Introduction
Microglia are innate immune cells resident in the
central nervous system; notably, microglia have a role in the
inflammatory defense system stimulated by infection and
environmental pathogens (1,2). Microglia interact with neurons,
astrocytes and blood vessels in their quiescent state, and
constantly scavenge damaged neurons and pathogens. Microglia can be
activated to transition from the quiescent to the active state in
response to brain injury or inflammatory stimuli, such as systemic
infection, cerebral ischemia, hemorrhagic stroke and traumatic
brain injury (3,4). In their pro-inflammatory state (M1),
microglia release various cytokines (IL-6, IL-1β and TNF-α) that
combat pathogenic infection; however, prolonged activation of the
M1 state can cause inflammatory damage to peripheral cells and lead
to neuronal cell death (5).
Microglia have a neuroprotective function in their
anti-inflammatory state (M2) via the expression of
anti-inflammatory cytokines and other factors (6). Therefore, the induction of M1 to M2
phenotype transition may be an effective strategy for combating
neurological damage.
To assess the initiation of inflammatory processes,
the expression levels of key inflammatory molecules can be
evaluated. The inflammasome is a multicomponent protein complex
that triggers innate immune responses by activation of specific
proteins, such as NLR family pyrin domain-containing (NLRP)1,
NLRP3, NLR family CARD domain-containing 4 and absent in melanoma 2
(7); among these proteins, the
NLRP3 inflammasome has received considerable attention due to its
unique role in the inflammatory response and associated types of
cell death (8,9). The NLRP3 inflammasome can be activated
by numerous pathogen-associated molecular patterns (PAMPs) or
endogenous danger-associated molecular patterns (DAMPs) (10), such as reactive oxygen species (ROS)
(11), mitochondrial DNA and
aggregated peptide (12). A
previous study confirmed that lipopolysaccharide (LPS) can initiate
a priming step via induction of NF-κB and can trigger secondary
activation of the NLRP3 inflammasome (13).
Pyroptosis was initially identified as
caspase-1-dependent inflammasome-mediated programmed necrosis
(14). Pyroptosis is a lytic
process, which is distinct from apoptosis and other types of
programmed cell death (15), and is
associated with cell swelling and large bubbles emanating from the
plasma membrane (16). It has been
reported that pyroptosis is initiated by NLRP3 inflammasome
assembly and is terminated with gasdermin D (GSDMD) cleavage, which
is in turn accompanied by the release of mature IL-18 and IL-1β
(10,17). NLRP3 polymerizes with cleaved
caspase-1 via the apoptosis-associated speck-like
protein-containing CARD adaptor, leading to cleavage of the
inflammatory cytokines IL-18 and IL-1β into their mature forms
(18). Activated caspase-1
subsequently cleaves GSDMD (19),
thus releasing its N- and C-domains. The N-domain of GSDMD forms
membrane pores and releases mature IL-1β and IL-18 into the
extracellular space, causing a severe inflammatory cascade reaction
(20).
It has been reported that inflammatory stimuli
induce immune responsive gene 1 (Irg-1) expression, which in
turn catalyzes the production of itaconate from the tricarboxylic
acid (TCA) cycle. Notably, Irg-1 is highly expressed in
LPS-activated macrophages (21,22).
Itaconate affects macrophage metabolism and the inflammatory
response by inhibiting succinate dehydrogenase (SDH) (23–25).
Upon inhibition of SDH-derived ROS, itaconate exerts its
anti-inflammatory effects on macrophage activation following
ischemia-reperfusion injury. Using Irg−/− mice,
Lampropoulou et al (23)
demonstrated that itaconate was a major physiological regulator of
succinate metabolism, mitochondrial respiration and inflammatory
cytokine expression. Recently, it has been shown that itaconate and
its membrane-permeable derivative dimethyl itaconate (DI) can
selectively inhibit the expression of several cytokines (26,27),
including IL-6 and IL-12. However, the inhibition of SDH is not
sufficient to account for the prominent immunoregulatory effect of
DI (22,28). Mills et al (28) revealed that itaconate can directly
increase nuclear factor erythroid 2-related factor 2 (Nrf-2)
expression via alkylation of the cysteine residues of Kelch-like
ECH-associated protein 1 (KEAP1), a covalent inhibitor of Nrf-2.
This may consequently result in the induction of downstream
antioxidant and anti-inflammatory gene expression (29). However, the function of this
biochemical pathway in microglial cells and its precise role in
pyroptosis remains unknown. The present study evaluated the ability
of DI to prevent pyroptosis via modulation of the inflammatory
response, microglia polarization, and the regulation of Nrf-2
expression and autophagy.
Materials and methods
Reagents and antibodies
The cell culture reagents were purchased from Gibco;
Thermo Fisher Scientific, Inc. The antibodies against GSDMD
(1:1,000; cat. no. ab209845), caspase-1 (p20) (1:500; cat. no.
ab1872), IL-1β (1:1,000; cat. no. ab9722), autophagy-related
(ATG)-5 (1:1,000; cat. no. ab199560), Beclin-1 (1:1,000; cat. no.
ab62557), heme oxygenase-1 (HO-1; 1:1,000; cat. no. ab137749) and
β-actin (1:1,000; cat. no. ab8227) were purchased from Abcam. The
antibodies against NLRP3 (1:1,000; cat. no. 13158), P62 (1:1,000;
cat. no. 13121S), and LC3B (1:1,000; cat. no. 2775) were obtained
from Cell Signaling Technology, Inc. The antibodies against NF-κB
p65 (1:1,000; cat. no. 14220-1) and Nrf-2 (1:1,000; cat. no.
16396-1) were purchased from ProteinTech Group, Inc., and the
antibodies against phosphorylated (p)-NF-κB p65 (Ser536) (1:1,000;
cat. no. AF2006) and Nucleolin (1:1,000; cat. no. DF12542) were
purchased from Affinity Biosciences. DI was purchased from
Sigma-Aldrich; Merck KGaA. DAPI and other reagents were obtained
from Beyotime Institute of Biotechnology.
Cell culture
BV2 microglial cells were purchased from iCell
Bioscience Inc., (cat. no. iCell-m011). The cells were cultured in
DMEM (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
FBS (Gibco; Thermo Fisher Scientific, Inc.) and penicillin (100
U/ml)/streptomycin (100 U/ml) with 5% CO2 at 37°C. LPS,
VX765 and 3-methyladenine (3-MA) were obtained from MedChemExpress.
ATP was purchased from Beyotime Institute of Biotechnology. Sham
group were only treated with the same volume of PBS. To effectively
induce cellular inflammation, 6-h treatment at 37°C with LPS (1.5
µg/ml) was selected, and DI (0–200 µM) and VX765 (10 µM; inhibitor
of pyroptosis) were added simultaneously. ATP (3 mM) was added for
an additional 1 h prior to cell lysis or imaging to trigger
pyroptosis in vitro. If necessary, 5 mM 3-MA pretreatment
was performed 2 h prior to LPS treatment to inhibit autophagy.
Morphological analysis
BV2 microglial cells were digested and incubated in
a 6-well plate overnight. After treatment with LPS (1.5 µg/ml) and
DI (200 µM) at 37°C for 5 h, ATP (3 mM) was added for an additional
1 h to trigger pyroptosis in vitro. The microglia morphology
was analyzed using a fluorescence microscope (DMi8; Leica
Microsystems, Inc.).
Animals and ethics guidelines
A total of five male C57BL/6 mice (age, 1–3 days;
weight, 1–2 g) were obtained from two pregnant female mice, which
were purchased from the Animal Centre of Wenzhou Medical University
(Wenzhou, China). The pregnant mice were maintained in specific
pathogen-free conditions at room temperature with 60% relative
humidity, under a 12-h light/dark cycle with free access to food
and water. The present study was conducted in accordance with the
ARRIVE guidelines for the Care and Use of Laboratory Animals
(25), and the experimental
procedures were approved by the Ethics Committee of Wenzhou Medical
University (approval no. wydw2019-0598). All efforts were made to
minimize suffering. Newborn mice were euthanized via the
administration of CO2 at 30% volume displacement rate.
Lack of heartbeat and long-term unconsciousness were used to
confirm animal death. Subsequently, mice were placed in 75% ethyl
alcohol for disinfection and primary microglia extraction.
Primary microglia culture
Newborn (1–3 days old) C57BL/6 mice, after
euthanasia with CO2, were decapitated and the heads were
quickly placed into a petri dish with 3–5 ml ice-cold DMEM.
Following removal of the meninges and blood vessels, the tissues
were cut, digested and passed through a cell filter screen (40-µm
nylon mesh). The obtained samples were centrifuged at 300 × g at
4°C for 5 min, and the resulting cell pellet was reconstituted in
DMEM with 10% FBS and penicillin/streptomycin under standard
conditions. The microglial cells were allowed to proliferate for
10–12 days and reached 90% confluence. Subsequently, they were
isolated by mild trypsinization and seeded in new culture plates
for further experiments. For further verification of the
anti-pyroptosis activity of DI against ATP and LPS-induced
pyroptosis, microglia was treated with LPS (1.5 µg/ml) and DI (200
µM) for 5 h at 37°C, and 3 mM ATP was added for an additional 1 h
prior to cell lysis.
Cell viability assay
Cell viability was measured using the MTT assay.
Briefly, BV2 microglial cells (5×103/well) were digested
and incubated in a 96-well plate overnight prior to DI treatment
(50, 100, 150, 200 or 250 µM) for 6 or 12 h. Subsequently, 50 µl
MTT solution (1 mg/ml) was added to each well and was continuously
incubated at 37°C for 4 h. Following incubation, the supernatant
was removed carefully and the insoluble formazan was dissolved in
150 µl DMSO. The absorbance was measured at 570 nm to assess cell
viability.
TUNEL assay
TUNEL assays were performed using a Colorimetric
TUNEL Apoptosis Assay kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. The cells were fixed
with 4% paraformaldehyde at room temperature for 15 min, incubated
with 50 µl TUNEL detection mixture at 37°C for 1 h in the dark and
rinsed in PBS. Cyanine 3-labeled dUTP was applied to stain
apoptotic cells, and DAPI was used for nuclear staining. Images of
cells were captured using a fluorescence microscope (DMi8; Leica
Microsystems, Inc.).
Western blot analysis
The cells were collected and lysed using RIPA lysis
buffer (cat. no. 89901; Thermo Fisher Scientific, Inc.) to extract
total cellular proteins. The protein concentration was determined
with a BCA protein assay kit (cat. no. 23225; Pierce; Thermo Fisher
Scientific, Inc.). Equal amounts of protein (30 µg) were separated
by SDS-PAGE on 12% gels and were subsequently transferred onto PVDF
membranes (MilliporeSigma). After blocking with 5% milk at room
temperature for 2 h, the membranes were incubated with primary
antibodies at 4°C overnight. Subsequently, the membranes were
washed three times and incubated with HRP-linked anti-rabbit IgG
secondary antibody (cat. no. 7074s; 1:5,000; Cell Signaling
Technology, Inc.) at room temperature for 1 h. Finally, the bands
were visualized using ECL Reagents (Advansta, Inc.) and analyzed by
ImageJ software (version 1.51; National Institutes of Health).
Lactate dehydrogenase (LDH),
malondialdehyde (MDA) and SDH activity detection
To detect LDH release, the medium was collected from
the cell cultures following drug treatment and the OD was measured
at 450 nm using LDH detection kits (Beijing Solarbio Science &
Technology Co., Ltd.) according to the manufacturer's instructions.
To detect MDA content, the cells were lysed in RIPA buffer (Thermo
Fisher Scientific, Inc.) on ice for 30 min. Following
centrifugation at 12,000 × g at 4°C for 10 min, 100 µl supernatant
was fixed with 200 µl MDA working regent and boiled for 15 min. The
absorbance was detected at 532 nm using the Lipid Peroxidation MDA
Assay kit (Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. To detect SDH activity, cells were
lysed and centrifuged at 12,000 × g at 4°C for 10 min. The
supernatant was collected and the OD was detected at 600 nm using
the SDH activity detection kit (Beijing Solarbio Science &
Technology Co., Ltd.) according to the manufacturer's
instructions.
RNA extraction from microglial cells
and reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was isolated from BV2 cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and cDNA was obtained with a RevertAid First Strand cDNA
Synthesis kit (cat. no. K1622; Thermo Fisher Scientific, Inc.).
qPCR was performed with SYBR Premix Ex Taq (Bio-Rad Laboratories,
Inc.). The thermocycling conditions were set as follows: Initial
denaturation at 95°C for 30 sec, followed by annealing and
elongation for 40 cycles at 95°C for 15 sec, 60°C for 30 sec and
72°C for 30 sec, and a final extension at 72°C for 2 min. Relative
quantification was calculated using the 2−ΔΔCq method
(30). The primers used are listed
in Table I; primers were
synthesized by Sangon Biotech Co., Ltd. GAPDH was used as the
internal reference gene for comparison.
 | Table I.Primers used for
reverse-transcription quantitative PCR. |
Table I.
Primers used for
reverse-transcription quantitative PCR.
| Gene | Forward primer
5′-3′ | Reverse primer
5′-3′ |
|---|
| IL-1β |
GTGTCTTTCCCGTGGACCTTC |
TCATCTCGGAGCCTGTAGTGC |
| TNF-α |
CTCCAGGCGGTGCCTATGTCT |
CTCCTCCACTTGGTGGTTTGC |
| IL-18 |
TGTCAGAAGACTCTTGCGTCAA |
TATTCCGTATTACTGCGGTTGT |
| IL-4 |
TTGAACGAGGTCACAGGAGAAG |
CTTGGAAGCCCTACAGACGAG |
| iNOS |
AGGGAATCTTGGAGCGAGTTG |
CGTAATGTCCAGGAAGTAGGTGAG |
| Arg-1 |
TGCCAGACGGAAGGACCATT |
CCAGCACGAAGGTCTCGATGT |
| Irg-1 |
GATGGTATCATTCGGAGGAGC |
CTGGAGGTGTTGGAACTGTAGAT |
| MCP-1 |
TGCATCTGCCCTAAGGTCTTC |
AGTGCTTGAGGTGGTTGTGGA |
| NLRP3 |
GTGGATGGGTTTGCTGGGATA |
TGCGTGTAGCGACTGTTGAGG |
| NLRP1 |
CTCTTTATCTTGGTTCCCGCTAT |
GTGCCTCTTGTCTTTGATTTGC |
| AIM2 |
GAATCTAACCACGAAGTCCCA |
ACCAACACCTCCATTGTCCCT |
| GAPDH |
GCCAAGGCTGTGGGCAAGGT |
TCTCCAGGCGGCACGCAGA |
ELISA for determination of cytokine
levels
The levels of cytokines were determined using
specific ELISA kits. BV2 microglial cells were seeded into
25-cm2 culture flasks at a density of 1×105
cells/well. After treatment, the cells were collected and
homogenized in ice-cold PBS. Following centrifugation at 12,000 × g
at 4°C for 10 min, the supernatant was collected, and the samples
were analyzed to detect inducible nitric oxide synthase (iNOS)
(cat. no. BP-E20450; Shanghai Boyun Biotech Co., Ltd.), TNF-α (cat.
no. BMS607-2INST; Thermo Fisher Scientific, Inc.), IL-6 (cat. no.
BMS603-2; Thermo Fisher Scientific, Inc.), IL-18 (cat. no.
BMS618-3; Thermo Fisher Scientific, Inc.), IFN-γ (cat. no.
BMS606-2; Thermo Fisher Scientific, Inc.) and IL-10 (cat. no.
BMS614INST; Thermo Fisher Scientific, Inc.) levels in accordance
with the manufacturer's protocols. The OD was measured at 450 nm on
a microplate reader (Spectra MAX 190; Molecular Devices, LLC).
Immunofluorescence staining
The treated microglial cells were fixed with 4%
paraformaldehyde at room temperature for 30 min and subsequently
permeabilized with 0.2% Triton X-100. Following blocking with 10%
donkey serum (Sigma-Aldrich; Merck KGaA) and 0.2% Triton X-100 at
room temperature for 2 h, the samples were incubated at 4°C for 24
h with primary antibodies against IBA-1 (1:200; cat. no. ab5076;
Abcam), iNOS (1:200; cat. no. ab15323; Abcam), CD206 (1:200; cat.
no. ab8918; Abcam) and p-NF-κB p65 (Ser536) (1:200; cat. no.
AF2006; Affinity Biosciences), followed by incubation with
respective secondary antibodies at 37°C for 1 h, including
488-conjugated donkey anti-goat antibody (1:500; cat. no.
E032231-02; Canlifesci, Inc.), 594-conjugated donkey anti-mouse
antibody (1:500; cat. no. E032411-02; Canlifesci, Inc.), and
594-conjugated donkey anti-rabbit antibody (1:500; cat. no.
E032421-01; Canlifesci, Inc.). Finally, the cells were stained with
DAPI (cat. no. C1002; Beyotime Institute of Biotechnology) at room
temperature for 10 min and subsequently examined with a scanning
fluorescence microscope (DMi8; Leica Microsystems, Inc.).
Gene knockdown with small interfering
RNA (siRNA)
The cells were transfected using the riboFECT CP
transfection reagent (cat. no. C10511-05; Guangzhou RiboBio Co.,
Ltd.) according to the manufacturer's instructions. Briefly, BV2
cells were seeded into 6-well plates at a density of
1×106 cells/well. siRNA (1.25 µl) was diluted with 30 µl
riboFECT™ CP Buffer, and mixed with 3 µl riboFECT™ CP reagent to
form the transfection complex. Subsequently, the cells were
transfected with the mixture containing 100 nM siRNA-Nrf-2
(Guangzhou RiboBio Co., Ltd.) (three targeted sequences:
siRNA-Nrf-2-1, 5′-GCAGGAGAGGTAAGAATAA-3′; siRNA-Nrf-2-2,
5′-GCACAATGGAATTCAATGA-3′; and siRNA-Nrf-2-3,
5′-CGACAGAAACCTCCATCTA-3′) or 100 nM siRNA-NLRP3 (Guangzhou RiboBio
Co., Ltd.) (three targeted sequences: siRNA-NLRP3,
5′-GTACTTAAATCGTGAAACA-3′; siRNA-NLRP3-2,
5′-CAGCCAGAGTGGAATGACA-3′; siRNA-NLRP3-3,
5′-GGATGGCTTTGATGAGCTA-3′), and incubated in 2 ml DMEM for 48 h at
37°C. Scrambled siRNA (targeted sequence,
5′-GGCTCTAGAAAAGCCTATGC-3′; Guangzhou RiboBio Co., Ltd.) was used
as the negative control. The transfection efficiency of the
siRNA-Nrf-2 was assessed by western blot analysis, whereas the
transfection efficiency of the siRNA-NLRP3 was assessed by RT-qPCR.
The siRNA with the most effective silencing efficiency was used for
further experiments. The transfected cells were immediately treated
with drugs within 24 h for investigation.
Flow cytometric analysis of ROS
ROS production was determined using
2,7-dichlorodihydrofluorescein diacetate (DCFH-DA; cat. no. S0033S;
Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. After drug treatment, the culture medium
was discarded and the cells were incubated with 10 µM DCFH-DA for
10 min at 37°C. After washing away the unbound DCFH-DA, the cells
were collected and resuspended in DMEM for ROS detection. Cellular
ROS was detected using a flow cytometer (CytoFLEX LX; Beckman
Coulter, Inc.), and analyzed via FlowJo software (version 10;
FlowJo LLC).
Statistical analysis
All experiments were repeated three times and all
data were analyzed using GraphPad Prism 5.0 (GraphPad Software,
Inc.) or SPSS 22.0 (IBM Corporation) and presented as the mean ±
SD. An unpaired Student's t-test was applied to analyze the
comparisons between two groups. One-way ANOVA followed by Tukey's
post hoc test was used to compare data from the different groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
DI is not cytotoxic at specific
concentrations in BV2 microglial cells
DI is a novel membrane-permeable derivative of
itaconate (Fig. 1A and B). The
excessive production of itaconate may cause aberrant cell
metabolism. Prior to the investigation of the biochemical effects
of DI on microglial cells, its cytotoxicity was assessed using the
MTT assay. Cell viability was assessed following DI treatment
(50–250 µM) for 6 or 12 h (Fig. 1C and
D). The results demonstrated that cell viability was not
significantly different in the treated groups compared with that in
the sham group (P<0.05). These findings indicated that DI did
not exert cytotoxic effects against BV2 microglia at a
concentration <200 µM. Based on these findings, DI was used in
subsequent experiments at a concentration range between 0 and 200
µM.
DI protects BV2 cells against
NLRP3-dependent canonical pyroptosis
Prior to investigation of the anti-inflammatory
effects of DI in active microglia, the induction of Irg-1
expression was assessed. The Irg-1 gene encodes an enzyme,
which catalyzes endogenous itaconate production under inflammatory
conditions (in the present study following LPS stimulation). The
results revealed that the expression levels of the Irg-1
transcript were significantly increased following LPS and ATP
stimulation (Fig. S1), which
suggested the involvement of this enzyme in inhibition of
inflammation.
Pyroptosis is a lytic, cell-swelling process, which
is accompanied by GSDMD-mediated pore formation and secretion of
the cell contents (16). The TUNEL
assay indicated a higher number of apoptotic cells following LPS
stimulation and ATP treatment of the cells. These effects were
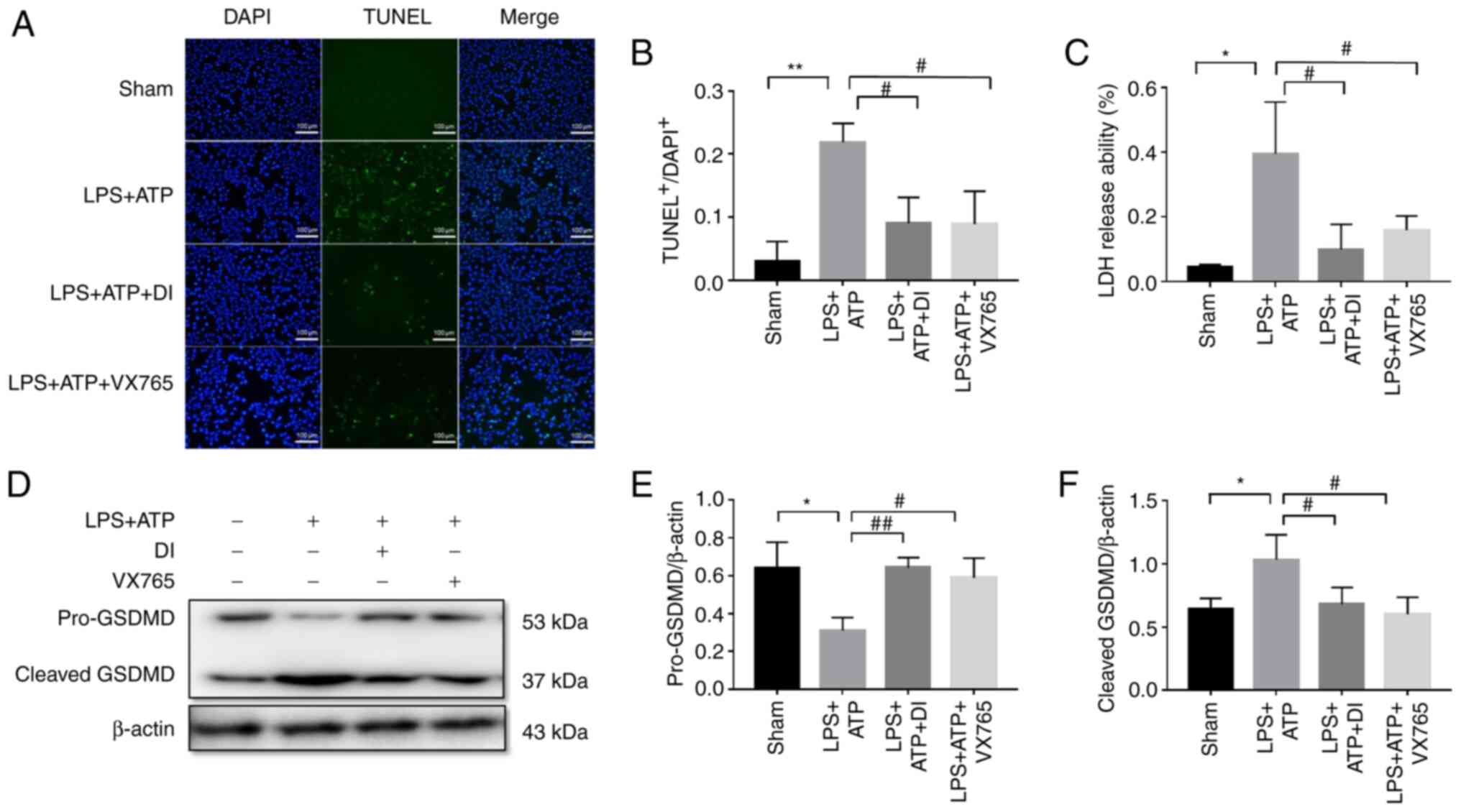
accompanied by increased LDH secretion and GSDMD cleavage (Fig. 2A-F). These findings confirmed
membrane perforation and GSDMD-mediated pyroptosis. Subsequently,
the anti-pyroptotic effect of DI was investigated. Cotreatment with
DI markedly reduced the induction of cell apoptosis, LDH secretion
and GSDMD cleavage, reflecting the inhibitory effect of DI on
GSDMD-mediated pyroptosis (Fig.
2A-F).
Pyroptosis inhibition was confirmed following DI
administration; therefore, the intrinsic mechanism was examined.
The detection of the transcriptional levels of NLRs indicated the
inhibitory activities of DI on NLRP3 transcription (Fig. S2), which suggested the involvement
of NLRP3-associated pathways in this process. The expression levels
of NLRP3, cleaved caspase-1, pro-IL-1β and cleaved IL-1β were
markedly increased following stimulation of the cells with LPS and
ATP, while co-treatment with DI significantly decreased expression
of NLRP3, cleaved caspase-1, pro-IL-1β and cleaved IL-1β (Fig. 3A-E). These findings indicated that
DI inhibited the expression of NLRP3 and pro-IL-1β, and prevented
the cleavage of caspase-1, which eventually resulted in decreased
cleaved IL-1β. In addition, knockdown of NLRP3 by siRNA
interference significantly inhibited pro-IL-1β expression and GSDMD
cleavage compared with in the LPS+ATP group (Fig. S3A-F), suggesting that DI regulated
pyroptosis in the BV2 cell line via the
NLRP3-caspase-1-GSDMD-dependent canonical pathway.
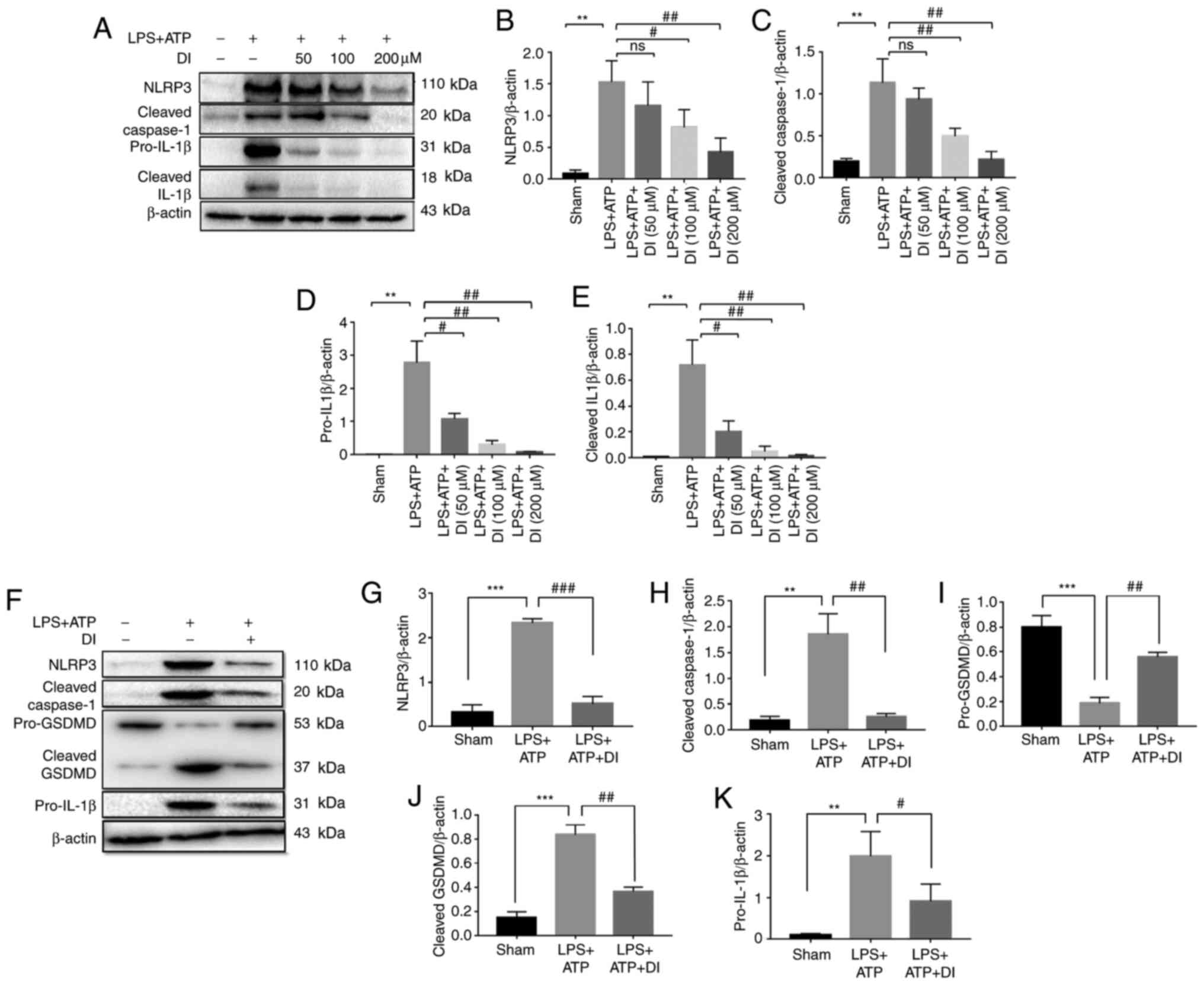 | Figure 3.DI inhibits NLRP3-dependent
pyroptosis in BV2 and primary microglia cells. (A) Western blot
analysis. Semi-quantification of (B) NLRP3, (C) cleaved caspase-1
(p20), (D) pro-IL1β and (E) cleaved IL1β. (F) Western blot
analysis. Semi-quantification of (G) NLRP3, (H) cleaved caspase-1,
(I) pro-GSDMD, (J) cleaved GSDMD and (K) pro-IL-1β. Data are
presented as the mean ± standard error of the mean (n=3).
**P<0.01, ***P<0.001 vs. sham group; #P<0.05,
##P<0.01, ###P<0.001 vs. LPS+ATP group.
DI, dimethyl itaconate; LPS, lipopolysaccharide; NLRP3, NLR family
pyrin domain-containing 3; GSDMD, gasdermin D. |
In order to further confirm the anti-pyroptotic
activity of DI, primary microglial cells were extracted and
cultured. DI administration effectively inhibited NLRP3-mediated
pyroptosis by reducing the expression levels of NLRP3 and
pro-IL-1β, and preventing cleavage of caspase-1 and GSDMD (Fig. 3F-K). This suggested the
anti-pyroptotic activity of this compound in primary microglia.
DI regulates microglia polarization
and cellular inflammation by inducing NF-κB expression
To assess the effects of DI on microglia
polarization, the present study examined the transcripts of M1 and
M2 markers. M1-polarized microglia cells are iNOS-positive and
induce pro-inflammatory mediator expression (5), whereas M2-polarized microglia cells
are CD206-positive and induce anti-inflammatory mediator
expression. The present study revealed that 100 µM DI reduced the
transcription of iNOS, TNF-α and monocyte chemoattractant
protein 1, which was increased following stimulation of the cells
with LPS and ATP. Concomitantly, the transcription of arginase 1
and IL-4 was increased by DI (Fig. 4A), indicating inhibition of M1
polarization and induction of M2 polarization. Furthermore, a
double-immunofluorescence staining assay was performed and the
results indicated that the number of
Iba-1+/iNOS+ cells in the DI-treated group
was significantly lower than that in the LPS and ATP group
(Fig. 4B and C). These findings
suggested that DI inhibited LPS and ATP-induced M1 polarization in
BV2 cells. Moreover, DI treatment significantly increased the
number of Iba-1+/CD206+ positive cells
(Fig. 4D and E), which indicated
that microglia were polarized to the M2 phenotype. Through
detecting cellular morphology (Fig.
S4A-C), it was demonstrated that LPS and ATP-stimulated
microglia appeared with long synapses, whereas DI-treated microglia
exhibited shorter synapses; however, more microglia were detected
in the DI-treated group. These findings were in accordance with the
classic morphology of microglia polarization, suggesting that DI
may be a critical regulator of the transition from pro-inflammatory
M1 to anti-inflammatory M2 polarization.
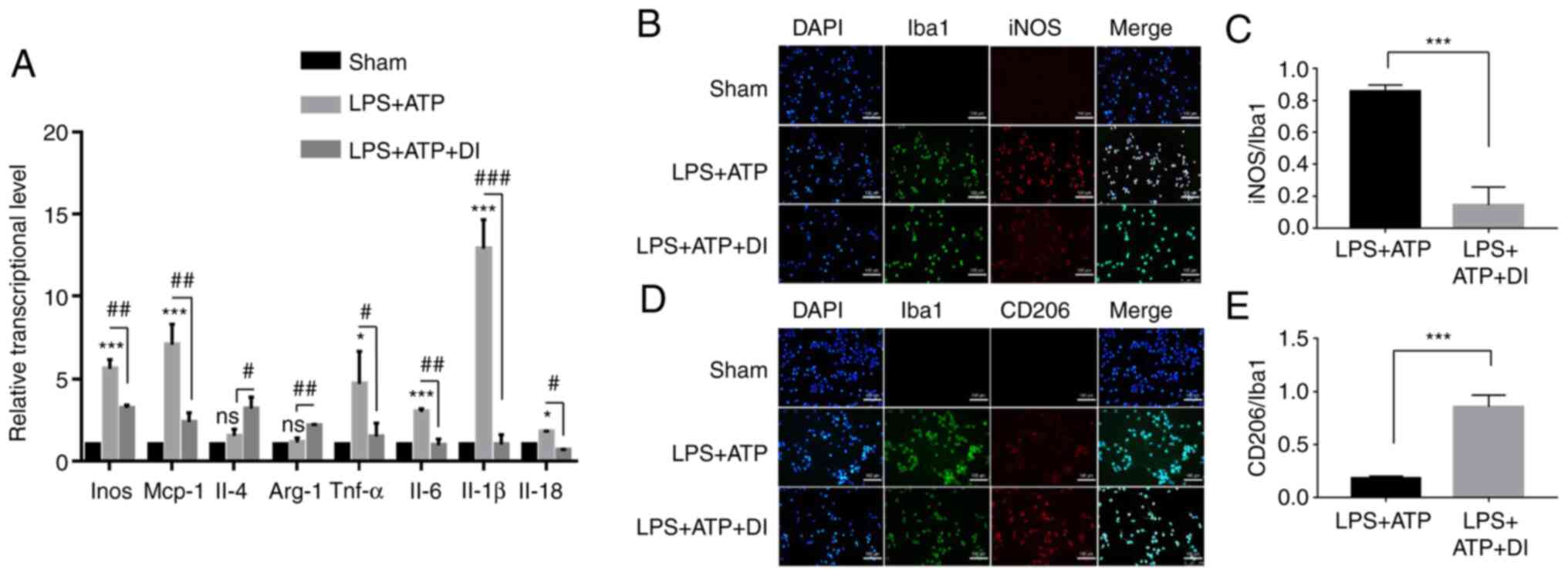 | Figure 4.Regulatory role of DI on microglia
polarization. (A) mRNA expression levels of Inos, Mcp-1, Il-4,
Arg-1, Tnf-α, Il-6, Il-1β and Il-18 as determined by
reverse transcription-quantitative PCR. (B and C) Microglia M1
polarization (Iba-1+/iNOS+) and (D and E) M2
polarization (Iba-1+/CD206+) were determined
by double-immunofluorescence assay. Scale bar, 100 µm. Data are
presented as the mean ± standard error of the mean (n=3).
*P<0.05, ***P<0.001 vs. sham group or as indicated;
#P<0.05, ##P<0.01,
###P<0.001 vs. LPS+ATP group. Arg-1, arginase 1; DI,
dimethyl itaconate; iNOS, inducible nitric oxide synthase; LPS,
lipopolysaccharide; Mcp-1, monocyte chemoattractant protein
1. |
To elucidate the precise anti-inflammatory profile
of DI, the levels of pro-inflammatory and anti-inflammatory
mediators were detected. The levels of pro-inflammatory mediators,
including iNOS, TNF-α, IL-6, IFN-γ and IL-18, which were induced by
LPS and ATP stimulation, were significantly inhibited by DI
(Fig. 5A-E), while
anti-inflammatory cytokine IL-10 was largely induced by DI
treatment (Fig. 5F). NF-κB
possesses a key priming role in LPS-induced inflammation (6). By detecting the phosphorylation of
NF-κB using immunofluorescence staining, it was revealed that LPS
and ATP treatment promoted NF-κB nuclear translocation, whereas DI
administration effectively reduced these effects (Fig. 5G). In addition, western blotting was
employed to assess the induction of NF-κB phosphorylation following
stimulation of the cells with LPS and ATP, it was found that
cellular p-NF-κB and nuclear p-NF-κB were significantly increased
(Fig. 5H-K). These effects were
markedly inhibited by treatment of the cells with DI, suggesting
that this compound regulated the inflammatory response by
inhibiting NF-κB phosphorylation and nuclear translocation.
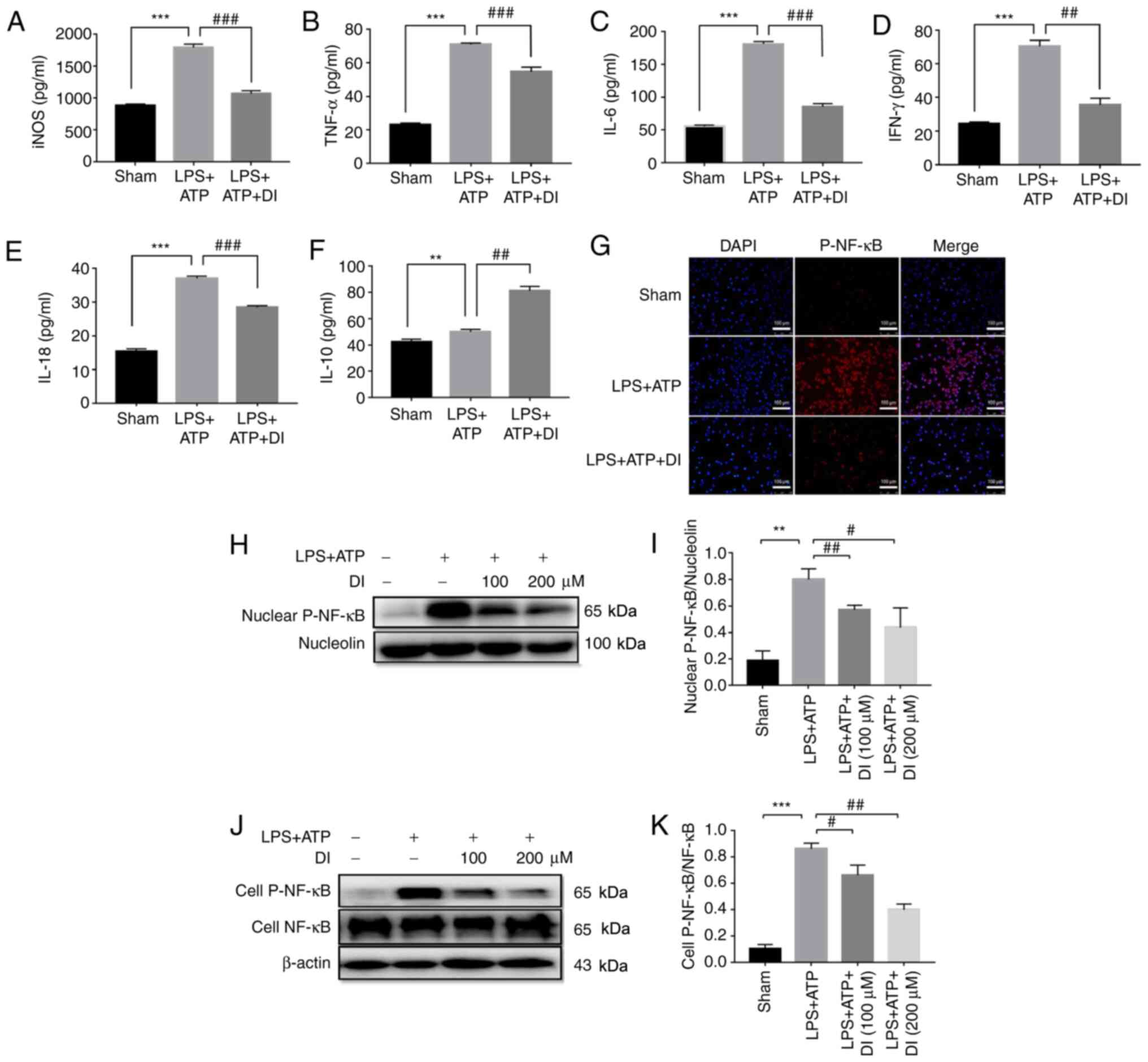 | Figure 5.DI regulates microglia inflammatory
profile via inhibiting NF-κB phosphorylation and nuclear
translocation. Levels of cytokines, including (A) iNOS, (B) TNF-α,
(C) IL-6, (D) IFN-γ, (E) IL-18 and (F) IL-10, were determined by
ELISA. (G) Nuclear translocation of p-NF-κB was determined by
immunofluorescence staining. Scale bar, 100 µm. (H and I) p-NF-κB
expression in the nucleus were determined by western blotting. (J
and K) Ratio of whole cell p-NF-κB/total NF-κB expression was
determined by western blotting. Data are presented as the mean ±
standard error of the mean (n=3). **P<0.01, ***P<0.001 vs.
sham group; #P<0.05, ##P<0.01,
###P<0.001 vs. LPS+ATP group. DI, dimethyl itaconate;
iNOS, inducible nitric oxide synthase; LPS, lipopolysaccharide; p-,
phosphorylated. |
DI exerts cytoprotective effects via
the Nrf-2/HO-1 pathway
Nrf-2 is the main transcription factor regulating
the cytoprotective responses to oxidative and inflammatory stress.
Following induction of oxidative and inflammatory stress, Nrf-2 is
released from KEAP1, leading to its translocation to the nucleus,
which initiates the expression of the downstream effector HO-1
(31). DI treatment significantly
upregulated Nrf-2 and HO-1 expression levels in a dose-dependent
manner compared with those in the sham group, indicating that the
Nrf-2/HO-1 pathway contributed to cellular antioxidant activity
(Fig. 6A-C).
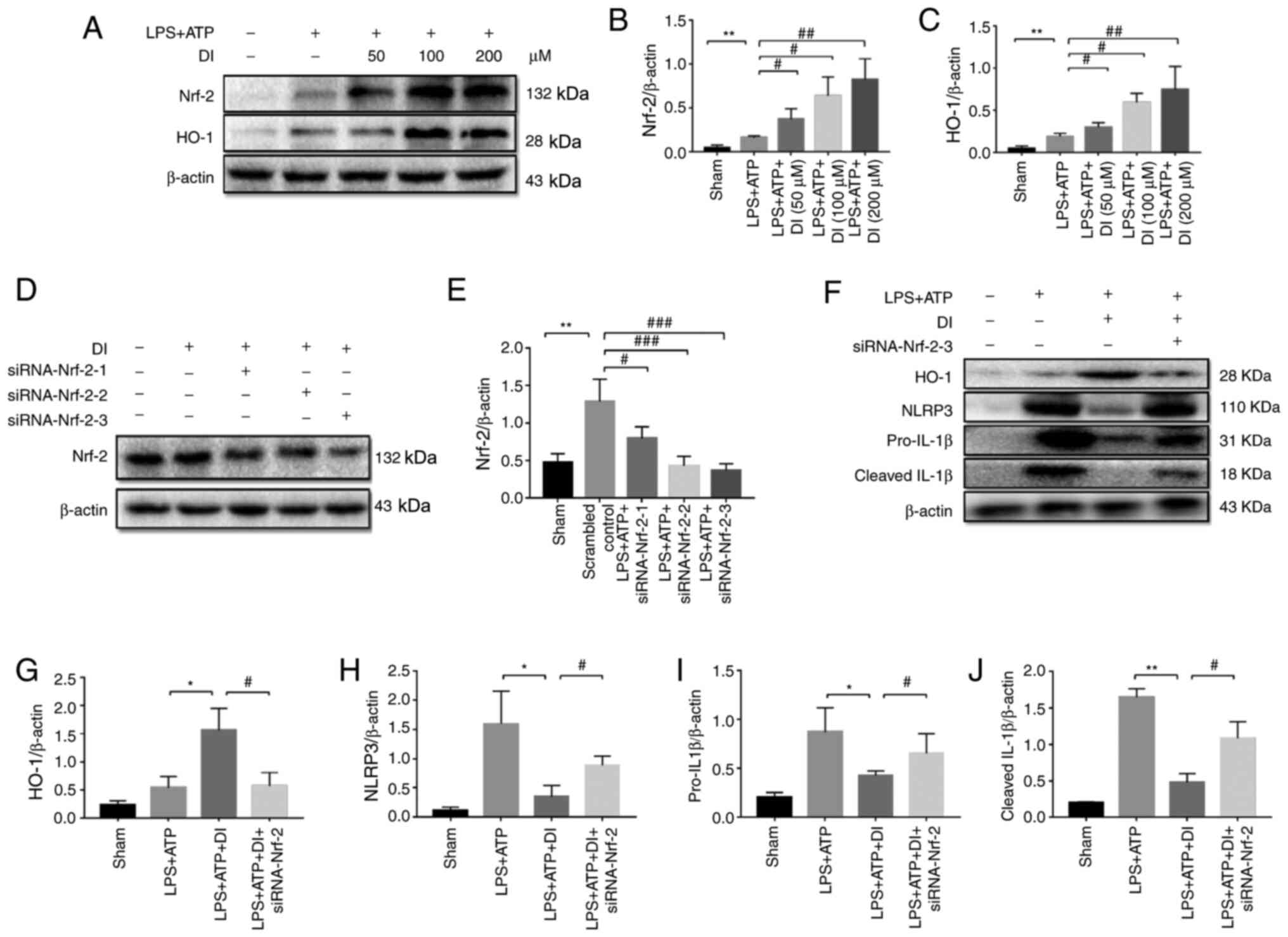 | Figure 6.DI positively regulates the
Nrf-2/HO-1 pathway. (A) BV2 microglia cells were treated with LPS
(1.5 µg/ml) together with various concentrations of DI (50, 100 or
200 µM) for 5 h, with an additional treatment with ATP (3 mM) for 1
h, and western blotting was performed. The protein expression
levels of (B) Nrf-2 and (C) HO-1 were semi-quantified. (D) BV2
cells were transfected with siRNA directed against Nrf-2 and
western blotting was performed. (E) Protein expression levels of
Nrf-2 were semi-quantified. (F) Western blotting was performed
after siRNA transfection. Protein expression levels of (G) HO-1,
(H) NLRP3, (I) pro-IL-1β and (J) cleaved IL-1β upon siRNA-Nrf-2
transfection were semi-quantified. Data are presented as the mean ±
standard error of the mean (n=3). *P<0.05, **P<0.01 vs. sham
group or as indicated; #P<0.05,
##P<0.01, ###P<0.001 vs. LPS+ATP group
or as indicated. DI, dimethyl itaconate; HO-1, heme oxygenase-1;
LPS, lipopolysaccharide; NLRP3, NLR family pyrin domain-containing
3; Nrf-2, nuclear factor erythroid 2-related factor 2; siRNA, small
interfering RNA. |
To explore the association between the Nrf-2/HO-1
pathway and NLRP3-mediated pyroptosis, the expression of Nrf-2 was
knocked down using siRNA interference (Fig. 6D and E). Treatment of the cells with
DI induced the expression of HO-1; however, this effect was
suppressed by the knockdown of Nrf-2 expression via transfection
with siRNA (Fig. 6F and G). NLRP3,
pro-IL-1β and cleaved IL-1β expression levels were reduced by
treatment with DI, which was reversed by the knockdown of Nrf-2
expression (Fig. 6H-J), thus
suggesting the protective effect of the Nrf-2/HO-1 pathway against
inflammation.
ROS has been identified as a source of DAMPs, which
trigger pyroptosis (32). Flow
cytometric detection with DCFH-DA verified that DI could
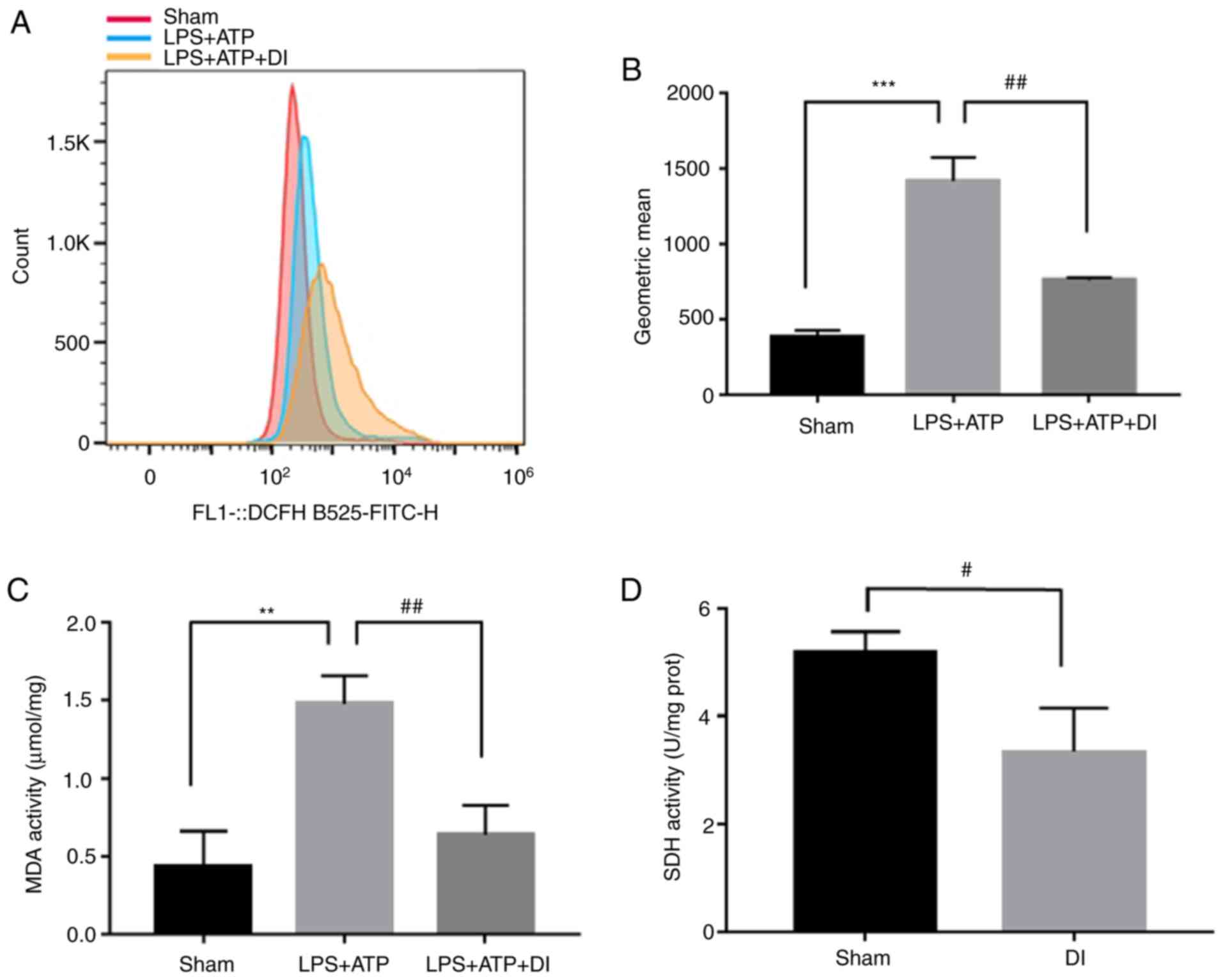
effectively inhibit LPS and ATP-induced ROS production (Fig. 7A and B), alongside its inhibitory
effect on MDA and SDH activity (Fig. 7C
and D). These results suggested that DI may exert its
anti-pyroptotic effects by regulating ROS production.
DI inhibits inflammasome-mediated
pyroptosis via induction of autophagy
A previous study reported the regulatory effect of
autophagy on microglia polarization and the inflammatory process
(33). To determine whether DI
interfered with autophagy, the protein expression levels of LC3B,
ATG-5, Beclin-1 and P62 were analyzed. LC3B-II expression was
increased in a concentration-dependent manner and the
LC3B-II/LC3B-I ratio was significantly increased following
treatment of the cells with 200 µM DI compared with the LPS and ATP
group. Similarly, Beclin-1 and ATG-5 expression levels were
increased, whereas P62 exhibited a tendency toward downregulation
following DI treatment (Fig. 8A-E).
These data indicated that DI may be involved in the regulation of
autophagy.
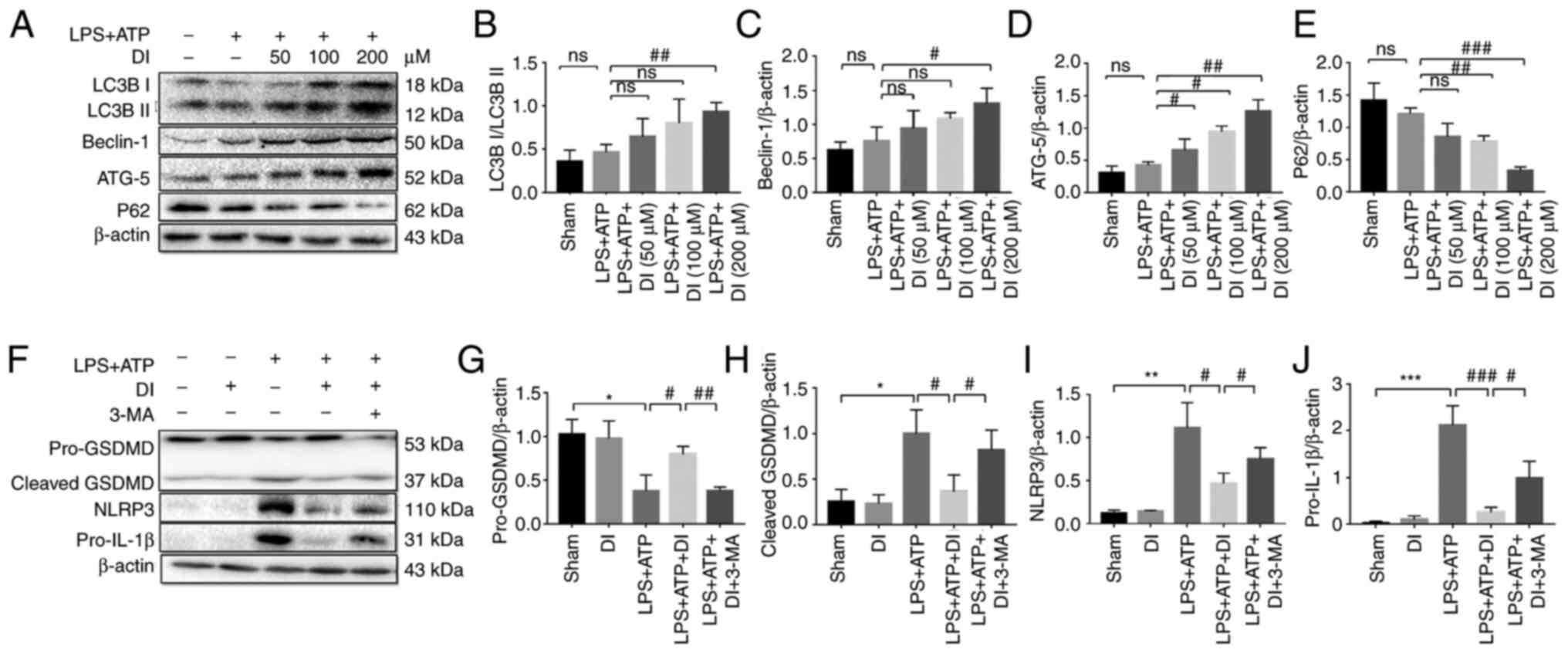 | Figure 8.DI inhibits inflammasome-mediated
pyroptosis through autophagy. (A) Autophagy markers were detected
by western blotting. Protein expression levels of (B) LC3B, (C)
Beclin-1, (D) ATG-5 and (E) P62 were semi-quantified. (F)
Association between DI-induced autophagy and inflammation was
investigated by western blotting. Protein expression levels of (G)
GSDMD, (H) cleaved GSDMD, (I) NLRP3 and (J) pro-IL-1β were
semi-quantified. Data are presented as the mean ± standard error of
the mean (n=3). *P<0.05, **P<0.01, ***P<0.001 vs. sham
group; #P<0.05, ##P<0.01,
###P<0.001 vs. LPS+ATP group or as indicated. 3-MA,
3-methyladenine; ATG-5, autophagy-related 5; DI, dimethyl
itaconate; GSDMD, gasdermin D; LPS, lipopolysaccharide; NLRP3, NLR
family pyrin domain-containing 3. |
To confirm the regulatory effect of autophagy on
anti-inflammatory activity, the autophagy inhibitor, 3-MA, was used
to suppress this process. Cotreatment of the cells with DI and the
autophagy inhibitor 3-MA reduced the effects of DI on the
expression levels of NLRP3, cleaved GSDMD and IL-1β, suggesting
that DI may regulate cellular anti-inflammatory activity via
autophagy (Fig. 8F-J). Taken
together, these data indicated that DI exerted anti-inflammatory
effects via the induction of autophagy.
Discussion
The present study indicated that DI inhibited the
inflammatory response and regulated microglia polarization
following stimulation with LPS and ATP. The functions ascribed to
DI include inhibition of the NLRP3 inflammasome, stimulation of the
antioxidative transcription factor Nrf-2 and regulation of
autophagy. The experiments aimed to determine the role of DI in
LPS- and ATP-induced pyroptotic microglia. DI effectively
attenuated NLRP3 inflammasome assembly and subsequently restrained
caspase-1-associated canonical pyroptosis in vitro. These
data confirmed that DI may activate the Nrf-2/HO-1 signaling
pathway to induce antioxidant and anti-inflammatory activities.
Notably, inhibition of autophagy reversed the anti-inflammatory
function of DI. These results suggested that DI effectively
decreased inflammation and NLRP3-assciated pyroptosis by modulating
the Nrf-2/HO-1 pathway and the induction of autophagy.
Metabolic products affect crucial biological
functions. Itaconate has recently been reported to function as an
immunomodulator in macrophages (34). Following induction of inflammation
by a specific stimulus, Irg-1 is activated and promotes the
production of itaconate from the TCA cycle by decarboxylating
cis-aconitase (35). Macrophage
models of inflammation have effectively demonstrated the
anti-inflammatory activity of itaconate and increased itaconate
synthesis has been shown to occur during macrophage activation.
Irg1−/− bone marrow-derived macrophages, which
lack the ability to synthesize endogenous itaconate, can release
higher levels of cytokines in response to LPS stimulation (23). Notably, itaconate is known to exert
its antioxidant activity by targeting SDH, thereby reducing the
production of ROS derived from succinate oxidation (23,24);
however, using the SDH inhibitor dimethyl malonate did not inhibit
IL-1β release in an NLRP3 inflammasome assay (35), indicating that SDH inhibition by
itaconate was unlikely to be the main mechanism underlying its
anti-inflammatory ability. Several cell-permeable derivatives have
been synthesized to achieve efficient intracellular delivery of
itaconate (36); DI is produced by
esterification of a carboxyl group and reduces the negative charge
of itaconate, which increases its electrophilicity and derivative
functions (23). Microglia are
unique resident immune cells in the brain that comprise the first
and most important line of immune defense in the central nervous
system (5). The induction of genes
that are induced most rapidly by LPS stimulation are known as the
primary response, such as TNF-α; while the induction of secondary
response genes require chromatin remodeling as a prerequisite for
transcription factor binding and the recruitment of
histone-modifying enzymes and the general transcription initiation
machinery (37). The findings of
the present study indicated that treatment of microglia with DI
inhibited their primary and secondary transcriptional response to
LPS. Moreover, LPS-stimulated microglia M1 polarization was reduced
with DI treatment. However, exogenous addition of the itaconate
derivative did not result in metabolic production of endogenous
itaconate (38). As a more powerful
version of itaconate, the dimethyl derivative exhibits increased
electrophilicity that may not truly reflect the innate biological
function of the endogenous compound. Irg-1 is the key protein that
produces intracellular itaconate, so knockdown of Irg-1
could fundamentally impair itaconate production, which could be
helpful to investigate the true biological function of itaconate
(25). Therefore, the
Irg-1−/− microglia model should be utilized for
subsequent investigations.
It is now generally accepted that NLRP3 inflammasome
signaling serves a critical role in the pathogenesis of several
neurological diseases, including traumatic brain injury, ischemic
stroke and neurodegenerative diseases (39,40).
Following stimulation by PAMPs and/or DAMPs, microglia are
activated and release a series of inflammatory mediators, including
NLR-inflammasome members and specific cytokines (7). Hooftman et al (35) revealed that the metabolite itaconate
specifically suppressed NLRP3 inflammasome activation by reducing
the NLRP3-NEK7 interaction and modulating C548 on NLRP3, eventually
resulting in blockage of IL-1β release, which may account for the
intrinsic molecular mechanism of its anti-inflammatory ability
(35). In the present study,
external administration of DI effectively reduced NLRP3
inflammasome activation and subsequently inhibited NLRP3-mediated
GSDMD cleavage, further emphasizing the inhibitory effect of DI on
pyroptosis. The previous studies performed by our group highlighted
the importance of inhibiting the NLRP3 inflammasome in order to
achieve neural functional recovery from traumatic injury and
ischemia stroke (41–43). DI may possess significant potential
as a neuropathological drug for promoting neurological functional
recovery due to its strong inhibitory activity against
NLRP3-mediated pathways.
NLRP3 mediates the canonical pyroptotic pathway in
microglia, which is characterized by cellular membrane perforation
and disintegration, and eventually results in cell lysis (44). Canonical pyroptosis elicits NLRP3
inflammasome assembly, activation of caspase-1, cleavage and
formation of GSDMD pores, and release of mature IL-18 and IL-1β
(45). The formation of the GSDMD
pore leads to disrupted osmotic potential and cell swelling
(13,20). In the present study, LPS- and
ATP-mediated stimulation of microglia was used to trigger
intracellular pyroptosis in vitro. LDH release occurs
following the activation of GSDMD expression, which is accompanied
by increased expression of cleaved caspase-1 and subsequent
inhibition of GSDMD cleavage (46).
Simultaneously, LPS- and ATP-mediated stimulation of microglia also
induced M1 polarization, which released pro-inflammatory cytokines
and increased NLRP3 expression. This triggered NLRP3-mediated
pyroptosis. DI effectively facilitated the transition from M1 to
M2, which in turn blocked NLRP3-mediated canonical pyroptosis.
To further elucidate the mechanism by which DI
attenuated inflammation, the Nrf-2/HO-1 pathway was assessed. It is
widely recognized that Nrf-2 is a transcription factor that
regulates the inflammatory response and the induction of oxidative
stress (28). Nrf-2 induces the
expression of numerous proteins that are released in response to
cytotoxic effects, oxidative stress and inflammation. Xu et
al (47) reported that Nrf-2
ameliorated inflammation by inducing HO-1 expression, which
resulted in downregulation of TNF-α expression. Moreover, Nrf-2 can
directly bind to the promoter region of IL-6 and IL-1β. Under
normal conditions, Nrf-2 is covalently bound to KEAP1, whereas
itaconate can effectively reduce the inhibitory effect on Nrf-2
(28), thus indicating the
intrinsic anti-inflammatory mechanism of itaconate. The present
study confirmed that DI caused an upregulation in Nrf-2 expression
and inhibited LPS-induced inflammatory cytokine levels in
microglia. Furthermore, DI inhibited the NF-κB signaling pathway,
which may regulate pro-inflammatory cytokine production. Several
studies have shown that activation of the Nrf-2/HO-1 pathway is
associated with the NF-κB signaling pathway (48–50),
indicating the anti-inflammatory effect of DI on microglia.
The activation of autophagy can cause or prevent
neurological damage. It has been reported that the induction of
autophagy can decrease cell death and promote recovery of
neurological function (51,52). Therefore, the induction of autophagy
may protect microglial cells against LPS- and ATP-induced damage.
Previous studies have demonstrated the association between
autophagy and inflammatory processes. Wang et al (33) demonstrated that macrophage
polarization was regulated via LC3-dependent autophagy in
Brucella. In addition, several reports have concluded that
activation of autophagy can prevent pyroptosis (53,54).
Notably, the NLRP3 inflammasome can be selectively degraded by
autophagy, limiting cytokine secretion and reducing pyroptosis
(55,56). In the present study, DI caused a
marked increase in LC3B-II, ATG-5 and Beclin-1 expression levels,
indicating that it augmented intracellular autophagy levels.
Furthermore, a suppressive effect of DI was noted on LPS-induced
NLRP3, IL-1β and cleaved GSDMD expression; this effect was reversed
by 3-MA, thus confirming that DI exerted its anti-inflammatory
effects partly by activating autophagy.
There are some limitations to the present study.
Firstly, apart from NLRs-driven inflammasome assembly, the present
study did not assess TLRs, which also trigger the initiation of
inflammation and pyroptosis. Secondly, the esterification of a
carboxyl group increases the electrophilicity of itaconate, which
hides the true biological function of itaconate; therefore,
application of lower electrophilic derivatives, such as 4-octyl
itaconate, or an Irg-1−/− microglia model in
further studies would intuitively present the biological function.
Thirdly, in the present study, groups were set and experiments were
repeated three times; according to acknowledged rules, the sample
size (n) should be more than three in in vitro cell tests
and more than five in in vivo animal tests (57). Expanding the sample size would make
the statistical analysis more reliable. Despite these limitations,
the present study discovered a novel itaconate derivative with
prominent anti-inflammatory and anti-pyroptotic activity. Future
research may focus on its anti-inflammatory effect in
vivo.
In conclusion, the results of the present study
indicated that DI exerted anti-inflammatory and anti-pyroptotic
effects on LPS- and ATP-stimulated microglia. This protective
mechanism of DI was associated with inhibition of production of the
NLRP3 inflammasome and suppression of inflammatory cytokine
release. These effects were mediated by activation of the
Nrf-2/HO-1 pathway and the regulation of autophagy. The findings of
the present study emphasize the therapeutic potential of DI for
neurological diseases. The potential anti-inflammatory and
anti-pyroptotic mechanisms of DI in microglia are shown in Fig. 9.
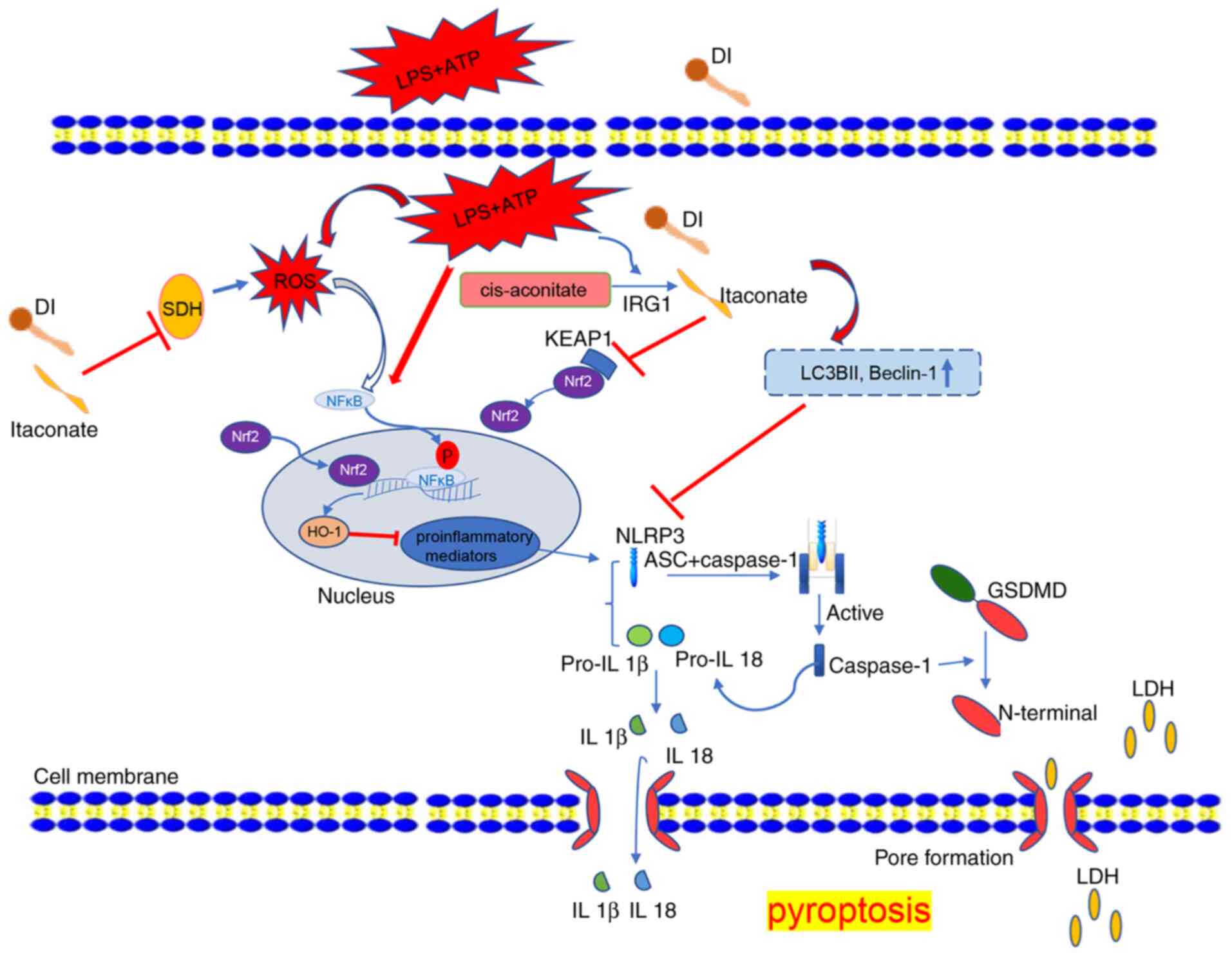 | Figure 9.Anti-inflammatory and anti-pyroptotic
function of DI in microglia. LPS and ATP induced inflammation and
NLRP3 inflammasome-dependent pyroptosis, which also triggered ROS
production, NF-κB phosphorylation and inflammatory mediator
expression. DI inhibited SDH activity and induced Nrf2 expression,
which subsequently induced HO-1 to inhibit pro-inflammatory
mediator induction. DI inhibited NLRP3 inflammasome and triggered
autophagy to inhibit NLRP3 inflammasome assembly, eventually
blocking GSDMD pore formation and IL-1β secretion. DI, dimethyl
itaconate; GSDMD, gasdermin D; HO-1, heme oxygenase-1; IRG1, immune
responsive gene 1; KEAP1, Kelch-like ECH-associated protein 1; LDH,
lactate dehydrogenase; LPS, lipopolysaccharide; NLRP3, NLR family
pyrin domain-containing 3; ROS, reactive oxygen species; SDH,
succinate dehydrogenase. |
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This research was funded by grants from the National
Natural Science Foundation of China (grant nos. 81820108011 and
81771262) and the Wenzhou Municipal Science and Technology Bureau
Project (grant nos. Y20190566 and Y20180153).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SY and LH conceived and designed the experiments,
performed the experiments, analyzed the data, contributed
reagents/materials/analytical tools, wrote the manuscript, prepared
figures and reviewed the manuscript. XZ, XiangL, YZ, XC, HZ and
XiaoL performed the experiments, analyzed the data, contributed
reagents/materials/analysis tools and prepared figures. QZ
conceived and designed the experiments and provided funding. SY and
LH confirm the authenticity of all the raw data. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Wenzhou Medical University (approval no.
wydw2019-0598).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dheen ST, Kaur C and Ling EA: Microglial
activation and its implications in the brain diseases. Curr Med
Chem. 14:1189–1197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Walter L and Neumann H: Role of microglia
in neuronal degeneration and regeneration. Semin Immunopathol.
31:513–525. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hu X, Leak RK, Shi Y, Suenaga J, Gao Y,
Zheng P and Chen J: Microglial and macrophage polarization-new
prospects for brain repair. Nat Rev Neurol. 11:56–64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lan X, Han X, Li Q, Yang QW and Wang J:
Modulators of microglial activation and polarization after
intracerebral haemorrhage. Nat Rev Neurol. 13:420–433. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee E, Eo JC, Lee C and Yu JW: Distinct
features of brain-resident macrophages: Microglia and
non-parenchymal brain macrophages. Mol Cells. 44:281–291. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xiong XY, Liu L and Yang QW: Functions and
mechanisms of microglia/macrophages in neuroinflammation and
neurogenesis after stroke. Prog Neurobiol. 142:23–44. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mathur A, Hayward JA and Man SM: Molecular
mechanisms of inflammasome signaling. J Leukoc Biol. 103:233–257.
2018.PubMed/NCBI
|
|
8
|
Gritsenko A, Green JP, Brough D and
Lopez-Castejon G: Mechanisms of NLRP3 priming in inflammaging and
age related diseases. Cytokine Growth Factor Rev. 55:15–25. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang L and Hauenstein AV: The NLRP3
inflammasome: Mechanism of action, role in disease and therapies.
Mol Aspects Med. 76:1008892020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu YG, Chen JK, Zhang ZT, Ma XJ, Chen YC,
Du XM, Liu H, Zong Y and Lu GC: NLRP3 inflammasome activation
mediates radiation-induced pyroptosis in bone marrow-derived
macrophages. Cell Death Dis. 8:e25792017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sorbara MT and Girardin SE: Mitochondrial
ROS fuel the inflammasome. Cell Res. 21:558–560. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gurung P, Lukens JR and Kanneganti TD:
Mitochondria: Diversity in the regulation of the NLRP3
inflammasome. Trends Mol Med. 21:193–201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi J, Gao W and Shao F: Pyroptosis:
Gasdermin-mediated programmed necrotic cell death. Trends Biochem
Sci. 42:245–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fink SL and Cookson BT: Apoptosis,
pyroptosis, and necrosis: Mechanistic description of dead and dying
eukaryotic cells. Infect Immun. 73:1907–1916. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sarhan M, Land WG, Tonnus W, Hugo CP and
Linkermann A: Origin and consequences of necroinflammation. Physiol
Rev. 98:727–780. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:347–364. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
He Y, Hara H and Nunez G: Mechanism and
regulation of NLRP3 inflammasome activation. Trends Biochem Sci.
41:1012–1021. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weigt SS, Palchevskiy V and Belperio JA:
Inflammasomes and IL-1 biology in the pathogenesis of allograft
dysfunction. J Clin Invest. 127:2022–2029. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kovacs SB and Miao EA: Gasdermins:
Effectors of pyroptosis. Trends Cell Biol. 27:673–684. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Zhang Z, Ruan J, Pan Y, Magupalli
VG, Wu H and Lieberman J: Inflammasome-activated gasdermin D causes
pyroptosis by forming membrane pores. Nature. 535:153–158. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dempsey LA: Immunoregulatory itaconate.
Nat Immunol. 19:5112018. View Article : Google Scholar
|
|
22
|
Yu XH, Zhang DW, Zheng XL and Tang CK:
Itaconate: An emerging determinant of inflammation in activated
macrophages. Immunol Cell Biol. 97:134–141. 2019.PubMed/NCBI
|
|
23
|
Lampropoulou V, Sergushichev A,
Bambouskova M, Nair S, Vincent EE, Loginicheva E,
Cervantes-Barragan L, Ma X, Huang SC, Griss T, et al: Itaconate
links inhibition of succinate dehydrogenase with macrophage
metabolic remodeling and regulation of inflammation. Cell Metab.
24:158–166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bordon Y: Itaconate charges down
inflammation. Nat Rev Immunol. 18:360–361. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cordes T, Wallace M, Michelucci A,
Divakaruni AS, Sapcariu SC, Sousa C, Koseki H, Cabrales P, Murphy
AN, Hiller K and Metallo CM: Immunoresponsive gene 1 and itaconate
inhibit succinate dehydrogenase to modulate intracellular succinate
levels. J Biol Chem. 291:14274–14284. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao C, Jiang P, He Z, Yuan X, Guo J, Li
Y, Hu X, Cao Y, Fu Y and Zhang N: Dimethyl itaconate protects
against lippolysacchride-induced mastitis in mice by activating
MAPKs and Nrf2 and inhibiting NF-kappaB signaling pathways. Microb
Pathog. 133:1035412019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shan Q, Li X, Zheng M, Lin X, Lu G, Su D
and Lu X: Protective effects of dimethyl itaconate in mice acute
cardiotoxicity induced by doxorubicin. Biochem Biophys Res Commun.
517:538–544. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mills EL, Ryan DG, Prag HA, Dikovskaya D,
Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E,
et al: Itaconate is an anti-inflammatory metabolite that activates
Nrf2 via alkylation of KEAP1. Nature. 556:113–117. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Perico L, Wyatt CM and Benigni A: A new
BEACON of hope for the treatment of inflammation? The endogenous
metabolite itaconate as an alternative activator of the KEAP1-Nrf2
system. Kidney Int. 94:646–649. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tang C, Wang X, Xie Y, Cai X, Yu N, Hu Y
and Zheng Z: 4-Octyl itaconate activates Nrf2 signaling to inhibit
pro-inflammatory cytokine production in peripheral blood
mononuclear cells of systemic lupus erythematosus patients. Cell
Physiol Biochem. 51:979–990. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abais JM, Xia M, Zhang Y, Boini KM and Li
PL: Redox regulation of NLRP3 inflammasomes: ROS as trigger or
effector? Antioxid Redox Signal. 22:1111–1129. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang Y, Li Y, Li H, Song H, Zhai N, Lou L,
Wang F, Zhang K, Bao W, Jin X, et al: Brucella dysregulates
monocytes and inhibits macrophage polarization through
LC3-dependent autophagy. Front Immunol. 8:6912017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li R, Zhang P, Wang Y and Tao K:
Itaconate: A metabolite regulates inflammation response and
oxidative stress. Oxid Med Cell Longev. 2020:54047802020.PubMed/NCBI
|
|
35
|
Hooftman A, Angiari S, Hester S, Corcoran
SE, Runtsch MC, Ling C, Ruzek MC, Slivka PF, McGettrick AF, Banahan
K, et al: The immunomodulatory metabolite itaconate modifies NLRP3
and inhibits inflammasome activation. Cell Metab. 32:468–478.e7.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sano M, Tanaka T, Ohara H and Aso Y:
Itaconic acid derivatives: Structure, function, biosynthesis, and
perspectives. Appl Microbiol Biotechnol. 104:9041–9051. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Medzhitov R and Horng T: Transcriptional
control of the inflammatory response. Nat Rev Immunol. 9:692–703.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun KA, Li Y, Meliton AY, Woods PS, Kimmig
LM, Cetin-Atalay R, Hamanaka RB and Mutlu GM: Endogenous itaconate
is not required for particulate matter-induced NRF2 expression or
inflammatory response. Elife. 9:e548772020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fusco R, Siracusa R, Genovese T, Cuzzocrea
S and Di Paola R: Focus on the role of NLRP3 inflammasome in
diseases. Int J Mol Sci. 21:2020. View Article : Google Scholar
|
|
40
|
Song L, Pei L, Yao S, Wu Y and Shang Y:
NLRP3 Inflammasome in Neurological Diseases, from Functions to
Therapies. Front Cell Neurosci. 11:632017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang K, Sun Z, Ru J, Wang S, Huang L, Ruan
L, Lin X, Jin K, Zhuge Q and Yang S: Ablation of GSDMD improves
outcome of ischemic stroke through blocking canonical and
non-canonical inflammasomes dependent pyroptosis in microglia.
Front Neurol. 11:5779272020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang K, Ru J, Zhang H, Chen J, Lin X, Lin
Z, Wen M, Huang L, Ni H, Zhuge Q and Yang S: Melatonin enhances the
therapeutic effect of plasma exosomes against cerebral
ischemia-induced pyroptosis through the TLR4/NF-κB pathway. Front
Neurosci. 14:8482020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sun Z, Nyanzu M, Yang S, Zhu X, Wang K, Ru
J, Yu E, Zhang H, Wang Z, Shen J, et al: VX765 attenuates
pyroptosis and HMGB1/TLR4/NF-kappaB pathways to improve functional
outcomes in TBI mice. Oxid Med Cell Longev. 2020:78796292020.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Orning P, Lien E and Fitzgerald KA:
Gasdermins and their role in immunity and inflammation. J Exp Med.
216:2453–2465. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gaidt MM and Hornung V: Pore formation by
GSDMD is the effector mechanism of pyroptosis. EMBO J.
35:2167–2169. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Xu M, Jiang P, Sun H, Yuan X, Gao S, Guo
J, Zhao C, Hu X, Liu X and Fu Y: Dimethyl itaconate protects
against lipopolysaccharide-induced endometritis by inhibition of
TLR4/NF-kappaB and activation of Nrf2/HO-1 signaling pathway in
mice. Iran J Basic Med Sci. 23:1239–1244. 2020.PubMed/NCBI
|
|
48
|
Shalaby YM, Menze ET, Azab SS and Awad AS:
Involvement of Nrf2/HO-1 antioxidant signaling and NF-kappaB
inflammatory response in the potential protective effects of
vincamine against methotrexate-induced nephrotoxicity in rats:
Cross talk between nephrotoxicity and neurotoxicity. Arch Toxicol.
93:1417–1431. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim HN, Park GH, Park SB, Kim JD, Eo HJ,
Son HJ, Song JH and Jeong JB: Sageretia thea inhibits inflammation
through suppression of NF-κB and MAPK and activation of Nrf2/HO-1
signaling pathways in RAW264.7 cells. Am J Chin Med. 47:385–403.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Subedi L, Lee JH, Yumnam S, Ji E and Kim
SY: Anti-inflammatory effect of sulforaphane on LPS-activated
microglia potentially through JNK/AP-1/NF-κB inhibition and
Nrf2/HO-1 activation. Cells. 8:1942019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Harris H and Rubinsztein DC: Control of
autophagy as a therapy for neurodegenerative disease. Nat Rev
Neurol. 8:108–117. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cadwell K: Crosstalk between autophagy and
inflammatory signalling pathways: Balancing defence and
homeostasis. Nat Rev Immunol. 16:661–675. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tu Y, Guo C, Song F, Huo Y, Geng Y, Guo M,
Bao H, Wu X and Fan W: Mild hypothermia alleviates diabetes
aggravated cerebral ischemic injury via activating autophagy and
inhibiting pyroptosis. Brain Res Bull. 150:1–12. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li MY, Zhu XL, Zhao BX, Shi L, Wang W, Hu
W, Qin SL, Chen BH, Zhou PH, Qiu B, et al: Adrenomedullin
alleviates the pyroptosis of Leydig cells by promoting autophagy
via the ROS-AMPK-mTOR axis. Cell Death Dis. 10:4892019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mehto S, Jena KK, Nath P and Chauhan S,
Kolapalli SP, Das SK, Sahoo PK, Jain A, Taylor GA and Chauhan S:
The Crohn's disease risk factor IRGM limits NLRP3 inflammasome
activation by impeding its assembly and by mediating its selective
autophagy. Mol Cell. 73:429–445 e427. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Shi CS, Shenderov K, Huang NN, Kabat J,
Abu-Asab M, Fitzgerald KA, Sher A and Kehrl JH: Activation of
autophagy by inflammatory signals limits IL-1β production by
targeting ubiquitinated inflammasomes for destruction. Nat Immunol.
13:255–263. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Marino MJ: The use and misuse of
statistical methodologies in pharmacology research. Biochem
Pharmacol. 87:78–92. 2014. View Article : Google Scholar : PubMed/NCBI
|