Introduction
Diabetic kidney disease (DKD) is a chronic renal
condition and the most common cause of end-stage renal disease
(ESRD) worldwide (1). Controlling
blood glucose and blood pressure levels can appropriately delay the
onset of ESRD, and several hyperglycemic drugs with renal
protection have been reported, including sodium-glucose
co-transporter-2 inhibitors and glucagon-like peptide 1 receptor
agonists (2). However, due to the
high morbidity and complicated pathogenesis of CKD, further studies
on novel drugs and their mechanisms of action are required to
prevent the progression of DKD.
Epithelial-mesenchymal transition (EMT) is a process
where epithelial cells acquire mesenchymal properties, as
characterized by increased expression of vimentin and N-cadherin,
and decreased expression of specific epithelial cell markers, such
as nephrin, zonula occludens-1 (ZO-1) and E-cadherin. Glomerular
podocytes are essential for the correct function of the glomerular
filtration barrier. Podocyte injury and loss contribute to the
progression of DKD (3). Previous
studies have proposed that podocytes undergo EMT stimulated by high
glucose (4,5), transforming growth factor-β (TGF-β)
(6–9), adriamycin (8) and homocysteine (10), leading to podocyte detachment or
dysfunction, which ultimately results in glomerular filtration
dysfunction. Numerous studies have reported that podocytes are
phenotypically altered in the early stages of a rodent model of DKD
induced by high glucose and TGF-β, with increased mesenchymal
markers (desmin) and decreased epithelial markers (nephrin)
(5,7,9). In
addition to the findings in animal studies, a marked reduction in
nephrin and ZO-1 expression has been observed in the glomeruli of
patients with DKD (5,11–12).
Furthermore, podocyte EMT has been reported to participate in the
loss of podocytes in CKD through induction of podocyte detachment
or apoptosis (11). Accumulating
evidence suggests that EMT may be a potential pathway that
contributes to the progression of DKD. Therefore, it may be
utilized as a novel route and potential drug target for therapeutic
interventions in DKD.
Autophagy is a highly regulated lysosomal pathway
involved in cytoplasm recycling, and removal of excess or damaged
organelles. It is essential for cell survival, differentiation,
development and homeostasis. Changes in autophagy are detected by
microtubule-associated protein 1 light chain 3 (LC3) and
sequestosome 1 (SQSTM1/P62). LC3 is located on the autophagosome
membrane and consists of cytoplasmic LC3 I and membrane-bound LC3
II (13). Previous studies have
reported that autophagy is a protective mechanism of podocytes, and
autophagy dysfunction is the major risk factor for podocyte injury,
including apoptosis (14) and EMT
(15).
Twist1 regulates the occurrence of cellular EMT by
controlling the transcription of EMT-associated genes through
promoter activation or repression; it downregulates epithelial
phenotype-related genes, such as E-cadherin, and upregulates
mesenchymal cell phenotype-related genes, such as vimentin
(16). Twist1 has been shown to
promote EMT in endometrial and liver cancer through vimentin
regulation (17,18), indicating that it is a key gene in
EMT and cancer. While the specific role of Twist1 in DKD remains
unclear, Qiang and He (19)
reported that autophagy deficiency may inhibit the degradation of
Twist1 through SQSTM1/p62 accumulation to promote cellular EMT,
thus proposing a novel mechanism for the regulation of autophagy
and EMT.
Tripterygium glycoside (TG) is a fat-soluble mixture
(composed of diterpene lactone, alkaloid and triterpenoid)
extracted from the root xylem of Tripterygium wilfordii Hook
F, which has anti-inflammatory, immunosuppressive, immunomodulatory
and anti-tumor effects (20). TG
has been reported to markedly attenuate renal injury and to
regulate immune-inflammatory responses in DKD animal models
(21,22). The present study aimed to explore
the effect of TG on podocyte autophagy, apoptosis and EMT. It was
hypothesized that TG could restore autophagy to alleviate EMT and
apoptosis via the mTOR/Twist1 signaling pathway, resulting in the
improvement of DKD.
Materials and methods
Patient selection and renal
biopsies
All patients with DKD were diagnosed based on renal
biopsies carried out at the Department of Nephrology, Zhejiang
Provincial People's Hospital (Hangzhou, China). The patients were
selected using the Mayo Clinic/Renal Pathology Society Consensus
Report on Pathological Classification, Diagnosis, and Reporting of
GN (23). The demographic and
clinical data of these patients, including age, systolic blood
pressure (SBP), blood urea nitrogen (BUN), serum creatinine (Scr),
fasting plasma glucose (FPG), glycosylated hemoglobin A1c (HbA1c),
low-density lipoprotein cholesterol (LDL-C) and high-density
lipoprotein cholesterol (HDL-C) were obtained. A total of 20
patients with DKD (11 male patients, nine female patients; age
range, 36–87 years) and 10 normal human controls (six men, four
women; age range, 22–44 years) were enrolled in the study. All
protocols concerning the use of patient samples in the present
study were approved by the Human Subjects Committee of Zhejiang
Provincial People's Hospital. Written informed consent was obtained
from all donors.
Animals
A total of 16 mice, including male spontaneous
diabetic nephropathy mice (C57BL/KsJ db/db; n=12; age, 8 weeks;
weighing, 16–20 g) and male healthy control mice (C57BL/KsJ db/m;
n=4; age, 8 weeks; weighing, 16–20 g) were provided by Changzhou
Cavans Laboratory Animal Co., Ltd., [license no. SCX (Su)
2016-0010]. The mice were housed in a pathogen-free facility under
a 12-h light/dark cycle, with 50–65% humidity at 22–25°C. Mice were
supplied with continuous access to drinking water and a normal
diet, and were observed weekly. At 13 weeks of age, blood samples
were collected from the tail vein of mice (total volume collected,
1.5 ml; volume collected from each mouse, ~100 µl), and the blood
samples were coagulated for 20–30 min before being centrifuged at
2,000 × g for 20 min at 4°C. The serum was removed from the
centrifuged samples and was stored at −20°C until use. All sera
used were thawed and heat-inactivated at 56°C before being used in
subsequent cell culture experiments (24). At the end of the experimental
period, the mice (n=16) were sacrificed by cervical dislocation.
Death was confirmed by checking whether the heart and respiration
of the mice had stopped completely and the pupils were dilated. The
experiments were carried out in strict accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (25). All protocols
involving animals were approved by the Institutional Animal Care
and Use Committee of Zhejiang Provincial People's Hospital.
Cell culture
The mouse podocyte MPC5 cell line was obtained from
the Shanghai Institutes for Biological Sciences, Chinese Academy of
Sciences. Podocytes were cultured and expanded at 33°C in RPMI-1640
medium (HyClone; Cytiva) supplemented with 10% FBS (cat. no.
SH30084.03; HyClone; Cytiva), a low concentration of glucose (11 nM
D-glucose) and 20 U/ml γ-IFN (cat. no. CSB-E04578m; Cusabio
Technology LLC). Podocyte MPC5 cells were aspirated and passaged at
a ratio of 1:4. Podocytes were cultured at 37°C and 5% CO2 for
maturation. Before podocytes were passaged, type IV collagen (1.5
ml/25 cm2; cat. no. CSB-E08884m; Cusabio Technology LLC)
was placed in the culture flask and cells were incubated for 1 h.
After washing the bottom of the culture flask with PBS (3 ml/25
cm2), the cells were aspirated and passaged at a ratio
of 1:2. To induce differentiation, MPC5 cells were grown under
restrictive conditions at 37°C for 10 days, then MPC5 cells were
treated simultaneously for 24 h with 0.2% FBS in 5 mM D-glucose
RPMI-1640. Mature and differentiated podocytes were collected and
seeded uniformly in a 6-well plate, and the concentration of
podocytes was adjusted to 1×106 cells/ml. A total of 0.5
ml podocyte suspension and 1.5 ml RPMI-1640 medium containing 10%
FBS were added to each well (6-well plate) and were cultured at
37°C in a 5% CO2 incubator. MPC5 cells were treated with 10% DKD
mice serum (C57BLKS/J db/db) or 10% control mice serum (C57BLKS/J
db/m) at 37°C for 24 h.
Drugs
TG (cat. no. 14002219121) was purchased from
Zhejiang DND Pharmaceutical Co., Ltd. Podocytes were treated with
TG (1.25 µg/ml) at 37°C for 72 h. The effective concentration of TG
was verified in our preliminary experiments (26). 3-benzyl-5-((2-nitrophenoxy)
methyl)-dihydrofuran-2(3H)-one (3BDO; cat. no. S8317) was purchased
from Selleck Chemicals. Podocytes were treated with 3BDO (60 µM) at
37°C for 24 h (27).
ELISA
Levels of p62, Twist1 and E-cadherin in human serum
samples were detected using p62 ELISA kit (cat. no. NBP2-61300;
Novus Biologicals, LLC), Twist1 ELISA kit (cat. no. NBP3-06809;
Novus Biologicals, LLC) and E-cadherin ELISA kit (cat. no. KA0433;
Abnova), respectively. Serum N-cadherin and vimentin levels were
detected using N-cadherin ELISA kit (cat. no. CSB-E09718h; Cusabio
Technology LLC) and vimentin ELISA kit (cat. no. CSB-E08982h;
Cusabio Technology LLC). All ELISA kits were used according to the
manufacturer's protocols.
Urine protein content
determination
Mice (n=16) were raised to 13 weeks of age in a
metabolic cage. Urine was then collected for 24 h and the protein
levels in urine were determined using a BCA kit (cat. no. P0010;
Beyotime Institute of Biotechnology). Protein concentrations were
calculated from the standard curve and the sample volume used.
Immunofluorescence staining
Cells cultured on coverslips were washed three times
with PBS and fixed with 4% paraformaldehyde for 15 min at room
temperature. Cells were then treated with 0.1% Triton X-100 for 10
min and blocked with normal goat serum (cat. no. C0265; Beyotime
Institute of Biotechnology) for 30 min at room temperature. Cells
were then incubated with specific primary antibodies against LC3
(1:50; cat. no. 14600-1-AP; ProteinTech Group, Inc.) and vimentin
(1:50; cat. no. bs-8533R; BIOSS) at 4°C overnight in a wet box.
Subsequently, the samples were incubated with the corresponding
secondary antibody for 1 h at room temperature [Alexa Fluor
594-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L); 1:100; cat.
no. 111585003; Jackson ImmunoResearch Laboratories, Inc.; and Alexa
Fluor 488-conjugated AffiniPure Goat Anti-Rabbit IgG (H+L); 1:100;
cat. no. 111545003; Jackson ImmunoResearch Laboratories, Inc.].
Finally, cells were stained with DAPI hydrochloride to visualize
the nuclei. Slides were visualized under a fluorescence microscope
(DM6000B; Leica Microsystems, Inc.).
Western blot analysis
Cells were washed three times with ice-cold PBS and
lysed with cell lysis RIPA buffer (cat. no. P0013B; Beyotime
Institute of Biotechnology) for 20 min. The cell pellet and lysate
were collected and centrifuged at 14,000 × g for 4 min at 4°C. The
supernatant was collected and the protein concentration was
determined using a BCA kit. An appropriate volume of 5X loading mix
was added to the samples and heated for 5 min at 100°C to denature
the proteins. Samples and pre-stained markers were added as
required. The protein samples (30 µg) were mixed with loading
buffer and subjected to SDS-PAGE on 12% gels. The
electrically-transformed PVDF membrane was taken out and washed
with TBS containing 0.1% Tween-20 (TBST) and blocked with 5% milk
in TBST on a shaker at room temperature for 1 h. Subsequently,
primary antibodies against E-cadherin (1:1,000; cat. no.
20874-1-AP; ProteinTech Group, Inc.), N-cadherin (1:1,000; cat. no.
22018-1-AP; ProteinTech Group, Inc.), Twist1 (1:500; cat. no.
25465-1-AP; ProteinTech Group, Inc.), LC3 (1:1,000; cat. no.
14600-1-AP; ProteinTech Group, Inc.); phosphorylated (p)-mTOR
(1:1,000; cat. no. 5536T; Cell Signaling Technology, Inc.), p62
(1:1,000; cat. no. 184201-AP; ProteinTech Group, Inc.); mTOR
(1:1,000; cat. no. 20657-1-AP; ProteinTech Group, Inc.) and β-actin
(1:3,000; cat. no. 4970; Cell Signaling Technology, Inc.) were
added and incubated at 4°C overnight. After washing the membranes
with PBS containing 0.1% Tween-20 (PBST) three times, the membranes
were incubated with a horseradish peroxidase-labeled secondary
antibody (1:5,000; cat. no. A0208; Beyotime Institute of
Biotechnology) diluted in 5% milk/PBST for 1 h at room temperature
on a shaker. The PVDF membrane was exposed to the luminescent
reagent for coloring, and after 1.5–2 min, it was observed using a
gel imaging system (ChemiDoc™ XRS+; Bio-Rad Laboratories, Inc.).
ImageJ software (version 1.49p; National Institutes of Health) was
used to calculate the gray value and analysis of the protein
bands.
Flow cytometry
The supernatant treated as aforementioned was
aspirated and transferred to a microtube for storage. Cells were
washed twice with PBS and collected. Cells were then digested with
0.25% trypsin (without EDTA) at 37°C for 2–3 min. A pre-mixed 1X
Annexin V Binding Solution was added to the cell suspension at a
final concentration of 1×106 cells/ml. A total of 5 µl
Annexin V-FITC conjugate and 5 µl propidium iodide (cat. no.
KGA108; Nanjing KeyGen Biotech Co., Ltd.) were then added to the
cell suspension and incubated for 10 min at room temperature in the
dark. Finally, flow cytometry (cytoFLEX; Beckman Coulter, Inc.) was
conducted within 1 h of adding 400 µl 1X Annexin V Binding
Solution. The apoptotic rate of cells (including early and late
apoptotic cells) was analyzed using CytExpert version 2.0 software
(Beckman Coulter, Inc.).
Twist1 small interfering RNA (siRNA)
synthesis and transfection
siRNA sequences [three specific interference
sequences and one negative control (NC) sequence] were designed and
synthesized (GenScript) to target mouse Twist1. MPC5 cells were
inoculated into a six-well plate at 5×105 cells/well and
cultured at 37°C for 24 h in a 5% CO2 incubator. After cell
attachment, transfection was performed with
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Briefly, one Eppendorf (EP) tube (A tube) containing 5 µl
Lipofectamine 2000 diluted with 250 µl Opti-MEM (Thermo Fisher
Scientific, Inc.) was incubated at room temperature for 5 min,
whereas another EP tube (B tube) contained 2 µl siRNA diluted with
250 µl Opti-MEM. The A and B tubes were gently mixed and allowed to
stand at room temperature for 20 min. The siRNA-Lipofectamine 2000
mixture was then added to a six-well plate (5×105
cells/well) containing 1.5 ml complete medium. The medium was
changed after 4–6 h of transfection. Subsequent experiments were
performed at 48 h post-transfection. The siRNA sequences were as
follows: siRNA-NC (sense, 5′-UUCUCCGAACGUGUCACGUTT-3′ and
antisense, 5′-ACGUGACACGUUCGGAGAATT-3′), siRNA1-Twist1 (sense,
5′-CCUCUGCAUUCUGAUAGAATT-3′ and antisense,
5′-UUCUAUCAGAAUGCAGAGGTT-3′), siRNA2-Twist1 (sense,
5′-GUGUCUAAAUGCAUUCAUATT-3′ and antisense,
5′-UAUGAAUGCAUUUAGACACTT-3′) and siRNA3-Twist1 (sense,
5′-GGUACAUCGACUUCCUGUATT-3′ and antisense,
5′-UACAGGAAGUCGAUGUACCTT-3′).
Construction of Twist1 overexpression
(OE) plasmid vector
Plasmids pcDNA3.1-Hygro(+) and pUC57-Mus Twist1
(both from GenScript) were double digested with XhoI and
NotI (both from Takara Bio, Inc.) at 37°C overnight, and the
vector and fragment were recovered and purified on an agarose gel
by double digestion. The recovered and purified target fragment was
ligated with the recovered and purified vector at 4°C overnight.
The thawed DH5α competent cells and plasmid were thoroughly mixed
and stabilized on ice for 30 min. Then, the mixture was put in a
water tank at 42°C for 90 sec. Subsequently, 400 µl Luria-Bertani
medium (1% tryptone, 0.5% yeast extract, 1% NaCl; purchased from
Merck KGaA) was added, and the culture was shaken at 37°C for 1 h.
Subsequently, 1 ml Luria-Bertani medium broth was added to the
mixture, which was then incubated at 37°C for 1 h. The transformed
DH5α competent cells were plated onto Luria Bertani-containing
Petri dishes with 100 µg/ml ampicillin and incubated overnight at
37°C. Several monoclonal colonies were selected from the Petri
dish, inoculated in ampicillin-resistant Luria Bertani culture
medium, cultured at 37°C at overnight and subjected to colony PCR.
A single colony was carefully picked up and placed in the EP tube
containing 20 µl ddH2O and then denatured at 100°C for 2 min. A 1
µl suspension was used as a template in PCR reactions. PCR
reactions were performed in a 20 µl volume containing 2.5 µl 10X
Taq DNA Polymerase buffer, 2 µl dNTP, 0.5 µM of each primer and 0.5
µl Taq DNA polymerase. The reaction protocols were as follows:
Initial denaturation at 94°C for 10 min; 30 cycles of 94°C for 30
sec, 60°C for 30 sec and 72°C for 1 min. The primer sequences were
as follows: CMV-Forward: 5′-CGCAAATGGGCGGTAGGCGTG-3′ and
BGH-Reverse: 5′-TAGAAGGCACAGTCGAGG-3′. Cell transfection was
performed using Lipofectamine 2000. MPC5 cells were seeded into a
six-well plate at 5×105 cells/well and cultured
overnight to 90% coverage, then transfected with Twist1
overexpression vector (4 µg) or empty vector at 37°C for 72 h.
Untransfected cells were used as the control cells. Cells
transfected with empty vector were used as NC cells.
Statistical analysis
All experiments were repeated three times. All
statistical analyses were performed with SPSS 19.0 software (IBM
Corp.). Data are presented as the mean ± standard deviation.
Comparisons between multiple groups were analyzed by one-way ANOVA
followed by Tukey's post-hoc test and comparisons between two
groups were analyzed by unpaired Student's t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
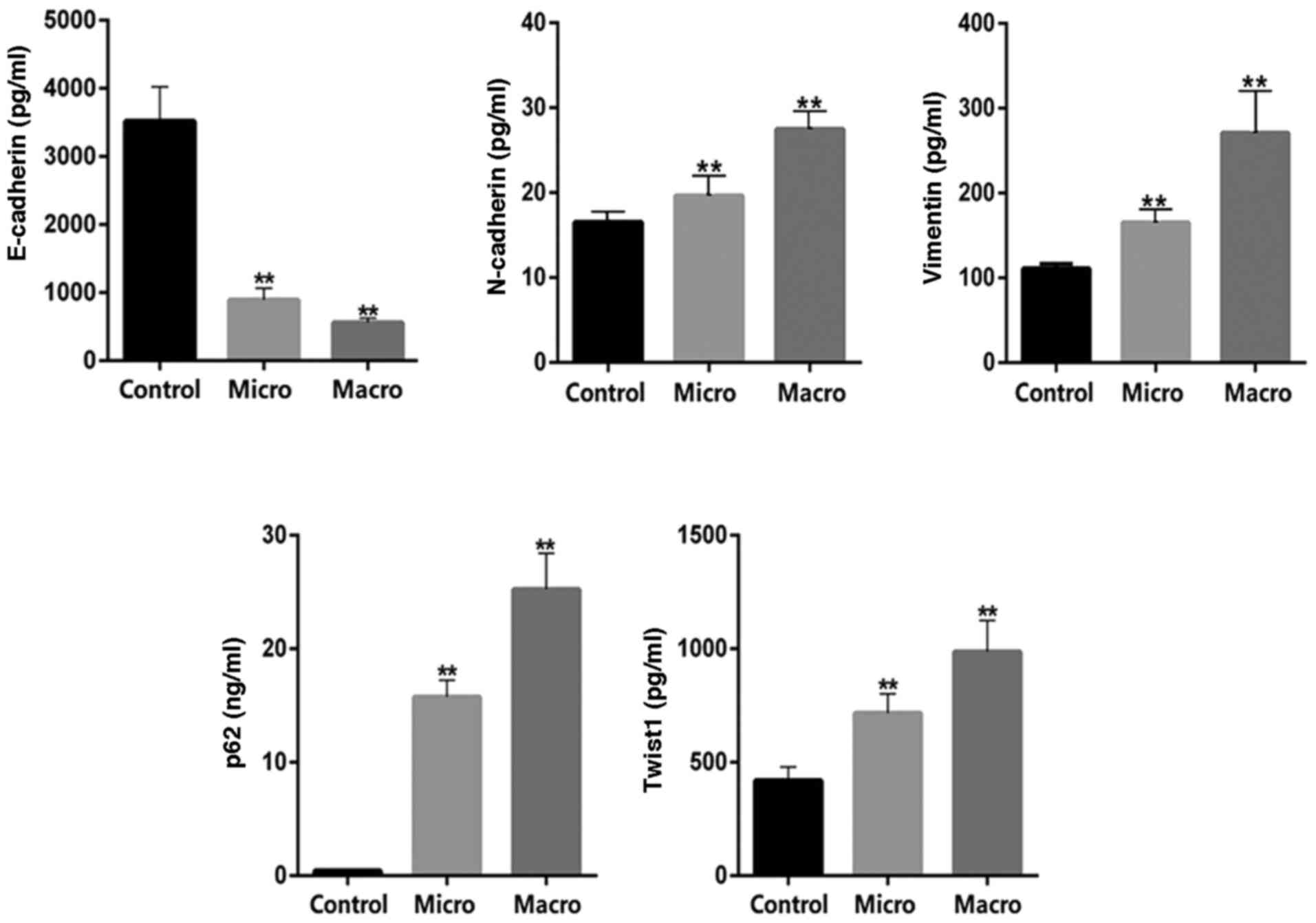
Determination of serum E-cadherin,
N-cadherin, vimentin, p62 and Twist1 levels in patients with
DKD
Serum samples from healthy controls (n=10) and
patients with DKD (n=20) were collected for biochemical index
detection. According to the urinary albumin to creatinine ratio
(UACR), patients with DKD were divided into two groups:
DKD-microalbuminuria (micro; UACR, 30–300 mg/g) and
DKD-macroalbuminuria (macro; UACR, >300 mg/g). The demographic
and clinical data for these patients are listed in Table SI. The results indicated that age,
SBP, BUN, Scr, FPG and HbA1c were significantly higher, whereas
HDL-C was significantly decreased in the macro group compared with
the control group. Similarly, compared with the control group, the
age, SBP, FPG, and HbA1c of the micro group were significantly
increased, whereas HDL-C was significantly decreased. ELISA was
used to detect the levels of autophagy and EMT-related proteins. In
patients with DKD, a significant decrease in serum E-cadherin
levels, and a significant increase in serum N-cadherin, vimentin,
p62 and Twist1 levels were observed compared with those in the
healthy controls (Fig. 1).
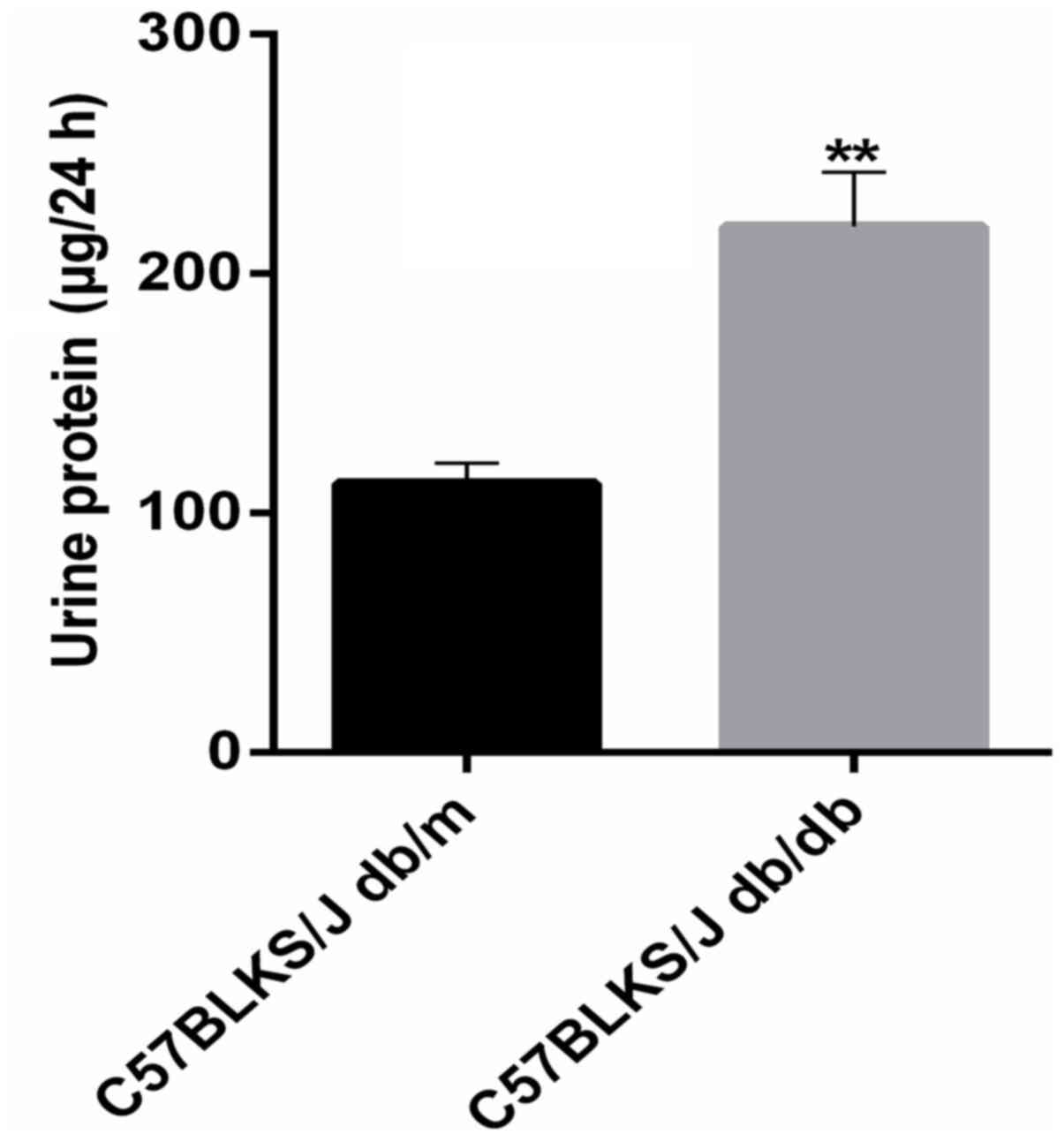
Quantitative determination of urinary
protein in mice
The present study raised 8-week-old mice until they
were 13 weeks of age. To confirm that the mice had diabetic
nephropathy, urine was collected for 24 h to determine the protein
content. The results revealed that the 24-h urinary protein content
in mice with diabetic nephropathy was significantly higher than
that in control mice (Fig. 2),
indicating that the model was reliable and could be used for serum
preparation.
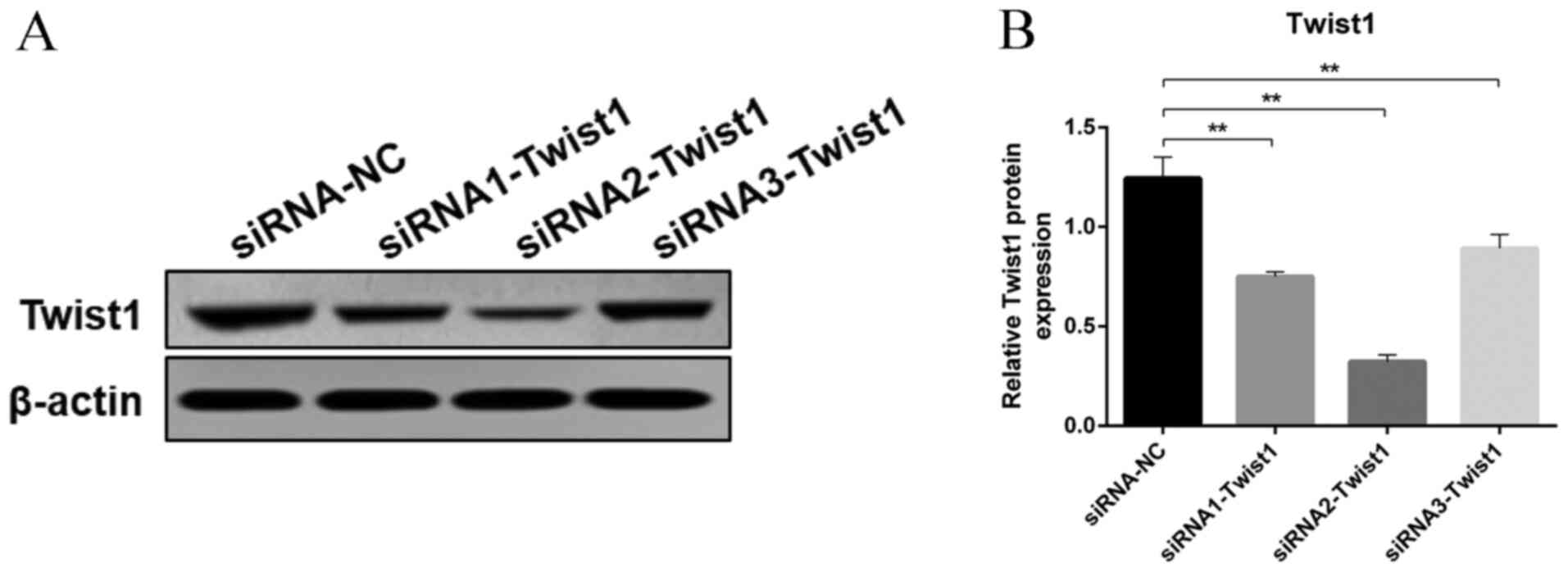
Effects of Twist1 on DKD-induced
podocyte EMT and apoptosis
Since Twist1 is known to serve a vital role in EMT,
the present study investigated whether Twist1 was able to regulate
EMT and apoptosis of podocytes induced by serum of DKD mice. Three
interference sequences were designed for cell transfection of MPC5
cells, and the most effective siRNA (siRNA2-Twist1) was selected
for subsequent experiments (Fig. 3A and
B). The immunofluorescence results showed that, compared with
those in the control group, the protein expression levels of
vimentin were significantly increased in the DKD group, but were
inhibited after siRNA2-Twist1 transfection (Fig. 4A and B). The western blotting
results revealed that the protein expression levels of Twist1 and
N-cadherin were significantly increased, whereas the expression
levels of E-cadherin were significantly decreased in the DKD group
compared with those in the control group. After siRNA2-Twist1
transfection, compared with those in the DKD + siRNA-NC group, the
expression levels of Twist1 and N-cadherin were significantly
decreased, whereas the expression levels of E-cadherin were
markedly increased (Fig. 4C and D).
Both immunofluorescence and western blotting results demonstrated
that silencing Twist1 alleviated DKD-induced podocyte EMT.
Furthermore, the results of flow cytometry demonstrated that, after
siRNA-Twist1 transfection, the apoptosis of podocytes in the DKD
group was inhibited compared with those in the DKD + siRNA-NC group
(Fig. 4E and F), which indicated
that silencing Twist1 could reduce podocyte apoptosis and EMT.
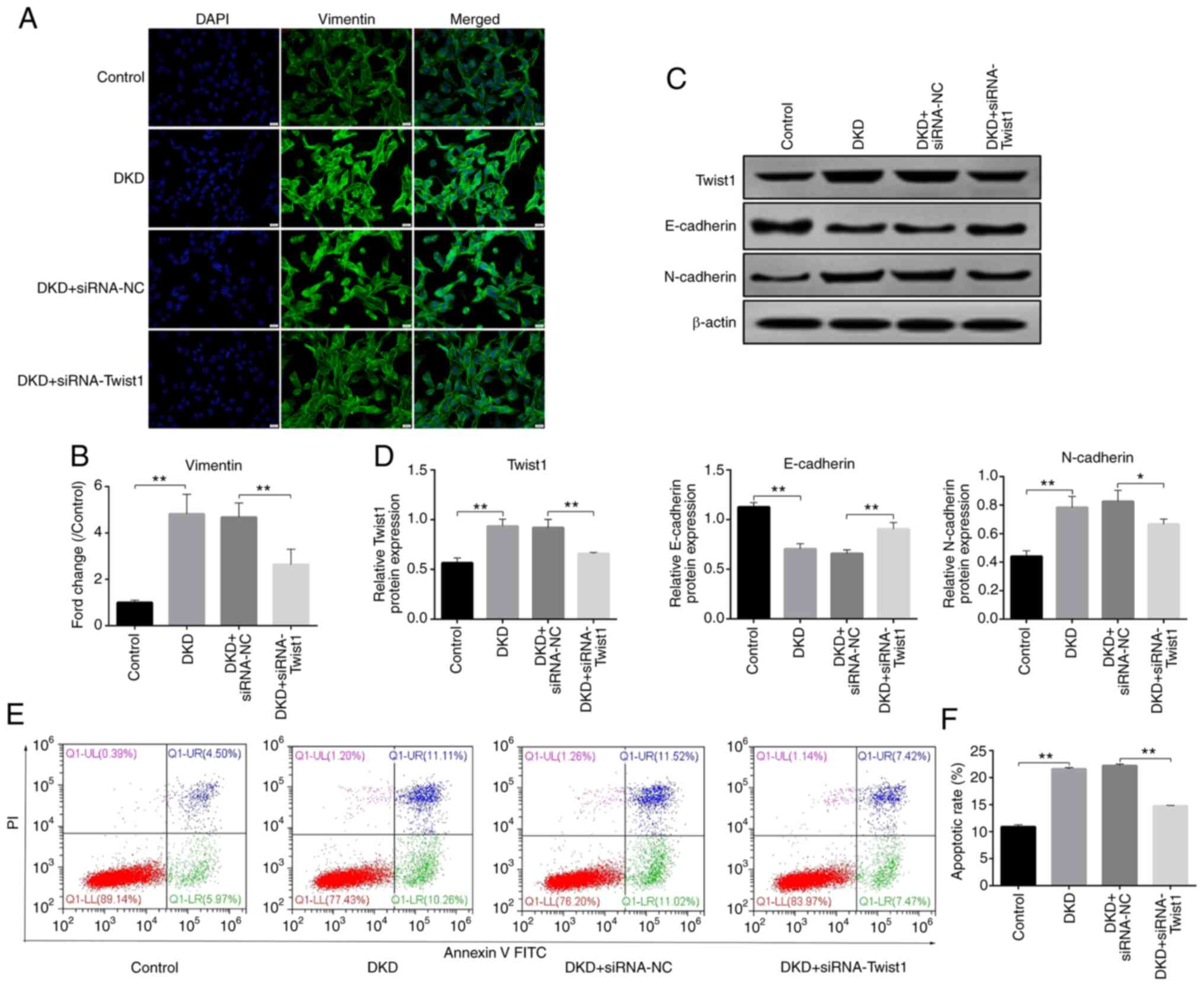 | Figure 4.Twist1 interference inhibits
epithelial-mesenchymal transition and apoptosis. (A)
Immunofluorescence of vimentin expression and localization. (Scale
bar, 20 µm; magnification, ×400). (B) Semi-quantitative evaluation
of vimentin, as determined by immunofluorescence. Control values
were set at 1. (C) Representative western blots of Twist1,
N-cadherin and E-cadherin expression in the different groups. (D)
Densitometric analysis of Twist1, N-cadherin and E-cadherin
expression, as determined by western blotting. (E) Cellular
apoptosis, as detected by flow cytometry, in each group. (F)
Apoptotic rates in each group. Control, cells treated with serum
from control mice (C57BLKS/J db/m) for 24 h; DKD, cells treated
with serum from DKD mice (C57BLKS/J db/db) for 24 h; DKD +
siRNA-NC, cells treated with serum from DKD mice (C57BLKS/J db/db)
for 24 h, and then transfected with siRNA-NC for 48 h; DKD +
siRNA-Twist1, cells treated with serum from DKD mice (C57BLKS/J
db/db) for 24 h, and then transfected with siRNA-Twist1 for 48 h.
Data analysis was performed by one-way ANOVA followed by Tukey's
post-hoc test. Data are presented as the mean ± standard deviation.
**P<0.01, *P<0.05. DKD, diabetic kidney disease; siRNA, small
interfering RNA; NC, negative control. |
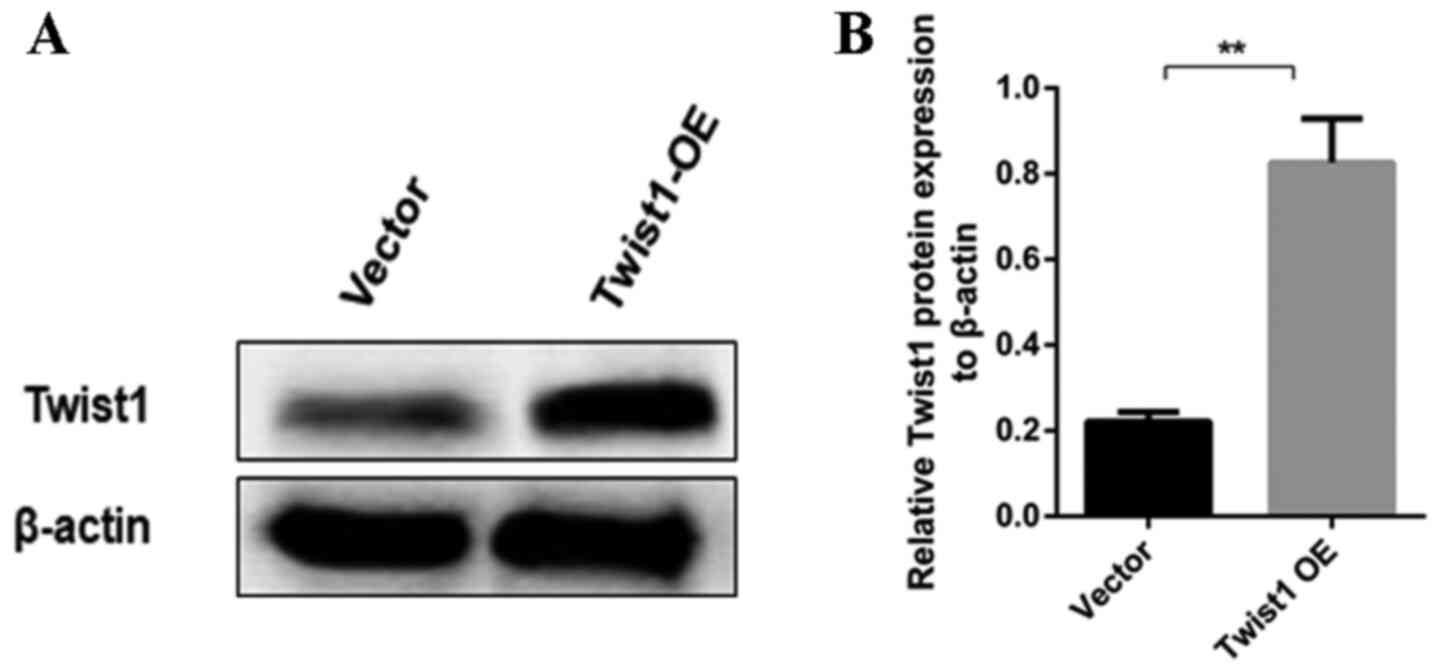
TG inhibits DKD-induced podocyte
apoptosis through Twist1
After determining the role of Twist1 in EMT and
apoptosis, the present study aimed to determine the effect of TG on
Twist1 by constructing a Twist1 OE vector. The expression of Twist1
increased significantly after transfection with Twist1 OE vector in
MPC5 cells, indicating that the Twist1 OE vector was successfully
constructed (Fig. 5A and B). As
shown in Fig. 6A and B, flow
cytometry revealed that, compared with that in the control group,
the apoptotic rate was significantly increased in podocytes in the
DKD group. There was no significant difference in the rate of
apoptosis between the DKD group and the DKD + Vector group,
indicating that transfection with an empty vector did not affect
the apoptosis of podocytes. Furthermore, compared with that in the
DKD + Vector group podocyte apoptosis was significantly increased
in the DKD + Twist1 OE group, indicating that Twist1 overexpression
increased podocyte apoptosis. In the DKD + TG + Vector group, the
apoptotic rate was inhibited by TG compared with that in the DKD +
Vector group, indicating that TG could inhibit podocyte apoptosis.
In addition, compared with that in the DKD + TG + Vector group, the
apoptotic rate of podocytes was significantly increased in the DKD
+ TG + Twist1 OE group. These results indicated that TG inhibited
podocyte apoptosis through Twist1.
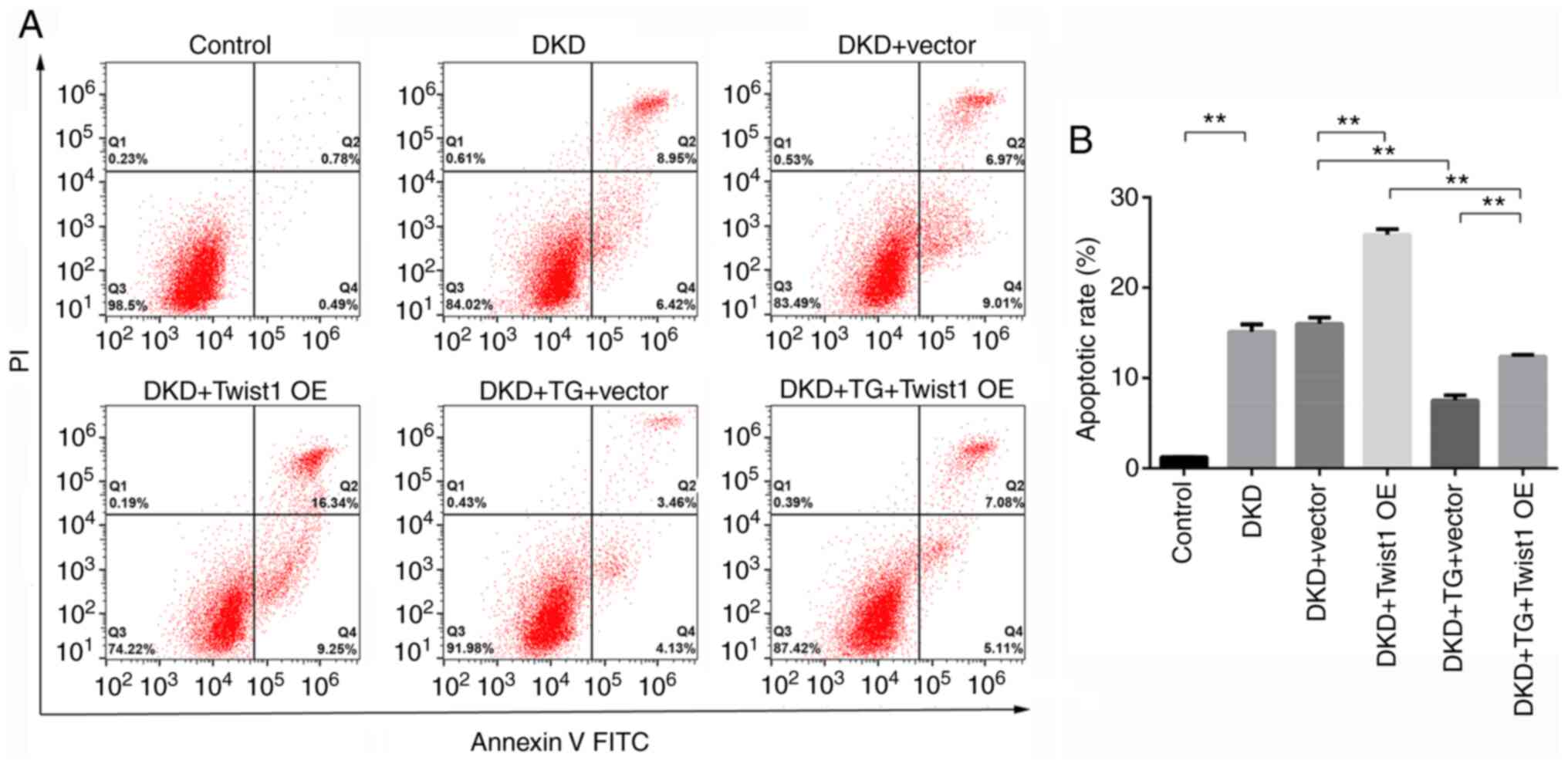 | Figure 6.TG affects podocyte apoptosis through
Twist1. (A) Cellular apoptosis, as detected by flow cytometry, in
each group. (B) Apoptotic rates in each group. Control, cells
treated with serum from control mice (C57BLKS/J db/m) for 24 h;
DKD, cells treated with serum from DKD mice (C57BLKS/J db/db) for
24 h; DKD + Vector, cells treated with serum from DKD mice
(C57BLKS/J db/db) for 24 h, and then transfected with an empty
vector for 72 h; DKD + Twist1 OE, cells treated with serum from DKD
mice (C57BLKS/J db/db) for 24 h, and then transfected with the
Twist1 OE vector for 72 h; DKD + TG + Vector, cells treated with
serum from DKD mice (C57BLKS/J db/db) for 24 h and 1.25 µg/ml TG
for 72 h, and then transfected with an empty vector for 72 h; DKD +
TG + Twist1 OE, cells treated with serum from DKD mice (C57BLKS/J
db/db) for 24 h and 1.25 µg/ml TG for 72 h, and then transfected
with the Twist1 OE vector for 72 h. Data are presented as the mean
± standard deviation. Data analysis was performed by one-way ANOVA
followed by Tukey's post-hoc test. **P<0.01. DKD, diabetic
kidney disease; TG, tripterygium glycoside; OE, overexpression. |
TG alleviates podocyte EMT and
apoptosis by upregulating autophagy through the mTOR/Twist1
signaling pathway
3BDO, a mTOR activator, was applied to confirm that
TG alleviates podocyte EMT and apoptosis through the autophagy
pathway. As shown in Fig. 7A and B,
the fluorescent signals of vimentin were significantly increased in
the DKD group, but decreased in the DKD + TG group. The fluorescent
signals of LC3II were reduced in the DKD group, but increased in
the DKD + TG group (Fig. 7A and B).
These results indicated that TG may alleviate EMT and restore
autophagy. After addition of the mTOR activator 3BDO, autophagy was
inhibited and the effects of TG on EMT were impaired. The results
were further verified by western blotting (Fig. 7C and D), which revealed that the
expression levels of p62, N-cadherin, Twist1 and the ratio of
p-mTOR to mTOR were significantly increased, whereas the expression
levels of E-cadherin and the ratio of LC3II to LC3I were
significantly decreased in the DKD group compared with those in the
control group. By contrast, after TG treatment, the protein
expression levels of p62, Twist1, N-cadherin and the ratio of
p-mTOR to mTOR were markedly reduced, whereas those of E-cadherin
and the ratio of LC3II to LC3I were significantly increased,
demonstrating that TG may upregulate autophagy, alleviate EMT and
inhibit Twist1 expression. Following the addition of 3BDO, the
therapeutic effect of TG was suppressed, indicating that TG may
upregulate autophagy through the mTOR signaling pathway to reduce
EMT and Twist1 expression. The flow cytometry results (Fig. 7E and F) revealed a significant
increase in the podocyte apoptotic rate of the DKD group, which was
reversed with TG treatment. Upon addition of 3BDO, a reduction in
the protective effects of TG was noted, indicating that TG may
prevent podocyte apoptosis by upregulating autophagy through the
mTOR signaling pathway.
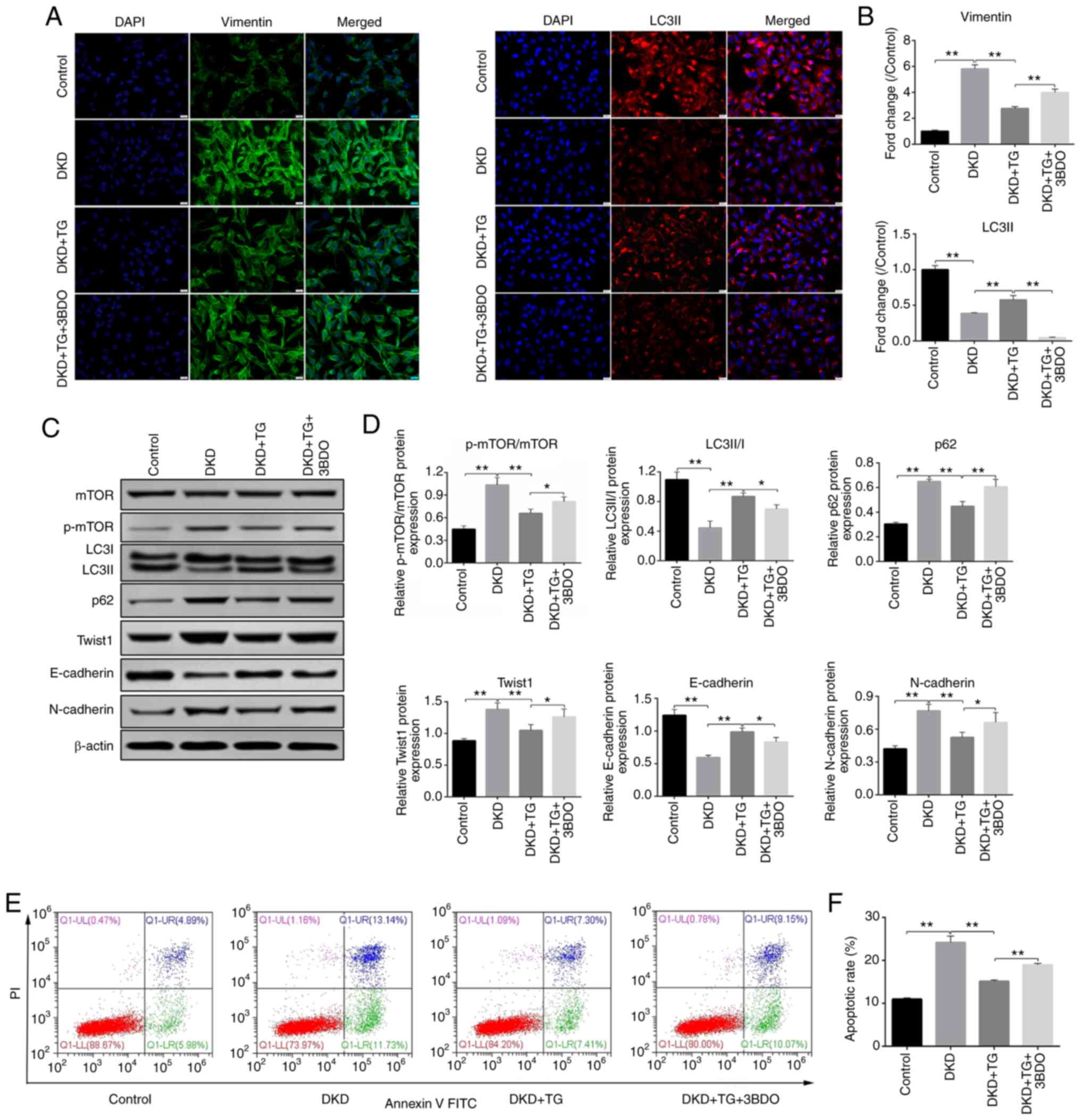 | Figure 7.TG inhibits podocyte
epithelial-mesenchymal transition and apoptosis through the
mTOR/Twist1 signaling pathway. (A) Immunofluorescence of vimentin
and LC3II (scale bar, 20 µm; magnification, ×400). (B)
Semi-quantitative evaluation of vimentin and LC3II, as determined
by immunofluorescence. Control values were set at 1. (C)
Representative western blots of p-mTOR, LC3, p62, Twist1,
E-cadherin and N-cadherin expression in the different groups. (D)
Densitometric analysis of p-mTOR/mTOR, LC3II/I, p62, Twist1,
E-cadherin and N-cadherin expression from different groups, as
determined by western blotting. (E) Cellular apoptosis in each
group was detected by flow cytometry. (F) Apoptotic rates in each
group. Control, cells treated with serum from control mice
(C57BLKS/J db/m) for 24 h; DKD, cells treated with serum from DKD
mice (C57BLKS/J db/db) for 24 h; DKD + TG, cells treated with serum
from DKD mice (C57BLKS/J db/db) for 24 h, and then treated with
1.25 µg/ml TG for 72 h; DKD + TG + 3BDO, cells treated with serum
from DKD mice (C57BLKS/J db/db) for 24 h and 1.25 µg/ml TG for 72
h, and then incubated with 60 µM 3BDO for 24 h. Data analysis was
performed by one-way ANOVA followed by Tukey's post-hoc test. Data
are presented as the mean ± standard deviation. *P<0.05,
**P<0.01. TG, tripterygium glycoside; p-, phosphorylated; LC3,
microtubule-associated protein 1 light chain 3; DKD, diabetic
kidney disease; 3BDO, 3-benzyl-5-((2-nitrophenoxy)
methyl)-dihydrofuran-2(3H)-one. |
Collectively, these results suggested that after
culturing podocytes with serum from mice with DKD, EMT and
apoptosis were significantly increased, whereas autophagic activity
was significantly decreased. TG potentially inhibited the
occurrence of DKD-induced podocyte EMT and apoptosis by
downregulating mTOR phosphorylation, which promoted autophagy and
regulated Twist1 expression.
Discussion
The role of EMT in renal disease has been
intensively investigated in previous years. Multiple studies have
examined renal EMT in different animal models of chronic kidney
disease and human kidney biopsies (28,29),
and increasing evidence has suggested that EMT may contribute to
the development and progression of renal fibrosis in DKD (28,30–33).
While autophagy deficiency in podocytes is known to serve a role in
promoting EMT (15,19), Li et al (34) reported that autophagy could be
restored with TG treatment. The present study revealed that TG
provided protection against podocyte EMT induced by serum from mice
with DKD, and that this effect was mediated by concomitant
activation of autophagy and downregulation of Twist1.
Serum E-cadherin, N-cadherin, vimentin, p62 and
Twist1 levels from 20 patients with diabetic nephropathy were
measured and compared with levels from healthy controls (n=10).
Patients with DKD exhibited decreased autophagy levels (as
indicated by p62 expression) and increased EMT levels (as indicated
by E-cadherin, N-cadherin and vimentin expression). In addition,
the changes in EMT and autophagy level were greater in patients
with macroalbuminuria compared with in those with microalbuminuria,
which further confirmed the significance of EMT in the progression
of DKD.
While intracellular signal transduction pathways,
including integrin-linked kinase TGF-β/Smad and Wnt/β-catenin
signaling pathways, have been widely studied in the regulation of
renal EMT (35), relatively few
studies have explored the involvement of the EMT-inducing factor
Twist1 (32). The present study
demonstrated that the levels of Twist1 were significantly increased
in the serum of patients with DKD. Furthermore, siRNA2-Twist1 was
designed and it was revealed that Twist1 inhibition significantly
reduced DKD-induced podocyte EMT and apoptosis. By contrast, Twist1
OE increased podocyte apoptosis. In DKD, EMT may occur as cells
attempt to evade apoptosis due to exposure to pathophysiological
stimuli. EMT and apoptosis are considered to be the major cause of
podocyte injury in DKD (36). Thus,
the aforementioned findings indicated that Twist1 may act as an
important regulatory molecule involved in podocyte injury in
DKD.
TG represents a novel, effective and safe drug for
treating DKD in patients with proteinuria (37–39) by
targeting pathways associated with renal inflammation and oxidative
stress (38,40). Furthermore, TG has been reported as
a therapeutic target of podocyte autophagy (26,34,41–44),
and autophagy activation was shown to alleviate human podocyte
injury induced by high glucose (45). In the present study, the flow
cytometry results demonstrated that TG treatment could inhibit
podocyte apoptosis by targeting Twist1. Further experiments
indicated that TG could regulate Twist1 expression, relieve
podocyte EMT and decrease podocyte apoptosis through the autophagy
pathway. Autophagy deficiency is known to prevent Twist1 and
SQSTM1/p62 degradation via the autophagy pathway, leading to
aggregation of Twist1 and SQSTM1/p62. SQSTM1/p62 aggregates and
subsequently binds to Twist1, thus preventing its degradation
through the ubiquitination pathway, which ultimately promotes EMT
(19). These previous findings are
consistent with the current observations that Twist1 expression and
EMT levels were increased, but autophagy levels were significantly
reduced, in the serum of patients with DKD. Consistent with this,
the present study revealed that TG restored podocyte autophagy,
which may promote Twist1 protein degradation and alleviate EMT.
3BDO, a well-known mTOR activator, attenuated the
therapeutic effects of TG, indicating that autophagy is a
therapeutic target of TG and that TG restores autophagy via the
mTOR signaling pathway. The PI3K/Akt/mTOR signaling pathway has
been identified as a critical signaling pathway in the regulation
of cellular autophagy and apoptosis, and targeting this signaling
pathway has been suggested as a prospective strategy for cancer
treatment (46). Furthermore, TG
inhibited the proliferation of glomerular mesangial cells to
prevent diabetic glomerulosclerosis through the Akt/mTOR signaling
pathway (47). The PI3K/Akt/mTOR
signaling pathway has also been shown to participate in the
regulation of cellular EMT and cellular autophagy (48,49).
To determine the contribution of the mTOR signaling pathway in the
TG-mediated anti-apoptotic effect on podocytes, the present study
examined the phosphorylation and expression of mTOR in podocytes
cultured with DKD mice serum. The results revealed that p-mTOR and
p62 were upregulated, whereas LC3II was downregulated. Upon TG
treatment, autophagy was activated, and significant downregulation
of p-mTOR was observed. However, no significant differences in
total mTOR expression levels were noticed between the TG-treated
and untreated groups. These findings suggested that TG may inhibit
the phosphorylation of mTOR while mediating autophagy in podocytes,
rather than inhibiting mTOR formation.
Although Twist1 serves a key role in EMT, studies
exploring its role in molecular-targeted therapy remain limited. To
the best of our knowledge, the present study is the first to
confirm the involvement of Twist1 in DKD progression, suggesting
that Twist1 may have the potential to act as a molecular target for
DKD treatment. The present study also demonstrated that TG
treatment could inhibit podocyte apoptosis via the Twist1 pathway
in DKD. However, the effect of TG targeting the Twist1 pathway on
podocyte EMT and autophagy was not explored, and further research
is required to clarify the effect of Twist1 on podocyte autophagy
regulation.
In conclusion, the present study demonstrated that
TG may effectively prevent podocyte EMT in DKD, which was mediated,
at least partly, by downregulating mTOR phosphorylation and
increasing autophagy, thus resulting in a decrease in Twist1
expression. These findings provide novel insights into the
molecular mechanisms underlying the protective effects of TG in
DKD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
Natural Science Foundation of Zhejiang Province (grant nos.
LY16H050005, LQ15H050002 and Y18H050024), the Project of Scientific
Research Foundation of Chinese Medicine (grant nos. 2016ZA023
2017ZA008 and 2015ZZ002), and the General Project of the Medical
and Health of Zhejiang Province (grant no. 2016KYA015). The funding
bodies had no role in the design of the study, or in the
collection, analysis and interpretation of the data, or in writing
the manuscript.
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
QH and JJ designed the present study and provided
administrative support. MT collected samples and clinical
information. XL, DW, KH, DZ and MT analyzed and interpreted the
data. MT and JJ were involved in drafting the manuscript. QH and JJ
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study involving patient samples was
approved by the local Ethics Committee of Zhejiang Provincial
People's Hospital (Hangzhou, China). The present study was
performed in accordance with the ethical standards of the 1964
Declaration of Helsinki. All the enrolled patients provided written
informed consent for renal biopsy and participation in research
before the renal biopsy was performed. All protocols involving
animals were approved by the Institutional Animal Care and Use
Committee of Zhejiang Provincial People's Hospital.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TG
|
tripterygium glycoside
|
|
3BDO
|
3-benzyl-5-((2-nitrophenoxy)
methyl)-dihydrofuran-2(3H)-one
|
|
EMT
|
epithelial-mesenchymal transition
|
|
DKD
|
diabetic kidney disease
|
|
ESRD
|
end-stage renal disease
|
References
|
1
|
Saran R, Robinson B, Abbott KC, Agodoa
LYC, Albertus P, Ayanian J, Balkrishnan R, Bragg-Gresham J, Cao J,
Chen JL, et al: US Renal Data System 2016 Annual Data Report:
epidemiology of kidney disease in the United States. Am J Kidney
Dis. 69 (Suppl 1):A7–A8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matoba K, Takeda Y, Nagai Y, Kawanami D,
Utsunomiya K and Nishimura R: Unraveling the role of inflammation
in the pathogenesis of diabetic kidney disease. Int J Mol Sci.
20:33932019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stitt-Cavanagh E, MacLeod L and Kennedy C:
The podocyte in diabetic kidney disease. ScientificWorldJournal.
9:1127–1139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo J, Xia N, Yang L, Zhou S, Zhang Q,
Qiao Y and Liu Z: GSK-3beta and vitamin D receptor are involved in
beta-catenin and snail signaling in high glucose-induced
epithelial-mesenchymal transition of mouse podocytes. Cell Physiol
Biochem. 33:1087–1096. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li Y, Kang YS, Dai C, Kiss LP, Wen X and
Liu Y: Epithelia-to-mesenchymal transition is a potential pathway
leading to podocyte dysfunction and proteinuria. Am J Pathol.
172:299–308. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu L, Fu W, Xu J, Shao L and Wang Y:
Effect of BMP7 on podocyte transdifferentiation and Smad7
expression induced by hyperglycemia. Clin Nephrol. 84:95–99. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dai HY, Zheng M, Tang RN, Ni J, Ma KL, Li
Q and Liu BC: Effects of angiotensin receptor blocker on phenotypic
alterations of podocytes in early diabetic nephropathy. Am J Med
Sci. 341:207–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kang YS, Li Y, Dai C, Kiss LP, Wu C and
Liu Y: Inhibition of integrin-linked kinase blocks podocyte
epithelial-mesenchymal transition and ameliorates proteinuria.
Kidney Int. 78:363–373. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jin J, Zhang Z, Chen J, Liu Y, Chen Q and
Wang Q: Jixuepaidu Tang-1 inhibits epithelial-mesenchymal
transition and alleviates renal damage in DN mice through
suppressing long non-coding RNA LOC498759. Cell Cycle.
18:3125–3136. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li CX, Xia M, Han WQ, Li XX, Zhang C,
Boini KM, Liu XC and Li PL: Reversal by growth hormone of
homocysteine-induced epithelial-to-mesenchymal transition through
membrane raft-redox signaling in podocytes. Cell Physiol Biochem.
27:691–702. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamaguchi Y, Iwano M, Suzuki D, Nakatani
K, Kimura K, Harada K, Kubo A, Akai Y, Toyoda M, Kanauchi M, et al:
Epithelial-mesenchymal transition as a potential explanation for
podocyte depletion in diabetic nephropathy. Am J Kidney Dis.
54:653–664. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Doublier S, Salvidio G, Lupia E,
Ruotsalainen V, Verzola D, Deferrari G and Camussi G: Nephrin
expression is reduced in human diabetic nephropathy: Evidence for a
distinct role for glycated albumin and angiotensin II. Diabetes.
52:1023–1030. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yasuda-Yamahara M, Kume S, Tagawa A,
Maegawa H and Uzu T: Emerging role of podocyte autophagy in the
progression of diabetic nephropathy. Autophagy. 11:2385–2386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li G, Li CX, Xia M, Ritter JK, Gehr TW,
Boini K and Li PL: Enhanced epithelial-to-mesenchymal transition
associated with lysosome dysfunction in podocytes: role of
p62/Sequestosome 1 as a signaling hub. Cell Physiol Biochem.
35:1773–1786. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shibue T and Weinberg RA: EMT, CSCs, and
drug resistance: The mechanistic link and clinical implications.
Nat Rev Clin Oncol. 14:611–629. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng J, Chen S, Han JX, Qian B, Wang XR,
Zhong WL, Qin Y, Zhang H, Gao WF, Lei YY, et al: Twist1 regulates
vimentin through Cul2 circular RNA to promote EMT in hepatocellular
carcinoma. Cancer Res. 78:4150–4162. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu W, Zhang B, Xu N, Wang MJ and Liu Q:
miR-326 regulates EMT and metastasis of endometrial cancer through
targeting TWIST1. Eur Rev Med Pharmacol Sci. 21:3787–3793.
2017.PubMed/NCBI
|
|
19
|
Qiang L and He YY: Autophagy deficiency
stabilizes TWIST1 to promote epithelial-mesenchymal transition.
Autophagy. 10:1864–1865. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cui J, Chen X and Su JC: Advanced progress
of main pharmacology activities of triptolide. Zhongguo Zhongyao
Zazhi. 42:2655–2658. 2017.(In Chinese). PubMed/NCBI
|
|
21
|
Zhang H, Sun W, Wan Y, Che X, He F, Pu H
and Dou C: Preventive effects of multi-glycoside of Tripterygium
wilfordii on glomerular lesions in experimental diabetic
nephropathy. Zhongguo Zhongyao Zazhi. 35:1460–1465. 2010.(In
Chinese). PubMed/NCBI
|
|
22
|
Ma RX, Zhao N and Zhang W: The effects and
mechanism of Tripterygium wilfordii Hook F combination with
irbesartan on urinary podocyte excretion in diabetic nephropathy
patients. Zhonghua Nei Ke Za Zhi. 52:469–473. 2013.(In Chinese).
PubMed/NCBI
|
|
23
|
Sethi S, Haas M, Markowitz GS, D'Agati VD,
Rennke HG, Jennette JC, Bajema IM, Alpers CE, Chang A, Cornell LD,
et al: Mayo clinic/renal pathology society consensus report on
pathologic classification, diagnosis, and reporting of GN. J Am Soc
Nephrol. 27:1278–1287. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
de Cabo R, Fürer-Galbán S, Anson RM,
Gilman C, Gorospe M and Lane MA: An in vitro model of caloric
restriction. Exp Gerontol. 38:631–639. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
National Research Council Committee for
the Update of the Guide for the C and Use of Laboratory A, . The
National Academies Collection: Reports funded by National
Institutes of Health. Guide for the Care and Use of Laboratory
Animals National Academies Press Copyright© 2011.
National Academy of Sciences; Washington, DC: 2011
|
|
26
|
Zhan H, Jin J, Liang S, Zhao L, Gong J and
He Q: Tripterygium glycoside protects diabetic kidney disease mouse
serum-induced podocyte injury by upregulating autophagy and
downregulating β-arrestin-1. Histol Histopathol. 34:943–952.
2019.PubMed/NCBI
|
|
27
|
Xia M, Conley SM, Li G, Li PL and Boini
KM: Inhibition of hyperhomocysteinemia-induced inflammasome
activation and glomerular sclerosis by NLRP3 gene deletion. Cell
Physiol Biochem. 34:829–841. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rastaldi MP, Ferrario F, Giardino L,
Dell'Antonio G, Grillo C, Grillo P, Strutz F, Müller GA, Colasanti
G and D'Amico G: Epithelial-mesenchymal transition of tubular
epithelial cells in human renal biopsies. Kidney Int. 62:137–146.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zeisberg M and Kalluri R: The role of
epithelial-to-mesenchymal transition in renal fibrosis. J Mol Med
(Berl). 82:175–181. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Reidy K and Susztak K:
Epithelial-mesenchymal transition and podocyte loss in diabetic
kidney disease. Am J Kidney Dis. 54:590–593. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Srivastava SP, Koya D and Kanasaki K:
MicroRNAs in kidney fibrosis and diabetic nephropathy: Roles on EMT
and EndMT. BioMed Res Int. 2013:1254692013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Loeffler I and Wolf G:
Epithelial-to-mesenchymal transition in diabetic nephropathy: Fact
or Fiction? Cells. 4:631–652. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hills CE and Squires PE: The role of TGF-β
and epithelial-to mesenchymal transition in diabetic nephropathy.
Cytokine Growth Factor Rev. 22:131–139. 2011.PubMed/NCBI
|
|
34
|
Li XY, Wang SS, Han Z, Han F, Chang YP,
Yang Y, Xue M, Sun B and Chen LM: Triptolide restores autophagy to
alleviate diabetic renal fibrosis through the
miR-141-3p/PTEN/Akt/mTOR pathway. Mol Ther Nucleic Acids. 9:48–56.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu Y: New insights into
epithelial-mesenchymal transition in kidney fibrosis. J Am Soc
Nephrol. 21:212–222. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dai H, Liu Q and Liu B: Research progress
on mechanism of podocyte depletion in diabetic nephropathy. J
Diabetes Res. 2017:26152862017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, Chen N, Fang L, Feng Z, Li G,
Mucelli A, Zhang X and Zhou X: A systematic review about the
efficacy and safety of Tripterygium wilfordii Hook.f.
preparations used for the management of rheumatoid arthritis. Evid
Based Complement Alternat Med. 2018:15674632018.PubMed/NCBI
|
|
38
|
Gao Q, Shen W, Qin W, Zheng C, Zhang M,
Zeng C, Wang S, Wang J, Zhu X and Liu Z: Treatment of db/db
diabetic mice with triptolide: A novel therapy for diabetic
nephropathy. Nephrol Dial Transplant. 25:3539–3547. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ge Y, Xie H, Li S, Jin B, Hou J, Zhang H,
Shi M and Liu Z: Treatment of diabetic nephropathy with
Tripterygium wilfordii Hook F extract: A prospective,
randomized, controlled clinical trial. J Transl Med. 11:134. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guo H, Pan C, Chang B, Wu X, Guo J, Zhou
Y, Liu H, Zhu Z, Chang B and Chen L: Triptolide Improves Diabetic
Nephropathy by Regulating Th Cell Balance and Macrophage
Infiltration in Rat Models of Diabetic Nephropathy. Exp Clin
Endocrinol Diabetes. 124:389–398. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen ZH, Qin WS, Zeng CH, Zheng CX, Hong
YM, Lu YZ, Li LS and Liu ZH: Triptolide reduces proteinuria in
experimental membranous nephropathy and protects against
C5b-9-induced podocyte injury in vitro. Kidney Int. 77:974–988.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gong J, Jin J, Zhao L, Li Y, Li Y and He
Q: Tripterygium glycoside protects against puromycin amino
nucleoside-induced podocyte injury by upregulating autophagy. Int J
Mol Med. 42:115–122. 2018.PubMed/NCBI
|
|
43
|
Chan SF, Chen YY, Lin JJ, Liao CL, Ko YC,
Tang NY, Kuo CL, Liu KC and Chung JG: Triptolide induced cell death
through apoptosis and autophagy in murine leukemia WEHI-3 cells in
vitro and promoting immune responses in WEHI-3 generated leukemia
mice in vivo. Environ Toxicol. 32:550–568. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao F, Huang W, Zhang Z, Mao L, Han Y,
Yan J and Lei M: Triptolide induces protective autophagy through
activation of the CaMKKβ-AMPK signaling pathway in prostate cancer
cells. Oncotarget. 7:5366–5382. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Xin W, Li Z, Xu Y, Yu Y, Zhou Q, Chen L
and Wan Q: Autophagy protects human podocytes from high
glucose-induced injury by preventing insulin resistance.
Metabolism. 65:1307–1315. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Rodon J, Dienstmann R, Serra V and
Tabernero J: Development of PI3K inhibitors: Lessons learned from
early clinical trials. Nat Rev Clin Oncol. 10:143–153. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Han F, Xue M, Chang Y, Li X, Yang Y, Sun B
and Chen L: Triptolide suppresses glomerular mesangial cell
proliferation in diabetic nephropathy is associated with inhibition
of PDK1/Akt/mTOR pathway. Int J Biol Sci. 13:1266–1275. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lamouille S and Derynck R: Cell size and
invasion in TGF-beta-induced epithelial to mesenchymal transition
is regulated by activation of the mTOR pathway. J Cell Biol.
178:437–451. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang F, Li H, Yan XG, Zhou ZW, Yi ZG, He
ZX, Pan ST, Yang YX, Wang ZZ, Zhang X, et al: Alisertib induces
cell cycle arrest and autophagy and suppresses
epithelial-to-mesenchymal transition involving PI3K/Akt/mTOR and
sirtuin 1-mediated signaling pathways in human pancreatic cancer
cells. Drug Des Devel Ther. 9:575–601. 2015.PubMed/NCBI
|