Introduction
JNK belongs to the family of MAPKs and encompasses
three encoded genes, JNK1, JNK2 and JNK3 (1). JNK1 and JNK2 are widely
expressed in various tissues, whereas JNK3 is primarily
expressed in the brain and heart (1,2). JNK
is also known as stress-activated kinase, which is involved in the
regulation of the response of the body to external or internal
stimuli, inflammation and cell apoptosis (3). As a response to a specific
stimulation, MAPKs [such as MAPK kinase (MKK) 4 and MKK7] activate
JNK via phosphorylation (4), and
subsequently, regulate the phosphorylation and activity of several
downstream factors, such as activating transcription factor-2
(5), ETS transcription factor
(6) and nuclear factor of activated
T-cells (7). Among these factors,
c-jun/activator protein-1 (AP-1) has been clearly elucidated with
respect to the JNK regulatory pathway. JNK can activate AP-1 via
phosphorylation, followed by the regulation of cell proliferation
and apoptosis (8). Several
pathological studies (9,10) have reported the abnormal activation
of the JNK signaling pathway and its involvement in the modulation
of cell apoptosis, and inhibition of this pathway can reduce the
proportion of apoptotic cells under pathological conditions
(9). Furthermore, JNK can be
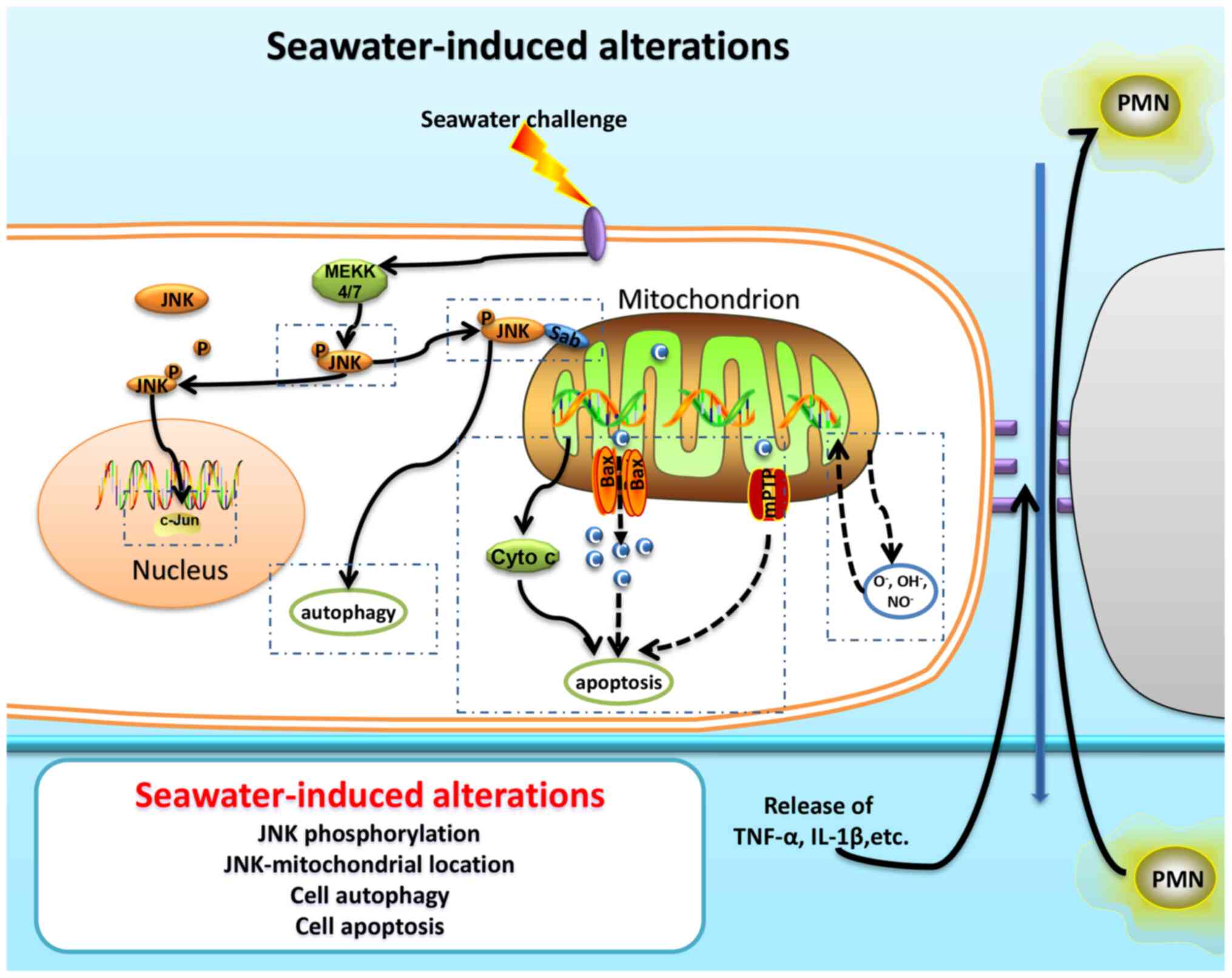
activated via phosphorylation, and bind to the outer membrane
mitochondrial protein mitochondrial SH3BP5 (also known as Sab),
thereby regulating the function of this organelle (11). The localization of p-JNK to
mitochondria can lead to its functional disorder, resulting in a
decrease in the energy supply and membrane potential, as well as an
increase in reactive oxygen species (ROS) production and the
occurrence of apoptosis (8,9,12).
Previous studies (13–15)
have shown that JNK was involved in the occurrence of acute lung
injury (ALI) and served an important role in the regulation of
apoptosis in lung tissue. Experimental studies have revealed that
the activation of the JNK pathway was one of the crucial factors
resulting in injury; this pathway also interacts with the NF-κB
pathway, intercellular tight junction proteins and TGF-β to
participate in the development of lung injury (13,16).
The pathological changes associated with ALI are partly due to the
abnormal regulation of mitochondria, such as the production of ROS,
induced autophagy and apoptosis (17–19).
Moreover, maintaining the stability of mitochondrial function is
vital to ameliorate ALI/acute respiratory distress syndrome (ARDS)
(20,21). Mitochondria, as intracellular energy
sources, serve a crucial role in cell survival and the normal
physiological function of organelles, and maintaining mitochondrial
stability is essential for cells, tissues and organs (22). Activation of the JNK pathway has
been reported to disrupt the function of mitochondria in myocardial
ischemia-reperfusion injury, and as a result, decreases the
function and survival of myocardial cells (8). However, activation of JNK-mediated
mitochondrial function abnormalities is rarely reported with
respect to the occurrence and progression of ALI/ARDS. In the
present study, we hypothesized that abnormal activation of the
JNK-mitochondrial (mitoJNK) pathway could significantly disrupt the
normal physiological function of lung cells, resulting in the
occurrence of ALI/ARDS. Moreover, the underlying role of the
mitoJNK pathway in ALI remains unknown, and requires further
study.
Materials and methods
Reagents
Antibodies (Ab) against cytochrome c (cat. no.
ab133504), Bcl-2 (cat. no. ab196495), cytochrome c oxidase IV (COX
IV) (cat. no. ab202554) and GAPDH (cat. no. ab181602) were
purchased from Abcam. Phosphorylated (p)-Bcl-2 (Ser70) rabbit
monoclonal (m)Ab (cat. no. #2827), p-JNK (Thr183/Tyr185) rabbit mAb
(cat. no. #4668) and JNK rabbit mAb (cat. no. #9258) were obtained
from Cell Signaling Technology, Inc. LC3 rabbit mAb (cat. no.
sc-398822) was purchased from Santa Cruz Biotechnology, Inc. The
malondialdehyde (MDA) Assay kit (TBA method) (cat. no. A003-1) and
H2O2 Assay kit (cat. no. A064-1) were
obtained from Nanjing Jiancheng Bioengineering Institute. An In
Situ Cell Death Detection kit was obtained from Roche
Diagnostics GmbH. Dexamethasone (DXM) was purchased from
Sigma-Aldrich (Merck KGaA). Tat-SabKIM1 (GFE SLS VPS PLD
LSG PRV VAPPRRRQRRKKRG-NH2) was purchased from
NeoPeptide. Seawater (osmolality 1,300 mmol/l; pH 8.2; 26.518 g/l
NaCl; 3.305 g/l MgSO4; 2.447 g/l MgCl2; 1.141
g/l CaCl2; 0.725 g/l KCl; 0.202 g/l NaHCO3;
0.083 g/l NaBr) was prepared according to the major composition of
the East China Sea provided by the Chinese Ocean Bureau (23).
Animal procedures
The animal procedures in this study were approved by
the Animal Care and Use Committee of The Fourth Military Medical
University (approval no. TDLL20160193), and were carried out in
accordance with the Declaration of the National Institutes of
Health Guide for Care and Use of Laboratory Animals (24). A total of 32 male Sprague-Dawley
(SD) rats (weight, 180–220 g; age, 6 weeks) were maintained on a
light/dark cycle of 12:12-h with free access to food and water. The
rats were maintained in an atmosphere with an ambient temperature
of 18–26°C and relative humidity of 40–70%. The SD rats were
randomly divided into the following experimental groups (n=8): i)
Control group; ii) seawater inhalation groups; iii) DXM
pre-treatment group, in which the rats were pre-treated
intraperitoneally with DXM (2.5 mg/kg body weight) 30 min before
modelling; and iv) Tat-SabKIM1 pre-treatment group, in
which the rats were pre-treated with Tat-SabKIM1 (2
mg/kg body weight) via tracheal injection 10 min before
modelling.
Seawater inhalation-induced ALI/ARDS was established
through the following procedures. First, the rats were anesthetized
with sodium pentobarbital (45 mg/kg weight) intraperitoneally.
Then, the animals were placed in the supine position with the head
elevated at an angle of 30° during the experiments. A 1-ml syringe
was gently inserted into the trachea at ~1.5 cm above the carina.
Subsequently, 4 ml/kg body weight seawater was instilled into both
lungs within 4 min. The lungs were then harvested after the rats
were sacrificed via intraperitoneal injection of 200 mg/kg sodium
pentobarbital at the predetermined time (6 h).
Western blot analysis
The protein extract prepared from the lung tissue
harvested 6 h after seawater instillation was subjected to western
blot analysis, as described previously (25). Briefly, the protein was obtained
using RIPA lysis buffer (Beyotime Institute of Biotechnology) and
protein concentration was quantified using a Bradford kit (Beyotime
Institute of Biotechnology) according to the manufacturer's
instructions. Total proteins (20 µg) were separated by SDS-PAGE on
12% gels and transferred to nitrocellulose membranes (EMD
Millipore). Then, the membranes were blocked with 10% non-fat dry
milk in TBS at room temperature for 30 min and probed at 4°C
overnight with primary Abs, including anti-p-JNK (1:1,000),
anti-JNK (1:1,000), anti-GAPDH (1:2,500), anti-COX IV (1:1,000),
anti-LC3 (1:1,000), anti-p-Bcl-2 (1:1,000), anti-Bcl-2 (1:1,000) or
anti-cytochrome c (1:10,000). Subsequently, the membranes were
washed with TBS-0.3%Tween-20 and then incubated with an appropriate
HRP-conjugated secondary Ab (1:10,000) at room temperature for 2 h.
The immunoreactive target proteins were detected using an ECL
detection system (Thermo Fisher Scientific, Inc.). Band intensities
were semi-quantified using Image Lab 4.1 (Bio-Rad Laboratories,
Inc.).
Histopathological evaluation
The lung tissue was harvested and fixed with 4%
paraformaldehyde at room temperature for 24 h, embedded in paraffin
and cut into 5-µm sections that were stained with H&E at room
temperature for 3 min. Sections were examined with an optical
microscope (magnification, ×100).
Preparation of mitochondrial and
cytosolic/nuclear proteins
Mitochondrial and cytosolic/nuclear proteins were
prepared by isolating mitochondria from the cells, as described
previously (9). Briefly, isolation
buffer (210 mM mannitol; 70 mM sucrose; 5 mM HEPES; 1 mM EGTA; 0.5
mg/ml BSA (Merck KGaA; pH=7.4) was used to wash and homogenize the
rat lungs. Then, the homogenate was centrifuged at 1,000 × g for 10
min at 4°C. The supernatant was collected and centrifuged at 10,000
× g for 10 min at 4°C. This second supernatant was used as the
soluble cytosolic/nuclear fraction with excluded mitochondria, and
the sedimentation pellet was resuspended in lysis buffer for
western blot analysis of the mitochondrial proteins according to
the aforementioned steps. COX IV was used as an internal
mitochondrial control, and GAPDH served as the control for other
organelles.
Lung wet-to-dry (W/D) weight
ratio
The W/D weight ratio of the lung tissue is commonly
used to reflect the severity of pulmonary oedema (25). Briefly, the rats were sacrificed at
the predetermined time points (6 h after seawater inhalation), and
the chests were quickly opened. The same part of the left lung from
each rat was weighed as the wet weight after removal of the other
tissues. Then, each lung was placed in an oven for baking at 80°C
for 72 h to obtain a constant weight, denoted as the dry weight.
The W/D weight ratio was obtained by dividing the wet weight by the
dry weight of the left lung.
Determination of
H2O2 concentrations and MDA levels
H2O2 and MDA concentrations in
the lung tissues were detected using the H2O2
Assay kit and MDA Assay kit, respectively, according to the
manufacturer's instructions. Briefly, the lung tissue samples were
homogenized in cold normal saline (lung tissue to normal saline
ratio, 1:9). Then, the homogenate was examined according to the
protocol of the kit. To detect the H2O2
concentration, the rate of change in absorbance was measured with a
spectrophotometer at 405 nm. For detecting the MDA concentration,
the rate of change in absorbance was measured with a
spectrophotometer at 532 nm.
Assessment of lung cells
apoptosis
In order to quantify cell apoptosis in the injured
rat lungs, a TUNEL assay was conducted using an In Situ Cell
Death Detection kit according to the manufacturer's instructions.
Briefly, lung tissues were fixed with 4% paraformaldehyde for 24 h
at room temperature. The lung tissues were then embedded in
paraffin and sectioned into 5-µm sections. After dewaxing, the
tissue sections were incubated with TUNEL working solution for 1 h
at 37°C to label apoptotic cells, and then nuclei were stained with
DAPI (Merck KGaA) for 5 min at room temperature. The slides were
mounted with 50% glycerol (Merck KGaA). Finally, the sections were
visualized using a fluorescence microscope (magnification, ×200;
Olympus Corporation). A total of 10 images from three slides from
every group were randomly selected, and the number of cells
exhibiting positive staining for apoptosis per field were counted
and analysed.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 8 (GraphPad Software, Inc.). The graphical data are presented
as the mean ± SEM and experiments were repeated four times. The
experimental groups were compared using a one-way ANOVA and
Bonferroni's multiple comparison tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
mitoJNK signaling is activated during
seawater inhalation-induced ALI/ARDS
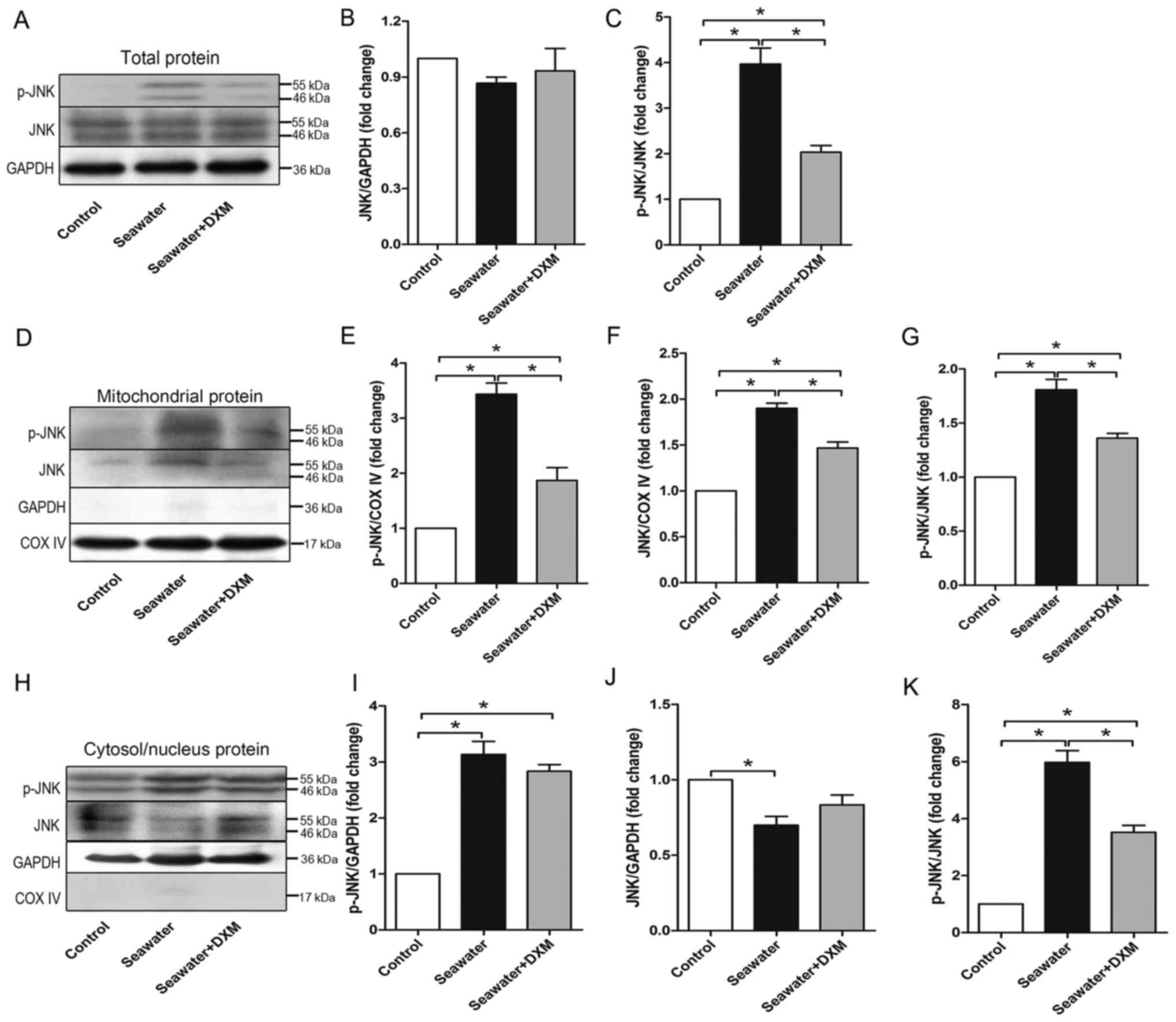
To elucidate the role of JNK, especially mitoJNK, in
ALI/ARDS, the current study assessed the expression levels of p-JNK
and JNK in the total protein, mitochondrial protein and
cytosol/nucleus protein of the lung via western blot analysis. As
shown in Fig. 1A, B and C, seawater
inhalation significantly phosphorylated JNK, thereby activating the
JNK pathway in the lung. Furthermore, it was found that the
phosphorylation level of JNK was elevated significantly in both the
mitochondria (Fig. 1D, E and G) and
cytosol/nucleus (Fig. 1H, I and K)
after seawater challenge. In addition, the total JNK content in the
mitochondria was also elevated (Fig. 1D
and F), whereas that in the cytosol/nucleus (Fig. 1H and J) was decreased, indicating
JNK translocation from the cytosol/nucleus to mitochondria during
ALI/ARDS.
The protective role of DXM in ALI/ARDS was also
investigated. DXM pre-treatment significantly alleviated the high
level of phosphorylated JNK (Fig. 1A
and C), in both the mitochondria (Fig. 1D and G) and cytosol/nucleus
(Fig. 1H and K).
Protective effects of the
mitoJNK-inhibiting peptide Tat-SabKIM1 against
ALI/ARDS
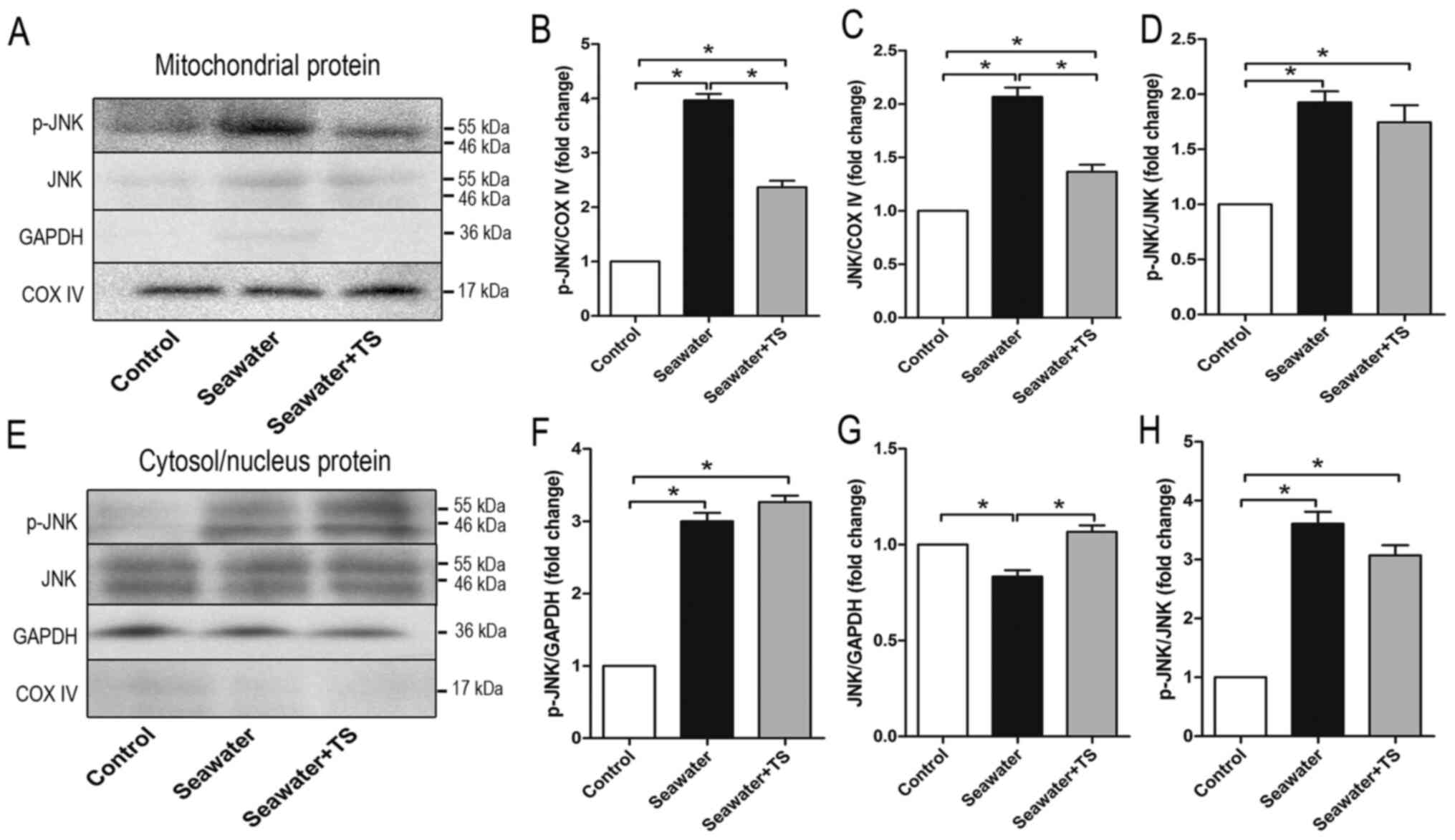
To further examine the role of mitoJNK activation in
ALI/ARDS, the peptide Tat-SabKIM1, which can act on the
SabKIM1 domain and be expressed in mitochondria
(6), was used. The peptide can also
mediate the localization of JNK by selectively blocking JNK
translocation to mitochondria both in vitro and in
vivo (12,26). However, Tat-SabKIM1 does
not exhibit any effect on the translocation of JNK to the nucleus
(12). Tracheal injection of
Tat-SabKIM1 10 min before modelling significantly
decreased the phosphorylation level of JNK in mitochondria
(Fig. 2A and B) and total JNK
expression in mitochondria when compared with the seawater
inhalation group (Fig. 2A and C);
however, it did not influence the ratio of p-JNK/JNK when compared
with the seawater inhalation group (Fig. 2D). p-JNK expression in the
cytosol/nucleus was elevated in both the seawater inhalation group
and Tat-SabKIM1 pre-treatment group compared with that
in the control group (Fig. 2E and
F). The ratio of p-JNK/JNK in the cytosol/nucleus was also
elevated after seawater inhalation compared with that in the
control group (Fig. 2E and H). In
addition, as presented in Fig. 2E and
G, Tat-SabKIM1 increased total JNK expression in the
cytosol/nucleus when compared with the seawater inhalation group.
These results indicated that Tat-SabKIM1 could
specifically inhibit JNK localization to the mitochondria and the
activation of mitoJNK signaling, without any impact on the ratio of
p-JNK/JNK or the cytosolic/nuclear JNK activation.
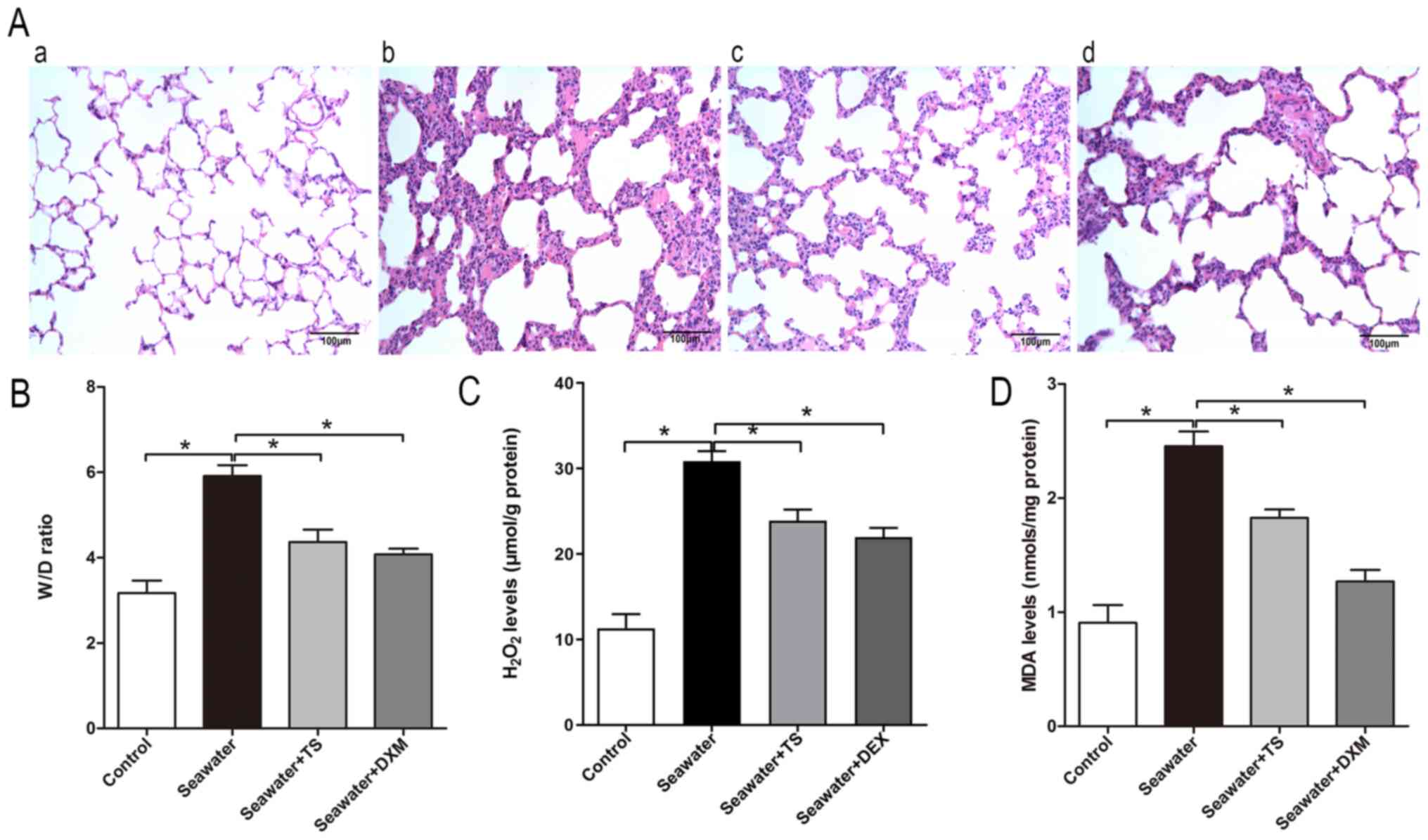
As Tat-SabKIM1 could selectively inhibit
mitoJNK, the effects of mitoJNK activation on the progression of
ALI/ARDS were further evaluated. Tat-SabKIM1-mediated
inhibition of JNK localization to the mitochondria alleviated the
seawater inhalation-induced destruction of lung tissue structure
(Fig. 3A) and pulmonary oedema
(Fig. 3B). In addition, the
inhibitory effects slightly improved the levels of the markers of
peroxidation and oxidative stress, H2O2 and
MDA, respectively, compared with the seawater inhalation group
(Fig. 3C and D). These results
suggested that the inhibition of mitoJNK activation exerted a
protective effect against ALI/ARDS. DXM pre-treatment could inhibit
JNK phosphorylation in both the mitochondria and cytosol/nucleus
compared with that in the seawater group, thus indicating that it
did not selectively inhibit mitoJNK (Fig. 1C and G). As shown in Fig. 3A, DXM could also alleviate seawater
inhalation-induced lung injury. In addition, the pulmonary oedema
and oxidative stress of the DXM pre-treatment group were
ameliorated compared with in the seawater inhalation group
(Fig. 3B-D).
Effects of mitoJNK activation on the
occurrence of autophagy
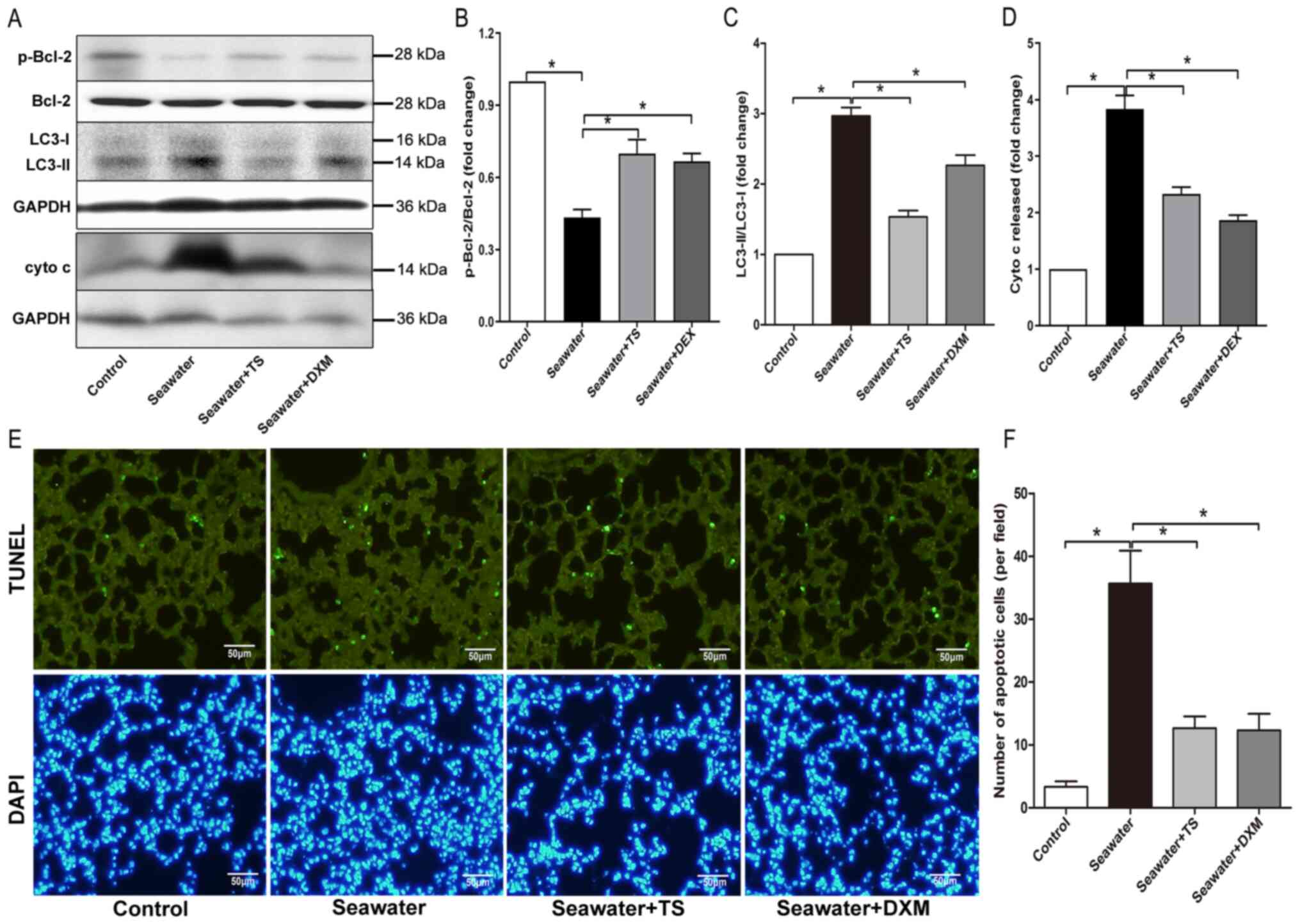
Autophagy has been reported to participate in the
development of ALI/ARDS, as per the conversion of LC3, i.e., LC3-I
to LC3-II (27). Bcl-2 may also
modulate autophagy via an interaction with the autophagy protein,
Beclin 1 (28). Seawater inhalation
decreased the expression level of p-Bcl-2 and increased the
conversion of LC3 to its active form LC3-II, while the inhibition
of mitoJNK by Tat-SabKIM1 restored the expression levels
of p-Bcl-2 and LC3 (Fig. 4A-C),
thereby indicating that the inhibition blocked the autophagy
induced by ALI/ARDS.
MitoJNK activation contributes to
mitochondria-mediated apoptosis
Next, mitochondria-mediated apoptosis during
ALI/ARDS was examined. In the seawater inhalation group, cytochrome
c released from mitochondria into the cytosol was significantly
increased compared with the control group (Fig. 4A and D). Furthermore, treatment with
Tat-SabKIM1 or DXM reduced the amount of cytochrome c
released from the mitochondria. The findings observed for cell
apoptosis, as evaluated via TUNEL staining, were in line with the
aforementioned results. The inhibition of mitoJNK signaling had an
anti-apoptotic effect (Fig. 4E and
F), as Tat-SabKIM1 treatment significantly decreased
the number of TUNEL-positive cells. These results indicated that
mitoJNK signaling participated in mitochondria-mediated apoptosis
during ALI/ARDS (Fig. 5).
Discussion
ARDS is one of the leading causes of morbidity and
mortality in critically ill patients, and in the USA, ~75,000
individuals die due to ARDS every year (29). Although research and clinical trials
have been conducted, the mortality of ARDS continues to be 25–40%,
and few pharmacological interventions can improve the mortality
rate (30). The pathological
changes associated with ALI/ARDS are partly due to the abnormal
regulation of mitochondria, including the production of ROS,
induced autophagy and apoptosis (17–19).
Moreover, maintaining the stability of mitochondrial function is
vital to ameliorate ALI/ARDS (20,21).
The main aim of the present study was to confirm
that the activation of mitoJNK signaling was involved in the
development of ALI/ARDS. By inhibiting the interaction of JNK and
Sab using a selective peptide, Tat-SabKIM1, JNK could
not be localized to mitochondria, thereby rendering an opportunity
to examine the underlying role of mitoJNK in the development of
ALI/ARDS. The present results demonstrated that JNK was
phosphorylated and activated during ALI/ARDS, not only in the
routine JNK pathway but also in the mitoJNK pathway. Furthermore,
the mitoJNK signal was activated during seawater inhalation-induced
ALI/ARDS, as shown by JNK translocation from the cytosol/nucleus to
mitochondria. Tat-SabKIM1 can specifically inhibit JNK
localization to mitochondria and the activation of the mitoJNK
signaling without any impact on the ratio of p-JNK/JNK or the
cytosolic/nuclear JNK activation. Thus, the present study used the
Tat-SabKIM1 peptide to inhibit mitoJNK, which
demonstrated a protective effect on seawater inhalation-induced
ALI/ARDS. In addition, the protective effect was associated with
mitoJNK. For example, Bcl-2-regulated autophagy and
mitochondria-mediated apoptosis were inhibited by
Tat-SabKIM1 pretreatment.
Several studies (31–33)
have reported that JNK translocates to the nucleus after its
activation and is involved in cellular functions via the
phosphorylation of transcription factors in the nucleus. However,
some studies (11,34–36)
have suggested that JNK not only serves a role in the nucleus, but
also in the localization and regulation in other parts of the cells
by binding to a specific protein, such as Sab (11). The carboxyl terminus of the Sab
protein harbors a kinase interaction motif (KIM), which is similar
to c-Jun, and JNK can bind to Sab through this motif (11,37).
Moreover, the carboxyl terminus of Sab also harbors four
serine/-proline residues, which can also be a site that is
phosphorylated by JNK (11).
Therefore, Sab can not only interact with JNK but also act as the
phosphorylated substrate of JNK (11). Several conformational studies
(8,11,26,34,35,37)
have reported that Sab was localized to the mitochondria, and that
a certain amount of JNK can translocate to the mitochondria by
binding to the organelle. The activation of JNK and its interaction
with mitochondria are involved in the regulation of mitochondrial
functions (12,34). The current experimental results also
suggested the occurrence of mitoJNK activation in ALI, which may
inhibit the energy production of mitochondria, decrease the
mitochondrial membrane potential and lead to mitochondrial
dysfunction. Therefore, inhibiting the translocation of JNK to the
mitochondria could be used to repair damage by protecting the
normal physiological function of the organelle. Previous studies
(8,34) have shown that inhibiting Sab
expression using gene silencing technology can maintain the normal
mitochondrial membrane potential and reduce the apoptosis of cells.
Moreover, Chambers et al (8,37)
revealed that the presence of KIM1 was an essential factor for the
binding and interaction between JNK and Sab. The synthesis of the
KIM1-specific binding peptide Tat-SabKIM1 may easily and
effectively block the interaction between Sab and JNK, thereby
inhibiting the localization of JNK to mitochondria (6,37). In
addition, previous studies (6,37)
demonstrated that Tat-SabKIM1 could successfully reach
the cytoplasm via the cell membrane, and its concentration was
stable; the concentration after 24 h in the cells could reach up to
90% of the initial concentration (37). Therefore, Tat-SabKIM1 may
be used to block the binding of Sab and JNK.
There are some limitations of the present study.
Firstly, as ARDS has numerous causes, further studies are required
to determine whether Tat-SabKIM1 is effective against
numerous causes of ARDS. Secondly, the protective mechanism
underlying blocking JNK-mitochondria interaction needs to be fully
studied.
In conclusion, as anticipated, during ALI/ARDS, the
activation of JNK can disrupt the normal physiological functions of
the mitochondria. This disorder leads to the accumulation of ROS
and increased cell apoptosis, which is a key event during ALI/ARDS.
Through selective inhibition of JNK and Sab binding using
Tat-SabKIM1, an effective and stable blocker of
JNK-mitochondria interaction, the normal function of mitochondria
can be maintained and the deterioration of from ALI/ARDS is
blocked.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 81800076, 81900083 and
81600053).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
CL and LB designed the study. LB and YL performed
the experiments. WL and FJ performed the statistical analysis and
drafted the manuscript. CL and LB confirm the authenticity of all
the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Musi CA, Agro G, Santarella F, Iervasi E
and Borsello T: JNK3 as therapeutic target and biomarker in
neurodegenerative and neurodevelopmental brain diseases.
Cells-Basel. 9:21902020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tan J, Gao W, Yang W, Zeng X, Wang L and
Cui X: Isoform-specific functions of c-Jun N-terminal kinase 1 and
2 in lung ischemia-reperfusion injury through the c-Jun/activator
protein-1 pathway. J Thorac Cardiovasc Surg. 2020. View Article : Google Scholar
|
|
3
|
Chen M, Sun J, Lu C, Chen X, Ba H, Lin Q,
Cai J and Dai J: The impact of neuronal Notch-1/JNK pathway on
intracerebral hemorrhage-induced neuronal injury of rat model.
Oncotarget. 7:73903–73911. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang H, Zhong L, Mi S, Song N, Zhang W and
Zhong M: Tanshinone IIA prevents platelet activation and
down-regulates CD36 and MKK4/JNK2 signaling pathway. BMC Cardiovasc
Disord. 20:812020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Desai S, Laskar S and Pandey BN: Autocrine
IL-8 and VEGF mediate epithelial-mesenchymal transition and
invasiveness via p38/JNK-ATF-2 signalling in A549 lung cancer
cells. Cell Signal. 25:1780–1791. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ngoei KR, Catimel B, Church N, Lio DS,
Dogovski C, Perugini MA, Watt PM, Cheng HC, Ng DC and Bogoyevitch
MA: Characterization of a novel JNK (c-Jun N-terminal kinase)
inhibitory peptide. Biochem J. 434:399–413. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Matsui Y, Kuwabara T, Eguchi T, Nakajima K
and Kondo M: Acetylation regulates the MKK4-JNK pathway in T cell
receptor signaling. Immunol Lett. 194:21–28. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chambers JW, Pachori A, Howard S, Iqbal S
and LoGrasso PV: Inhibition of JNK mitochondrial localization and
signaling is protective against ischemia/reperfusion injury in
rats. J Biol Chem. 288:4000–4011. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xu J, Qin X, Cai X, Yang L, Xing Y, Li J,
Zhang L, Tang Y, Liu J, Zhang X and Gao F: Mitochondrial JNK
activation triggers autophagy and apoptosis and aggravates
myocardial injury following ischemia/reperfusion. Biochim Biophys
Acta. 1852:262–270. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yabu T, Shiba H, Shibasaki Y, Nakanishi T,
Imamura S, Touhata K and Yamashita M: Stress-induced ceramide
generation and apoptosis via the phosphorylation and activation of
nSMase1 by JNK signaling. Cell Death Differ. 22:258–273. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wiltshire C, Matsushita M, Tsukada S,
Gillespie DA and May GH: A new c-Jun N-terminal kinase
(JNK)-interacting protein, Sab (SH3BP5), associates with
mitochondria. Biochem J. 367:577–585. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Win S, Than TA, Le BH, Garcia-Ruiz C,
Fernandez-Checa JC and Kaplowitz N: Sab (Sh3bp5) dependence of JNK
mediated inhibition of mitochondrial respiration in palmitic acid
induced hepatocyte lipotoxicity. J Hepatol. 62:1367–1374. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li Y, Zeng Z, Li Y, Huang W, Zhou M, Zhang
X and Jiang W: Angiotensin-converting enzyme inhibition attenuates
lipopolysaccharide-induced lung injury by regulating the balance
between angiotensin-converting enzyme and angiotensin-converting
enzyme 2 and inhibiting mitogen-activated protein kinase
activation. Shock. 43:395–404. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hui Z, Jie H and Fan GH: Expression of
DUSP12 reduces lung vascular endothelial cell damage in a murine
model of lipopolysaccharide-induced acute lung injury via the
apoptosis signal-Regulating Kinase 1 (ASK1)-Jun N-Terminal Kinase
activation (JNK) pathway. Med Sci Monit. 27:e9304292021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang ZK, Zhou Y, Cao J, Liu DY and Wan
LH: Rosmarinic acid ameliorates septic-associated mortality and
lung injury in mice via GRP78/IRE1alpha/JNK pathway. J Pharm
Pharmacol. Mar 16–2021.(Epub ahead of print). View Article : Google Scholar
|
|
16
|
Liu SH, Lu TH, Su CC, Lay IS, Lin HY, Fang
KM, Ho TJ, Chen KL, Su YC, Chiang WC and Chen YW: Lotus leaf
(Nelumbo nucifera) and its active constituents prevent inflammatory
responses in macrophages via JNK/NF-kappaB signaling pathway. Am J
Chin Med. 42:869–889. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu J, Wang Y, Li Z, Dong S, Wang D, Gong
L, Shi J, Zhang Y, Liu D and Mu R: Effect of Heme Oxygenase-1 on
Mitofusin-1 protein in LPS-induced ALI/ARDS in rats. Sci Rep.
6:365302016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Suresh K, Servinsky L, Reyes J, Baksh S,
Undem C, Caterina M, Pearse DB and Shimoda LA: Hydrogen
peroxide-induced calcium influx in lung microvascular endothelial
cells involves TRPV4. Am J Physiol Lung Cell Mol Physiol.
309:L1467–L1477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang K, Chen Y, Zhang P, Lin P, Xie N and
Wu M: Protective features of autophagy in pulmonary infection and
inflammatory diseases. Cells Basel. 8:1232019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jackson MV, Morrison TJ, Doherty DF,
McAuley DF, Matthay MA, Kissenpfennig A, O'Kane CM and
Krasnodembskaya AD: Mitochondrial transfer via tunneling nanotubes
is an important mechanism by which mesenchymal stem cells enhance
macrophage phagocytosis in the in vitro and in vivo models of ARDS.
Stem Cells. 34:2210–2223. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Islam MN, Das SR, Emin MT, Wei M, Sun L,
Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S and
Bhattacharya J: Mitochondrial transfer from bone-marrow-derived
stromal cells to pulmonary alveoli protects against acute lung
injury. Nat Med. 18:759–765. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gomzikova MO, James V and Rizvanov AA:
Mitochondria donation by mesenchymal stem cells: Current
understanding and mitochondria transplantation strategies. Front
Cell Dev Biol. 9:6533222021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang M, Gao Y, Zhao W, Yu G and Jin F:
ACE-2/ANG1-7 ameliorates ER stress-induced apoptosis in seawater
aspiration-induced acute lung injury. Am J Physiol Lung Cell Mol
Physiol. 315:L1015–L1027. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
25
|
Li C, Liu M, Bo L, Liu W, Liu Q, Chen X,
Xu D, Li Z and Jin F: NFAT5 participates in seawater
inhalationinduced acute lung injury via modulation of NF-kappaB
activity. Mol Med Rep. 14:5033–5040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanawa N, Shinohara M, Saberi B, Gaarde
WA, Han D and Kaplowitz N: Role of JNK translocation to
mitochondria leading to inhibition of mitochondria bioenergetics in
acetaminophen-induced liver injury. J Biol Chem. 283:13565–13577.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li X, Wang Y, Xiong Y, Wu J, Ding H, Chen
X, Lan L and Zhang H: Galangin induces autophagy via deacetylation
of LC3 by SIRT1 in HepG2 cells. Sci Rep. 6:304962016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xu HD and Qin ZH: Beclin 1, Bcl-2 and
autophagy. Adv Exp Med Biol. 1206:109–126. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan E, Brodie D and Slutsky AS: Acute
respiratory distress syndrome: Advances in diagnosis and treatment.
JAMA. 319:698–710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Farkhondeh T, Mehrpour O, Buhrmann C,
Pourbagher-Shahri AM, Shakibaei M and Samarghandian S:
Organophosphorus compounds and MAPK signaling pathways. Int J Mol
Sci. 21:42582020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han X, Liu C, Zhang K, Guo M, Shen Z, Liu
Y, Zuo Z, Cao M and Li Y: Calpain and JNK pathways participate in
isoflurane-induced nucleus translocation of apoptosis-inducing
factor in the brain of neonatal rats. Toxicol Lett. 285:60–73.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li Y, Xu M, Ding X, Yan C, Song Z, Chen L,
Jian Y, Tang G, Tang C, Di Y, et al: Protein kinase C controls
lysosome biogenesis independently of mTORC1. Nat Cell Biol.
18:1065–1077. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Win S, Than TA, Fernandez-Checa JC and
Kaplowitz N: JNK interaction with Sab mediates ER stress induced
inhibition of mitochondrial respiration and cell death. Cell Death
Dis. 5:e9892014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chambers JW, Howard S and LoGrasso PV:
Blocking c-Jun N-terminal kinase (JNK) translocation to the
mitochondria prevents 6-hydroxydopamine-induced toxicity in vitro
and in vivo. J Biol Chem. 288:1079–1087. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao Y and Herdegen T: Cerebral ischemia
provokes a profound exchange of activated JNK isoforms in brain
mitochondria. Mol Cell Neurosci. 41:186–195. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chambers JW, Cherry L, Laughlin JD,
Figuera-Losada M and Lograsso PV: Selective inhibition of
mitochondrial JNK signaling achieved using peptide mimicry of the
Sab kinase interacting motif-1 (KIM1). ACS Chem Biol. 6:808–818.
2011. View Article : Google Scholar : PubMed/NCBI
|