Introduction
Cutaneous basal cell carcinoma (BCC) is a common
malignant skin tumor originating from the basal layer of the
epidermis and its appendages, accounting for 65–70% of all skin
tumors (1). Currently, the specific
biomarkers useful for identifying BCC are still lacking and most of
the studies are focused on analyses at the protein level (2–4). A
small number of studies have recently reported that mRNAs may be
involved in the process of BCC, but research on lncRNAs remains
scarce (5–7).
Long non-coding RNA (lncRNA) is a functional RNA
molecule containing >200 nucleotides, which serves an important
regulatory role in gene transcription, post-transcriptional
translation and epigenetic levels. Studies have suggested that
lncRNAs are extensively involved in the biological process of
cancer (8–12), but few lncRNA studies have clarified
the roles of lncRNA in BCC. Therefore, it is a priority to explore
the possible biological effects of the lncRNAs involved in BCC.
Since lncRNAs have been considered regulators of mRNAs (13,14),
the present study constructed a lncRNA-mRNA co-expression network
for BCC with the presumption that the roles of these lncRNAs are
affected by the function of related mRNAs. The lncRNA-mRNA
co-expression network established bridges between lncRNAs and mRNAs
to reveal the potential functional involvement of lncRNAs in BCC
pathobiology. The interaction network is based on the hypothesis
that once the differential expression levels of two transcripts are
linearly related, a bridge can be established to demonstrate that
the two are co-expressed. In this case, the two transcripts
consisted of one lncRNA and one mRNA.
Overall, determining the important roles of lncRNAs
in tumors is essential. Therefore, it is particularly important to
conduct a whole-genome analysis of BCC tissues and construct a
valid lncRNA-mRNA co-expression network, exploring the
differentially expressed (DE) lncRNAs and making conjectures on
their biological functions.
Materials and methods
Patients and samples
The study protocol was conducted in accordance with
the Declaration of Helsinki and was approved by the Institutional
Ethical Review Board of Peking Union Medical College (approval no.
2016-KY013). The present study recruited thirteen patients (6 male
patients and 7 female patients; age range, 50–65 years) and
thirteen health volunteers (6 male volunteers and 7 female
volunteers; age range, 50–65 years) between February 2017 and
November 2017. A total of thirteen basal cell carcinoma samples and
thirteen normal tissue samples were collected from patients who
underwent biopsy operation at the Institute of Dermatology, Chinese
Academy of Medical Sciences. All thirteen BCC samples were
pathologically confirmed and all participants provided written
informed consent.
RNA extraction
Total cellular RNA was isolated from three BCC
samples and three normal skin tissue samples using an RNeasy Mini
kit (Qiagen GmbH) in accordance with the manufacturer's protocol
and then quantified through NanoDrop ND-1000 (Thermo Fisher
Scientific, Inc.). Total RNA was assessed by standard denaturing
agarose gel electrophoresis.
Microarray assay
GeneChip® Human Transcriptome Array 2.0
(HTA2.0, Affymetrix; Thermo Fisher Scientific, Inc.), reportedly
covers more than 245,000 coding and 40,000 non-coding RNAs of the
human genome from NCBI RefSeq, UCSC, RNAdb, Ensembl, lncRNAs, UCSC,
NONCODE and related literature (15). Each transcript was represented by
using 1–5 probes.
The microarray hybridization was performed according
to the manufacturer's standard protocols (Agilent Technologies,
Inc.) including sample labeling, transcribing into double-stranded
complementary (c)DNAs, cRNAs and second cycle cDNAs and then
hybridizing onto the microarray. Finally, the hybridized slides
were washed, fixed and scanned to images by the Affymetrix Scanner
3000 (Affymetrix; Thermo Fisher Scientific, Inc.). The data was
collected by Affymetrix GeneChip Command Console (version 4.0;
Affymetrix; Thermo Fisher Scientific, Inc.) and normalized by the
Robust Multichip Average algorithm through the Expression Console
(version 1.3.1, Affymetrix; Thermo Fisher Scientific, Inc.)
software. All data were imported into Genespring software (version
12.5; Agilent Technologies, Inc.) for further analysis. The
statistical significance of differentially expressed lncRNAs and
mRNAs was identified via Volcano plot filtering with the threshold
set of fold change ≥2.0 and P-value <0.05. Hierarchical
clustering analysis was performed to reveal relationships between
transcripts.
Reverse transcription-quantitative
(RT-q) PCR
A total of ten BCC samples and ten normal tissues
were used for RT-qPCR validation. Total RNA was isolated with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse transcribed into cDNA using the SuperScript III
First Strand Synthesis System (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Subsequently, qPCR was performed using SYBR-Green (Takara Bio,
Inc.) quantitative PCR and the ABI 7900HT Fast Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used for qPCR: 40 cycles at 95°C for
2 sec, 60°C for 20 sec and 70°C for 10 sec. β-actin was used as an
internal control. The primers used for qPCR are listed in Table I. For quantitative results, the
relative expression level of each lncRNA was calculated using
2−ΔΔCq method (16) and
then statistically analyzed.
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| lncRNA | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
|
XR_428611.1 |
GGCTCGCTCTCCACTCCATCC |
TTACACACCCCTTCTCCCTTCCAG |
|
XR_428612.1 |
TGCTGCTGGCGAGAAGAATGC |
ATGTTGAAGACCGGGGCTTTCTG |
|
ENST00000566225.1 |
GGAAGGGCACCACTTCTACC |
AATTACCTGTGAAGGCCCCG |
|
ENST00000430816.1 |
CAGCCAACCACGCAGACCTG |
TTCCAGAACAGCACGAAGTCCAC |
|
NR_002160.1 |
TGACCGTCAACTTGGGATTGT |
TTAAACAAGCCAGCCAAGCG |
|
ENST00000444265.1 |
CTGCCAAGAGGAATCCAGCA |
TCTGGGTTTTCCATGCGTGT |
|
HIT000067334 |
TGCATGTAAGTGGCAAAGATGA |
TGCTTCTCCAGAATGGCTTGA |
|
ENST00000598252.1 |
GGAGGCAGATTCAGTCAGACG |
TGGACAAAAACTCCAGTGTGC |
|
uc003hcu.1 |
GTTTTCCCCCTCACCAAGCA |
GCGACCAAAGCCACAATAGC |
|
ENST00000548135.1 |
TGCCCAGGAAGCTGAGGTGAG |
AGCTGCTGGAAGGCGAGGAG |
| β-actin |
TCCTTCCTGGGCATGGAGT |
AGCACTGTGTTGGCGTACAG |
Analysis of lncRNA-mRNA co-expression
network
To predict the functional roles of lncRNAs, the top
10 were associated with direct regulated expression of target mRNAs
using co-expression network. Pearson's correlation Coefficient
(PCC) between each lncRNA-mRNA pair was calculated and those with
|PCC| >0.99 and P<0.05 were selected. The co-expression
network was visually represented using Cytoscape software (version
3.6.1; cytoscape.org), in which each mRNA
corresponded to a node and two RNAs linked by an edge indicated a
high correlation.
Gene ontology (GO) and kyoto
encyclopedia of genes and genomes (KEGG) pathway analyses
GO analysis was used to identify the potential
functions of significant DE genes, which covered three realms,
including biological process, molecular function and cellular
component (http://www.geneontology.org). Furthermore, the KEGG
database (http://www.genome.ad.jp/kegg/) was conducted to
harvest pathway information.
Statistical analysis
All statistical analyses were performed using the
SPSS version 17.0 software (SPSS, Inc.). Unpaired Student's t-tests
were performed to generate P-values. P<0.05 was considered to
indicate a statistically significant difference. Data are
representative of three independent experiments.
Results
Microarray analysis of lncRNAs and
mRNAs in BCC
To obtain the expression profiles of lncRNAs and
mRNAs in BCC, three normal skin tissue samples and three BCC tissue
samples were examined. The results revealed 32,904 lncRNAs and
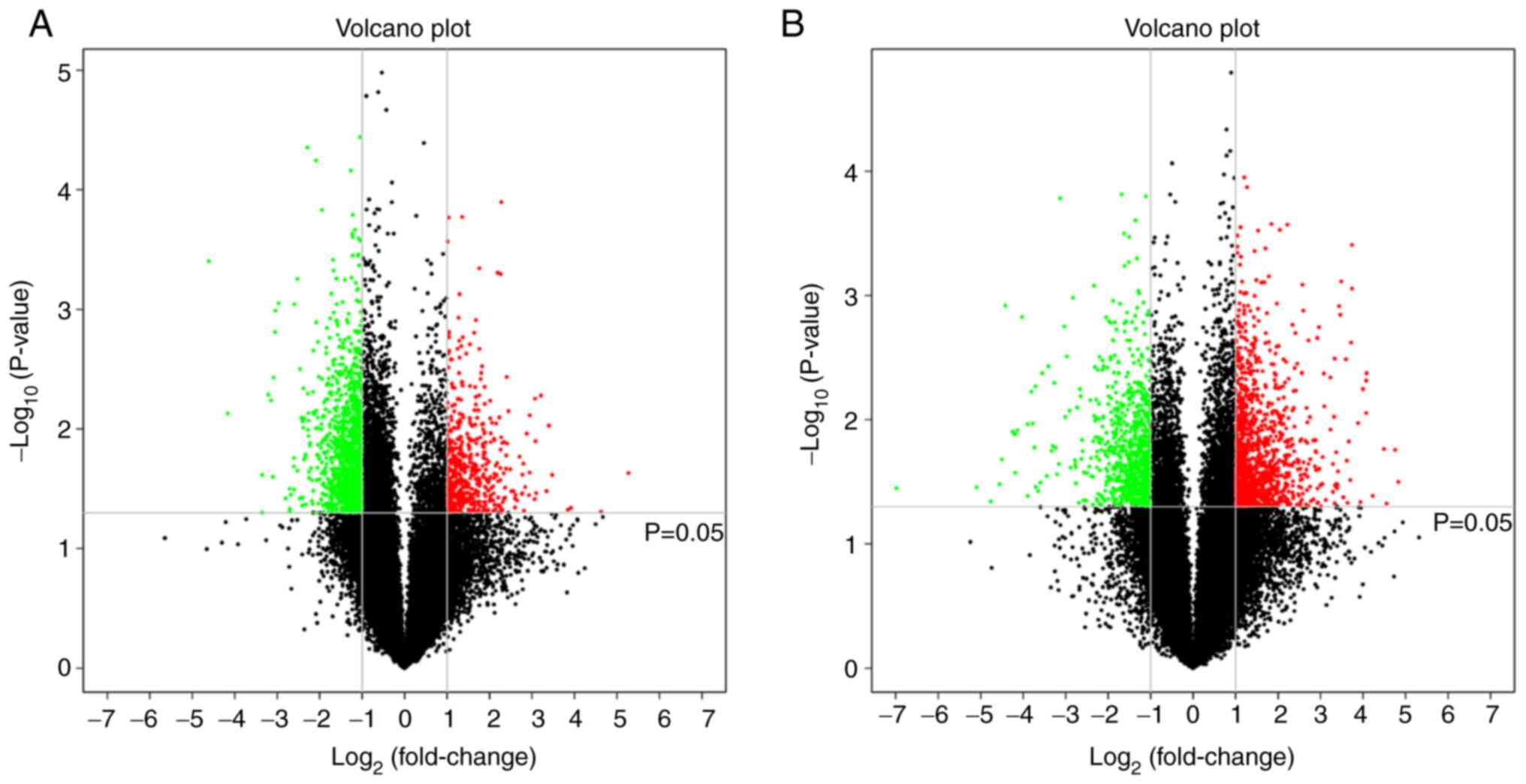
32,552 mRNAs in total, with the results are shown in Table SI. Compared with normal samples,
524 lncRNAs (red dots in Fig. 1A)
and 1,207 mRNAs (red dots in Fig.
1B) were upregulated (P<0.05; fold change >2) in BCC. In
contrast, 1,314 lncRNAs (green dots in Fig. 1A) and 803 mRNAs (green dots in
Fig. 1B) were significantly
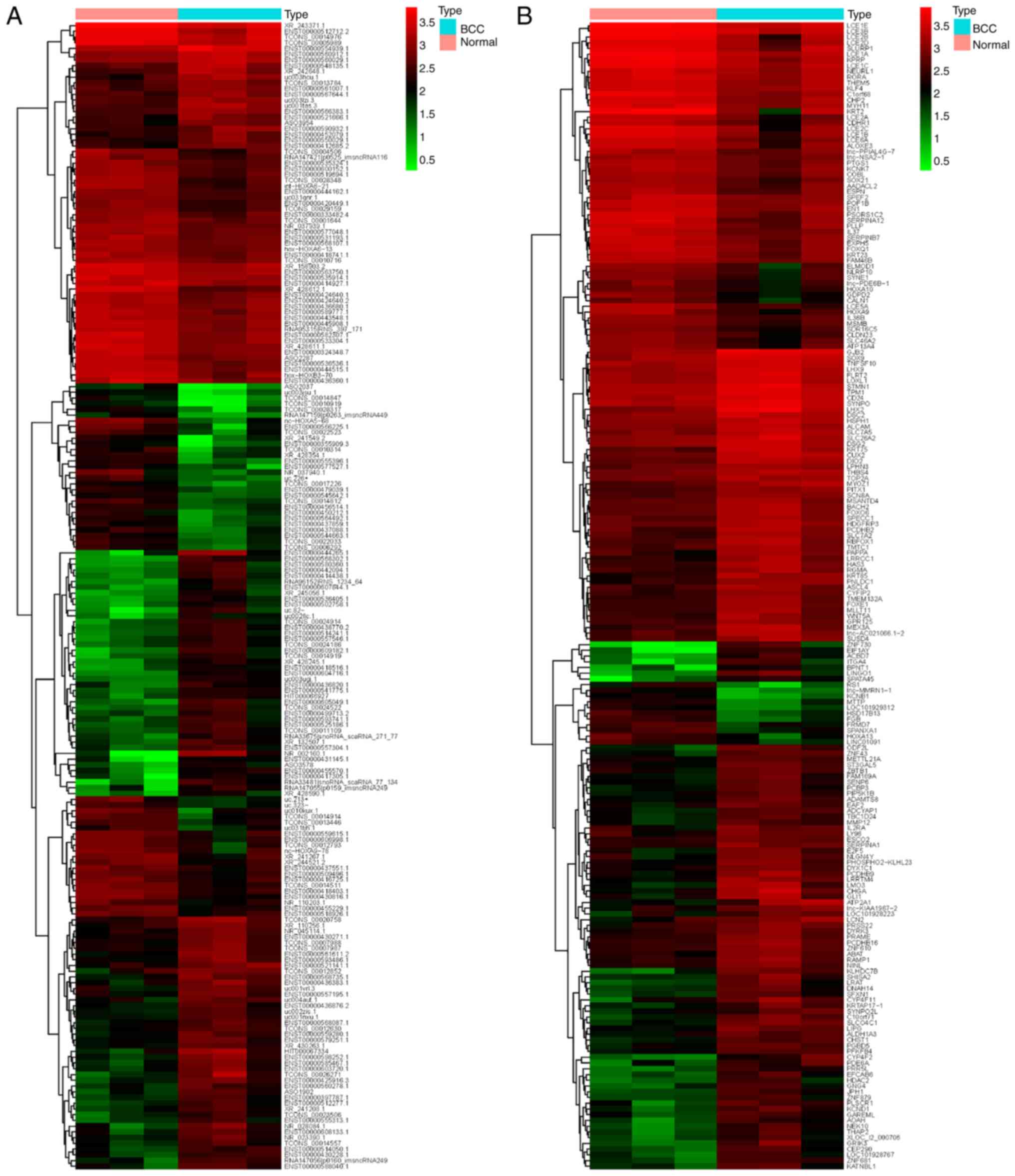
downregulated (P<0.05; fold change >2). The top 200
differentially expressed lncRNAs and mRNAs are presented in the
hierarchical cluster analysis heat map (Fig. 2A and B), in which ‘Normal’
represents three normal skin tissue samples and ‘BCC’ represents
three BCC tissue samples.
RT-qPCR validation of differentially
expressed lncRNAs
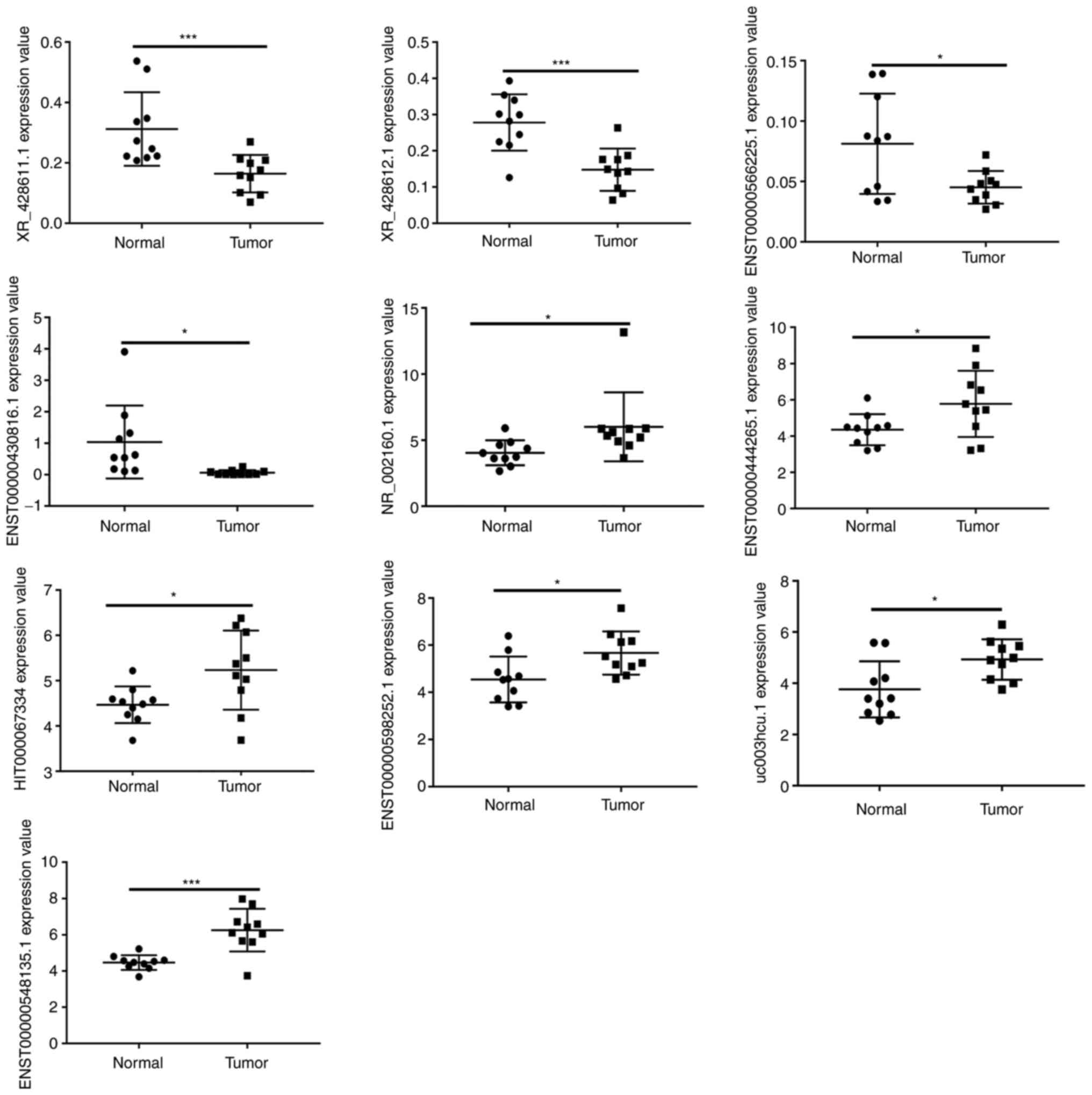
The top 10 DE lncRNAs were selected for RT-qPCR
analysis to verify the microarray results in ten BCC tissue samples
and ten normal tissue samples. The RT-qPCR results demonstrated
that four lncRNAs (XR_428611.1, XR_428612.1,
ENST00000566225.1 and ENST00000430816.1) were
downregulated in BCC and six lncRNAs (NR_002160.1,
ENST00000444265.1, HIT000067334, ENST00000598252.1 and
uc003hcu.1, ENST00000548135.1) were upregulated in BCC. As
shown in Fig. 3, the relative
values of the expression levels detected by RT-qPCR were found to
be consistent with the microarray data. This result suggests that
the transcript identification and abundance estimates were highly
reliable.
Construction of a lncRNA-mRNA
co-expression network
To construct the lncRNA-mRNA co-expression network
in BCC, the correlation of the expression levels of the top 10
lncRNAs and individual mRNAs in the samples were investigated by
Pearson correlation coefficient (PCC) and tested the reliability of
PCC by P-value. A strict positive correlation and a negative
correlation is indicated by a PCC equal to 1 or −1 (17). The present study limited the PCC
value to >0.99 or <-0.99 and set the P-value to <0.05,
which is a strict standard used to establish a correlation
(18).
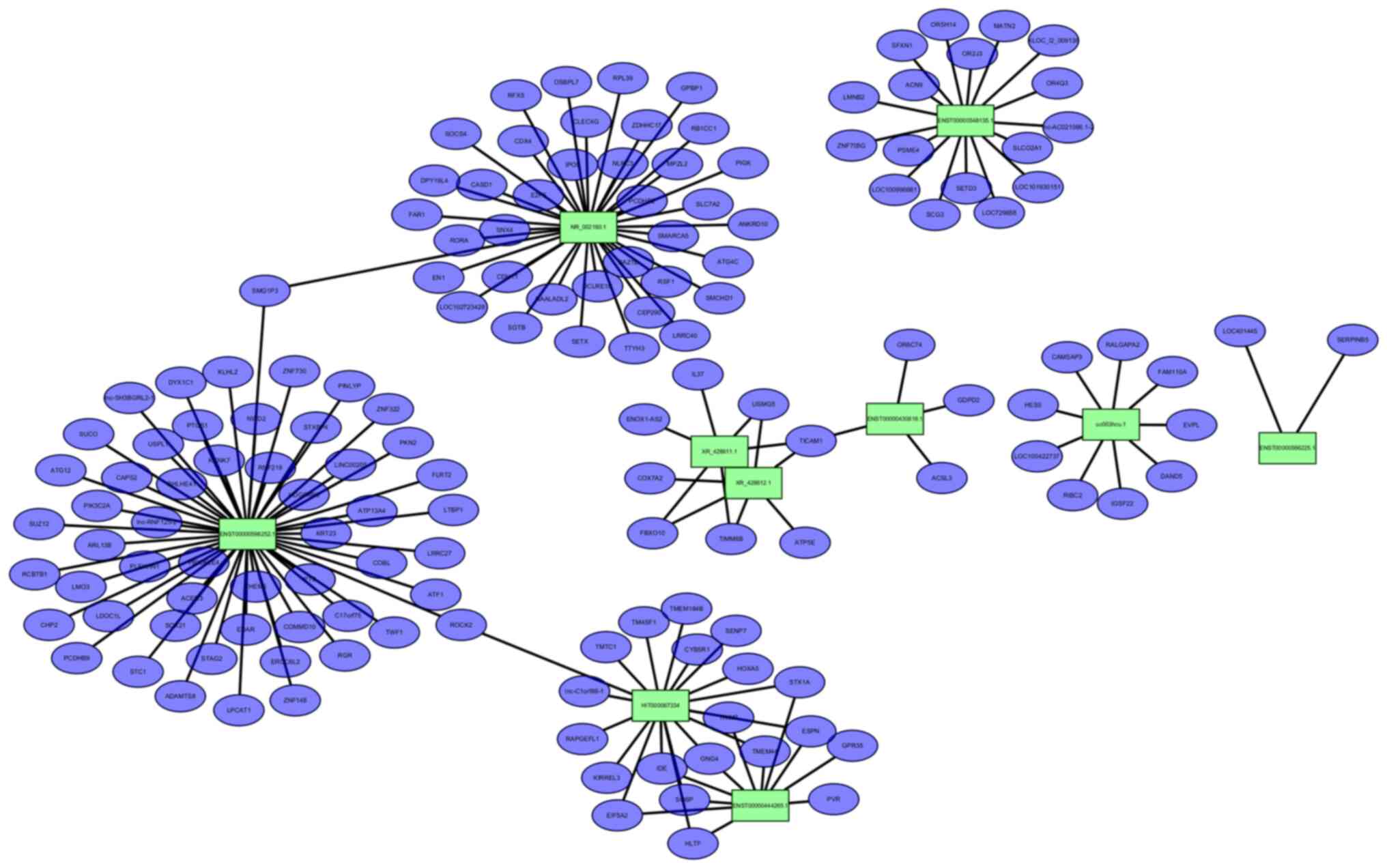
The lncRNA-mRNA network we constructed consisted of
10 lncRNA nodes (green rectangles in Fig. 4), 150 mRNA nodes (purple dots in
Fig. 4) and 166 edges. Different
edge types represent different interrelationships. As shown in the
figure, ENST00000598252.1 is co-expressed with 54 of the 150
mRNAs identified, the most of all identified DE lncRNAs. The next
most abundant, NR_002160.1, was co-expressed with as many as
38 mRNAs. Moreover, TICAM1 mRNA was co-expressed with the maximum
number (three) among the 10 DE lncRNAs.
GO enrichment and pathway
analysis
To further explore potential targets of the DE
lncRNAs in BCC progression, GO enrichment and KEGG pathway analyses
were performed on the DE mRNAs (Fig.
5).
Numerous GO terms were targeted by these coding
mRNAs, including 3,625 biological processes, 635 molecular
functions and 446 cellular components. The target genes and
detailed information are represented in Table SII. GO analysis is widely used to
identify a set of gene products in three categories: Biological
processes, cellular components and molecular functions (19). For the DE mRNAs in BCC identified by
microarray, the most significant emerging themes as determined by
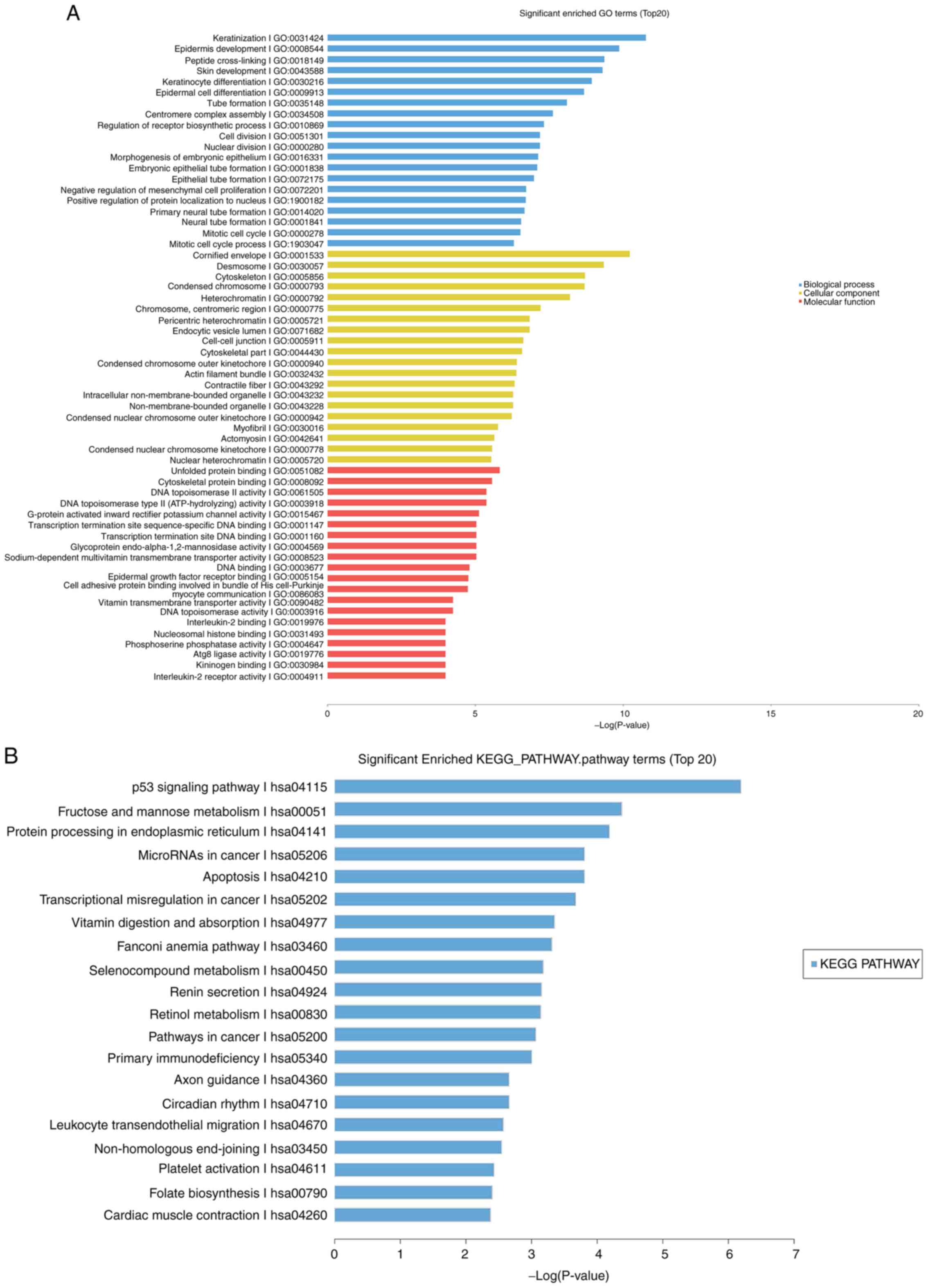
P-value were ‘keratinization’ (biological process; Fig. 5A blue), ‘cornified envelope’
(cellular component; Fig. 5A
yellow) and ‘unfolded protein binding’ (molecular function;
Fig. 5A red).
In addition, 152 KEGG pathways enriched with the DE
mRNAs were detected. For KEGG pathway analysis, the top 20 of the
152 identified KEGG pathways were selected and analyzed. The
majority of the KEGG pathways most enriched included the p53
signaling pathway, fructose and mannose metabolism, protein
processing in the endoplasmic reticulum, microRNAs in cancer and
apoptosis (Fig. 5B).
Discussion
The role of lncRNAs in cancer has been recognized
and a number of studies have been conducted with lung cancer,
breast cancer, liver cancer, gastrointestinal tumors and skin
cancer (20–22). LncRNAs can be upregulated or
downregulated in a variety of cancer processes, including cutaneous
tumors, affecting upstream and downstream target genes, signaling
pathways and metabolic pathways and ultimately promoting tumor
proliferation and invasion (23).
As a common cutaneous tumor, BCC has also been studied to determine
its possible pathogenesis. It is currently hypothesized that
abnormal activation of the Hedgehog pathway induced by the
PTCH1 gene serves a major role in BCC (24). In addition, abnormal upregulation of
the SMO gene, loss of tumor suppressor p53 and activation of
the Hippo-Yes-associated protein pathway are considered to be
involved in the progression of BCC. Some oncogenes, such as
ARID1A, CASP8, CSMD1, GRIN2A, KRAS, NOTCH1, NOTCH2, NRAS,
PIK3CA, PREX2 and RAC1, are also mutated in BCC
(25,26). In summary, lncRNAs are currently
important for cancer study, but their role in BCC is seldom
mentioned and remains unclear.
To explore the potential role of lncRNAs in the
processes of BCC, the present study performed whole-genome
identification of lncRNAs using an array to detect the expression
profiles of lncRNAs and mRNAs in three normal tissue samples and
three BCC tissue samples. A total of 32,904 lncRNA probes and
32,552 mRNA probes were detected and a number of RNAs were found to
be differentially expressed, including 1,838 lncRNAs and 2,010
mRNAs. The selection criteria, fold change >2 and P<0.05,
ensured the significance of the differential expression data. The
top 10 DE lncRNAs, including four downregulated lncRNAs and six
upregulated lncRNAs, were verified through RT-qPCR. The RT-qPCR
results showed a trend in expression levels consistent with those
of the lncRNA array analysis.
Some of the DE lncRNAs identified in the present
study have been reported to be involved in carcinogenesis; for
example, the p27963 probe detected lncRNA nc-HOXA5-68, one
of the top 10 downregulated DE lncRNAs (27). As suggested by its name it has a
regulatory effect on the proto-oncogene HOXA5. HOXA5
can affect various systemic systems and can serve a pivotal role in
the cancer process (28). The first
studied lncRNA HOTAIR can also be regulated to induce cancer
progression, such as liver cancer, cervical cancer and cutaneous
melanoma (29,30).
Although lncRNAs do not encode proteins directly,
they can be involved in various biological processes including the
progression of cancer by interacting with other biomolecules such
as transcription factors, miRNAs, mRNAs and RNA-binding proteins to
form gene regulatory networks (31). Several studies have been conducted
to obtain lncRNA biomarkers associated with tumor development and
prognosis by constructing lncRNA-mRNA networks, such as thyroid
cancer, hepatocellular carcinoma and ovarian cancer (32–34).
To further explore the potential role and mechanism of lncRNAs in
BCC, the present study constructed a lncRNA-mRNA co-expression
network focusing on the top 10 specific DE lncRNAs. The most
striking example involved the largest downregulation of lncRNA,
XR_428612.1, in BCC. This lncRNA has not been studied, but
the co-expression network demonstrated that it has a co-expression
relationship with six mRNAs, TICAM1, USMG5, COX7A2, FBXO10,
ATP5E and TIMM8B. Notably, all these mRNAs are reported
to be associated with mitochondrial dysfunction and glucose
metabolism (35–41). In the co-expression network of the
present study, lncRNA XR_428612.1 was positively correlated
with TIR domain-containing adaptor molecule-1 (TICAM1). It
has been reported that TICAM1 downregulation is an essential
step in Toll-like receptor 3 (TLR3) activation and stops
TLR3-mediated IFN production (35).
Activated TLR3 induces the upregulation of transactivating p63
isoform α (TAp63α), a p53-related protein that downregulates the
expression of Bcl-2, an anti-apoptosis molecule and induces cell
apoptosis in a caspase-dependent manner through death receptors and
mitochondria (36). The present
study noted that TICAM1 was co-expressed with the maximum
number (three) of lncRNAs, which indicated that it may serve an
important role in the pathogenesis of BCC. In addition,
FBXO10 is also thought to control the degradation of Bcl-2
and to regulate cell apoptosis (37). TIMM8B, a mitochondrial ribosomal
protein (MRP), has been reported to be upregulated in colon mucosa
carcinogenesis mediated by diabetes (28). MRPs may be affected by glucose
metabolism and participate in proliferation, metastasis, or
cellular migration in malignancies (38). In addition, diabetes-associated
protein in insulin-sensitive tissues (DAPIT) encoded by
USMG5 (39), ATP synthase
epsilon subunit (ATP5E) and cytochrome c oxidase subunit 7A2
(COX7A2) are partial components of ATP synthase and participate in
the interconversion of mitochondrial oxidative phosphorylation
(OXPHOS) and glycolysis, both of which are sources of ATP (40,41).
The upregulation of these transcripts clearly contribute to
modulating tumor anabolism, redox and calcium homeostasis,
apoptosis and metastasis by governing mitochondrial function.
In the present study, GO and KEGG pathway analyses
showed significant changes among the DE mRNAs. KEGG analysis found
that the ‘p53 signaling pathway’, ‘fructose and mannose
metabolism’, ‘protein processing in the endoplasmic reticulum’,
‘microRNAs in cancer’ and ‘apoptosis’ were the top five
significantly changed pathways. P53 has been reported to be
overexpressed in BCC (42). It
interferes with Bcl-2 in mitochondria, which results in the release
of cytochrome c and promotes apoptosis by activating the
mitochondrial pathway (42,43). In fact, three of the five top
significant KEGG pathways, including the ‘p53 signaling pathway’,
‘fructose and mannose metabolism’ and ‘apoptosis’, were aligned
with the carcinogenic mechanisms of the mRNAs co-expressed with
lncRNA XR_428612.1, as aforementioned. Since the
construction of lncRNA-mRNA co-expression network and KEGG analysis
relationships are independent, this outcome greatly increases the
reliability of the present study. Therefore, it was hypothesized
that mitochondrial dysfunction involving OXPHOS and glycolysis
modulated by lncRNA XR_428612.1 may serve a considerable
role in the progression of BCC and deserves to be the focused of a
follow-up study.
In conclusion, the present study provided a
comprehensive analysis of lncRNA-mRNA co-expression profiles of
patients with cutaneous basal cell carcinoma. GO and KEGG analyses
further revealed the function of mRNAs and suggested the possible
biological effects of lncRNAs. The results indicated that lncRNA
XR_428612.1 may serve an important role in mitochondrial
dysfunction and the progression of BCC by modulating TICAM1,
USMG5, COX7A2, FBXO10, ATP5E and TIMM8B. These data
argue for further intensive research to provide a more
comprehensive understanding of the molecular mechanisms underlying
the metabolism of BCC.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by CAMS Innovation
Fund for Medical Sciences (grant no. CIFMS-2017-I2M-1-017), the
National Natural Science Foundation of China (grant nos. 81703152
and 81872545) and the Jiangsu Province Natural Science Foundation
(grant no. BK20170161).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JZ, XZ, DH, MJ, CL and KC conceived and designed the
study. DH, YH, RL and SJ collected the samples and acquired the
data. JZ, XZ and CZ interpretated or analyzed data. XZ, CZ, YH, RL
and SJ prepared the manuscript, which was revised for important
intellectual content by JZ, CL and KC. All authors reviewed and
approved the final manuscript. CL and KC confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
The study protocol was conducted in accordance with
the Declaration of Helsinki and was approved by the Institutional
Ethical Review Board of Peking Union Medical College (approval no.
2016-KY013). All participants provided written informed
consent.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Puig S and Berrocal A: Management of
high-risk and advanced basal cell carcinoma. Clin Transl Oncol.
17:497–503. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pellegrini C, Maturo MG, Di Nardo L,
Ciciarelli V, Gutierrez Garcia-Rodrigo C and Fargnoli MC:
Understanding the molecular genetics of basal cell carcinoma. Int J
Mol Sci. 18:24852017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ciążyńska M, Bednarski IA, Wódz K, Kolano
P, Narbutt J, Sobjanek M, Woźniacka A and Lesiak A: Proteins
involved in cutaneous basal cell carcinoma development. Oncol Lett.
16:4064–4072. 2018.
|
|
4
|
Sălan AI, Mărăşescu PC, Camen A, Ciucă EM,
Matei M, Florescu AM, Pădureanu V and Mărgăritescu C: The
prognostic value of CXCR4, α-SMA and WASL in upper lip basal cell
carcinomas. Rom J Morphol Embryol. 59:839–849. 2018.
|
|
5
|
Sun H and Jiang P: MicroRNA-451a acts as
tumor suppressor in cutaneous basal cell carcinoma. Mol Genet
Genomic Med. 6:1001–1009. 2018. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vand-Rajabpour F, Sadeghipour N, Saee-Rad
S, Fathi H, Noormohammadpour P, Yaseri M, Hesari KK, Bagherpour Z
and Tabrizi M: Differential BMI1, TWIST1, SNAI2 mRNA expression
pattern correlation with malignancy type in a spectrum of common
cutaneous malignancies: Basal cell carcinoma, squamous cell
carcinoma, and melanoma. Clin Transl Oncol. 19:489–497. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sand M, Hessam S, Amur S, Skrygan M,
Bromba M, Stockfleth E, Gambichler T and Bechara FG: Expression of
oncogenic miR-17-92 and tumor suppressive miR-143-145 clusters in
basal cell carcinoma and cutaneous squamous cell carcinoma. J
Dermatol Sci. 86:142–148. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang KC and Chang HY: Molecular mechanisms
of long noncoding RNAs. Mol Cell. 43:904–914. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohammad F, Pandey GK, Mondal T, Enroth S,
Redrup L, Gyllensten U and Kanduri C: Long noncoding RNA-mediated
maintenance of DNA methylation and transcriptional gene silencing.
Development. 139:2792–2803. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu X, Zheng H, Tse G, Chan MT and Wu WK:
Long non-coding RNAs in melanoma. Cell Prolif. 51:e124572018.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
McCarthy N: Epigenetics. Going places with
BANCR. Nat Rev Cancer. 12:4512012. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li R, Zhang L, Jia L, Duan Y, Li Y, Bao L
and Sha N: Long non-coding RNA BANCR promotes proliferation in
malignant melanoma by regulating MAPK pathway activation. PLoS One.
9:e1008932014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang KC, Yang YW, Liu B, Sanyal A,
Corces-Zimmerman R, Chen Y, Lajoie BR, Protacio A, Flynn RA, Gupta
RA, et al: A long noncoding RNA maintains active chromatin to
coordinate homeotic gene expression. Nature. 472:120–124. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Panda AC, Abdelmohsen K and Gorospe M:
SASP regulation by noncoding RNA. Mech Ageing Dev. 168:37–43. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koczan D, Fitzner B, Zettl UK and Hecker
M: Microarray data of transcriptome shifts in blood cell subsets
during S1P receptor modulator therapy. Sci Data. 5:1801452018.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cao L, Zhang P, Li J and Wu M: LAST, a
c-Myc-inducible long noncoding RNA, cooperates with CNBP to promote
CCND1 mRNA stability in human cells. Elife. 6:e304332017.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Vila-Casadesús M, Gironella M and Lozano
JJ: MiRComb: An R package to analyse miRNA-mRNA interactions.
Examples across five digestive cancers. PLoS One. 11:e01511272016.
View Article : Google Scholar
|
|
19
|
Lan X, Zhang H, Wang Z, Dong W, Sun W,
Shao L, Zhang T and Zhang D: Genome-wide analysis of long noncoding
RNA expression profile in papillary thyroid carcinoma. Gene.
569:109–117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Prensner JR and Chinnaiyan AM: The
emergence of lncRNAs in cancer biology. Cancer Discov. 1:391–407.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Esteller M: Non-coding RNAs in human
disease. Nat Rev Genet. 12:861–874. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hulstaert E, Brochez L, Volders PJ,
Vandesompele J and Mestdagh P: Long non-coding RNAs in cutaneous
melanoma: Clinical perspectives. Oncotarget. 8:43470–43480. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Botchkareva NV: The molecular revolution
in cutaneous biology: Noncoding RNAs: New molecular players in
dermatology and cutaneous biology. J Invest Dermatol.
137:e105–e111. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Biehs B, Dijkgraaf GJ, Piskol R, Alicke B,
Boumahdi S, Peale F, Gould SE and de Sauvage FJ: A cell identity
switch allows residual BCC to survive Hedgehog pathway inhibition.
Nature. 562:429–433. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lobl M, Hass B, Clarey D, Higgins S,
Sutton A and Wysong A: Basal cell carcinoma gene mutations differ
between Asian, hispanic, and caucasian patients: A pilot study. J
Drugs Dermatol. 20:504–510. 2021.PubMed/NCBI
|
|
26
|
Stacey SN, Helgason H, Gudjonsson SA,
Thorleifsson G, Zink F, Sigurdsson A, Kehr B, Gudmundsson J, Sulem
P, Sigurgeirsson B, et al: New basal cell carcinoma susceptibility
loci. Nat Commun. 6:68252015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Saijo S, Kuwano Y, Tange S, Rokutan K and
Nishida K: A novel long non-coding RNA from the HOXA6-HOXA5 locus
facilitates colon cancer cell growth. BMC Cancer. 19:5322019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jeannotte L, Gotti F and Landry-Truchon K:
Hoxa5: A key player in development and disease. J Dev Biol.
4:132016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Aalijahan H and Ghorbian S: Long
non-coding RNAs and cervical cancer. Exp Mol Pathol. 106:7–16.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Abbastabar M, Sarfi M, Golestani A and
Khalili E: lncRNA involvement in hepatocellular carcinoma
metastasis and prognosis. EXCLI J. 17:900–913. 2018.PubMed/NCBI
|
|
31
|
Zhang J, Le TD, Liu L and Li J: Inferring
and analyzing module-specific lncRNA-mRNA causal regulatory
networks in human cancer. Brief Bioinform. 20:1403–1419. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Du Y, Xia W, Zhang J, Wan D, Yang Z and Li
X: Comprehensive analysis of long noncoding RNA-mRNA co-expression
patterns in thyroid cancer. Mol Biosyst. 13:2107–2115. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lim LJ, Jin Y, Yang H, Chung AYF, Goh BKP,
Chow PKH, Chan CY, Blanks WK, Cheow PC, Lee SY, et al: Network of
clinically-relevant lncRNAs-mRNAs associated with prognosis of
hepatocellular carcinoma patients. Sci Rep. 10:111242020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo Q, Cheng Y, Liang T, He Y, Ren C, Sun
L and Zhang G: Comprehensive analysis of lncRNA-mRNA co-expression
patterns identifies immune-associated lncRNA biomarkers in ovarian
cancer malignant progression. Sci Rep. 5:176832015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tao S, Zhu L, Lee P, Lee WM, Knox K, Chen
J, Di YP and Chen Y: Negative control of TLR3 signaling by TICAM1
down-regulation. Am J Respir Cell Mol Biol. 46:660–667. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sun R, Zhang Y, Lv Q, Liu B, Jin M, Zhang
W, He Q, Deng M, Liu X, Li G, et al: Toll-like receptor 3 (TLR3)
induces apoptosis via death receptors and mitochondria by
up-regulating the transactivating p63 isoform alpha (TAP63alpha). J
Biol Chem. 286:15918–15928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chiorazzi M, Rui L, Yang Y, Ceribelli M,
Tishbi N, Maurer CW, Ranuncolo SM, Zhao H, Xu W, Chan WC, et al:
Related F-box proteins control cell death in Caenorhabditis elegans
and human lymphoma. Proc Natl Acad Sci USA. 110:3943–3948. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Del Puerto-Nevado L, Santiago-Hernandez A,
Solanes-Casado S, Gonzalez N, Ricote M, Corton M, Prieto I, Mas S,
Sanz AB, Aguilera O, et al: Diabetes-mediated promotion of colon
mucosa carcinogenesis is associated with mitochondrial dysfunction.
Mol Oncol. 13:1887–1897. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kontro H, Cannino G, Rustin P, Dufour E
and Kainulainen H: DAPIT over-expression modulates glucose
metabolism and cell behaviour in HEK293T cells. PLoS One.
10:e01319902015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Huang YJ, Jan YH, Chang YC, Tsai HF, Wu
AT, Chen CL and Hsiao M: ATP synthase subunit epsilon
overexpression promotes metastasis by modulating AMPK signaling to
induce epithelial-to-mesenchymal transition and is a poor
prognostic marker in colorectal cancer patients. J Clin Med.
8:10702019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deng S, Li Y, Yi G, Lei B, Guo M, Xiang W,
Chen Z, Liu Y and Qi S: Overexpression of COX7A2 is associated with
a good prognosis in patients with glioma. J Neurooncol. 136:41–50.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Adamkov M, Halasova E, Rajcani J, Bencat
M, Vybohova D, Rybarova S and Galbavy S: Relation between
expression pattern of p53 and survivin in cutaneous basal cell
carcinomas. Med Sci Monit. 17:BR74–BR80. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chipuk JE, Kuwana T, Bouchier-Hayes L,
Droin NM, Newmeyer DD, Schuler M and Green DR: Direct activation of
Bax by p53 mediates mitochondrial membrane permeabilization and
apoptosis. Science. 303:1010–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|