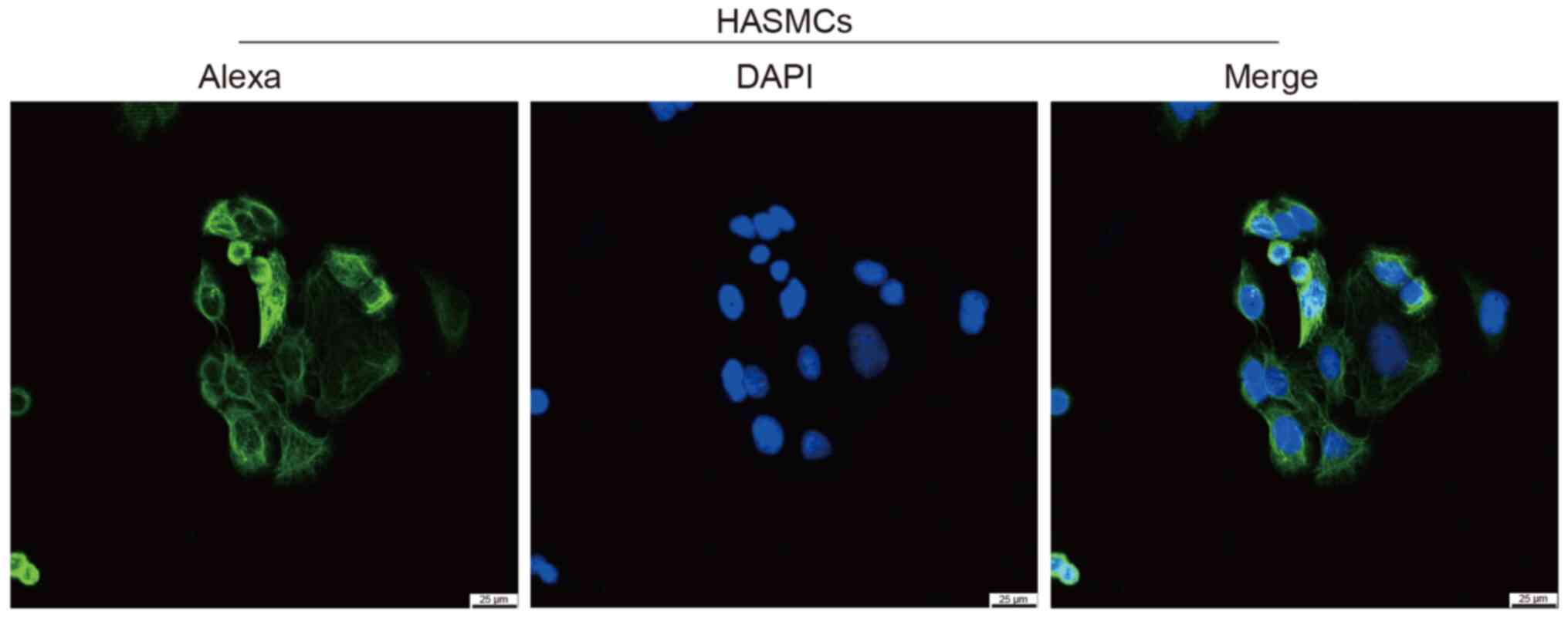
Identification of HASMCs by
immunofluorescence analysis of α-actin
In the present study, HASMCs were identified by
immunofluorescence analysis of α-actin. As shown in Fig. 3, α-actin (smooth muscle) protein is
located in the cytoskeleton, cytoplasm and cytoplasm, which is
consistent with the characteristics of smooth muscle cells.
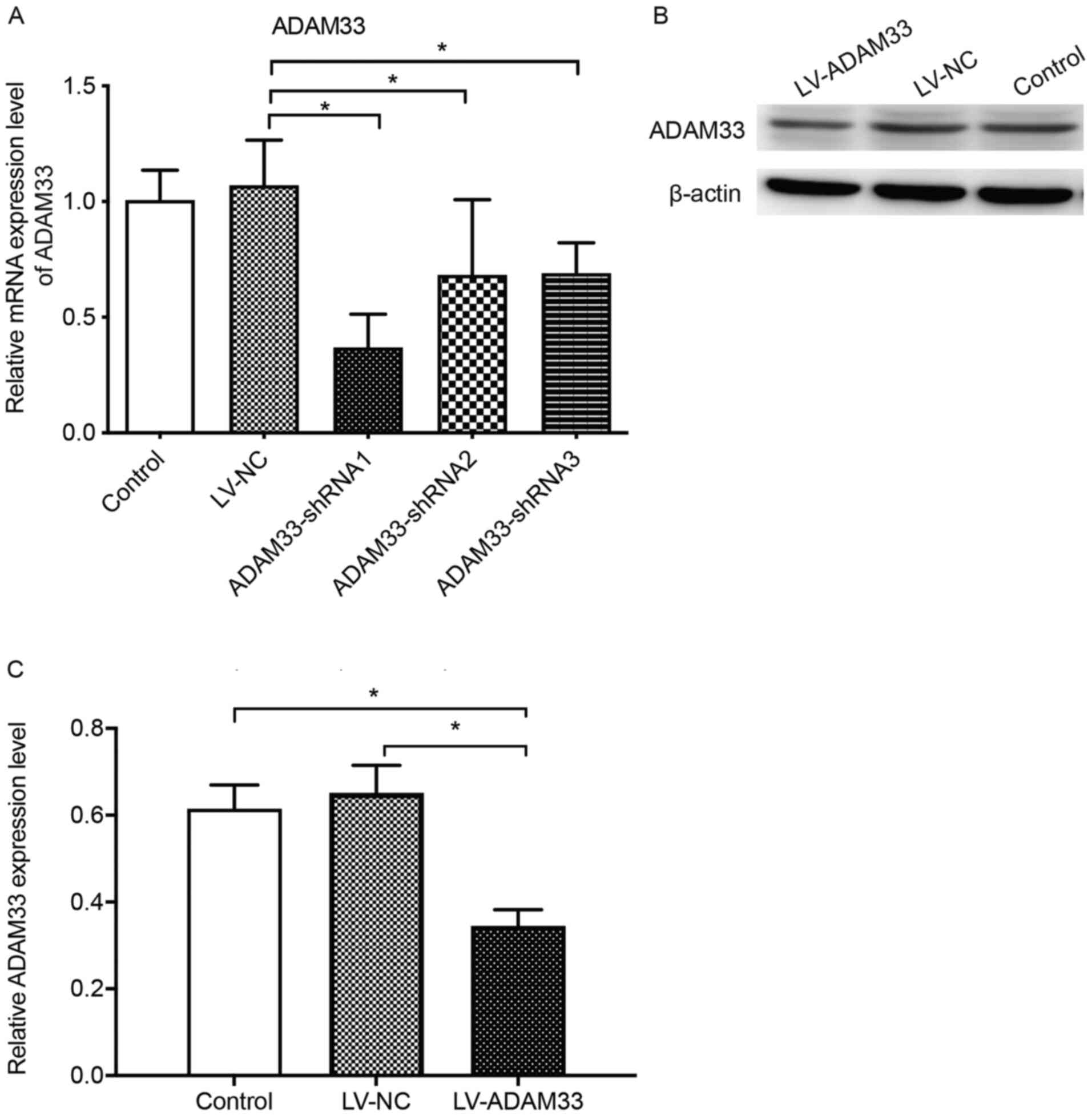
Efficiency of LV-ADAM33-shRNA in
silencing ADAM33 in HASMCs
The influence of LV-ADAM33-shRNA on mRNA and protein
expression of ADAM33 in HASMCs are shown in Fig. 4. Results of RT-qPCR showed that
compared with the control group with no treatment, treatment with
LV-NC did not significantly affect the mRNA expression of ADAM33 in
HASMCs (P=0.616; Fig. 4A). Compared
with the HASMCs transfected with LV-NC, HASMCs transfected with
LV-ADAM33-shRNA1, LV-ADAM33-shRNA2 and LV-ADAM33-shRNA3 had
significantly reduced mRNA levels of ADAM33 (P=0.000, P=0.006 and
P=0.007; Fig. 4A). Among them,
LV-ADAM33-shRNA1 was associated with most notable inhibitory effect
on ADAM33 in HASMCs. Therefore, LV-ADAM33-shRNA1 was selected for
subsequent studies. Further analyses with western blotting showed
that LV-ADAM33-shRNA1 transfection significantly reduced the
protein level of ADAM33 in HASMCs compared with the blank control
group and negative control group with LV-NC (P=0.001 and P=0.000;
Fig. 4B and C).
Influence of ADAM33 silencing on
proliferation of HASMCs
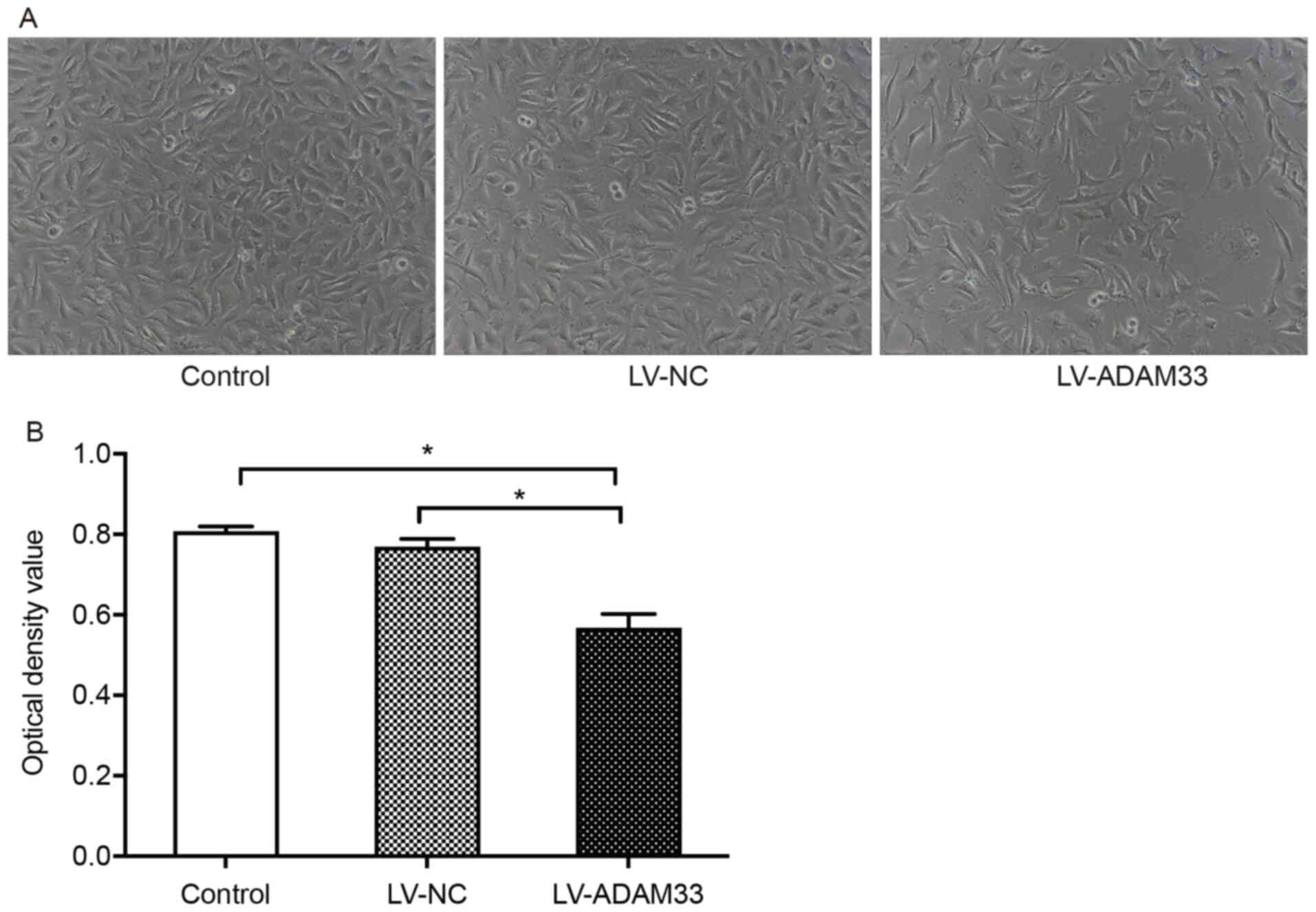
The effects of ADAM33 silencing with LV-ADAM33-shRNA
transfection on the proliferation of HASMCs were analyzed with the
CCK-8 proliferation method. Under the microscope, HASMCs in the
blank control group and negative control group were elongated and
spindle shaped, arranged in bundles and covered the whole field of
vision. However, in HASMCs with ADAM33 silencing by
LV-ADAM33-shRNA, the cells clusters were dispersed and the density
of the cellular distribution was notably reduced, suggesting that
the proliferation of HASMCs was limited (Fig. 5A). Quantitative analyses showed that
the OD value for cell growth of HASMCs after ADAM33 silencing was
significantly reduced compared with those in the blank control and
negative control groups (both P=0.000; Fig. 5B).
Influence of ADAM33 silencing on
apoptosis and cell cycle distribution of HASMCs
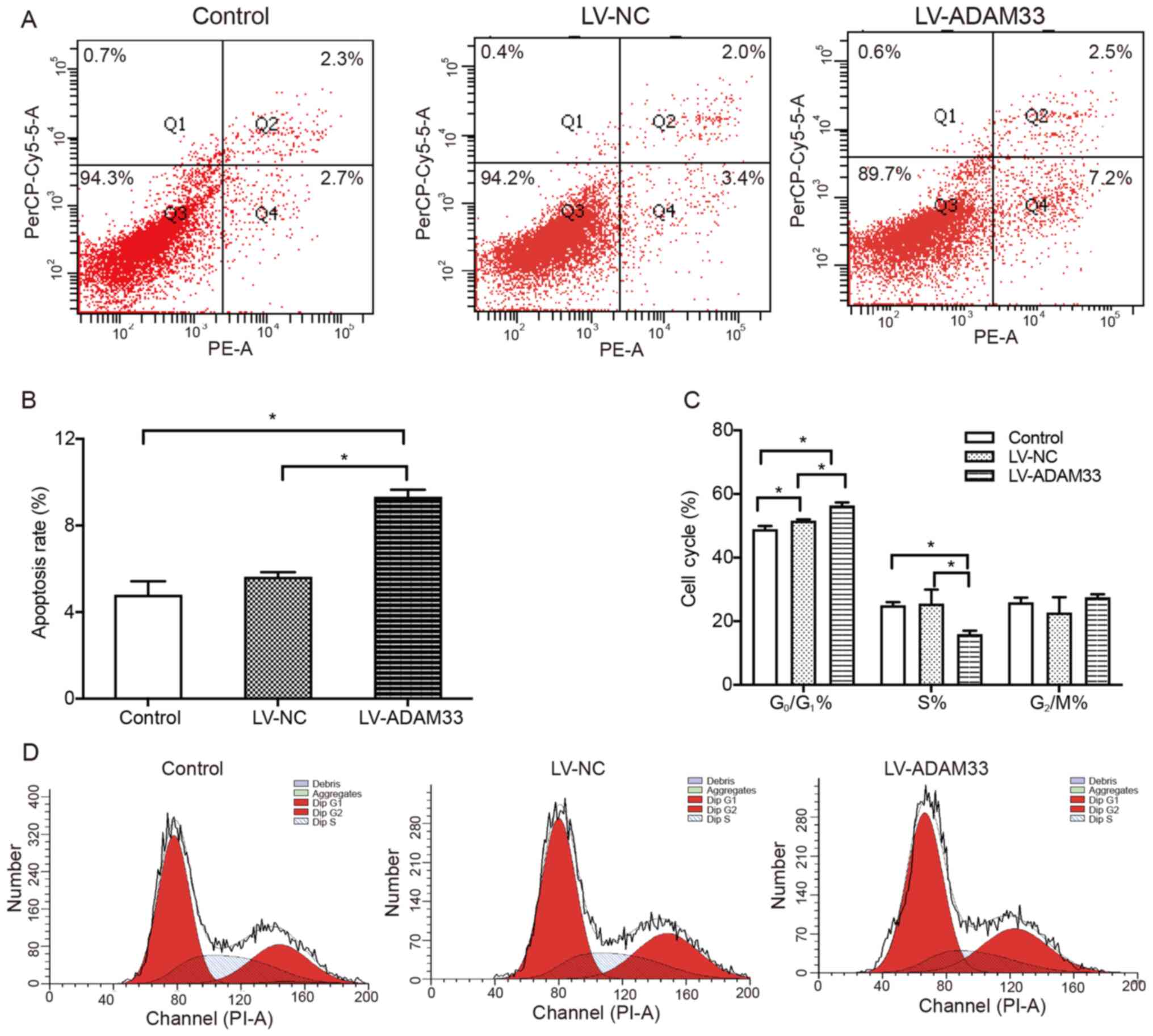
Subsequently, the influence of ADAM33 silencing on
apoptosis and cell cycle distribution in HASMCs was evaluated by
flow cytometry. It was found that the apoptotic rates of HASMCs
were similar between the blank control group and negative control
group (P=0.356; Fig. 6A and B).
However, ADAM33 silencing by LV-ADAM33-shRNA transfection
significantly increased the rate of apoptotic cells (9.333±0.321
vs. 4.800±0.625% and 5.633±0.208%; P=0.001 and P=0.005; Fig. 6A and B) compared with HASMCs in the
blank control group and negative control group. In addition, it was
also found that the percentage of G0/G1 phase
cells in the negative control group was higher than that in the
blank control group (P=0.007; Fig. 6C
and D). Compared with HASMCs in the blank control group and
negative control group, the percentage of cells in the
G0/G1 phase in the ADAM33 gene silencing
group was significantly higher (P=0.000 and P=0.000; Fig. 6C), while the percentage of cells in
S phase was lower (P=0.006 and P=0.005; Fig. 6C) and there was no significant
difference in the percentage of cells in G2/M phase
(P=0.523 and P=0.091; Fig. 6C). The
results showed that ADAM33 gene silencing could inhibit the cell
cycle transition from G0/G1 to S phase and
promote cell apoptosis.
Influence of ADAM33 silencing on
PI3K/AKT/ERK pathway singling molecules
To further understand the underlying molecular
mechanisms of ADAM33 silencing on the proliferation of HASMCs, the
changes of signaling molecules involved in PI3K/AKT/ERK pathway
were evaluated following ADAM33 silencing. Results showed that
ADAM33 silencing significantly reduced the mRNA level of PI3K
(P=0.002 and P=0.003), while changes of mRNA levels of AKT and ERK
were not significant compared with HASMCs in the blank control
group and negative control group (P=0.259 and P=0.100; P=0.134 and
P=0.470; Fig. 7A-C). Furthermore,
western blotting analyses showed that ADAM33 silencing
significantly reduced the protein level of PI3K compared with
HASMCs in the blank control group and negative control group
(P=0.000 and P=0.000; Fig. 7D and
E). In addition, although protein levels of total AKT and
ERK1/2 were not significantly changed, ADAM33 silencing
significantly reduced the protein levels of p-AKT and p-ERK
(P=0.011 and P=0.011; P=0.035 and P=0.019; Fig. 7F-I). The ratios of p-AKT/AKT and
p-ERK/ERK1/2 were reduced (P=0.013 and P=0.020; P=0.044 and
P=0.011; Fig. 7J and K). Taken
together, these results suggested that ADAM33 silencing may
attenuate the proliferation of HASMCs via inhibiting the
PI3K/AKT/ERK pathways.
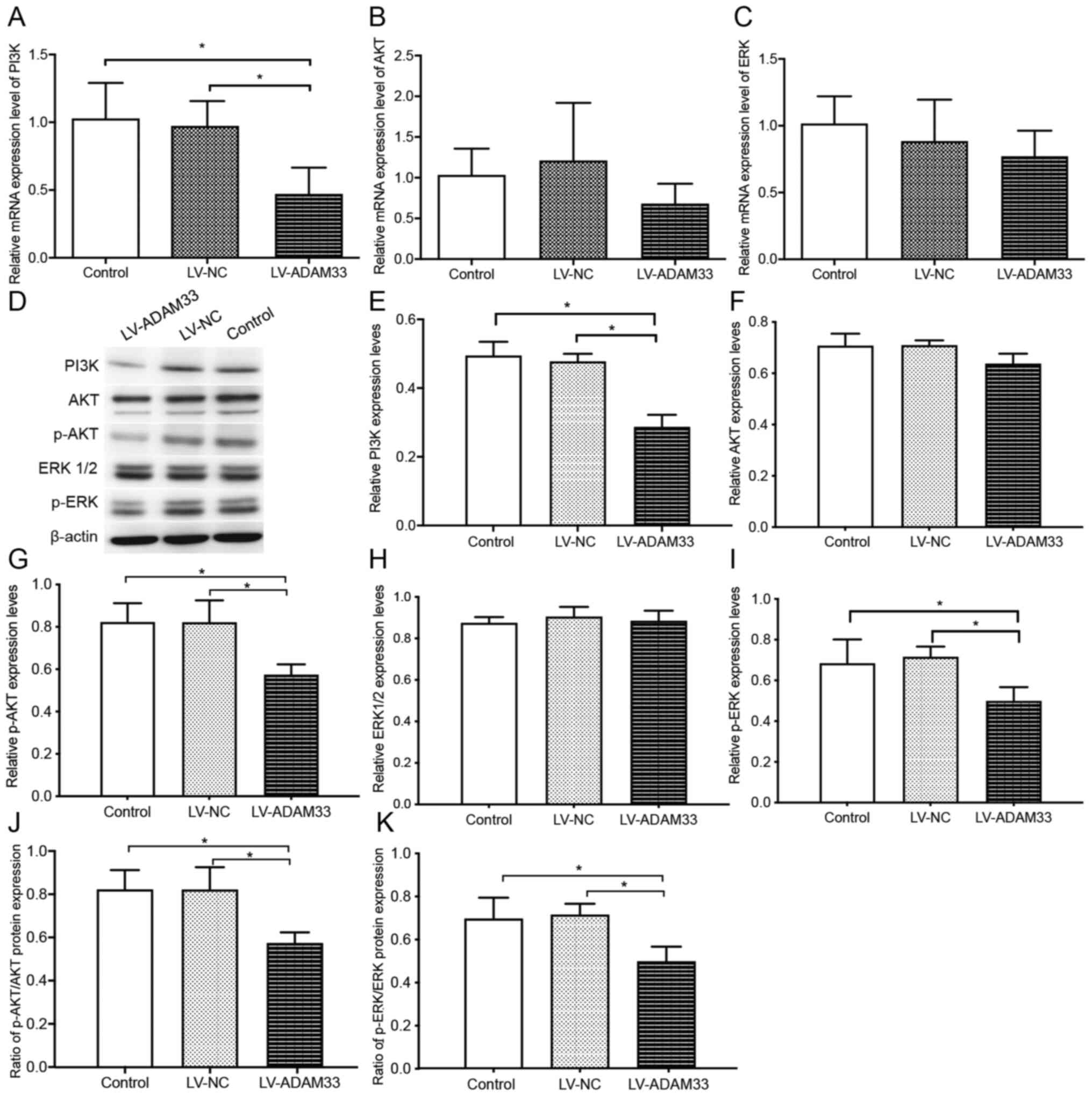 | Figure 7.Influence of ADAM33 silencing on the
PI3K/AKT/ERK pathway singling molecules. Reverse
transcription-quantitative PCR for the mRNA levels of (A) PI3K, (B)
AKT and (C) ERK of HASMCs in the blank control, LV-NC and
LV-ADAM33-shRNA groups. (D) Representative images of western
blotting for the protein levels of PI3K, AKT, p-AKT, ERK1/2 and
p-ERK of HASMCs in the blank control, LV-NC and LV-ADAM33-shRNA
groups. Semi-quantitative analysis of the protein levels of (E)
PI3K, (F) AKT, (G) p-AKT, (H) ERK1/2 and (I) p-ERK of HASMCs in the
blank control, LV-NC and LV-ADAM33-shRNA groups evaluated by
western blotting. Ratio of (J) p-AKT/AKT and (K) p-ERK/ERK1-2 of
HASMCs in the blank control, LV-NC and LV-ADAM33-shRNA groups.
*P<0.05. ADAM33, a disintegrin and metalloproteinase-33; HASMCs,
human aortic smooth muscle cells; LV, lentiviral vector; NC,
negative control; shRNA, short-hairpin RNA; p-, phosphorylated. |
Influence of ADAM33 silencing on Bcl-2
and Bax apoptotic molecules
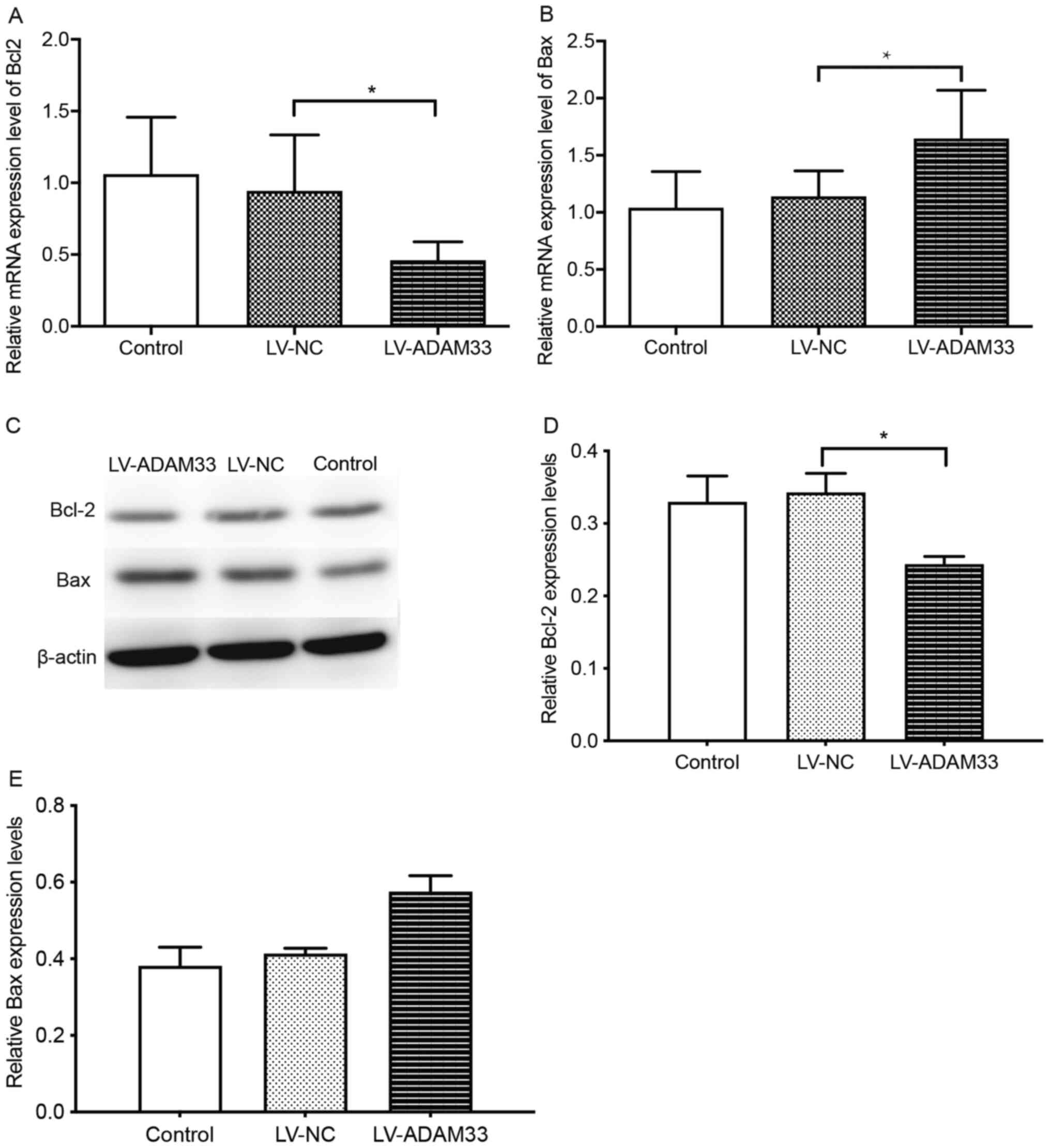
To elucidate the underlying mechanisms for the role
of ADAM33 silencing on the apoptosis of HASMCs, the changes in
Bcl-2 and Bax expression levels following ADAM33 silencing was
evaluated. Results showed that ADAM33 silencing in HASMCs was
associated with reduced mRNA expression of Bcl-2, but increased
mRNA expression of Bax compared with HASMCs in the blank control
group and negative control groups (P=0.014 and P=0.039; P=0.013 and
P=0.032; Fig. 8A and B).
Furthermore, results from western blotting also showed that ADAM33
silencing reduced the protein expression of Bcl-2, but increased
the protein expression of Bax (P=0.007 and P=0.004; P=0.001 and
P=0.002; Fig. 8C-E). Taken
together, these results demonstrated that ADAM33 silencing may
upregulate HASMCs apoptosis via regulation of Bax/Bcl-2
apoptotic-related protein expression.
Discussion
The present study, using in vitro HASMCs and
lentiviral vectors carrying shRNA for ADAM3, found that
constitutive expression of ADAM33 in VSMCs is important to maintain
a proliferative phenotype of VSMCs, probably via regulation of the
PI3K/AKT/ERK pathways. In addition, silencing of ADAM33 inhibited
the proliferation and induced the apoptosis of HASMCs, which was
accompanied by the inhibition of PI3K/AKT/ERK pathways and
regulation of the expression levels of Bcl-2 and Bax proteins
towards a pro-apoptotic ratio. These findings, together with the
findings of previous studies (25,26)
and immunohistochemical result in this present study, which showed
constitutive expression of ADAM33 in airway VSMCs in patients with
asthma, suggested an important role of ADAM33 in airway vascular
remodeling. These findings also suggested potential therapeutic
significance of ADAM33 inhibition in airway remodeling-related
diseases.
In the field of asthma research, ADAM33 is an
important susceptibility gene, as revealed by whole genome scanning
(27). Accordingly, further
clarification of its function is important in translational medical
research. Previous studies by our team have shown that cytokines of
T cells, including IFN-γ, IL-4 and IL-13, may act on the airway
wall vascular smooth muscle cells and affect airway wall vascular
remodeling by regulating ADAM33 expression (18,28).
The present study was designed for further understanding the
molecular mechanism (signaling pathway) of abnormal ADAM33 gene
expression on airway vascular remodeling. During the present study,
a large amount of biopsy tissues of human airway VSMCs would be
needed for cell isolation and culture and these technical
difficulties greatly challenged the use of cells derived from
biopsy. Therefore, HASMCs were selected.
Composed of 22 exons and 8 encoding regions, the
biological functions of ADAM33 mainly involve protein hydrolysis,
molecular modification, molecular release, intercellular and
cellular matrix interaction, intracellular signal transduction and
intercellular communication (29).
Expression of ADAM33 in pulmonary tissues of patients with asthma
has been confirmed (14,26). Subsequent studies have evaluated the
role of ADAM33 in airway remodeling. A previous study in a human
bronchial epithelial cell line showed that TGFβ1 is associated with
enhanced expression of ADAM33, which subsequently induced
epithelial-mesenchymal transition of airway epithelial cells that
participate in airway remodeling in asthma (30). Notably, an early study showed that
TGFβ1 suppresses the expression of ADAM33 mRNA in normal or
asthmatic fibroblasts (31). By
using an ovalbumin-induced asthma model in rats, silencing of
ADAM33 has been shown to decrease the proliferation and increase
the apoptosis of airway smooth muscle cells (ASMCs), suggesting
that ADAM33 represents a potential investigative focus target
aiding allergic asthma (22).
Another study showed that vascular endothelial growth factor
enhances ADAM33 expression and cell proliferation of ASMCs by
activating the VEGFR2/ERK1/2 signaling pathway, which might be
involved in the pathogenesis of airway remodeling (32). A subsequent study showed that
1,25-dihydroxyvitamin D3 can inhibit VEGF-induced ASMCs
proliferation by suppressing VEGFR2 and ERK1/2 activation and
downregulating ADAM33 (33). In
addition, ADAM33 overexpression has been shown to alter the
mechanical behavior of ASMCs in vitro, promoting a
hypercontractile phenotype transition of ASMCs (34). A recent in vitro study in
human embryonic lung Mrc-5 fibroblasts sensitized with
Dermatophagoides farinae 1 showed that IFN-γ may participate
in airway remodeling in asthma by regulating the expression of
ADAM33 (35). It is evident that
previous studies evaluating the role of ADAM33 in airway remodeling
have focused on its role in bronchial epithelial cells, ASMCs and
lung fibroblasts. It remains to be elucidated whether ADAM33 is
expressed in airway VSMCs and whether it serves a role in the
pathogenesis of airway remodeling. The present study confirmed that
ADAM33 was expressed in VSMCs. Further in vitro experiments
showed that silencing the constitutive expression of ADAM33 in
HASMCs could significantly inhibit the proliferation of HASMCs and
induced HASMC apoptosis. In view of the importance of vascular
proliferation in the pathogenesis of airway remodeling-related
diseases (5,7), the findings of the present study
suggested that expression of ADAM33 in airway VSMCs may participate
in the process of airway remodeling in the pathogenesis of asthma
via stimulation of airway VSMC proliferation.
Activation of the PI3K/AKT signaling pathway has
been extensively involved in the regulation of cell growth,
proliferation and differentiation via phosphorylation of a variety
of downstream enzymes, kinases and transcription factors of the
pathway (36). A previous study
showed that PI3K/AKT signaling mediated hyperinsulinemia-induced
proliferation and collagen release of human ASMCs, therefore
promoting airway remodeling in asthma (37). Another study showed that inhibition
of the expression of p-PI3K and p-AKT was associated with
attenuated angiogenesis and vascular remodeling and alleviated
symptoms in a mouse model of asthma (38). The present study showed that
silencing of ADAM33 in HASMCs was associated with inhibited
cellular proliferation, which was accompanied by the inhibition of
the PI3K/AKT/ERK pathways. These findings suggested that ADAM33 may
stimulate proliferation of airway VSMCs via activation of the
PI3K/AKT/ERK pathways. Bcl-2 and Bax proteins are universally
expressed apoptosis regulatory proteins among various cells, which
exert anti-apoptotic and pro-apoptotic activities, respectively
(39). The present study found that
silencing of ADAM33 in HASMCs was associated with induced cellular
apoptosis, accompanied by the upregulation of Bax and
downregulation of Bcl-2. These findings further indicated that
constitutive expression of ADAM33 may inhibit the apoptosis of
airway VSMCs via regulation of Bcl-2/Bax expression. Previous
studies have also suggested the potential interactions between
PI3K/AKT/ERK pathway and Bcl-2 and Bax apoptotic molecules
(40,41). An early study in cultured airway
SMCs showed that leptin can significantly inhibit apoptosis in
airway SMCs apoptosis, at least partially via the activation of
PI3K/AKT signaling pathway and subsequent upregulation of Bcl-2 and
downregulation of Bax, towards an anti-apoptotic direction
(40). Another study in VSMCs
showed that overexpression of phosphatase and tensin homolog could
inhibit the PI3K/AKT/ERK pathway, accompanied with upregulation of
Bax and downregulation of Bcl-2, towards a pro-apoptotic direction
(41). These findings, together
with those of the present study, suggest that inhibition of
PI3K/AKT/ERK pathway may lead to apoptosis in SMCs by upregulation
of Bax and downregulation of Bcl-2, towards a pro-apoptotic
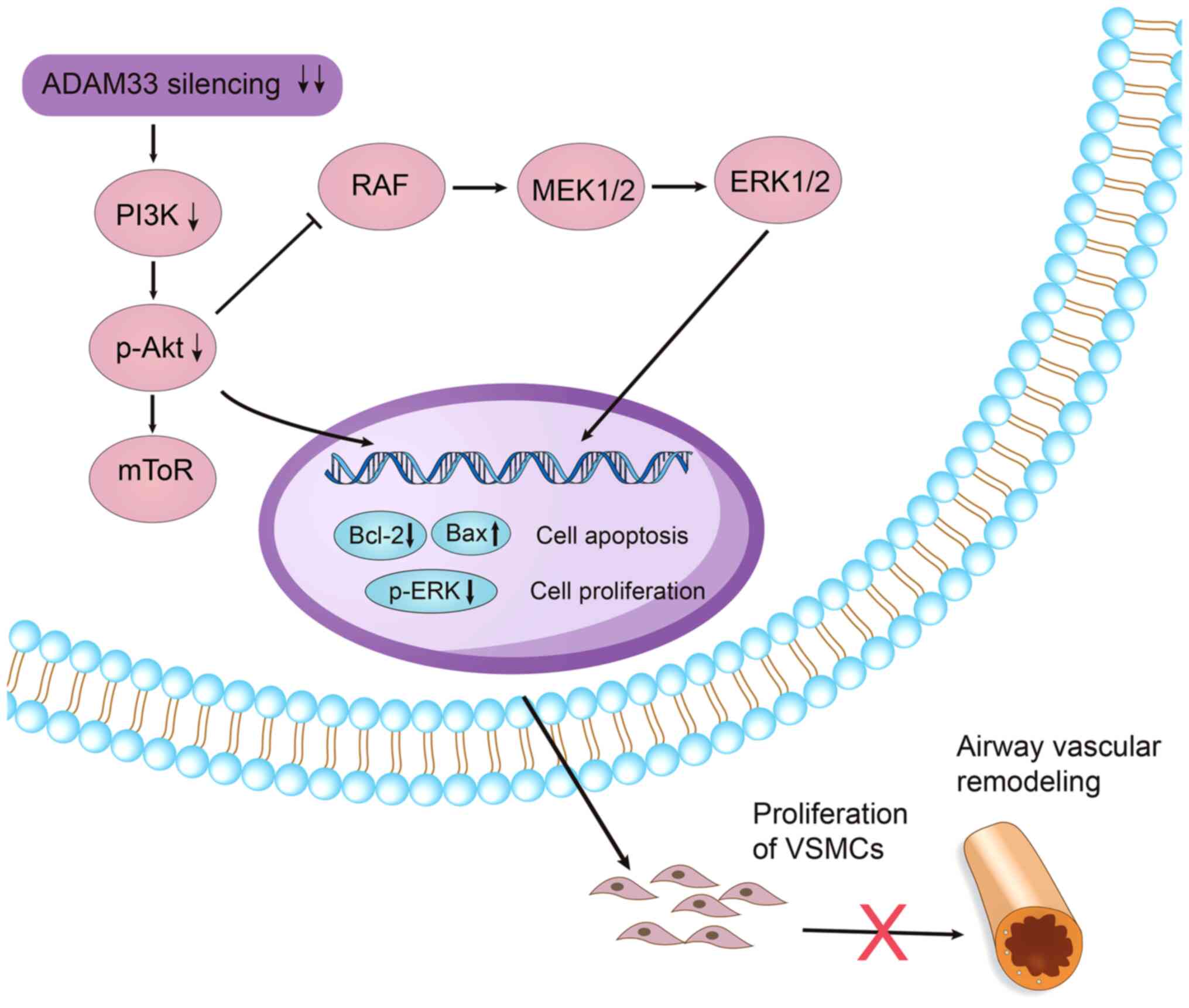
direction. Taken together, by integrating the results of the
aforementioned findings, it could be hypothesized that constitutive
expression of ADAM33 in VSMCs may maintain a proliferative
phenotype of VSMCs via regulation of the PI3K/AKT/ERK pathways.
Accordingly, silencing of ADAM33 inhibited proliferation and
induced the apoptosis of HASMCs via inhibition of PI3K/AKT/ERK
pathways and subsequent regulation of Bcl-2 and Bax protein
expression towards a pro-apoptotic ratio (Fig. 9). As we have previously shown
constitutive expression of ADAM33 in airway VSMCs in patients with
asthma, these findings may suggest the potential therapeutic
significance of ADAM33 inhibition in airway remodeling-related
diseases. It has to be mentioned that the findings of the present
study are based on observations in HASMCs rather than in isolated
airway VSMCs due to technical difficulties. These findings should
be validated in future studies in airway VSMCs. Furthermore,
although silencing ADAM33 appeared promising to inhibit the
proliferation of HASMCs, the influences on proliferation of VSMCs,
as well as in the process of airway vascular remodeling, should be
investigated using preclinical asthma models.
In conclusion, the present study demonstrated that
constitutive expression of ADAM33 may be important to maintain a
proliferative phenotype in HASMCs. The influences of ADAM33 on the
proliferation and apoptosis of HASMCs may involve the regulation of
PI3K/AKT/ERK and Bax/Bcl-2 pathways. These findings suggested an
important role of ADAM33 in airway vascular remodeling and a
potential therapeutic significance of ADAM33 inhibition in airway
remodeling-related diseases.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation Regional Fund Project (grant no. 81760005).
Availability of data and materials
The datasets generated and analyzed during the
current study are not publicly available due to patient privacy
regulations of the different countries, but are available from the
corresponding author on reasonable request.
Authors' contributions
JD and JW conceived and designed the experiments.
FY, YH and HS performed the experiments. XG analyzed the data. FY
and YH wrote the manuscript. All authors read and approved the
final manuscript. JD, JW and FY confirmed the authenticity of all
the raw data.
Ethics approval and consent to
participate
The present study was approved by Ethics Committee
of the First Affiliated Hospital of Xinjiang Medical University
(approval no. 20180130-01; Urumqi, China). All the patients
provided signed informed consent. The study complied with the
Declaration of Helsinki. All methods were carried out in accordance
with the ethical rules on clinical studies established by the World
Health Organization guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hough KP, Curtiss ML, Blain TJ, Liu RM,
Trevor J, Deshane JS and Thannickal VJ: Airway remodeling in
asthma. Front Med (Lausanne). 7:1912020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kardas G, Kuna P and Panek M: Biological
therapies of severe asthma and their possible effects on airway
remodeling. Front Immunol. 11:11342020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fehrenbach H, Wagner C and Wegmann M:
Airway remodeling in asthma: What really matters. Cell Tissue Res.
367:551–569. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhou-Suckow Z, Duerr J, Hagner M, Agrawal
R and Mall MA: Airway mucus, inflammation and remodeling: Emerging
links in the pathogenesis of chronic lung diseases. Cell Tissue
Res. 367:537–550. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Harkness LM, Kanabar V, Sharma HS,
Westergren-Thorsson G and Larsson-Callerfelt AK: Pulmonary vascular
changes in asthma and COPD. Pulm Pharmacol Ther. 29:144–155. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Olivieri D and Chetta A: Therapeutic
perspectives in vascular remodeling in asthma and chronic
obstructive pulmonary disease. Chem Immunol Allergy. 99:216–225.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Barbato A, Turato G, Baraldo S, Bazzan E,
Calabrese F, Panizzolo C, Zanin ME, Zuin R, Maestrelli P, Fabbri LM
and Saetta M: Epithelial damage and angiogenesis in the airways of
children with asthma. Am J Respir Crit Care Med. 174:975–981. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Alagappan VK, de Boer WI, Misra VK, Mooi
WJ and Sharma HS: Angiogenesis and vascular remodeling in chronic
airway diseases. Cell Biochem Biophys. 67:219–234. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bakakos P, Patentalakis G and Papi A:
Vascular biomarkers in asthma and COPD. Curr Top Med Chem.
16:1599–1609. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rzucidlo EM: Signaling pathways regulating
vascular smooth muscle cell differentiation. Vascular. 17 (Suppl
1):S15–S20. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klein T and Bischoff R: Active
metalloproteases of the A Disintegrin and Metalloprotease (ADAM)
family: Biological function and structure. J Proteome Res.
10:17–33. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dreymueller D, Uhlig S and Ludwig A:
ADAM-family metalloproteinases in lung inflammation: Potential
therapeutic targets. Am J Physiol Lung Cell Mol Physiol.
308:L325–L343. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li HF, Yan LP, Wang K, Li XT, Liu HX and
Tan W: Association between ADAM33 polymorphisms and asthma risk: A
systematic review and meta-analysis. Respir Res. 20:382019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee JY, Park SW, Chang HK, Kim HY, Rhim T,
Lee JH, Jang AS, Koh ES and Park CS: A disintegrin and
metalloproteinase 33 protein in patients with asthma: Relevance to
airflow limitation. Am J Respir Crit Care Med. 173:729–735. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Poon AH, Houseman EA, Ryan L, Sparrow D,
Vokonas PS and Litonjua AA: Variants of asthma and chronic
obstructive pulmonary disease genes and lung function decline in
aging. J Gerontol A Biol Sci Med Sci. 69:907–913. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hur GY and Broide DH: Genes and pathways
regulating decline in lung function and airway remodeling in
asthma. Allergy Asthma Immunol Res. 11:604–621. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kozlik P, Zuk J, Bartyzel S, Zarychta J,
Okon K, Zareba L, Bazan JG, Kosalka J, Soja J, Musial J and
Bazan-Socha S: The relationship of airway structural changes to
blood and bronchoalveolar lavage biomarkers, and lung function
abnormalities in asthma. Clin Exp Allergy. 50:15–28. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang L, Hi X, Yao L, J. W and W. Q: The
effect of interleukin4 on ADAM33 gene expression in airway wall
vascular smooth muscle cells. Journal of Xinjiang Medical
University. 39:265–269. 2016.PubMed/NCBI
|
|
19
|
Isenovic ER, Kedees MH, Tepavcevic S,
Milosavljevic T, Koricanac G, Trpkovic A and Marche P: Role of
PI3K/AKT, cPLA2 and ERK1/2 signaling pathways in insulin regulation
of vascular smooth muscle cells proliferation. Cardiovasc Hematol
Disord Drug Targets. 9:172–180. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chae IH, Park KW, Kim HS and Oh BH: Nitric
oxide-induced apoptosis is mediated by Bax/Bcl-2 gene expression,
transition of cytochrome c, and activation of caspase-3 in rat
vascular smooth muscle cells. Clin Chim Acta. 341:83–91. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin F, Song A, Wu J, Jiang X, Long J, Chen
J, Duan Y, Shi Y and Deng L: ADAM33 protein expression and the
mechanics of airway smooth muscle cells are highly correlated in
ovalbumin-sensitized rats. Mol Med Rep. 8:1209–1215. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou J, Bai W, Liu Q, Cui J and Zhang W:
Silencing of ADAM33 restrains proliferation and induces apoptosis
of airway smooth muscle cells in ovalbumin-induced asthma model. J
Cell Biochem. Nov 18–2018.(Epub ahead of print).
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tamhane AC: Statistical analysis of
designed experiments: Theory and applications. Higher Education
Press; Beijing: 2006
|
|
25
|
Dijkstra A, Postma DS, Noordhoek JA,
Lodewijk ME, Kauffman HF, ten Hacken NH and Timens W: Expression of
ADAMs (‘a disintegrin and metalloprotease’) in the human lung.
Virchows Arch. 454:441–449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Foley SC, Mogas AK, Olivenstein R, Fiset
PO, Chakir J, Bourbeau J, Ernst P, Lemière C, Martin JG and Hamid
Q: Increased expression of ADAM33 and ADAM8 with disease
progression in asthma. J Allergy Clin Immunol. 119:863–871. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Van Eerdewegh P, Little RD, Dupuis J, Del
Mastro RG, Falls K, Simon J, Torrey D, Pandit S, McKenny J,
Braunschweiger K, et al: Association of the ADAM33 gene with asthma
and bronchial hyperresponsiveness. Nature. 418:426–430. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wen J, Muyesha P, Gong X, Hu X, Hao Y, Yan
F, Zang L and Wang J: Effect of γ-interferon on the expression of
ADAM33 gene in human airway wall vascular smooth muscle cells.
Journal of Xinjiang Medical University. 42:965–970. 2019.
|
|
29
|
Tripathi P, Awasthi S and Gao P: ADAM
metallopeptidase domain 33 (ADAM33): A promising target for asthma.
Mediators Inflamm. 2014:5720252014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fang L, Wu J, Huang T, Zhang P, Xin X and
Shi Y: TGF-β1 stimulates epithelial-mesenchymal transition mediated
by ADAM33. Exp Ther Med. 15:985–992. 2018.PubMed/NCBI
|
|
31
|
Yang Y, Wicks J, Haitchi HM, Powell RM,
Manuyakorn W, Howarth PH, Holgate ST and Davies DE: Regulation of a
disintegrin and metalloprotease-33 expression by transforming
growth factor-β. Am J Respir Cell Mol Biol. 46:633–640. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pei QM, Jiang P, Yang M, Qian XJ, Liu JB,
Zheng H, Zhao LH and Kim SH: Upregulation of a disintegrin and
metalloproteinase-33 by VEGF in human airway smooth muscle cells:
Implications for asthma. Cell Cycle. 15:2819–2826. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kim SH, Pei QM, Jiang P, Yang M, Qian XJ
and Liu JB: Effect of active vitamin D3 on VEGF-induced ADAM33
expression and proliferation in human airway smooth muscle cells:
Implications for asthma treatment. Respir Res. 18:72017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Duan Y, Long J, Chen J, Jiang X, Zhu J,
Jin Y, Lin F, Zhong J, Xu R, Mao L and Deng L: Overexpression of
soluble ADAM33 promotes a hypercontractile phenotype of the airway
smooth muscle cell in rat. Exp Cell Res. 349:109–118. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li W, Liang R, Huang H, Wu B and Zhong Y:
Effects of IFN-gamma on cell growth and the expression of ADAM33
gene in human embryonic lung Mrc-5 fibroblasts in vitro. J Asthma.
55:15–25. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yu JS and Cui W: Proliferation, survival
and metabolism: The role of PI3K/AKT/mTOR signalling in
pluripotency and cell fate determination. Development.
143:3050–3060. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Singh S, Bodas M, Bhatraju NK, Pattnaik B,
Gheware A, Parameswaran PK, Thompson M, Freeman M, Mabalirajan U,
Gosens R, et al: Hyperinsulinemia adversely affects lung structure
and function. Am J Physiol Lung Cell Mol Physiol. 310:L837–L845.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Su X, Ren Y, Yu N, Kong L and Kang J:
Thymoquinone inhibits inflammation, neoangiogenesis and vascular
remodeling in asthma mice. Int Immunopharmacol. 38:70–80. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Edlich F: BCL-2 proteins and apoptosis:
Recent insights and unknowns. Biochem Biophys Res Commun.
500:26–34. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu WJ, Zhu SY, Chen YL, Wu X, Ni WJ, Chen
YF and Zhao L: The effects of leptin on apoptosis of airway smooth
muscle cells via the PI3K/Akt signaling pathway. Zhonghua Jie He He
Hu Xi Za Zhi. 35:915–918. 2012.(In Chinese). PubMed/NCBI
|
|
41
|
Wang S, Cheng Z and Chen X: Promotion of
PTEN on apoptosis through PI3K/Akt signal in vascular smooth muscle
cells of mice model of coronary heart disease. J Cell Biochem.
120:14636–14644. 2019. View Article : Google Scholar : PubMed/NCBI
|