Introduction
Sporotrichosis, an implantation mycosis caused
mainly by the dimorphic fungus Sporothrix schenckii
(S. schenckii), has gained attention over the two
last decades due to its broad geographic range and prevalence in
tropical and subtropical areas (1,2).
Sporotrichosis, which is caused by transcutaneous trauma, may
progress into chronic cutaneous, subcutaneous or even deeper
infections that may involve lymphatic tissue, fascia, muscles,
cartilage and bones (1,3). After S. schenckii
implants in a host via skin wounds, the marked changes in its
environment, including in the temperature, pH, osmotic pressure and
nutrients, exert pressure, causing it to adapt to the new
environment by transforming from the mycelium phase to the yeast
phase and settling down (4).
Dimorphic switching, which requires that the fungus sense and
respond to stimuli in the host environment, is necessary for
establishing its pathogenicity (5).
A number of signaling pathways, including the two-component and
heterotrimeric G-protein signaling systems, as well as Ras and cAMP
signaling and the downstream mitogen-activated protein kinase
(MAPK) cascades have been found to induce or influence the
dimorphic switch (6–11). Dozens of genes in these signaling
pathways in Histoplasma capsulatum, Talaromyces marneffei,
Blastomyces dermatitidis and Paracoccidioides
brasiliensis, four dimorphic pathogenic fungi with higher
morbidity and mortality rates than S. schenckii have
been investigated to identify the key determinants of pathogenicity
and dimorphic switching using mutagenesis or RNAi knockdown
techniques (6,10,12,13).
In our previous study, it was demonstrated that SsDRK1 in the
two-component system and SsSte20 in the Ras signaling pathway were
overexpressed during the early yeast stage, but not in the mycelial
stage, of S. schenckii using two-dimensional
electrophoresis (14). Furthermore,
it was demonstrated that SsDRK1 is essential for normal asexual
development, yeast-phase cell formation, cell wall composition and
integrity, and melanin synthesis using double-stranded RNA
interference mediated by Agrobacterium tumefaciens (14–17).
However, the details of the signaling pathways controlling
dimorphic switching remain unclear due to the limited literature
available regarding S. schenckii.
In the present study, the transcriptomics of the
48-h induced yeast and mycelial stages of S.
schenckii underwent transcriptome analysis. Signaling
pathways associated with the dimorphic switch, including the
two-component and heterotrimeric G-protein signaling systems, Ras,
and MAPK cascades were mapped using the Kyoto Encyclopedia of Genes
and Genomes (KEGG) based on comparative transcriptomic results
between 48-h induced yeast and mycelial cells. In addition, the
cell wall ultrastructural features of 48-h induced yeast cells were
compared with those of mycelial cells. The results provided novel
insights into the molecular mechanisms controlling the dimorphic
switch in S. schenckii.
Materials and methods
Fungal strain and culture
conditions
The strain of S. schenckii used,
ATCC10268, was maintained at the Research Center for Pathogenic
Fungi, Liaoning University, China. To obtain a mycelial culture,
the ATCC10268 isolate was inoculated onto Sabouraud dextrose agar
(SDA) solid medium (10 g/l tryptone, 40 g/l glucose) and incubated
at 25°C. The mycelial colonies subsequently obtained were
inoculated in liquid Sabouraud medium and cultured with shaking at
100 rpm at 25°C for 48 h. To induce the switch of S.
schenckii from the mycelial phase to the 48-h induced yeast
phase, mycelial culture was enriched and transferred to brain-heart
infusion (BHI) liquid medium (HyClone; GE Healthcare Life Sciences)
(18–23), which was incubated at 37°C and
shaken at 100 rpm for 48 h.
Transmission electron microscopy
(TEM)
S. schenckii cultures grown in SDA at
25°C or in BHI at 37°C for 48 h were collected, and the cell
suspensions were fixed at 4°C by addition of an equal volume of
fixing solution (0.2 M Na2HPO4, 0.2 M
NaH2PO4) for 2–4 h. Cells were transferred to
a centrifuge tube and spun at 12,000 × g for 15 min at 4°C to
obtain a cell pellet, which was embedded in 1% agarose and then
washed in 0.1 M PBS three times for 15 min each. Post-fixation
staining was carried out with 1% OsO4 in 0.1 M PBS (pH
7.4) for 2 h at room temperature, followed by removal of
OsO4, rinsing in 0.1 M PBS (pH 7.4) three times, and
dehydration. Following infiltration and embedding the cell pellet,
ultrathin sections (60–80 nm) were cut with an ultramicrotome.
Sections were stained with uranyl acetate in pure ethanol for 15
min at 4°C rinsed with distilled water, stained with lead citrate
for 15 min at 4°C, and rinsed with distilled water. The sections
were allowed to air-dry overnight and were then observed using TEM
(24). The thicknesses of the inner
and outer layers of the mycelial cell wall were measured using
Image-Pro Plus 6.0 (Media Cybernetics, Inc.).
Fluorescence staining of fungi
S. schenckii cultured in SDA at 25°C
or in BHI at 37°C for 48 h were smeared onto slides and stained
with fungal fluorescence dyes (Lifetime bio) that bind specifically
to chitin and cellulose for 15 min at 4°C. The slides were
visualized using a Nikon 2000 fluorescent microscope (Nikon
Instruments, Inc.) with a ×20 objective. For each slide, 10
microscopic fields were examined and the fields with median
fluorescent brightness selected for comparation. Images were
processed with NIS-Elements (version 4.10; Nikon Instruments, Inc.)
imaging software.
cDNA library construction and
sequencing
S. schenckii in mycelial and 48-h
induced yeast forms were collected for RNA extraction. Total RNA
was extracted using RNAiso™ plus (Takara Bio, Inc.) and treated
with RNase-free DNase I (Fermentas; Thermo Fisher Scientific, Inc.)
to remove residual DNA. The quantity of RNA was determined using
NanoDrop ND-1000 (Thermo Fisher Scientific, Inc.) and 1.2% agarose
gels. The integrity of the total RNA was assessed using an Agilent
2200 Tape Station (Agilent Technologies, Inc.), and each sample had
an RNA integrity number >7.5.
cDNA libraries were constructed using the
NEBNext® Ultra™ RNA Library Prep Kit for Illumina
(Illumina, Inc.) according to the manufacturer's protocols. After
the total RNA was extracted, the mRNA was purified using poly-T
oligo-attached magnetic beads and fragmented into small pieces in
fragmentation buffer from the kit. Using mRNA as a template,
first-strand cDNA was synthesized using random primers and reverse
transcriptase. Second-strand cDNA was synthesized using RNase H and
DNA polymerase I (Takara Bio, Inc.) (25). Subsequently, the cDNA was purified,
and subjected to end-repair, poly(A) addition, sequencing, adapter
connecting and fragment size selection. Finally, the purified cDNA
was PCR amplified and sequenced on the Illumina HiSeq™ Xten
platform (Guangzhou RiboBio Co., Ltd.).
Transcriptome de novo assembly and
annotation
After analyzing the base composition and quality
value, the sequencing data were filtered according to the raw data
analysis results to remove the adaptor sequences, contaminated
parts and low-quality reads to obtain clean reads. Next, clean
reads were de novo assembled using Trinity (version no.
v2013-02-25; Trinity Software, Inc.). Trinity software consists of
three independent software modules, Inchworm, Chrysalis and
Butterfly, which are used in turn to process large-scale RNA-Seq
read data and obtain unigene sequences.
After assembly, the unigene sequences were annotated
into the NCBI non-redundant (NR; http://www.ncbi.nlm.nih.gov/), Swiss-Prot (http://www.expasy.ch/spot/), Clusters of eukaryotic
Orthologous Group (KOG; http://www.ncbi.nlm.nih.gov/cog/) and Kyoto
Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg) databases using the
BLASTALL package (release 2.2.28) from NCBI with an E-value
≤10−5. Based on NR annotation, GO functional annotation
and further functional classification were performed using Blast2GO
(http://www.blast2go.com/b2ghome).
Identification of differentially
expressed genes (DEGs) and functional analysis
Bowtie2 (version no. 2.2.5) was used to map the
clean reads back to the unigenes, and the unigene mapping rate was
counted. The unigene expressed values and transcript levels were
calculated by the fragments per kilobase of transcript per million
mapped reads (FPKM) method using the mapping results (26). EdgeR (version no. 3.10.0) was used
to calculate the expression difference, and multiple hypothesis
testing was performed to correct the P-value of the difference
test. The threshold P-value was determined by controlling the false
discovery rate (FDR). When the FDR value of the difference test was
obtained, the differential expression multiples of the gene in
different samples were calculated according to the amount of gene
expression, using |log2FC| ≥1, and FDR <0.05 as the
threshold for screening differential genes (27).
The hypergeometric distribution test was used for GO
classification and KEGG analysis of the DEGs to understand the
functional properties of the genes and regulatory pathways
involved. The obtained GO categories with P<0.05 and pathways
with q-value ≤0.05 were defined as significantly enriched GO
classifications and KEGG pathways (28).
Statistical analysis
One-way analysis of variance was performed using
SPSS 17.0 (SPSS, Inc.). Differences were tested with Duncan's test,
unless otherwise specified.
Results
Cell wall structures
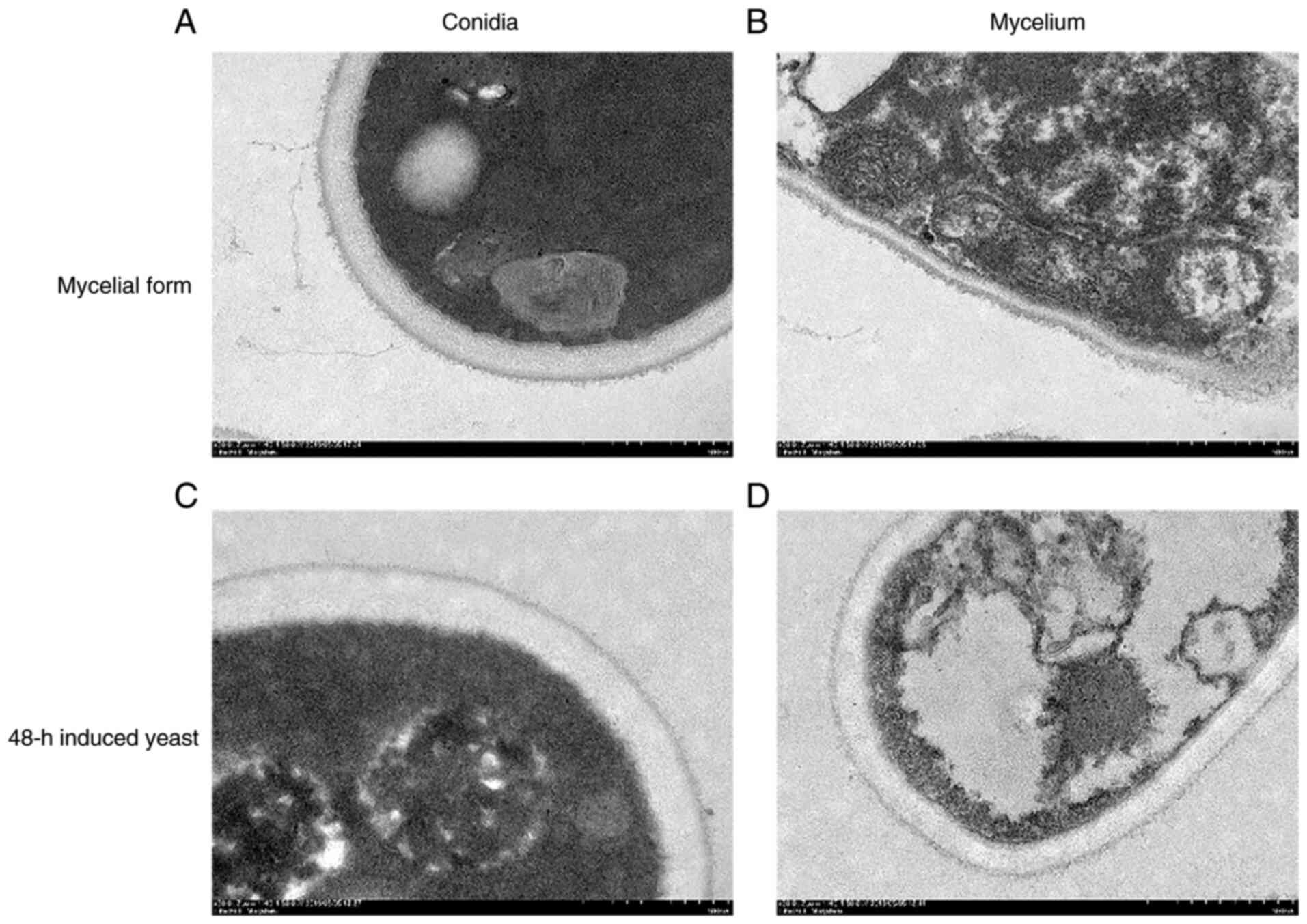
The structural organization of hyphal and conidial
cell walls of S. schenckii in the mycelial and 48-h
induced yeast forms was studied using high-pressure freezing TEM
(HPF-TEM). Fewer hyphal fragments and larger conidia were observed
when comparing the character of 48-h induced yeast cells with that
of mycelial cells scanned with HPF-TEM. The cell wall of S.
schenckii was observed as having a double-layered structure,
with a low electron-density inner layer and a high electron-density
outer layer, decorated with thin fibrils. The inner layer
thicknesses of the S. schenckii mycelial cell wall
was ~102 nm, thinner than the 151 nm in 48-h induced yeast cells,
which decreased the total thickness of the mycelial cell wall.
Meanwhile, compared with the 48-h induced yeast form, the mycelial
cell walls showed a thicker outer layer densely covered with fine
fibrils (Fig. 1).
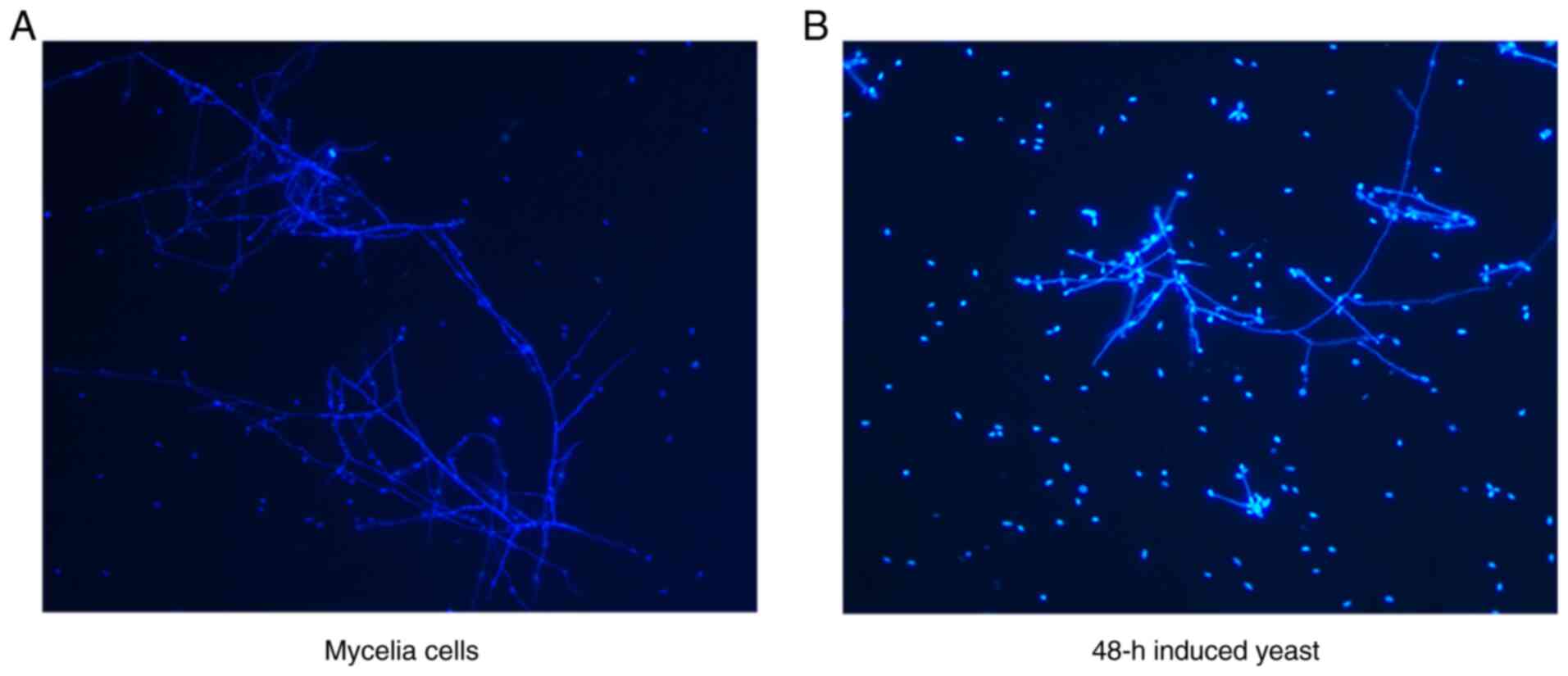
To confirm the difference in cell wall structure
between the mycelial and 48-h induced yeast forms, the cells were
stained with Calcofluor White, targeting the chitin in the cell
wall of S. schenckii. The fluorescence pattern
observed in 48-h induced yeast cells, compared with that in
mycelial cells, had stronger chitin and cellulose labeling patterns
on the 48-h induced yeast cell surface (Fig. 2).
Transcriptome sequencing data
processing and assembly result statistics
Following sequencing two samples of the mycelial and
48-h induced yeast phases using the Illumina platform, the mycelial
phase samples were sequenced to obtain 27,465,858 reads and 4.12 G
data were obtained, in which the Q20 and Q30 base proportions were
95.70 and 90.91%, respectively. The GC content proportion was
54.84% and the N ratio in sequencing was 0.33%. Sequencing the 48-h
induced yeast samples gave 23,746,248 reads and 3.56 G of data,
with Q20 and Q30 base ratios of 97.66 and 94.69%, respectively, a
GC content ratio of 46.80%, and N ratio in sequencing of 0.18%.
Subsequently, quality filtration of the raw data was performed.
Mycelial phase samples gave 24,904,510 clean reads, and 48-h
induced yeast phase samples gave 22,814,406 clean reads (Table I).
 | Table I.Sequencing data statistics. |
Table I.
Sequencing data statistics.
| Sample | Raw reads | Q20(%) | Q30(%) | Err(%) | GC% | Clean reads |
|---|
| Mycelial | 27,465,858 | 95.70 | 90.91 | 0.33 | 54.84 | 24,904,510 |
| 48-h induced
yeast | 23,746,248 | 97.66 | 94.69 | 0.18 | 46.80 | 22,814,406 |
Trinity was used to assemble high-quality sequences
in the samples. Following assembly, 31,779 unigene sequences were
obtained. The total number of sequences generated was 72.2 Mbp, and
the N50 and N90 lengths were 4,155 and 993 bp, respectively. The
Max-length and Min-length of the unigenes were 29,705 and 301 bp,
respectively, and the GC content was 52.98% (Table II). The transcriptomic data
supporting the results are available at NCBI under GEO accession
number GSE133322.
| Table II.Assembly result statistics. |
Table II.
Assembly result statistics.
| Total_sequence | Total_base | N50 | N90 | Max_length | Min_length | GC(%) |
|---|
| 31,779 | 72,164,889 | 4,155 | 993 | 29,705 | 301 | 52.98 |
Transcriptome data functional
annotation
For functional annotation analysis, BLAST was used
to align the assembled unigenes with five public databases.
Overall, 23,329 (73.41%) unigenes could be aligned in one or more
databases: 21,079 (66.33%) unigenes were similar to proteins in the
NR database and 17,398 (54.75%) were annotated in Swiss-Prot
(Table III). For GO annotation,
10,542 unigenes were assigned to the ‘cellular component’ category
and ‘integral membrane component’ (n=2,808), ‘nucleus’ (n=1,604)
and ‘cytosol’ (n=1442) were other main subcategories. In addition,
11,834 unigenes were classified as ‘biological processes’; the
three main categories were ‘metabolic process’ (n=1,373),
‘oxidation-reduction process’ (n=1,117) and ‘transmembrane
transport’ (706). Furthermore, 12,149 unigenes were assigned to the
‘molecular function’ category and genes assigned to ‘ATP binding’
(n=1,588) and ‘metal ion binding’ (747) accounted for the vast
majority of this category. In the KOG classification, 13,854
unigenes were categorized into 25 KOG functional groups; ‘General
function prediction only’ was the largest group, followed by
‘Post-translational modification, protein turnover, chaperones’ and
‘Translation, ribosomal structure and biogenesis’. In the KEGG
pathway analysis, 11,062 unigenes could be mapped to 342 metabolic
pathways and the largest category was ‘Carbon metabolism’
(n=424).
| Table III.Transcriptome data functional
annotation statistics. |
Table III.
Transcriptome data functional
annotation statistics.
| Annotation
Database | Number of
unigenes | Percentage (%) |
|---|
| Nr | 21,079 |
66.33 |
| Swissprot | 17,398 |
54.75 |
| KOG | 13,854 |
43.59 |
| KEGG | 11,062 |
34.81 |
| In at least one
database | 23,329 |
73.41 |
| Total unigenes | 31,779 | 100.0 |
Analysis of transcriptome expression
in the mycelium and 48-h induced yeast forms
The sequences of sample reads and unigenes were
compared using bowtie2; this aligned 22,315,744 (89.00%) reads of
the mycelial phase and 19,552,494 (85.00%) reads of the 48-h
induced yeast phase to unigene sequences.
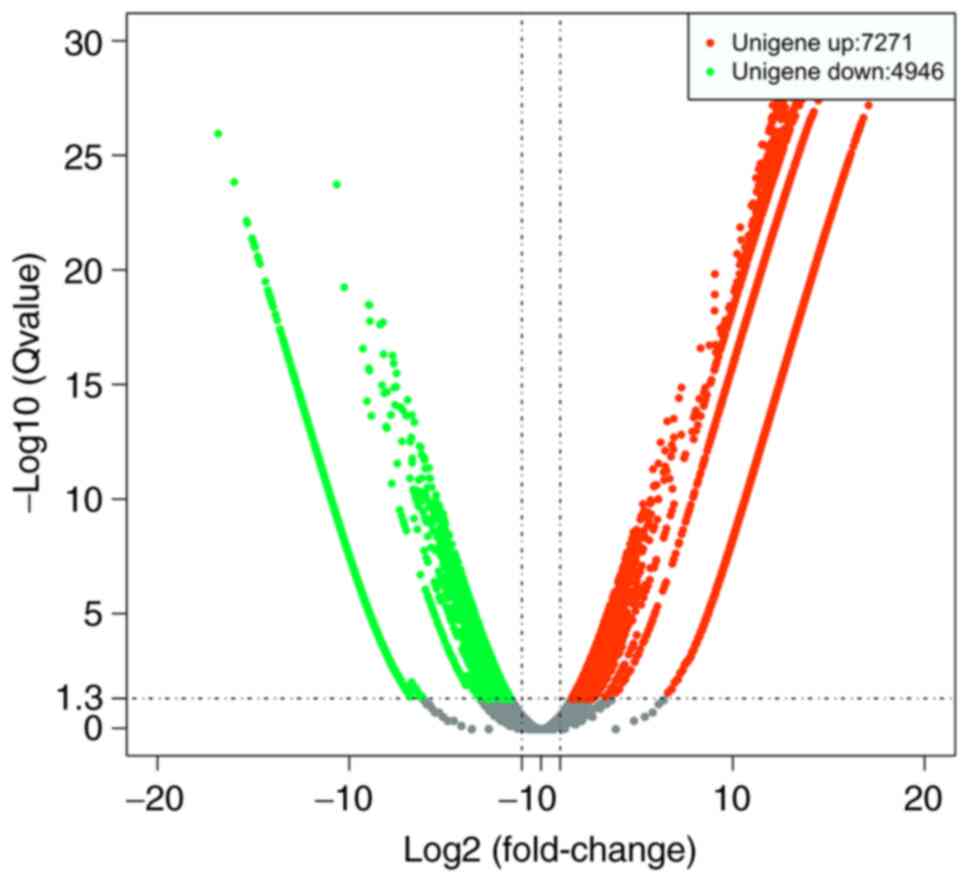
Next, DEGs were identified by comparing the FPKM
values for each gene between the mycelial and 48-h induced yeast
forms of S. schenckii, and DEGs between the two forms
were identified. The results demonstrated that 12,217 genes were
expressed differentially between the two forms, including 7,271
upregulated and 4,946 downregulated genes (Fig. 3).
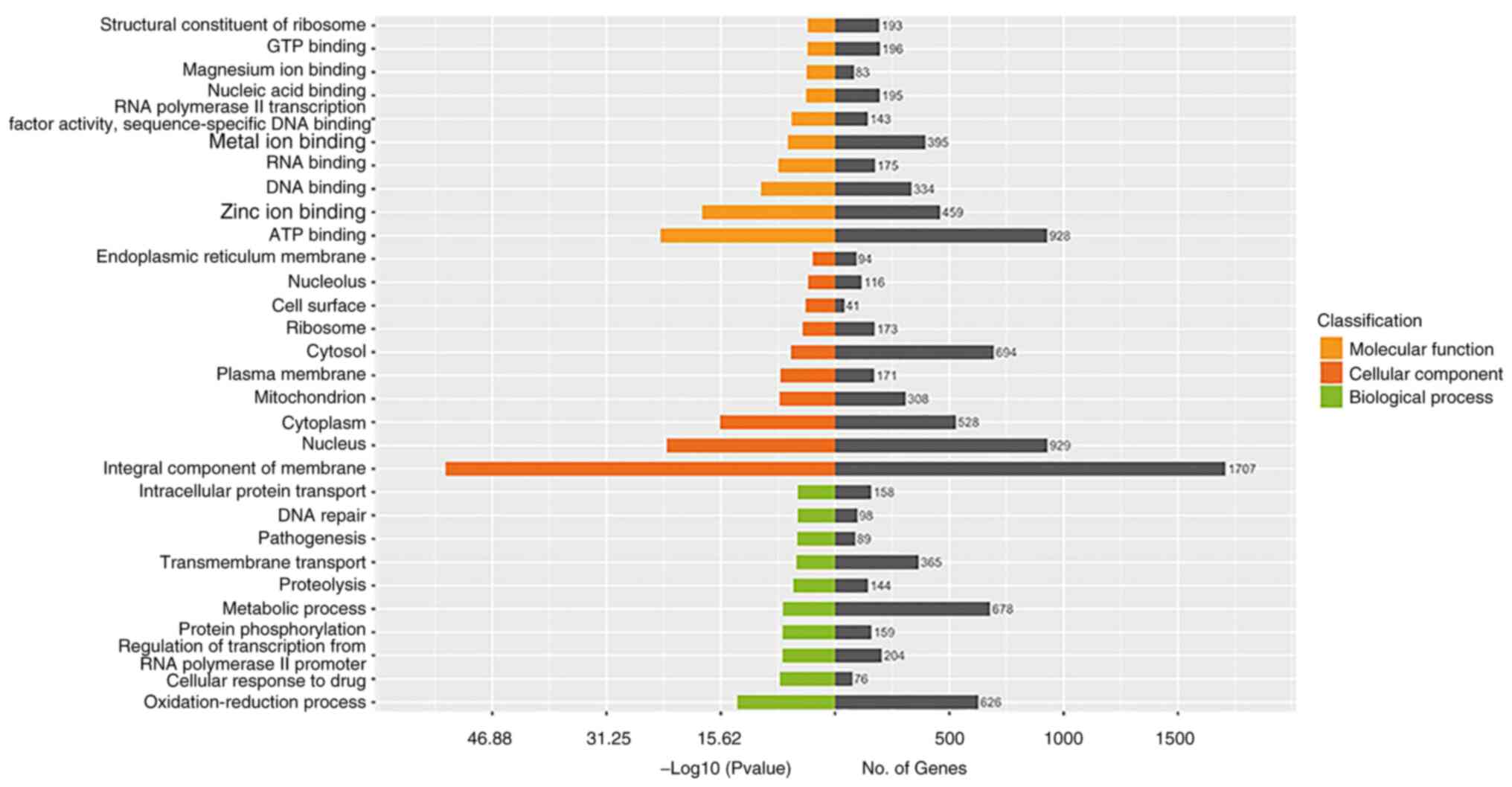
GO analysis of DEGs
To identify the major functional categories
represented by DEGs, with all unigene genes as background genes,
the P-value was calculated using hypergeometric distribution
method, and P<0.05 was taken as the threshold to obtain
significant heights relative to the background (29). Among DEGs between the mycelial and
48-h induced yeast forms, GOs associated with ATP binding, metal
ion binding and zinc ion binding in the molecular function
category; GOs associated with metabolic processes, oxidation
reduction reactions and transmembrane transport in the biological
process category; and particularly those associated with integral
components of the membrane, nucleus, cytosol, cytoplasm and
mitochondrion in the cellular component group were significantly
changed (Fig. 4).
During the process of switching, the form and
function of the cell change notably, and those changes must be
supported by changes in cell components, including cytoplasmic
proteins, nucleoproteins, regulatory proteins, toxic proteins and
cell wall component proteins. This led to the observation that
genes associated with the integral components of the membrane
changed most significantly. These changes require large amounts of
protein and energy for anabolism, as well as raw materials absorbed
from the environment; therefore, genes associated with metabolic
processes, oxidation-reduction processes, transmembrane transport,
and the steps of transcription and translation, including ATP
binding, metal ion binding, zinc ion binding and DNA binding,
changed markedly.
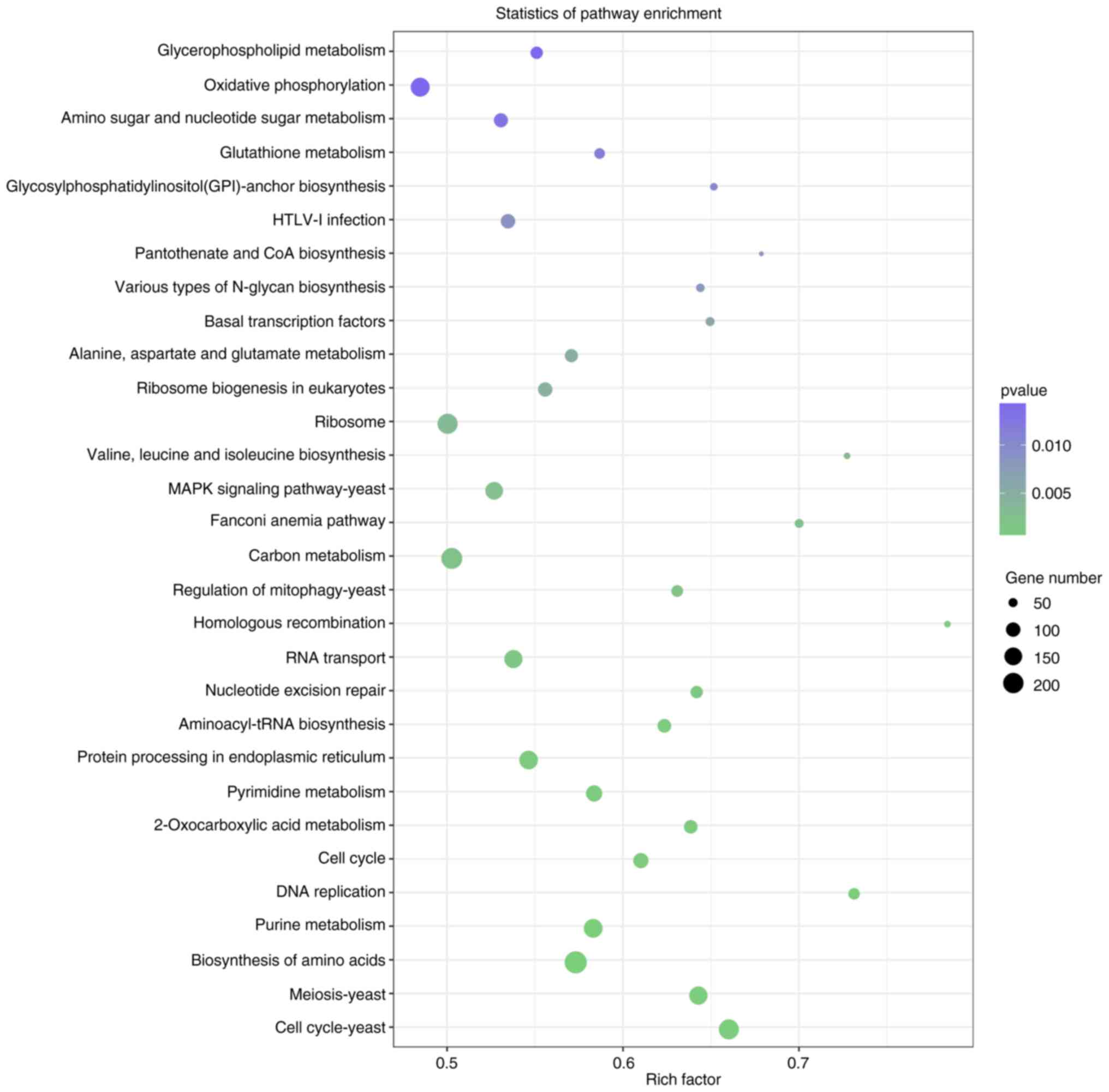
KEGG pathway enrichment of DEGs
To further elucidate the molecular interactions
among DEGs, KEGG analysis was performed (30). Among DEGs between the mycelial and
48-h induced yeast forms of S. schenckii, pathways
for oxidative phosphorylation, ribosome, the MAPK pathway, carbon
metabolism, RNA transport, protein processing in endoplasmic
reticulum, pyrimidine metabolism, purine metabolism, biosynthesis
of amino acids, meiosis and the cell cycle were enriched (Fig. 5). During the process of switching,
enormous changes must occur in the expression of the genes in these
pathways, which are necessary for transcription, translation,
biosynthesis and energy synthesis. Beyond those predictable
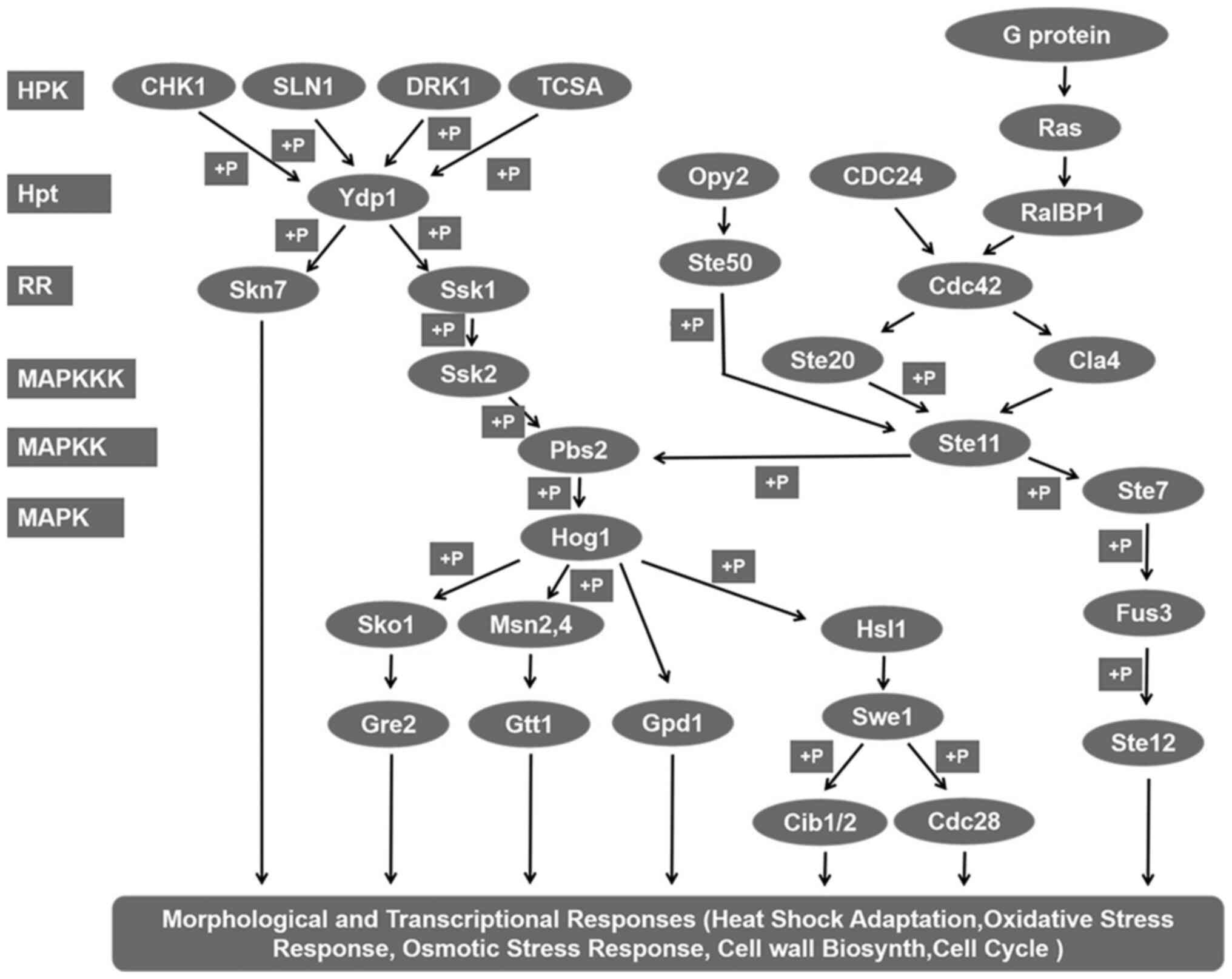
changes, it was also found that numerous genes, including DRK1,
Hog1, Skn7 and Ste11, which are involved in the two-component
system heterotrimeric G protein, cAMP and Ras-Hog1 signaling
pathways, were altered (Table IV;
Fig. 6). These signal transduction
pathways serve important roles during the initiation of the
dimorphic switch.
| Table IV.Genes in signaling pathways
associated with dimorphic switching identified in the transcriptome
of S. schenckii. |
Table IV.
Genes in signaling pathways
associated with dimorphic switching identified in the transcriptome
of S. schenckii.
| Gene | Log FC | P-value |
|---|
| CDC24 | 12.49721754 |
3.88×10−17 |
| cdc28 | −2.38905262 |
1.72×10−3 |
| cdc42 | 12.12659431 |
3.49×10−24 |
| chk1 | 10.97359207 |
5.54×10−24 |
| cla4 | 11.87930169 |
2.61×10−15 |
| drk1 | 8.876696836 |
5.43×10−07 |
| gpd1 |
10.2951049 |
9.25×10−11 |
| gre2 | 11.98573676 |
9.21×10−24 |
| hog1 | 13.35994596 |
1.06×10−19 |
| hsl1 | 12.78354566 |
5.48×10−18 |
| msn2,4 | 13.46928334 |
5.00×10−20 |
| opy2 | 10.87439311 |
2.16×10−12 |
| pbs2 | 10.66018548 |
8.56×10−20 |
| ralbp1 | 2.161074365 |
1.65×10−3 |
| skn7 | 3.663787304 |
3.75×10−06 |
| sko1 | 12.57430068 |
2.29×10−17 |
| sln1 | 14.1849827 |
3.60×10−22 |
| ssk1 | −3.84358211 |
3.69×10−05 |
| ssk2 | 12.08651326 |
1.00×10−26 |
| ste11 | 3.923507879 |
1.46×10−07 |
| ste12 | −3.680198317 |
5.13×10−07 |
| ste20 | 13.90389877 |
2.50×10−21 |
| ste50 | 11.67340917 |
1.49×10−27 |
| ste7 | 12.45175501 |
5.34×10−17 |
| swe1 | 9.460635913 |
1.83×10−08 |
| Tcsa | −2.369133725 |
1.06×10−2 |
| Ypd1 | 10.77251106 |
3.94×10−20 |
Discussion
To date, little is known regarding the factors
driving the pathogenesis of sporotrichosis despite its worldwide
prevalence, which hinders medical control of this disease (31). Similar to other pathogenic fungi,
host signals that trigger the dimorphic switch are transmitted via
signaling pathways and ultimately culminate in changes in gene
expression. However, the network of signaling pathways involved in
dimorphic switching in S. schenckii remain enigmatic. The
present study found that this species exhibits the morphological
and structural characteristics of the 48-h induced yeast phase when
cultured in BHI medium at 37°C for 48 h. Therefore, the
transcription profile of that culture was compared with that of the
same strain in the mycelial phase using an RNA-seq method. The
results of the present study revealed that more than 12,217 genes
were significantly upregulated or downregulated in the early yeast
phase. Among these genes, many encode signal transduction proteins
in the two-component and heterotrimeric G protein, Ras and cAMP
signaling pathways and the MAPK cascade, suggesting that these
signal transduction pathways serve important roles during the
initiation of the dimorphic switch. The results of the present
study provided a molecular basis for the development of S.
schenckii control strategies targeting genes in signaling
pathways associated with the dimorphic switch.
In fungi, the two-component system comprises a
membrane-associated histidine kinase (HK) and a cytoplasmic
response regulator (RR), also known as a hybrid HK (HHK). The HK
perceives an environmental stimulus and is autophosphorylated at a
conserved histidine in the kinase domain. Next, the phosphate group
is transferred to a conserved aspartate in the receiver domain of
the RR, and further phosphorelay occurs through an additional
phosphotransfer protein (HPt) and a second response regulator. To
date, 11 classes of HHKs (I–XI) in fungi have been identified that
are involved in signal phosphorylation and transmission to two RRs,
orthologous to Ssk1 and Skn7, via a single HPt, orthologous to Ypd1
(32). The results of the present
study revealed that four HKs, orthologous to CHK1 (class VIII),
Sln1 (class VI), DRK1 (class III) and TCSA (class IV), as well as
two RRs, orthologous to Ssk1 and Skn7, were significantly
upregulated or downregulated in the 48-h induced yeast stage
compared with the mycelial stage, indicating that they are involved
in inducing the dimorphic switch in S. schenckii. Further
KEGG analysis suggested that all four HHKs, orthologous to CHK1,
Sln1, DRK1 and TCSA, could phosphorylate Ssk1/Skn7 via Ypd1,
resulting in constitutive activation of the downstream Hog1 MAPK
pathway. Several studies, including our previous study focusing on
class III HHK (Os-1 ortholog) in human dimorphic pathogens, have
revealed conserved roles in fungicide resistance, osmotic stress
resistance and pathogenicity, in addition to roles in cell wall
integrity, asexual development and dimorphism (6,17,33,34).
Furthermore, SlnA (class VI) in T. marneffei, Chk1 (class
VIII) in C. albicans, and TcsA (class IV) in Aspergillus
nidulans were revealed to be associated with osmotic stress
resistance, HOG MAPK regulation, the dimorphic transition and
development under standard growth conditions (7,35). As
multiple histamine kinases may compensate for each other in S.
schenckii (Fig. 2), SsDRK1
interference is confirmed to inhibit infection but not kill the
pathogenic yeast (17). Therefore,
research efforts should focus on pivotal downstream genes,
including Pbs2 or Hog1, deletion of which may be lethal, as targets
for novel drug development. Our group are creating Pbs2 and Hog1
mutant strains to further investigate the function of vital genes
in controlling the dimorphic switch of S. schenckii
and will publish the results in a future study.
Heterotrimeric G proteins and the downstream Ras
signaling pathways have been demonstrated to influence dimorphic
switching and adaptation to oxidative stress in dimorphic fungi, in
addition to regulating asexual development and conidial germination
(8,9,13,36).
Canonical heterotrimeric G proteins comprise three subunits (α, ß
and γ) and transmit signals from cell surface receptors. The S.
schenckii Ssg-1 Gα subunit interacts with proteins that are
necessary for survival under oxidative stress conditions and for
iron acquisition, and the Ssg-2 Gα subunit interacts with cytosolic
phospholipase A 2, which stimulates the yeast-to-hyphal dimorphic
switch and prevents re-entry into the yeast cell cycle (8,9).
Several genes in the Ras signaling pathway, including Ras, Rho
GTPase Cdc42 and the p21 activated kinases Ste20 and Cla4, were
significantly upregulated in the yeast culture, suggesting that
signal transmission via these genes from heterotrimeric G proteins
occurs during the early stage of the dimorphic switch. In response
to stimulation from heterotrimeric G proteins, GTPase-activating
proteins convert GTPases of the Ras superfamily from an inactive
GDP-bound form to an active GTP-bound form. Further KEGG analysis
in the present study indicated that activated Ras activates the Rho
GTPase Cdc42, a member of the Ras GTPase superfamily, which
regulates the MAPK pathway via Ste20 or Cla4. The results of the
present study further support the proposal that Cdc42 serves a
conserved role in regulating morphogenesis by controlling
actin-mediated polarized growth and signaling pathways that are
required for morphological responses in a variety of fungi
(37–39).
Cell wall glycoconjugates of pathogenic fungi, known
as pathogen-associated molecular patterns, or PAMPs, are involved
in virulence and pathogenicity. Previous studies have reported that
the S. schenckii cell wall is composed of a
peptido-rhamnomannan component, β-glucans and chitin, which are
involved in the innate immune response, binding to the
corresponding pattern-recognition receptors and triggering the
secretion of specific cytokines (40–42).
In the present study, fluorescence microscopy of 48-h cultured
yeast cells labeled with calcofluor white to target chitin
exhibited a stronger fluorescence labeling pattern than that of
mycelial cells, which may indicate a thicker cell wall at the
ultrastructural level during the initial yeast phase (43). Chitin, a β (1,4)-linked
homopolymer of N-acetylglucosamine, is an essential component of
the cell walls and septa of all pathogenic fungi, and is therefore
considered an attractive target for antifungal therapies (44). A variable proportion of fungal
chitin is synthesized and then deacetylated to chitosan through the
action of one or more chitin deacetylases. Consistent with the
fluorescence labeling findings, the transcriptome analysis in the
present study indicated that several genes involved in chitin
synthesis metabolism were distinctly upregulated in 48-h induced
yeast cells. In addition, activation of the HOG MAPK cascade
contributed toward the increased level of chitin in S.
schenckii; previous studies have demonstrated that HOG MAPK
cascades regulate chitin synthase gene expression and chitin
synthesis in response to cell wall stresses in two other dimorphic
fungi, S. cerevisiae and C. albicans (45,46).
Taken together, the transcriptome data and analysis
in the present study lay the foundation for further research into
the molecular mechanisms controlling the dimorphic switch of
S. schenckii and support the development of
anti-S. schenckii strategies targeting genes
associated with signaling pathways.
The present study is limited by the fact that the
early yeast Sporothrix cells preparation was performed in the BHI
instead of in an animal model. Although the most frequently
reported yeast-like Sporothrix cells are grown in BHI, the
influence of culture media on the phenotypical trait cannot be
ignored.
Acknowledgements
Not applicable.
Funding
This study was supported by National Natural Science
Foundation of China (grant no. 81472891) and Shenzhen Science and
Technology Innovation Commission Project (grant no.
JCYJ20180306173356306) which were distributed to Hong Kong
University Shenzhen Hospital.
Availability of data and materials
The transcriptomic data supporting the results of
this article are available at NCBI under GEO with accession number
GSE133322 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE133322).
Authors' contributions
ZZ and FZ designed the study, wrote the article and
confirm the authenticity of all the raw data. WG, QC and YW
performed the experiments, and QZ, XJ and BH analyzed the data. All
the authors read and approved the final version of this
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chakrabarti A, Bonifaz A,
Gutierrez-Galhardo MC, Mochizuki T and Li S: Global epidemiology of
Sporotrichosis. Med Mycol. 53:3–14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopes-Bezerra LM, Mora-Montes HM, Zhang Y,
Nino-Vega G, Rodrigues AM, de Camargo ZP and de Hoog S:
Sporotrichosis between 1898 and 2017: The evolution of knowledge on
a changeable disease and on emerging etiological agents. Med Mycol.
56 (Suppl 1):S126–S143. 2018. View Article : Google Scholar
|
|
3
|
Arenas R, Sánchez-Cardenas CD,
Ramirez-Hobak L, Ruíz Arriaga LF and Vega Memije ME:
Sporotrichosis: From KOH to molecular biology. J Fungi (Basel).
4:622018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bruno VM, Wang Z, Marjani SL, Euskirchen
GM, Martin J, Sherlock G and Snyder M: Comprehensive annotation of
the transcriptome of the human fungal pathogen Candida
albicans using RNA-seq. Genome Res. 20:1451–1458. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Boyce KJ and Andrianopoulos A: Fungal
dimorphism: The switch from hyphae to yeast is a specialized
morphogenetic adaptation allowing colonization of a host. FEMS
Microbiol Rev. 39:797–811. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nemecek JC, Wüthrich M and Klein BS:
Global control of dimorphism and virulence in fungi. Science.
312:583–588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boyce KJ, Schreider L, Kirszenblat L and
Andrianopoulos A: The two-component histidine kinases DrkA
and SlnA are required for in vivo growth in the human
pathogen Penicillium marneffei. Mol Microbiol. 82:1164–1184.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Valentín-Berríos S, González-Velázquez W,
Pérez-Sánchez L, González-Méndez R and Rodríguez-Del Valle N:
Cytosolic phospholipase A2: A member of the signalling pathway of a
new G protein alpha subunit in Sporothrix schenckii. BMC
Microbiol. 9:1002009. View Article : Google Scholar
|
|
9
|
Pérez-Sánchez L, González E, Colón-Lorenzo
EE, González-Velázquez W, González-Méndez R and Rodríguez-del Valle
N: Interaction of the heterotrimeric G protein alpha subunit SSG-1
of Sporothrix schenckii with proteins related to stress
response and fungal pathogenicity using a yeast two-hybrid assay.
BMC Microbiol. 10:3172010. View Article : Google Scholar
|
|
10
|
Boyce KJ, Schreider L and Andrianopoulos
A: In vivo yeast cell morphogenesis is regulated by a p21-activated
kinase in the human pathogen Penicillium marneffei. PLoS
Pathog. 5:e10006782009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen D, Janganan TK, Chen G, Marques ER,
Kress MR, Goldman GH, Walmsley AR and Borges-Walmsley MI: The cAMP
pathway is important for controlling the morphological switch to
the pathogenic yeast form of Paracoccidioides brasiliensis.
Mol Microbiol. 65:761–779. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Almeida AJ, Cunha C, Carmona JA,
Sampaio-Marques B, Carvalho A, Malavazi I, Steensma HY, Johnson DI,
Leão C, Logarinho E, et al: Cdc42p controls yeast-cell shape and
virulence of Paracoccidioides brasiliensis. Fungal Genet
Biol. 46:919–926. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zuber S, Hynes MJ and Andrianopoulos A:
The G-protein alpha-subunit GasC plays a major role in germination
in the dimorphic fungus Penicillium marneffei. Genetics.
164:487–499. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Z, Hou B, Xin Y and Liu X: Protein
profiling of the dimorphic, pathogenic fungus, Sporithrix
schenckii. Mycopathologia. 173:1–11. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hou B, Zhang Z, Zheng F and Liu X:
Molecular cloning, characterization and differential expression of
DRK1 in Sporothrix schenckii. Int J Mol Med. 31:99–104.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Z, Hou B, Wu YZ, Wang Y, Liu X and
Han S: Two component histidine kinase DRK1 is required for
pathogenesis in Sporothrix schenckii. Mol Med Rep.
17:721–728. 2018.PubMed/NCBI
|
|
17
|
Zhang Z, Hou B, Zheng F, Yu X and Liu X:
Molecular cloning, characterization and differential expression of
a Sporothrix schenckii STE20-like protein kinase SsSte20.
Int J Mol Med. 31:1343–1348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lozoya-Pérez NE, Clavijo-Giraldo DM,
Martínez-Duncker I, García-Carnero LC, López-Ramírez LA, Niño-Vega
GA and Mora-Montes HM: Influences of the culturing media in the
virulence and cell wall of Sporothrix schenckii, sporothrix
brasiliensis, and sporothrix globosa. J Fungi (Basel). 6:3232020.
View Article : Google Scholar
|
|
19
|
de Almeida JRF, Jannuzzi GP, Kaihami GH,
Breda LCD, Ferreira KS and de Almeida SR: An immunoproteomic
approach revealing peptides from sporothrix brasiliensis that
induce a cellular immune response in subcutaneous sporotrichosis.
Sci Rep. 8:41922018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Della Terra PP, Rodrigues AM, Fernandes
GF, Nishikaku AS, Burger E and de Camargo ZP: Exploring virulence
and immunogenicity in the emerging pathogen sporothrix
brasiliensis. PLoS Negl Trop Dis. 11:e00059032017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kong X, Xiao T, Lin J, Wang Y and Chen HD:
Relationships among genotypes, virulence and clinical forms of
Sporothrix schenckii infection. Clin Microbiol Infect.
12:1077–1081. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Teixeira PAC, de Castro RA, Nascimento RC,
Tronchin G, Perez Torres A, Lazera M, de Almeida SR, Bouchara JP,
Loureiro Y, Penha CV and Lopes-Bezerra LM: Cell surface expression
of adhesins for fibronectin correlates with virulence in
Sporothrix schenckii. Microbiology (Reading). 155((Pt 11)):
3730–3738. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Brito MM, Conceição-Silva F, Morgado FN,
Raibolt PS, Schubach A, Schubach TP, Schäffer GM and Borba CM:
Comparison of virulence of different Sporothrix schenckii
clinical isolates using experimental murine model. Med Mycol.
45:721–729. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Garrison RG, Boyd KS and Mariat F:
Ultrastructural studies of the mycelium-to yeast transformation of
Sporothrix schenckii. J Bacteriol. 124:959–968. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang HY, Zhang JL, Yang JW and Ma HL:
Transcript profiling and gene identification involved in the
ethylene signal transduction pathways of creeping bentgrass
(Agrostis stolonifera) during ISR response induced by Butanediol.
Molecules. 23:7062018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang X, Gao B, Liu X, Dong X, Zhang Z, Fan
H, Zhang L, Wang J, Shi S and Tu P: Salinity stress induces the
production of 2-(2-phenylethyl)chromones and regulates novel
classes of responsive genes involved in signal transduction in
Aquilaria sinensis calli. BMC Plant Biol. 16:1192016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Qi X, Fang H, Yu X, Xu D, Li L, Liang C,
Lu H, Li W, Chen Y and Chen Z: Transcriptome analysis of JA signal
transduction, transcription factors, and monoterpene biosynthesis
pathway in response to methyl Jasmonate Elicitation in Mentha
canadensis L. Int J Mol Sci. 19:23642018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Han C, Li Q, Chen Q, Zhou G, Huang J and
Zhang Y: Transcriptome analysis of the spleen provides insight into
the immunoregulation of Mastacembelus armatus under Aeromonas
veronii infection. Fish Shellfish Immunol. 88:272–283. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Du ZY, Zang J, Tang XD and Guo W; Chinese
Orthopaedic Association Bone Oncology Group, : Experts' agreement
on therapy for bone metastases. Orthop Surg. 2:241–253. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Conceição-Silva F and Morgado FN:
Immunopathogenesis of human sporotrichosis: What we already know. J
Fungi (Basel). 4:892018. View Article : Google Scholar
|
|
32
|
Catlett NL, Yoder OC and Turgeon BG:
Whole-genome analysis of two-component signal transduction genes in
fungal pathogens. Eukaryot Cell. 2:1151–1161. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Boyce KJ, McLauchlan A, Schreider L and
Andrianopoulos A: Intracellular growth is dependent on tyrosine
catabolism in the dimorphic fungal pathogen Penicillium
marneffei. PLOS Pathog. 11:e10047902015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Vargas-Perez I, Sanchez O, Kawasaki L,
Georgellis D and Aguirre J: Response regulators SrrA and SskA are
central components of a phosphorelay system involved in stress
signal transduction and asexual sporulation in Aspergillus
nidulans. Eukaryot Cell. 6:1570–1583. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Maksimov V, Wäneskog M, Rodriguez A and
Bjerling P: Stress sensitivity of a fission yeast strain lacking
histidine kinases is rescued by the ectopic expression of Chk1 from
Candida albicans. Curr Genet. 63:343–357. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zuber S, Hynes MJ and Andrianopoulos A:
G-protein signaling mediates asexual development at 25 degree C but
has no effect on yeast-like growth at 37 degree C in the dimorphic
fungus Penicillium mameffei. Eukaryot Cell. 1:440–447. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Alspaugh JA, Cavallo LM, Perfect JR and
Heitman J: RAS1 regulates filamentation, mating and growth at high
temperature of Cryptococcus neoformans. Mol Microbiol.
36:352–365. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Leberer E, Harcus D, Dignard D, Johnson L,
Ushinsky S, Thomas DY and Schröppel K: Ras links cellular
morphogenesis to virulence by regulation of the MAP kinase and cAMP
signalling pathways in the pathogenic fungus Candida
albicans. Mol Microbiol. 42:673–687. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fortwendel JR, Zhao W, Bhabhra R, Park S,
Perlin DS, Askew DS and Rhodes JC: A fungus-specific ras homolog
contributes to the hyphal growth and virulence of Aspergillus
fumigatus. Eukaryot Cell. 4:1982–1989. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Erwig LP and Gow NA: Interactions of
fungal pathogens with phagocytes. Nat Rev Microbiol. 14:163–176.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Previato JO, Gorin PAJ, Haskins RH and
Travassos LR: Soluble and insoluble glucans from different cell
types of the human pathogen Sporothrix schenckii. Exp Mycol.
3:92–105. 1979. View Article : Google Scholar
|
|
42
|
Lloyd KO and Bitoon MA: Isolation and
purification of a peptido-rhamnomannan from the yeast form of
Sporothrix schenckii. Structural and immunochemical studies.
J Immunol. 107:663–671. 1971.PubMed/NCBI
|
|
43
|
Lopes-Bezerra LM, Walker LA, NiñoVega G,
Mora-Montes HM, Neves GWP, Villalobos-Duno H, Barreto L, Garcia K,
Franco B, Martínez-Álvarez JA, et al: Cell walls of the dimorphic
fungal pathogens Sporothrix schenckii and Sporothrix
brasiliensis exhibit bilaminate structures and sloughing of
extensive and intact layers. PLoS Negl Trop Dis. 12:e00061692018.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lenardon MD, Munro CA and Gow NA: Chitin
synthesis and fungal pathogenesis. Curr Opin Microbiol. 13:416–423.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bermejo C, Rodriguez E, Garcia R,
Rodriguez-Pena JM, Rodríguez de la Concepción ML, Rivas C, Arias P,
Nombela C, Posas F and Arroyo J: The sequential activation of the
yeast HOG and SLT2 pathways is required for cell survival to cell
wall stress. Mol Biol Cell. 19:1113–1124. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Munro CA, Selvaggini S, de Bruijn I,
Walker L, Lenardon MD, Gerssen B, Milne S, Brown AJ and Gow NA: The
PKC, HOG and Ca2+ signalling pathways co-ordinately regulate chitin
synthesis in Candida albicans. Mol Microbiol. 63:1399–1413.
2007. View Article : Google Scholar : PubMed/NCBI
|