Introduction
Mitochondria are essential subcellular organelles
that are involved in respiration, oxidative phosphorylation and
apoptosis. Balanced cycles of fusion and fission are essential for
maintaining mitochondrial morphology and activity (1). An aberrant balance between fission and
fusion is associated with a number of neurodegenerative disorders
and cardiac diseases (2,3). For example, peroxisome
proliferator-activated receptor-γ coactivator-1α (PGC-1α)
expression, which is decreased in postmortem brains of patients
with Parkinson's disease, protected against neurotoxicity induced
by 1-methyl-4-phenyl-1,2,3,6-tetrahydrodropyridine (4). Fission is regulated by dynamin-related
protein 1 (DRP1), which can assemble into multimeric ring-like
structures and wrap around the constriction sites of dividing
mitochondria (5). Mitochondrial
outer membrane dynamin-like guanosine triphosphatases (GTPases)
mitofusin (MFN)-1 and −2 orchestrate outer mitochondrial membrane
fusion (6), while optic atrophy 1
(OPA1) is required for inner mitochondrial membrane fusion
(7). OPA1 is encoded by eight mRNA
splice forms, which are produced by differential splicing. Cleavage
of OPA1 is a key regulatory step for coordinating fusion and
fission of mitochondria (8). There
are two mitochondrial inner membrane-embedded AAA proteases
(i-AAA), metalloendopeptidase OMA1, mitochondrial (OMA1) and
ATP-dependent zinc metalloprotease YME1L1 (YME1L), which can
convert long forms (L-OPA1) into short forms (S-OPA1). The
i-AAA protease YME1L regulates OPA1 cleavage at site S2,
whereas OMA1 mediates mitochondrial fragmentation by transformation
of L-OPA1 into S-OPA1 at site S1 (9,10). The
balance between the two forms of OPA1 maintains normal
mitochondrial morphology; fusion involves L-OPA1, whereas S-OPA1 is
associated with fission (11,12).
Furthermore, OPA1 serves an important role in maintaining
mitochondrial inner structure and crista remodeling (13).
Hypoxia is associated with heart disease, stroke and
tumor microenvironment (14). It
has also been demonstrated that hypoxia induces mitochondrial
fragmentation (15). The HIG1
hypoxia inducible domain (HIGD) family of genes, whose
expression is induced during hypoxia, is conserved throughout
evolution (16). There are five
human HIGD genes, HIGD-1A, −1B, −1C, −2A and
−2B. HIGD-1A is regulated by hypoxia-inducible
factor-1 (HIF1) under hypoxic conditions. HIGD-1A is a
survival factor that contains two transmembrane domains oriented in
a ‘N-terminal outside-C-terminal outside and loop inside’
conformation (17). As a
mitochondrial inner membrane protein, HIGD-1A knockdown
decreases cytochrome c oxidase activity, leading to
increased mitochondrial fission and cell death in response to
hypoxia (18). Ameri et al
(19) reported that HIGD-1A
promotes tumor cell survival by regulating AMPK activity and levels
of cellular reactive oxygen species (ROS) in vivo. In
addition, HIGD-1A prevents OPA1 cleavage and is required for
the functional integrity of mitochondria (20). Both HIGD-1A and
HIGD-2A promote cell survival in a number of cell lines in
response to hypoxia, suggesting that the HIGD gene family
may be a potential anti-apoptosis factor (21). HIGD-1B and HIGD-1A
share 42.4% homology and their similarity is highest in the
transmembrane domain. However, the role of HIGD-1B in
hypoxia-induced mitochondrial fragmentation and cell apoptosis is
not clearly understood. In the present study, the biological
function of HIGD-1B in cardiomyocytes was characterized and
the underlying mechanisms were investigated. Knockdown or
overexpression experiments were performed to examine the effects of
HIGD-1B on OPA1 expression. The interaction between HIGD-1B and
OPA1 was measured via co-immunoprecipitation (Co-IP) assays. The
effect of HIGD-1B on cell viability was determined via Cell
Counting Kit-8 (CCK-8) and apoptosis assays.
Materials and methods
Cell culture
AC16 human cardiomyocyte and 293T embryonic kidney
cell lines were purchased from American Type Culture Collection and
cultured in DMEM supplemented with 10% fetal bovine serum (both
Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin at 37°C in a humidified incubator with 5%
CO2. Hypoxia experiments were performed using a hypoxia chamber
(Billups-Rothenberg, Inc.). AC16 cells were cultured under hypoxic
conditions of 1% oxygen (5% CO2 and 94% N2). Small interfering
(si)RNA-mediated knockdown of HIGD-1B was performed using
specific siRNAs targeting HIGD-1B (cat. no. SR324184;
OriGene Technologies, Inc.) and scrambled siRNAs (cat. no. SR30004;
OriGene Technologies, Inc.) as a negative control.
Plasmid constructs
For the pB513B-HIGD-1B-Flag constructs, the coding
sequence of HIGD-1B (GenBank accession no. NM_016438) was
inserted into the pB513B-Flag (cat. no. 5619; BioVector NTCC, Inc.)
expression vector. The coding sequence of OPA1 (GenBank accession
no. NM_015560) was inserted into the pB513B-myc expression vector
to generate the pB513B-OPA1-myc vector. The coding sequence of
HIGD-1B was inserted into the pGEX-4T-1 vector to generate
the pGEX-HIGD-1B-GST vector.
In vitro cell viability assay
Cell viability was determined using a CCK-8 assay
(Beijing Solarbio Science & Technology Co., Ltd.), which was
performed according to the manufacturer's instructions in 96-well
plates. Briefly, AC16 cells were seeded (5×103
cells/well) in a 96-well plate and cultured at 37°C for 48 h. Cells
were incubated with the CCK-8 reagent for 1 h. Relative viability
of cells was assessed at 450 nm absorbance. For crystal violet (CV)
staining assay, cells were fixed with 4% formaldehyde at room
temperature for 15 min, followed by staining with a crystal violet
solution (crystal violet 0.2%, ethanol 2%) at room temperature for
10 min, and then the colonies were photographed using a dissection
microscope. For trypan blue exclusion assay, 0.1 ml trypan blue
stock solution (cat. no. 15250061; Thermo Fisher Scientific, Inc.)
was added to 1 ml cells at room temperature for 5 min, and then the
numbers of blue stained-cells and the number of total cells were
counted under a light microscope at ×10 magnification.
Immunofluorescence
AC16 cells were divided into the following four
groups: i) control + normoxia group, cells were cultured under
normoxic conditions without any treatment; ii) control + hypoxia
group, cells were cultured under hypoxic conditions without any
treatment; iii) Vector + hypoxia group, cells were cultured under
hypoxic conditions and transfected with empty vector; and iv)
HIGD-1B-Flag + hypoxia group, cells were cultured under hypoxic
conditions and transfected with HIGD-1B-Flag vector. Subsequently,
AC16 cells (5×103) were cultured on glass coverslips at
37°C for 48 h, and stained with 100 nM MitoTracker red CMXRos at
37°C for 30 min, then fixed with 4% formaldehyde at 4°C for 30 min.
After washing twice with ice-cold PBS, the samples were blocked
with 1% BSA in PBS with Tween-20 (0.05%) at 4°C for 1 h. Then, the
cells were incubated overnight at 4°C with anti-HIGD-1B antibody
(1:500; cat. no. ab238867; Abcam). Next, the samples were washed
with PBS and incubated at room temperature for 1 h with goat
anti-rabbit IgG H&L (1:1,000; cat. no. ab96899; DyLight 488;
Abcam). In order to determine the morphology of the mitochondria,
the cells were classified into three types under a fluorescence
microscope (magnification, ×400): Tubular, intermediate and
fragmented. ‘Tubular’ referred to the cells that exhibited the most
interconnected mitochondria, while ‘fragmented’ referred to cells
that primarily contained spherical mitochondrial segments.
Western blot analysis
Total protein was extracted from cells using RIPA
buffer (Beijing Solarbio Science & Technology Co., Ltd.) in the
presence of protease inhibitor cocktail and protein phosphatase
inhibitor (both Thermo Fisher Scientific, Inc.). Protein
concentration was quantified using a BCA kit (Takara Bio, Inc.).
The proteins (30 µg) were separated via SDS-PAGE on 8% gel, and
then electroblotted onto a PVDF membrane. Following blocking with
5% blocking reagent (cat. no. SW3015; Beijing Solarbio Science
& Technology Co., Ltd.) at room temperature for 1 h, the
membranes were incubated with the primary antibodies in 5% BSA
overnight at 4°C. Subsequently, the membranes were incubated with
secondary antibodies at room temperature for 1 h. Proteins were
visualized using ECL reagent (cat. no. WBKLS0050; EMD Millipore).
The blots were detected using ImageLab software (version 3.0;
Bio-Rad Laboratories, Inc.).
Reagents and antibodies
The following primary antibodies were used: HIGD-1B
(1:2,000; cat. no. ab238867; Abcam), Bcl-2 (1:2,000; cat. no.
sc-7382; Santa Cruz Biotechnology, Inc.), Bax (1:2,000; cat. no.
5023S; Cell Signaling Technology, Inc.), tubulin (1:5,000; cat. no.
2146S; Cell Signaling Technology, Inc.), heat shock protein 60
(1:3,000; cat. no. 12165S; Cell Signaling Technology, Inc.),
translocase of outer mitochondrial membrane (Tomm)20 (1:2,000; cat.
no. ab186735; Abcam), proliferating cell nuclear antigen (1:2,500;
cat. no. ab29; Abcam), YME1L (1:1,000; cat. no. ab234744; Abcam),
OMA1 (1:3,000; cat. no. ab154949; Abcam), MFN1 (1:2,000; cat. no.
ab221661; Abcam), MFN2 (1:2,500; cat. no. ab205236; Abcam), OPA1
(1:1,000; cat. no. 67589S; Cell Signaling Technology, Inc.), DRP1
(1:2,000; cat. no. ab184247; Abcam), phosphorylated (p)-DRP1
(1:2,000; cat. no. ab193216; Abcam), Flag-tag (1:3,000; cat. no.
14793S; Cell Signaling Technology, Inc.), Myc-tag (1:3,000; cat.
no. 2276S; Cell Signaling Technology, Inc.) and glutathione
S-transferase (GST)-tag (1:3,000; cat. no. ab138491; Abcam).
Secondary antibodies included HRP-conjugated anti-rabbit IgG
(1:5,000; cat. no. 7074P2; Cell Signaling Technology, Inc.) and
HRP-conjugated anti-mouse IgG (1:5,000; cat. no. 7076P2; Cell
Signaling Technology, Inc.). Caspase-3 and −9 activity was measured
with Fluorescent Assay kits (cat. nos. C1168S and C1157; Beyotime
Institute of Biotechnology). Carbonyl cyanide m-chlorophenyl
hydrazone (CCCP; cat. no. C2759) was purchased from Sigma-Aldrich
(Merck KGaA). For CCCP treatment, 10–50 µM CCCP was added to the
culture medium at 37°C for 0, 10, 15 or 30 min before harvesting.
Analysis of mitochondrial membrane potential was performed using a
mitochondrial membrane potential assay kit (cat. no. 13296S; Cell
Signaling Technologies, Inc.) according to the manufacturer's
instructions.
Flow cytometry for apoptosis
assay
For apoptosis analysis, AC16 cells were harvested
and incubated with Annexin V (5 µl) and PI dye (5 µl) for 30 min in
the dark using an Annexin V-FITC Apoptosis Detection kit (Beijing
Solarbio Science & Technology Co., Ltd.) according to the
manufacturer's instructions. The stained cells were analyzed using
a flow cytometer (FACSCanto II; BD Bioscience). Data was analyzed
using FlowJo version 10 (FlowJo, LLC).
Transfection and immunoprecipitation
(IP)
Transfection experiments were performed using
Effectene Transfection Reagent (Qiagen China Co., Ltd.) according
to the manufacturer's instructions. For knockdown or overexpression
experiments, AC16 cells (2×104) were transfected with 20
nM siRNA-ctrl (5′-GACUAC UGGUCGUUGAACU-3′), 20 nM siRNA-HIGD-1B
(5′-UAC GAAAUGUGUUAAAUCCUACUUG-3′), 1 µg empty vector or 1 µg
pB513B-HIGD-1B-Flag vector at room temperature for 48 h. Cells were
collected 48 h after transfection. For Co-IP experiments, 293T
cells (2×106) were transfected with the aforementioned
plasmids. After 24 h, cells were harvested and lysed in IP lysis
buffer (Thermo Fisher Scientific, Inc.). The supernatant (1 ml) was
incubated overnight at 4°C with HIGD-1B antibody or anti-Flag M2
affinity gel (Sigma-Aldrich; Merck KGaA). The protein A/G-sepharose
(100 µl) was added to the samples for 4 h. Following centrifugation
(600 × g) at 4°C for 10 min, the pellets were washed three times
with 1 ml lysis buffer before resuspension in 2X Laemmli SDS-PAGE
buffer, and then western blot analysis was performed according to
standard western blotting procedures as described above.
In vitro pull-down assay
Recombinant GST-fused HIGD-1B protein was expressed
and purified from Escherichia coli BL21 (DE3). The
purification of OPA1-Myc was performed using a EZview red
anti-c-Myc affinity gel (Sigma-Aldrich; Merck KGaA) according to
the manufacturer's instructions. GST pull-down assays were
performed by incubating HIGD-1B-GST, immobilized on
glutathione-sepharose resin (BD Biosciences) with OPA1-Myc at 4°C
for 3 h. The beads were washed three times to remove unbound
protein, and the bound proteins were eluted, separated using
SDS-PAGE and detected using western blot analysis according to
standard western blotting procedures as described above.
Subcellular fractionation
analysis
Subcellular fractionation was performed using a
Subcellular Protein Fractionation kit for Cultured Cells (Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, cells were harvested in cytoplasmic
extraction buffer at 4°C for 10 min. Cell suspension was
centrifuged 500 × g at 4°C for 5 min, and the supernatant was
transferred to a clean pre-chilled tube on ice. Membrane extraction
buffer was added to the pellet at 4°C for 10 min to isolate the
membrane extract, followed by addition of nuclear extraction buffer
at 4°C for 30 min to isolate the chromatin-bound nuclear extract.
Fraction purity was assessed via western blotting using antibodies
against tubulin, PCNA and Tomm20, as aforementioned. The
subcellular localization of HIGD-1B was predicted using the UniProt
Knowledge Database (UniProt, http://www.uniprot.org).
Mitochondrial membrane potential
measurement
Tetramethylrhodamine methyl ester (TMRE)
fluorescence mitochondrial membrane potential was measured using a
TMRM Assay Kit (cat. no. M20036; Thermo Fisher Scientific, Inc.).
After transfection, AC16 cells (1×105) were incubated
with 100 nM TMRE for 30 min in the dark, and then cells were
visualized with a fluorescence microscope (magnification,
×200).
Statistical analysis
The data are presented as the mean ± SEM (n≥3).
Statistical significance was analyzed using SPSS software v22.0
(IBM Corp.). Comparison between two groups was performed using an
unpaired Student's t-test, while ANOVA followed by Tukey's post hoc
test was used to analyze data in >two groups. P<0.05 was
considered to indicate a statistically significant difference.
Results
Knockdown of HIGD-1B induces
mitochondrial fragmentation
In order to identify the role of HIGD-1B, it was
investigated whether HIGD-1B was induced by hypoxia.
HIGD-1B protein expression levels increased gradually from 0
to 12 h in AC16 cells under hypoxic conditions (Fig. 1A). According to the UniProt
analysis, HIGD-1B was predicted to be a mitochondrial protein;
thus, the subcellular localization of HIGD-1B was investigated in
AC16 cells. HIGD-1B co-localized with MitoTracker-stained
mitochondria (Fig. 1B).
Consistently, subcellular fractionation analysis showed the
presence of HIGD-1B in the mitochondria (Fig. 1C). Next, the morphology of
mitochondria was assessed by staining with MitotTacker.
Downregulation of HIGD-1B via transfection with siRNA
resulted in fragmented mitochondria in 88.2±7.5% of cells (Fig. 1D). The effect of HIGD-1B on AC16
cell viability was also determined. Silencing of HIGD-1B
resulted in decreased cell viability compared with that of control
cells following hypoxia (Fig. 1E).
Furthermore, crystal violet staining demonstrated that there were
fewer viable cells observed in the si-HIGD-1B group compared with
the si-Ctrl group under hypoxic conditions for 48 h (Fig. 1F). Taken together, these data
suggested that HIGD-1B knockdown affected mitochondrial fusion and
decreased viability in AC16 cells.
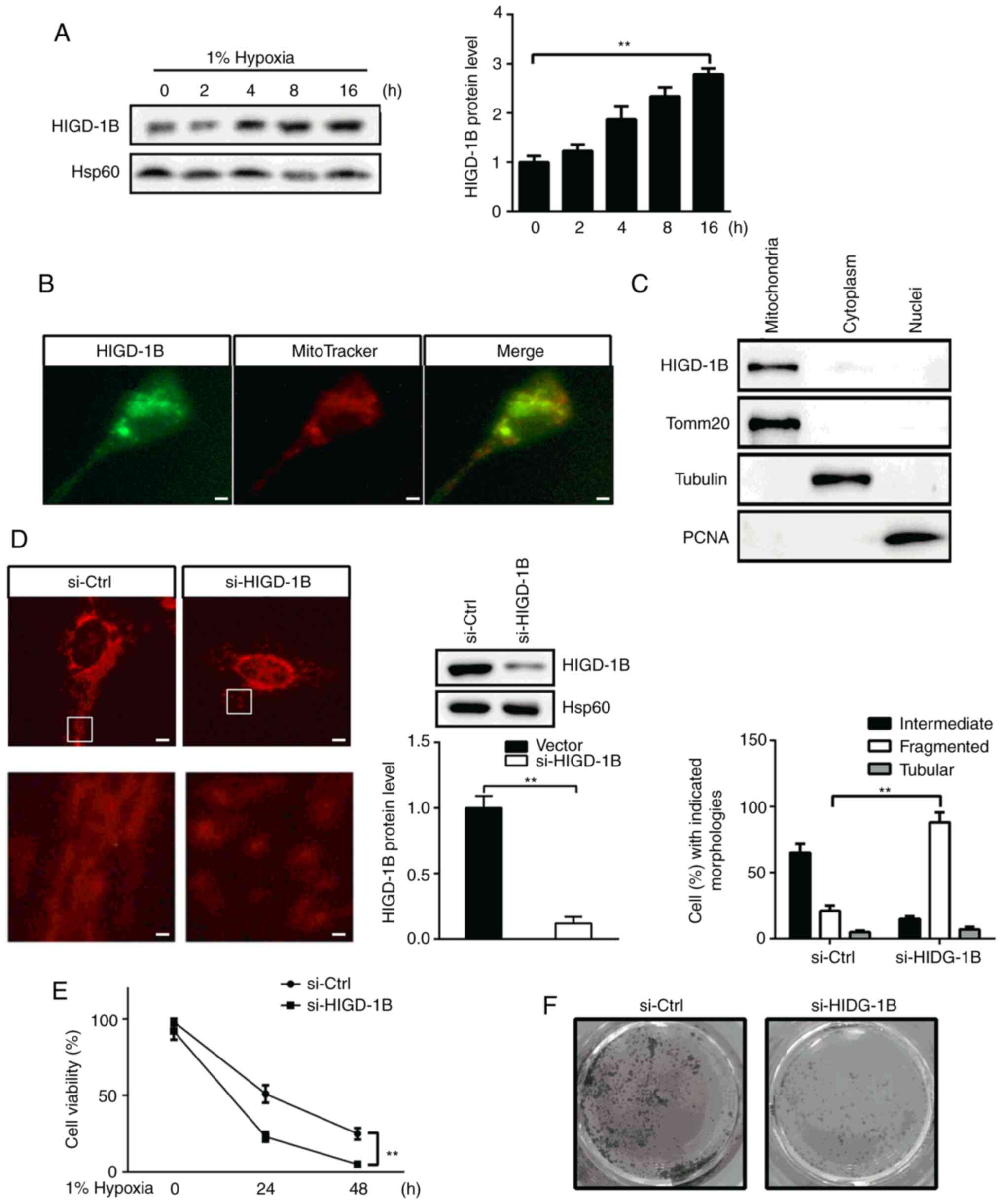 | Figure 1.Knockdown of HIGD-1B induces
mitochondrial fragmentation. (A) AC16 cells were cultured under
hypoxic conditions, then the protein expression levels of HIGD-1B
were measured using western blot analysis. (B) Representative
immunofluorescence image of anti-HIGD-1B antibody (green)-stained
AC16 cells. MitoTracker is a red fluorescent mitochondrial stain.
Scale bar, 10 µm. (C) Western blot analysis of HIGD-1B expression
levels in proteins extracted from the cytoplasm, mitochondria and
nucleus of AC16 cells under normoxic conditions. (D) AC16 cells
were transfected with si-Ctrl or si-HIGD-1B, then stained with
MitoTracker, fixed and mounted for microscopy. The illustration is
an enlarged view of the boxed area. In each group, >100 cells in
five fields of view were measured to determine the percentages of
the cells with tubular, intermediate and fragmented mitochondria.
Scale bar, 2 µm. (E) Cell Counting Kit-8 assay was used to evaluate
the viability of AC16 cells following transfection with si-Ctrl or
si-HIGD-1B under hypoxic conditions. (F) AC16 cells were
transfected with si-Ctrl or si-HIGD-1B, then cultured under hypoxic
conditions for 48 h. The cells were visualized using crystal violet
staining. The data are presented as the mean ± SEM. **P<0.01.
si, small interfering; Ctrl, control; HIGD-1B, HIG1 hypoxia
inducible domain family member 1B; Tomm20, translocase of outer
mitochondrial membrane 20; PCNA, proliferating cell nuclear
antigen. |
Silencing of HIGD-1B promotes
hypoxia-induced apoptosis
Next, the effect of HIGD-1B on hypoxia-induced
apoptosis was investigated. Annexin V/PI staining showed that
knockdown of HIGD-1B promoted hypoxia-induced apoptosis (Fig. 2A). Western blot analysis showed that
HIGD-1B knockdown increased the protein expression levels of Bcl-2
and decreased those of Bax under hypoxic conditions (Fig. 2B). Furthermore, HIGD-1B-knockdown
cells exhibited higher caspase-3 and −9 activity compared with that
control cells, suggesting that HIGD-1B regulated
mitochondrial-mediated apoptosis via the caspase signaling pathway
(Fig. 2C). Following hypoxia, AC16
cells exhibited decreased TMRE fluorescence compared with cells
treated under normoxic conditions. Silencing of HIGD-1B further
decreased the TMRE signal, indicating that HIGD-1B knockdown
decreased the mitochondrial membrane potential of AC16 cells under
hypoxic conditions (Fig. 2D). Taken
together, these data indicated that HIGD-1B silencing promoted
hypoxia-induced cell death by regulating caspase signaling in the
AC16 cells.
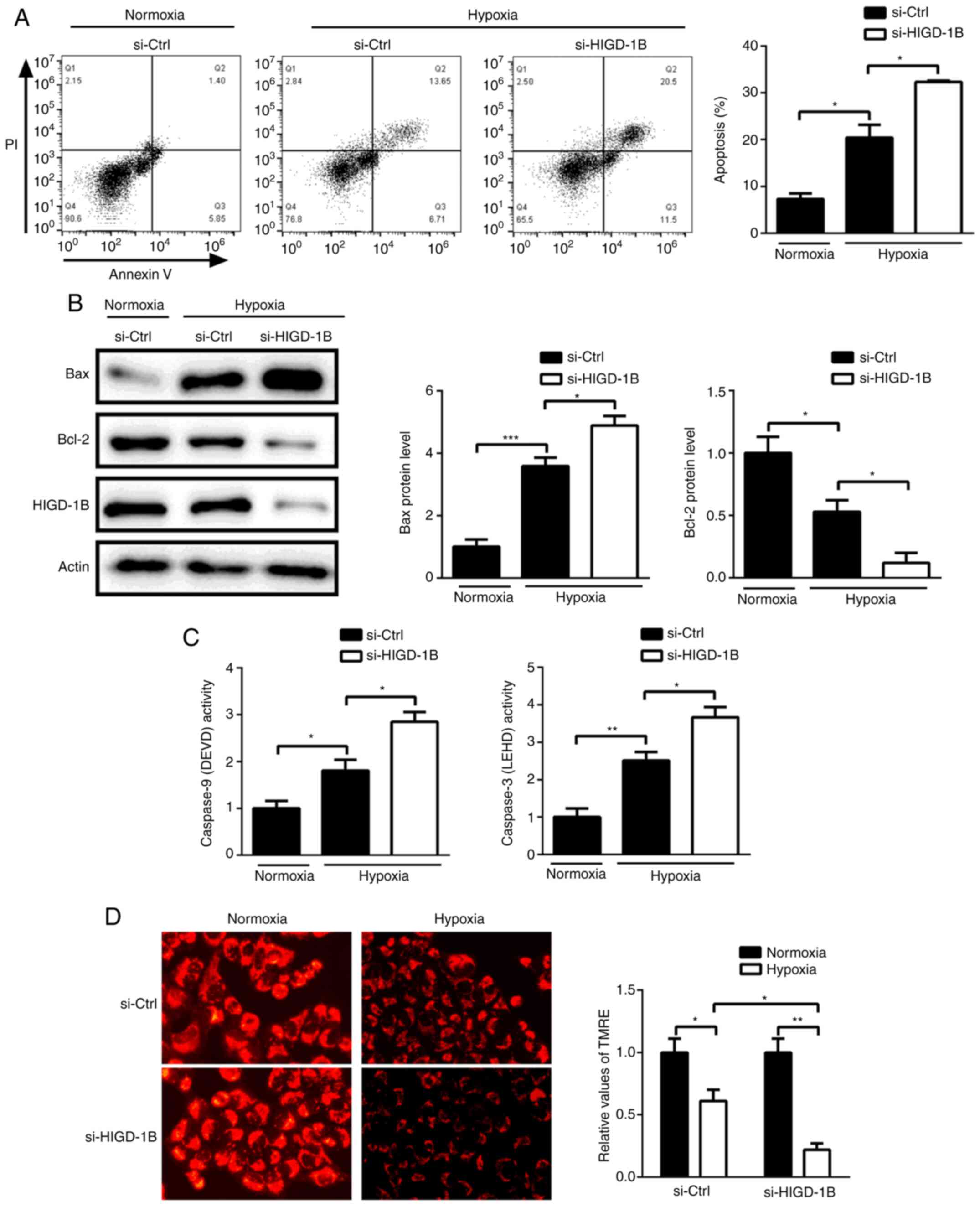 | Figure 2.Knockdown of HIGD-1B promotes
hypoxia-induced cell death. Following transfection with si-Ctrl or
si-HIGD-1B under normoxic or hypoxic conditions, (A) Annexin V/PI
staining and (B) detection of Bax, Bcl-2 and HIGD1B protein
expression levels were performed, and (C) caspase-3 and −9 activity
levels and (D) mitochondrial membrane potential were analyzed using
flow cytometry, western blot analysis, caspase assay and
tetramethylrhodamine ethyl ester perchlorate, respectively.
Caspase-9 activity was measured using luminogenic substrate of DEVD
and caspase-3 activity was measured using luminogenic substrate of
LEHD. Data are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001. si-, small interfering RNA, Ctrl,
control; HIGD-1B, HIG1 hypoxia inducible domain family member 1B;
DEVD, Ac-DEVD-AMC; LEHD, Z-LEHD-aminoluciferin; TMRE,
tetramethylrhodamine methyl ester. |
Silencing of HIGD-1B promotes
degradation of L-OPA1
The aforementioned results demonstrated that HIGD-1B
was required for optimal fusion of mitochondria; therefore, the
role of HIGD-1B in fusion- and fission-associated protein
expression levels was investigated. Protein expression levels of
OMA1, YME1L and outer mitochondrial membrane proteins MFN-1 and −2
were unchanged in HIGD-1B-knockdown cells, indicating that HIGD-1B
was not responsible for outer membrane fusion (Fig. 3A). OPA1 protein is produced by eight
different alternative splicing forms of mRNA, which are cleaved by
proteases (12). Therefore, five
different OPA1 bands (Fig. 3B) were
observed using western blot analysis. Notably, knockdown of HIGD-1B
resulted in a decrease in the expression levels of L-OPA1 and
increase in S-OPA1, whereas HIGD-1B overexpression increased the
expression levels of the larger band (Fig. 3B). In addition, activated DRP was
unchanged following knockdown or overexpression of HIGD-1B
(Fig. 3C). These results indicated
that mitochondrial fragmentation in HIGD-1B-knockdown cells could
change the isoform of OPA1.
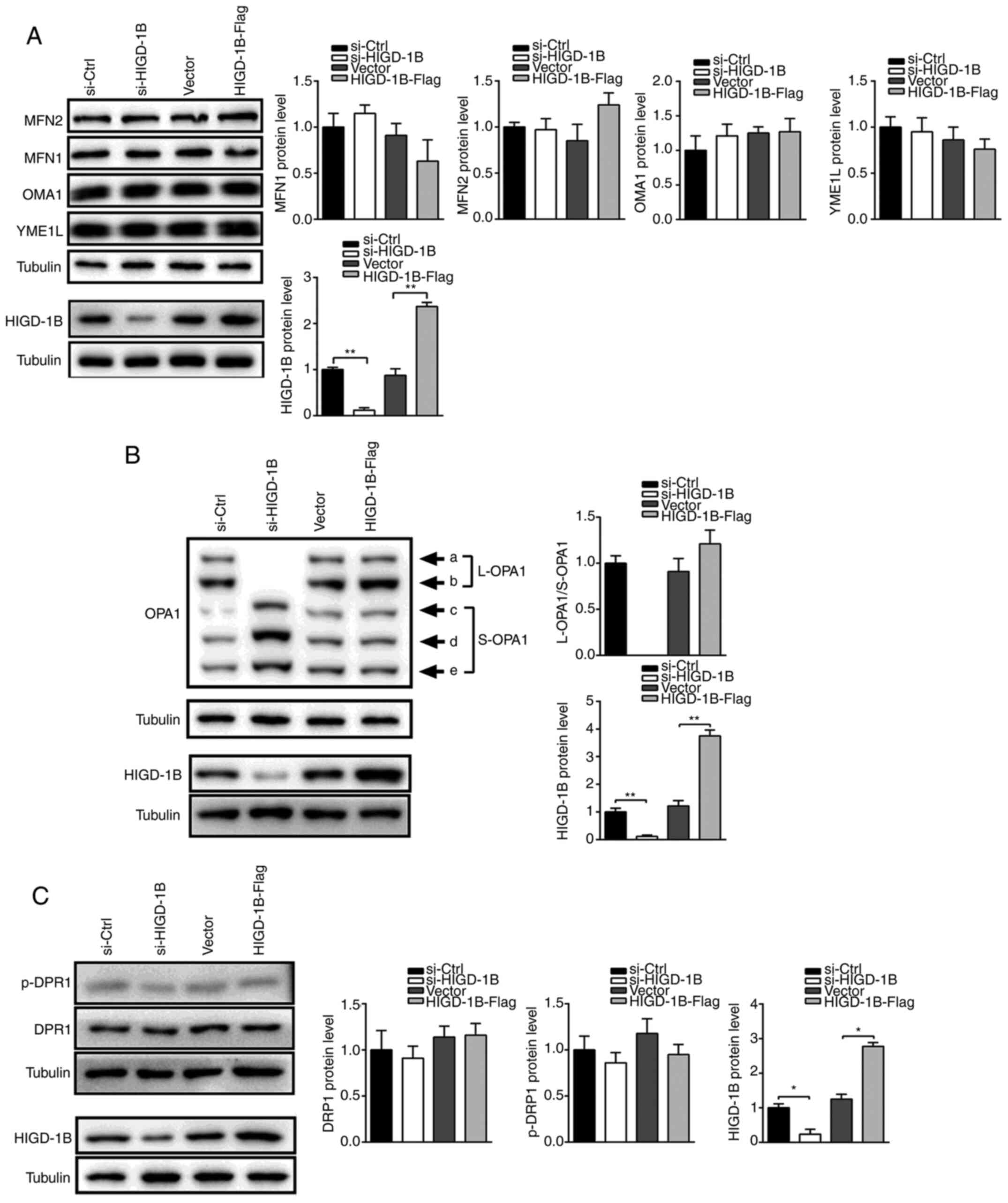 | Figure 3.HIGD-1B regulates OPA1 cleavage.
Western blot analysis was performed to evaluate the protein
expression levels of (A) HIGD-1B, MFN1, MFN2, OMA1 and YME1L.
Western blot analysis was performed to evaluate the protein
expression levels of (B) OPA1 and HIGD-1B. Western blot analysis
was performed to evaluate the protein expression levels of (C)
DRP1, p-DRP1 and HIGD-1B following transfection. The data are
presented as the mean ± SEM. *P<0.05, **P<0.01. p-,
phosphorylated; HIGD-1B, HIG1 hypoxia inducible domain family
member 1B; MFN, mitofusin; OMA1, metalloendopeptidase OMA1,
mitochondrial; YME1L, ATP-dependent zinc metalloprotease YME1L1;
OPA1, optic atrophy 1; DRP1, dynamin-related protein 1; L, long; S,
short; si-, small interfering RNA; Ctrl, control. |
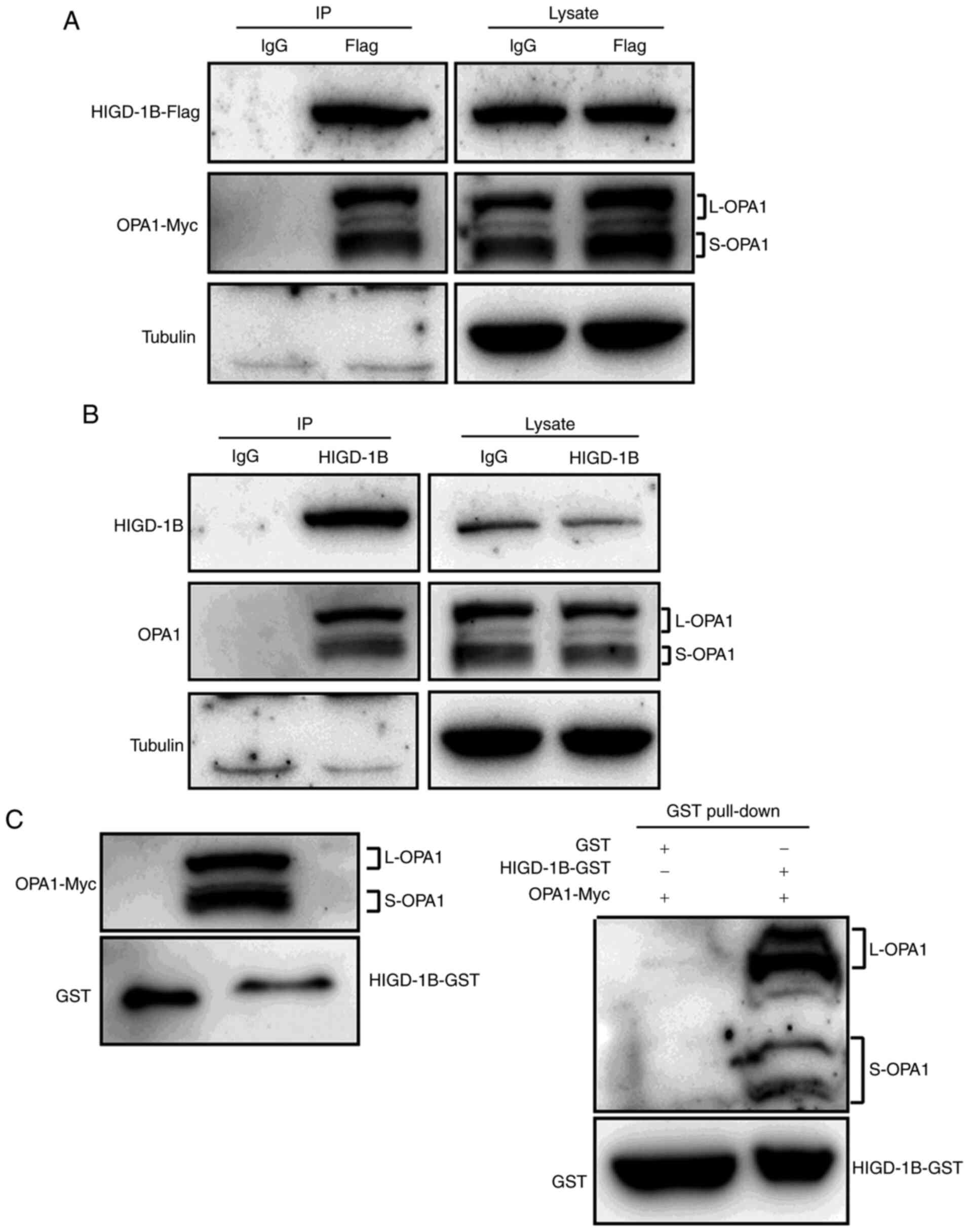
HIGD-1B physically interacts with
OPA1
In order to identify the molecular mechanism
underlying the association between HIGD-1B and OPA1, it was
determined whether HIGD-1B interacted with OPA1. Co-IP analysis
showed that exogenous HIGD-1B interacted with OPA1 in 293T cells
(Fig. 4A). In order to confirm this
result, endogenous IP assay was performed. OPA1 was present in
larger amounts in the immunoprecipitate with the anti-HIGD-1B
antibody compared with the control IgG group (Fig. 4B). The result was confirmed using an
in vitro pull-down assay. Myc-OPA1 was precipitated
specifically by recombinant GST-HIGD-1B, but not by GST-alone
(Fig. 4C). Taken together, these
results demonstrated that HIGD-1B directly interacted with OPA1 and
affected its cleavage.
HIGD-1B overexpression restricts the
cleavage of OPA1
In order to determine the function of HIGD-1B, it
was investigated whether overexpression of HIGD-1B inhibited
cleavage of OPA1. Under hypoxic conditions, the L-OPA1 isoform
gradually disappeared, whereas the S-OPA1 isoform accumulated.
Cleavage of the L-OPA1 bands was observed in cells transfected with
empty vector for 12 h, whereas overexpression of HIGD-1B delayed
cleavage for up to 24 h under hypoxic conditions (Fig. 5A). Treatment of AC16 cells with CCCP
also resulted in OPA1 cleavage, whereas HIGD-1B overexpression
stabilized the L-OPA1 isoform for up to 15 min (Fig. 5B). This indicated that HIGD-1B
delayed cleavage of OPA1 but did not restrict it completely.
HIGD-1B overexpression prevented hypoxia-induced mitochondrial
fragmentation (Fig. 5C).
Furthermore, HIGD-1B-Flag transfected cells exhibited improved
survival compared with control cells under hypoxic conditions
(Fig. 5D). Similarly, the number of
viable HIGD-1B-Flag transfected cells was 1-fold higher compared
with control cells following CCCP treatment (10–50 µM; Fig. 5E). Based on these findings, it was
concluded that HIGD-1B delayed the cleavage of OPA1 and stabilized
mitochondrial morphology, thus prolonging AC16 cell survival under
hypoxic conditions.
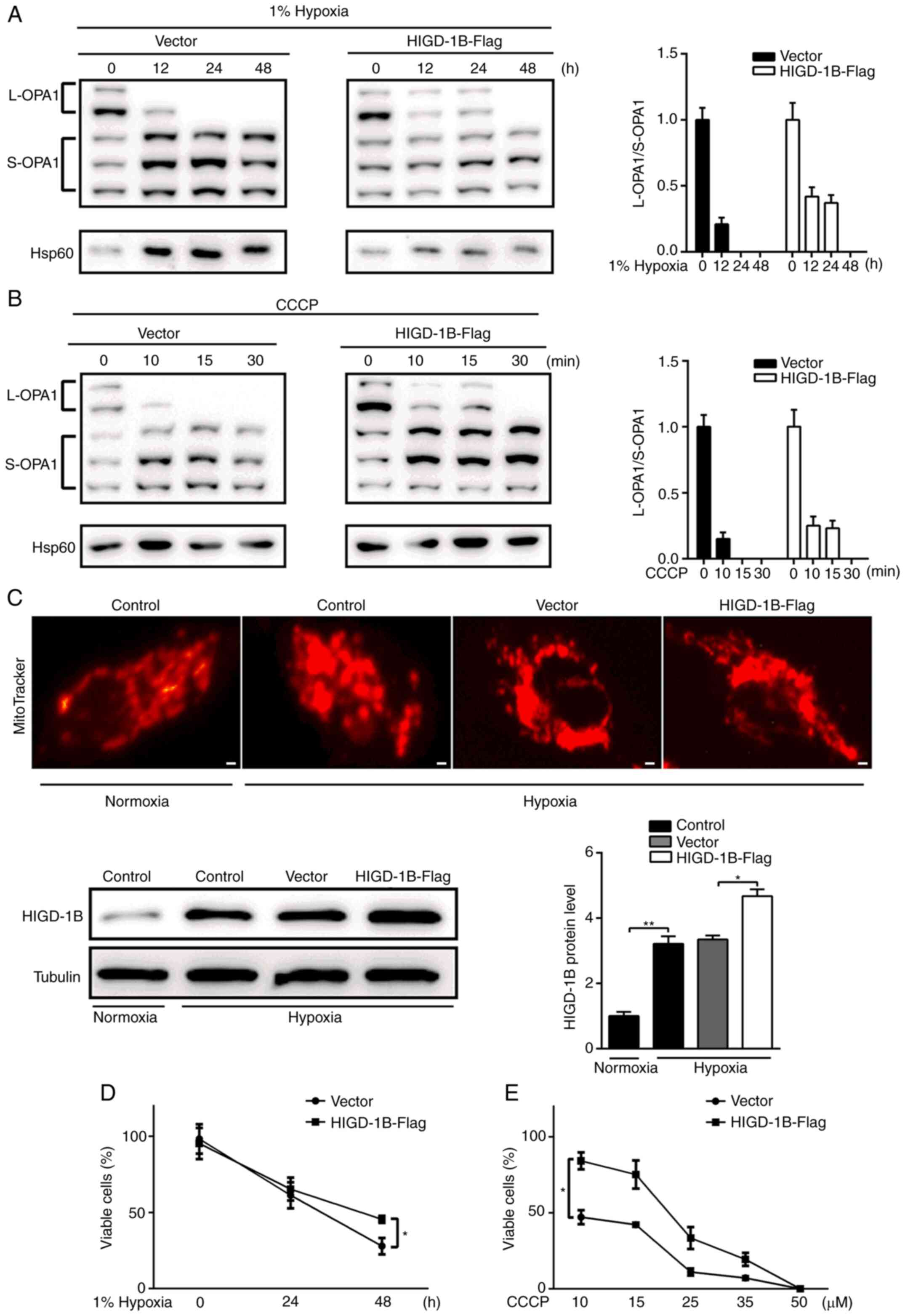 | Figure 5.HIGD-1B delays OPA1 cleavage. (A)
AC16 cells were transfected with empty or HIGD-1B-Flag vector, then
incubated under hypoxic conditions. (B) Transfected AC16 cells were
treated with CCCP. Total cell lysates were subjected to western
blot analysis using an anti-OPA1 and Hsp60 antibodies. (C) AC16
cells were transfected with HIGD-1B-Flag or empty vector, then
cultured under normoxic or hypoxic conditions. Western blotting was
performed to detect HIGD-1B expression in each group. The cells
were visualized under a fluorescent microscope following staining
with MitoTracker. Scale bar, 10 µm. (D) AC16 cells were transfected
with empty vector or HIGD-1B-Flag, then incubated under hypoxic
conditions. (E) Transfected AC16 cells were treated with 10–50 µM
CCCP for 1 h. Total viable cells were examined using trypan blue
exclusion. Data are presented as the mean ± SEM. *P<0.05,
**P<0.01. CCCP, carbonyl cyanide m-chlorophenyl hydrazone;
HIGD-1B, HIG1 hypoxia inducible domain family member 1B; OPA1,
optic atrophy 1; Hsp, heat shock protein; L, long; S, short. |
Discussion
OPA1 protein undergoes proteolytic processing, which
is key for mitochondrial fusion activity, cristae organization and
cell apoptosis (13,22). Cellular stress (such as hypoxia)
induces mitochondrial dysfunction, which activate sOPA1 cleavage,
resulting in increased conversion of L-OPA1 into S-OPA1 and
mitochondrial fragmentation (22).
Mitochondrial fusion is required for cardiomyocyte differentiation
and cardiac development, although it occurs infrequently. The key
role of OPA1 in normal cardiac function can be attributed to loss
of L-OPA1 (23,24). L-OPA1 is essential for maintaining
cardiac function, whereas accumulation of S-OPA1 and improper
fission are deleterious for the heart. In the present study, the
role of HIGD-1B in the mitochondria was identified; HIGD-1B
modulated mitochondrial fusion by preventing cleavage of OPA1.
Knockdown of HIGD-1B was found to enhance L-OPA1 cleavage into
S-OPA1. Therefore, mitochondrial fragmentation induced by HIGD-1B
knockdown may be caused by the accumulation of S-OPA1 in
cardiomyocytes. Hypoxia-induced mitochondrial network changes are
associated with processing of the OPA1 protein (25). For example, OPA1 is involved in
hypoxia-induced cardiomyocyte death by improving mitochondrial
quality control (26). Protein
expression of HIGD-1B was induced by hypoxia and overexpression of
HIGD-1B delayed cleavage of OPA1 induced by hypoxia or CCCP,
leading to decreased mitochondrial fragmentation and increased cell
viability. Accumulating evidence has revealed that different
stimuli regulate OPA1 processing by YME1L or OMA1, which allows
coordinated fusion and fission of mitochondria under various
physiological conditions (10).
Abnormal degradation of OMA1 and YME1L is involved in OPA1
dysfunction, OMA1 knockdown or YME1L overexpression can decrease
TNF-α-induced cell apoptosis in H9C2 cells (27). In addition, proteases, such as
presenilin-associated rhomboid-like protein mitochondrial and
mitochondrial-AAA, are involved in OPA1 cleavage (28,29).
Knockdown of YME1L activates OMA1, which increases conversion of
L-OPA1 into S-OPA1 and mitochondrial fragmentation (12,30).
Here, HIGD-1B did not affect the protein expression levels of YME1L
or OMA1, suggesting that HIGD-1B did not regulate the cleavage of
OPA1 via YME1L or OMA1, but by directly interacting with OPA1.
Furthermore, HIGD-1B was not responsible for outer membrane fusion
as HIGD-1B did not affect the protein expression levels of the
outer mitochondrial membrane proteins MFN-1 and −2. DRP1 is a
mitochondrial fission protein localized in the cytosol that is
recruited to mitochondria, and divides the outer and inner
membranes of the mitochondrion (31). Considering that HIGD-1B did not
alter the activity of DRP1, it was hypothesized that HIGD-1B is not
necessary for mitochondrial fission.
HIGD-1A and HIGD-2A both serve an anti-apoptotic
function in response to hypoxia; however, the regulatory mechanisms
involved are not identical. HIGD-2A is localized to the
mitochondrial network and the nucleus, whereas HIGD-1A is only
co-localized with mitochondria (32). HIGD-1A silencing has been found to
not alter mitochondria membrane potential or cellular ATP, whereas
knockdown of HIGD-2A in 293T cells can increase the mitochondrial
membrane potential (20,32). Unlike HIGD-1A, HIGD-2A is also
associated with OPA1; however, decreased HIGD-2A protein expression
has no effect on OPA1 cleavage (21,33).
Similar to HIGD-1A, HIGD-1B is primarily expressed in the brain,
heart and kidney (21). The results
from the present study showed that HIGD-1B co-localized with the
mitochondria and inhibited the cleavage of OPA1, suggesting that
its regulatory mechanism of mitochondrial fusion was similar to
HIGD-1A.
Bcl-2 family proteins are involved in
hypoxia-induced apoptosis. A previous report showed that inhibition
of Bcl-2 during hypoxia results in endothelial apoptosis, and this
is associated with the expression ratio of Bcl-2 and Bax (34). Furthermore, knockdown of Bax in mice
results in lower mortality when deprived of oxygen and
Bax−/− mice exhibit decreased apoptosis and caspase-3
activity following hypoxia-ischemia compared with wild-type mice
(35,36). In the present study, HIGD-1B may
have inhibited hypoxia-induced apoptosis by changing expression
levels of Bcl-2 family proteins: HIGD-1B-knockdown cells exhibited
significantly increased protein expression levels of Bax and
decreased expression levels of Bcl-2. Furthermore,
HIGD-1B-knockdown cells exhibited more caspase-3 and −9 activity.
Based on these observations, it was hypothesized that HIGD-1B
regulated the mitochondrial death pathway involving caspase. An
et al (20) reported that
knockdown of HIGD-1A in 293T cells does not induce apoptosis but
inhibits proliferation. The results from the present study revealed
that knockdown of HIGD-1B promoted apoptosis of cardiomyocytes
cells under hypoxic conditions. Furthermore, HIGD-1A was required
for optimal fusion of mitochondria in DRP1-silenced cells, whereas
HIGD-1B silencing did not alter DRP1 expression levels. This may be
due to different functions of the HIGD family in different cell
lines (37). Hypoxia is one of the
important hallmarks of tumor microenvironment (38). Thus, the role of HIGD family in
tumor cells warrants further study. Overexpression of HIGD-1B
delayed cleavage of OPA1, which contributed to inhibition of
hypoxia or CCCP-induced mitochondrial fragmentation and cell death.
In addition, the results from the present study demonstrated that
HIGD family proteins may have a wide effect in protecting
mitochondrial function. A previous study indicated that HIGD-1A in
the mitochondria directly binds to γ-secretase components and
attenuates hypoxia-induced γ-secretase activation on the
mitochondrial membrane, thus mitigating hypoxia-induced
mitochondrial dysfunction (32).
HIGD-1A is induced by hypoxia in a HIF-1-dependent manner.
Similarly, ROS accumulation in HepG2 cells promotes HIGD-1A
expression levels by upregulating HIF-1α and PGC-1α expression
levels under high-fat exposure (39). HIGD-1A inhibits the
pERK/p27KIP1/retinoblastoma protein signaling pathway, which leads
to cell cycle arrest (40).
Collectively, HIGD isomers serve vital roles against cell death in
response to stress. Given the important role of HIGD-1B in the cell
hypoxia response and maintenance of mitochondrial integrity,
understanding the protein interaction network underlying HIGD-1B
expression may allow identification of which proteins participate
in hypoxia-induced apoptosis and mitochondrial fragmentation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Nature Science Foundation of China (grant nos. 81260522 and
81673891).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YP, ZW, JuL, MT, HL and JiL performed data analysis
and interpretation and wrote the manuscript. ZZ and ZX designed the
present study and performed the literature review. YP, ZW and JiL
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hoppins S, Lackner L and Nunnari J: The
machines that divide and fuse mitochondria. Annu Rev Biochem.
76:751–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burté F, Carelli V, Chinnery PF and
Yu-Wai-Man P: Disturbed mitochondrial dynamics and
neurodegenerative disorders. Nat Rev Neurol. 11:11–24. 2015.
View Article : Google Scholar
|
|
3
|
Dorn GW II and Kitsis RN: The
mitochondrial dynamism-mitophagy-cell death interactome: Multiple
roles performed by members of a mitochondrial molecular ensemble.
Circ Res. 116:167–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bose A and Beal MF: Mitochondrial
dysfunction in Parkinson's disease. J Neurochem. 139 (Suppl
1):216–231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Smirnova E, Griparic L, Shurland DL and
van der Bliek AM: Dynamin-related protein Drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Koshiba T, Detmer SA, Kaiser JT, Chen H,
McCaffery JM and Chan DC: Structural basis of mitochondrial
tethering by mitofusin complexes. Science. 305:858–862. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cipolat S, Martins de Brito O, Dal Zilio B
and Scorrano L: OPA1 requires mitofusin 1 to promote mitochondrial
fusion. Proc Natl Acad Sci USA. 101:15927–15932. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Roy M, Reddy PH, Iijima M and Sesaki H:
Mitochondrial division and fusion in metabolism. Curr Opin Cell
Biol. 33:111–118. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Head B, Griparic L, Amiri M, Gandre-Babbe
S and van der Bliek AM: Inducible proteolytic inactivation of OPA1
mediated by the OMA1 protease in mammalian cells. J Cell Biol.
187:959–966. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rainbolt TK, Lebeau J, Puchades C and
Wiseman RL: Reciprocal degradation of YME1L and OMA1 adapts
mitochondrial proteolytic activity during stress. Cell Rep.
14:2041–2049. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tondera D, Grandemange S, Jourdain A,
Karbowski M, Mattenberger Y, Herzig S, Da Cruz S, Clerc P, Raschke
I, Merkwirth C, et al: SLP-2 is required for stress-induced
mitochondrial hyperfusion. EMBO J. 28:1589–1600. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Anand R, Wai T, Baker MJ, Kladt N, Schauss
AC, Rugarli E and Langer T: The i-AAA protease YME1L and OMA1
cleave OPA1 to balance mitochondrial fusion and fission. J Cell
Biol. 204:919–929. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Olichon A, Baricault L, Gas N, Guillou E,
Valette A, Belenguer P and Lenaers G: Loss of OPA1 perturbates the
mitochondrial inner membrane structure and integrity, leading to
cytochrome c release and apoptosis. J Biol Chem. 278:7743–7746.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Semenza GL: Oxygen sensing,
hypoxia-inducible factors, and disease pathophysiology. Annu Rev
Pathol. 9:47–71. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parra V, Bravo-Sagua R, Norambuena-Soto I,
Hernández-Fuentes CP, Gómez-Contreras AG, Verdejo HE, Mellado R,
Chiong M, Lavandero S and Castro PF: Inhibition of mitochondrial
fission prevents hypoxia-induced metabolic shift and cellular
proliferation of pulmonary arterial smooth muscle cells. Biochim
Biophys Acta Mol Basis Dis. 1863:2891–2903. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bedo G, Vargas M, Ferreiro MJ, Chalar C
and Agrati D: Characterization of hypoxia induced gene 1:
Expression during rat central nervous system maturation and
evidence of antisense RNA expression. Int J Dev Biol. 49:431–436.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang J, Cao Y, Chen Y, Chen Y, Gardner P
and Steiner DF: Pancreatic beta cells lack a low glucose and
O2-inducible mitochondrial protein that augments cell survival.
Proc Natl Acad Sci USA. 103:10636–10641. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hayashi T, Asano Y, Shintani Y, Aoyama H,
Kioka H, Tsukamoto O, Hikita M, Shinzawa-Itoh K, Takafuji K, Higo
S, et al: Higd1a is a positive regulator of cytochrome c oxidase.
Proc Natl Acad Sci USA. 112:1553–1558. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ameri K, Jahangiri A, Rajah AM, Tormos KV,
Nagarajan R, Pekmezci M, Nguyen V, Wheeler ML, Murphy MP, Sanders
TA, et al: HIGD1A regulates oxygen consumption, ROS production, and
AMPK activity during glucose deprivation to modulate cell survival
and tumor growth. Cell Rep. 10:891–899. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
An HJ, Cho G, Lee JO, Paik SG, Kim YS and
Lee H: Higd-1a interacts with Opa1 and is required for the
morphological and functional integrity of mitochondria. Proc Natl
Acad Sci USA. 110:13014–13019. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
An HJ, Shin H, Jo SG, Kim YJ, Lee JO, Paik
SG and Lee H: The survival effect of mitochondrial Higd-1a is
associated with suppression of cytochrome C release and prevention
of caspase activation. Biochim Biophys Acta. 1813:2088–2098. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ishihara N, Fujita Y, Oka T and Mihara K:
Regulation of mitochondrial morphology through proteolytic cleavage
of OPA1. EMBO J. 25:2966–2977. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Y, Liu Y and Dorn GW II:
Mitochondrial fusion is essential for organelle function and
cardiac homeostasis. Circ Res. 109:1327–1331. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Papanicolaou KN, Khairallah RJ, Ngoh GA,
Chikando A, Luptak I, O'Shea KM, Riley DD, Lugus JJ, Colucci WS,
Lederer WJ, et al: Mitofusin-2 maintains mitochondrial structure
and contributes to stress-induced permeability transition in
cardiac myocytes. Mol Cell Biol. 31:1309–1328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Baburamani AA, Hurling C, Stolp H, Sobotka
K, Gressens P, Hagberg H and Thornton C: Mitochondrial optic
atrophy (OPA) 1 processing is altered in response to neonatal
hypoxic-ischemic brain injury. Int J Mol Sci. 16:22509–22526. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xin T, Lv W, Liu D, Jing Y and Hu F: Opa1
reduces hypoxia-induced cardiomyocyte death by improving
mitochondrial quality control. Front Cell Dev Biol. 8:8532020.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu B, Li J, Ni H, Zhuang X, Qi Z, Chen Q,
Wen Z, Shi H, Luo X and Jin B: TLR4 activation promotes the
progression of experimental autoimmune myocarditis to dilated
cardiomyopathy by inducing mitochondrial dynamic imbalance. Oxid
Med Cell Longev. 2018:31812782018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ehses S, Raschke I, Mancuso G, Bernacchia
A, Geimer S, Tondera D, Martinou JC, Westermann B, Rugarli EI and
Langer T: Regulation of OPA1 processing and mitochondrial fusion by
m-AAA protease isoenzymes and OMA1. J Cell Biol. 187:1023–1036.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cipolat S, Rudka T, Hartmann D, Costa V,
Serneels L, Craessaerts K, Metzger K, Frezza C, Annaert W, D'Adamio
L, et al: Mitochondrial rhomboid PARL regulates cytochrome c
release during apoptosis via OPA1-dependent cristae remodeling.
Cell. 126:163–175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang K, Li H and Song Z: Membrane
depolarization activates the mitochondrial protease OMA1 by
stimulating self-cleavage. EMBO Rep. 15:576–585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mears JA, Lackner LL, Fang S, Ingerman E,
Nunnari J and Hinshaw JE: Conformational changes in Dnm1 support a
contractile mechanism for mitochondrial fission. Nat Struct Mol
Biol. 18:20–26. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hayashi H, Nakagami H, Takeichi M,
Shimamura M, Koibuchi N, Oiki E, Sato N, Koriyama H, Mori M,
Gerardo Araujo R, et al: HIG1, a novel regulator of mitochondrial
γ-secretase, maintains normal mitochondrial function. FASEB J.
26:2306–2317. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Salazar C, Elorza AA, Cofre G,
Ruiz-Hincapie P, Shirihai O and Ruiz LM: The OXPHOS supercomplex
assembly factor HIG2A responds to changes in energetic metabolism
and cell cycle. J Cell Physiol. 234:17405–17419. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matsushita H, Morishita R, Nata T, Aoki M,
Nakagami H, Taniyama Y, Yamamoto K, Higaki J, Yasufumi K and
Ogihara T: Hypoxia-induced endothelial apoptosis through nuclear
factor-kappaB (NF-kappaB)-mediated bcl-2 suppression: In vivo
evidence of the importance of NF-kappaB in endothelial cell
regulation. Circ Res. 86:974–981. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
McClintock DS, Santore MT, Lee VY,
Brunelle J, Budinger GR, Zong WX, Thompson CB, Hay N and Chandel
NS: Bcl-2 family members and functional electron transport chain
regulate oxygen deprivation-induced cell death. Mol Cell Biol.
22:94–104. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gibson ME, Han BH, Choi J, Knudson CM,
Korsmeyer SJ, Parsadanian M and Holtzman DM: BAX contributes to
apoptotic-like death following neonatal hypoxia-ischemia: Evidence
for distinct apoptosis pathways. Mol Med. 7:644–655. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Timón-Gómez A, Bartley-Dier EL, Fontanesi
F and Barrientos A: HIGD-driven regulation of cytochrome c oxidase
biogenesis and function. Cells. 9:92020. View Article : Google Scholar
|
|
38
|
Francis A, Venkatesh GH, Zaarour RF,
Zeinelabdin NA, Nawafleh HH, Prasad P, Buart S, Terry S and Chouaib
S: Tumor hypoxia: A key determinant of microenvironment hostility
and a major checkpoint during the antitumor response. Crit Rev
Immunol. 38:505–524. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li T, Xian WJ, Gao Y, Jiang S, Yu QH,
Zheng QC and Zhang Y: Higd1a protects cells from lipotoxicity under
High-Fat Exposure. Oxid Med Cell Longev.
2019:60512622019.PubMed/NCBI
|
|
40
|
An HJ, Ryu M, Jeong HJ, Kang M, Jeon HM,
Lee JO, Kim YS and Lee H: Higd-1a regulates the proliferation of
pancreatic cancer cells through a pERK/p27KIP1/pRB pathway. Cancer
Lett. 461:78–89. 2019. View Article : Google Scholar : PubMed/NCBI
|