Introduction
Overview of autophagy
Autophagy is a range of biological processes in
which the cell degrades parts of itself within the lysosome (or the
analogous organelle, such as the vacuole in yeast and plants),
followed by the release and reuse of the breakdown products. In a
sense, autophagy is a mechanism for cellular recycling. In
addition, different autophagy processes are extremely important in
cell and body function and homeostasis (1,2).
Autophagy usually occurs under basic conditions, and is stimulated
under different types of cellular stress, including endoplasmic
reticulum (ER) stress, oxidative stress, mitochondrial damage,
nutrition and growth factor starvation, hypoxia and some drug
treatments (3). At present,
according to the intracellular components transported to the
lysosome, the autophagy pathway can be divided into the following
three categories: Macroautophagy, microautophagy and molecular
chaperone-mediated autophagy (CMA) (1,4,5).
Macroautophagy is the most common and clinically significant form
of autophagy. During the process of macroautophagy, soluble
proteins in the cytoplasm and degenerative and necrotic organelles
are wrapped by a double-layer membrane structure derived from
non-lysosomal cells, namely autophagic bubbles, and is carried into
the lysosome by autophagic vesicles for degradation and processing
(1,6). Microautophagy is a pathway directly
degraded by lysosomes, in which lysosomal membranes directly invade
and encapsulate cytoplasmic components in double-membrane vesicles
(4). Chaperone-mediated autophagy
(CMA) has a cytoplasmic protein with a specific pentapeptide motif
(KFERQ) that recognizes and binds to 70 kDa heat shock homologous
protein (Hsc70) and is directly delivered to the lysosome for
degradation. The whole process does not require the participation
of vesicles, as the substrate of CMA is a soluble protein molecule
(5). Hence, the CMA degradation
pathway is selective when removing proteins, while macroautophagy
and microautophagy have no obvious selectivity. In view of the
multi-directional regulation of autophagy, cell autophagy has been
confirmed in recent years to be closely associated with metabolic
diseases, cardiovascular diseases, lung diseases and
neurodegenerative diseases and cancer (7). Therefore, a thorough understanding of
the autophagy mechanism is particularly important in the diagnosis
and treatment of diseases.
Autophagy refers to the process whereby damaged
organelles, misfolded proteins and protein aggregates are packed
into autophagosomes and transported to the lysosome for degradation
(8,9). It mainly includes four stages:
Initiation stage, autophagosome extension stage, autophagosome
maturation stage and autophagosome degradation stage, and the
regulation mechanism of each stage is complicated. The formation
and renewal of autophagosomes involves evolutionary conserved genes
called autophagy-related (ATG) genes. During the start-up phase,
mechanistic target of rapamycin complex 1 (mTORC1) activates the
ULK1 (also known as ATG1) complex [involving ULK1, ULK2, ATG13,
FIP200 (also known as RB1CC1) and ATG101], and AMP-activated
protein kinase AMPK. During the extension phase of autophagosomes,
the ULK1 complex phosphorylates and activates the Beclin-1-VPS34
complex. This complex includes Beclin-1, VPS34 [Class III
phosphatidylinositol 3-kinase (PI3K)] and other proteins, such as
VPS15, ATG14L and Beclin 1 modulator 1, depending on their
subcellular complexity the positioning (10). The ATG5-ATG12 complex binds to ATG16
in order to expand the autophagosome membrane, and members of the
LC3 and GABARAP protein families bind to the lipid
phosphatidylethanolamine and are subsequently recruited to the
membrane. ATG4 binds to ATG7, combining LC3-I and PE to form LC3-II
(also known as MAP1LC3B). LC3 is commonly used as an autophagy
marker (11). Eventually, the
autophagosome is fused with the lysosome, the contents are
degraded, and the macromolecular precursor is recovered or used to
promote metabolic pathways. Adaptor protein chelate 1 (also known
as p62) targeting specific substrates to autophagosomes and LC3II
is degraded together with other cargo proteins and can be used as a
measure of autophagy flux (11).
Overview of ion channels
The ion channel of the biomembrane is a pathway for
the transport of various inorganic ions across the membrane. It is
mainly composed of transmembrane proteins and forms pores in the
cell membrane to regulate the exchange of specific ions. The role
of ion channels is mainly to maintain the steady state of ions
inside and outside the cell and to transmit cell signals, while
regulating the volume, proliferation, apoptosis, migration,
invasion and adhesion of cells. Ion channels can be classified
according to the channel opening and closing or gating mechanism
(12). For example, ion channels
can control the changes in ions inside and outside the cell through
the combination of membrane voltage (voltage-gated), extracellular
ligands (ligand-gated), or a combination of intracellular secondary
messengers. In addition to the gate mechanism, ion channels can
also be classified according to their selectively permeable ions
(12). For example, there are
various types of calcium (Ca2+), potassium
(K+) and sodium (Na+) ion channels.
Calcium-permeable ion channels can regulate the levels of
mitochondria, ER, lysosomes and cytosolic calcium. The ion channels
are located on the intracellular organelle membrane or plasma
membrane (13,14). Calcium penetration channels mainly
include transient receptor potential (TRP) channels, storage
operation channels (SOC), voltage-gated calcium channels, 2-pore
segment channel (TPCN), mitochondrial permeability transition pores
(MPTP), and mitochondrial calcium unidirectional transporters
(MCU), IP3 and ryanodine receptors and other receptors. By
regulating the driving force of Ca2+, it provides
Ca2+ in and out pathways, thereby regulating the changes
in Ca2+ concentration intracellularly and
extracellularly, and thus affecting calcium-dependent processes
such as proliferation, apoptosis and autophagy (13–18).
As an example, several members of the TRP family of ion channels,
namely TRPC1, TRPC3, TRPC6, TRPV1, TRPV6, TRPM1, TRPM4, TRPM5,
TRPM7 and TRPM8, show altered expression in cancer cells. The
involvement of SOCs, MPTP, MCU and IP3 receptors and ryanodine
receptors in the regulation of cell death has also been
described.
Aside from Ca2+-permeable ion channels,
other ion channels also play a critical role in cell biological
behavior. Sodium channels are considered as integral membrane
proteins that mediate sodium influx into the cell or intracellular
organelles. According to the gating mechanism, sodium channels can
be divided into voltage-gated sodium channels family and epithelial
sodium channels (19,20). Voltage-gated sodium channels are
distributed in almost all cell types, while epithelial sodium
channels are mainly located in the skin and kidneys. Voltage-gated
sodium channels are responsible for membrane depolarization and
regulating cell invasion and migration. Potassium channels mediate
the flow of potassium ions down their electrochemical gradient.
K+ channels are widely distributed in all kinds of cell
types and are involved in the regulation of various physiological
processes including maintenance of membrane potential, regulating
cell proliferation and apoptosis. Based on structural criteria,
conductance properties as well as activation mechanisms, potassium
channels are divided into 4 classes: Voltage-gated,
calcium-activated, inward-rectifier and 2-pore-domain potassium
channels (21). Chloride channels
mediate the flow of chloride ions across cell membranes and reside
both in the plasma membrane and in intracellular organelles.
Chloride channels are ubiquitously expressed and regulate a variety
of fundamental cellular processes including apoptosis, volume
regulation, cell cycle and intracellular organelle acidification
(22). According to the gating
mechanism, chloride ion channels can be divided into chloride
voltage-gated channels, cystic fibrosis transmembrane conductance
regulator, calcium activated chloride channels, volume-regulated
anion channels and ligand gate control anion channel (22).
Role of ion channels in the regulation of
autophagy
Calcium-permeable channels in the
regulation of autophagy
In recent years, reports on ion channel-regulated
autophagy have focused on calcium-permeable ion channels (23). Calcium is a ubiquitous intracellular
messenger that affects almost every aspect of cellular life
(24). The calcium-permeable ion
channel, as a ‘calcium signal toolkit’, promotes the change in
cytosolic calcium concentration by providing a Ca2+
entry pathway and regulating the driving force of Ca2+
entry. It also provides a way for Ca2+ to circulate in
the cytoplasm and organelles. In this way, calcium-permeable
channels indirectly control various calcium-dependent cellular
processes, such as cell proliferation, apoptosis and autophagy. The
complex role of Ca2+ in the regulation of autophagy has
become apparent since 1993, when the first report on autophagy and
intracellularly sequestered calcium was published (25). It was demonstrated that both a
decrease and an increase in intracellular Ca2+ levels
can inhibit autophagy in rat hepatocytes (24). Afterwards, Høyer-Hansen et al
(26) demonstrated that
Ca2+ mobilizing agents, namely vitamin D3, thapsigargin,
ATP and ionomycin, stimulate autophagy via a signaling pathway
involving CAMKK2/CaMKKβ (calcium/calmodulin dependent protein
kinase 2), which directly activates AMPK to inhibit mTOR and induce
the accumulation of autophagosomes to prove that the increase in
free cytoplasmic calcium is an effective inducer of macroautophagy.
However, the ‘classical’ Ca2+-CAMKK2-AMPK pathway (both
MTOR-dependent and MTOR-independent) is not a unique pathway for
cytosolic calcium-induced autophagy. One study showed that
Ca2+ signaling can induce autophagy independently of
Ca2+-mediated AMPK activation (27). It is reported that exogenous
introduction of Ca2+ precipitated into mammalian cells
as calcium phosphate will induce BECN1, ATG5 and
phosphatidylinositol 3 kinase dependent autophagy (28). To date, the role of calcium in the
regulation of autophagy is quite complex and controversial. Several
reports indicate that calcium has an inhibitory effect on
autophagy, in contrary to the calcium stimulation effect reported
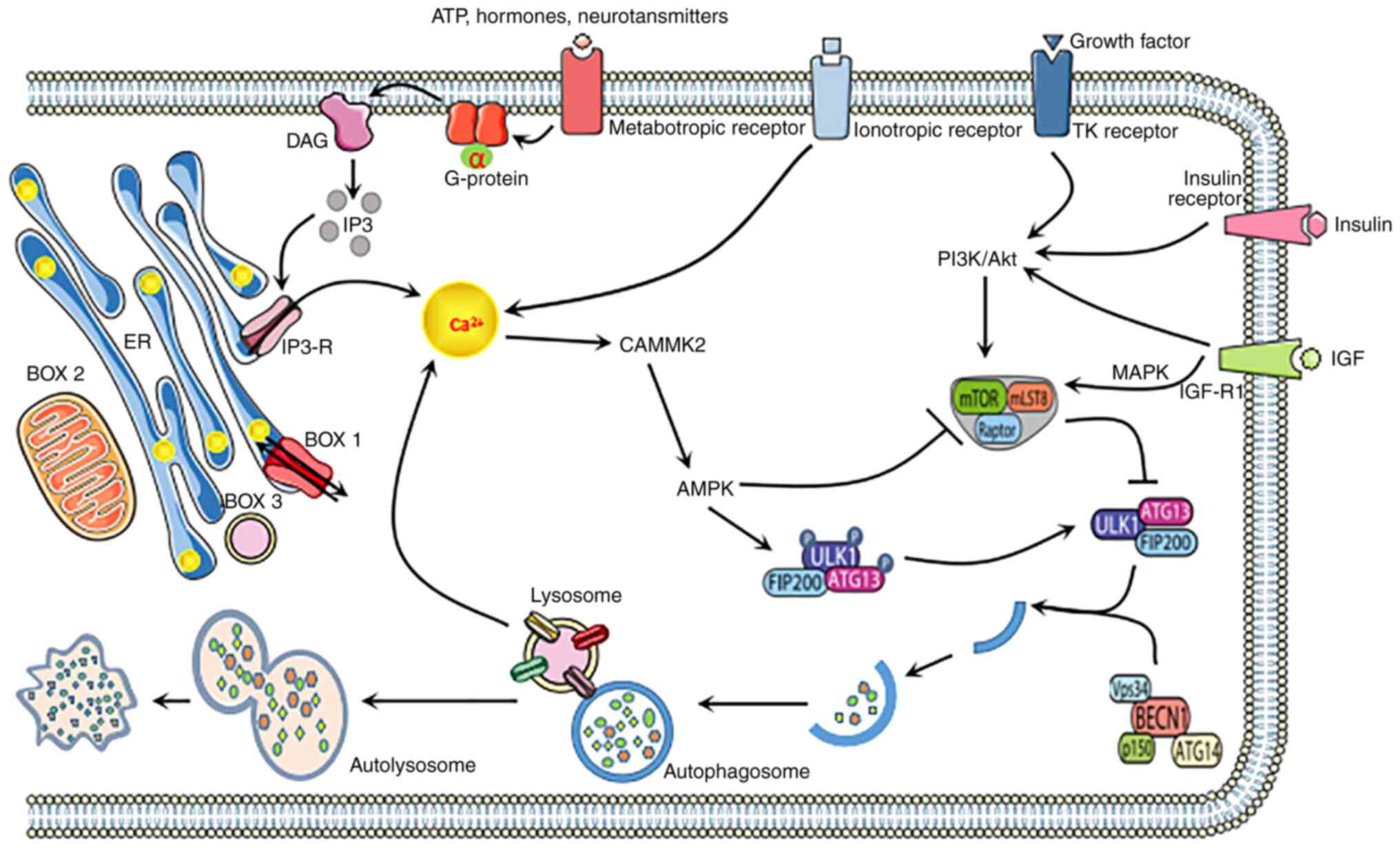
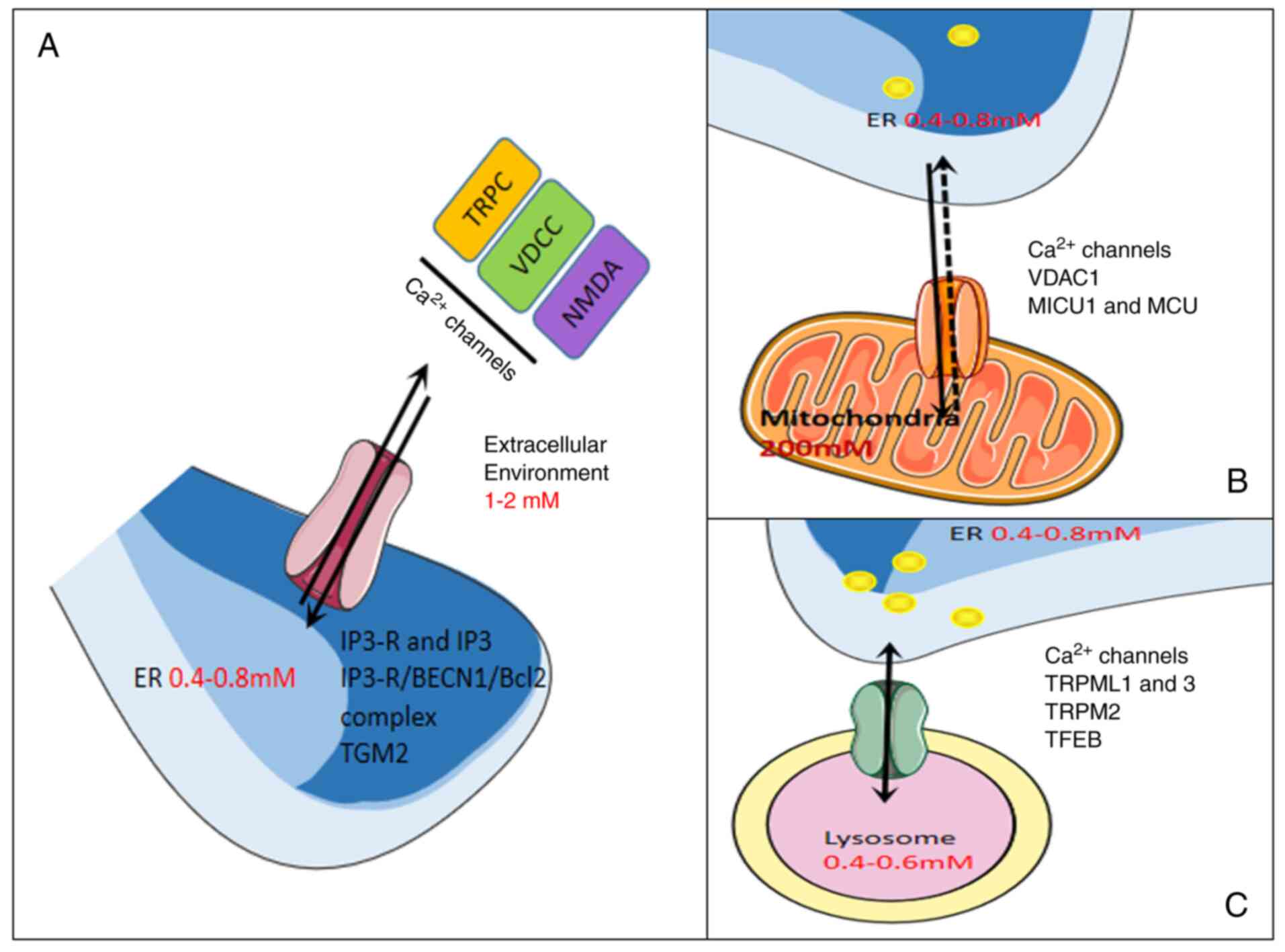
by others (29,30). A graphic overview of the mechanisms
of calcium-permeable channel in autophagy regulation is presented
in Figs. 1 and 2.
The ubiquitously expressed inositol
1,4,5-triphosphate receptor (ITPR) is the main intracellular
Ca2+ release channel and also the channel most reported
for calcium-permeable channels as autophagy regulators. It is
located on the membrane of the ER, mainly inositol
1,4,5-triphosphate (InsP3)-mediated calcium release from the ER
(31). These reports indicate that
the complex role of ITPR in the regulation of autophagy may lead to
the activation and inhibition of this process (32,33).
These complex effects mainly depend on the spatiotemporal
characteristics of Ca2+ signals, including the presence
of Ca2+ signal microdomains in calcium of
ER-mitochondria and ER-lysosomes. In DT40 cells, basal autophagy is
negatively regulated by ITPR-dependent Ca2+ signaling,
which maintains elevated MTORC1 activity through an
AMPK-independent pathway (34).
Besides, some researchers have proposed another MTOR-independent
mechanism, ITPR-mediated Ca2+ release from ER and
subsequent Ca2+ absorption by mitochondria to maintain
the basic needs of mitochondrial bioenergy and ATP production in
resting cells. The lack of Ca2+ transfer will lead to a
decrease in ATP production and AMPK activation, which in turn
activates survival autophagy in a MTOR-independent manner (35). The transfer of Ca2+ from
the ER to the mitochondria is proposed to occur through the ITPR on
the ER to the voltage-dependent anion channel 1 (VDAC1) on the
outer mitochondrial membrane. It then enters the matrix through the
mitochondrial inner membrane mediated by a highly selective,
low-affinity Ca2+ channel known as mitochondrial calcium
unidirectional transporter (MCU). By controlling mitochondrial
Ca2+ concentration, MCU plays a key role in regulating
aerobic metabolism and cell survival. Tomar et al (36) confirmed the important role of MCU
and MCUR1 in mitochondrial bioenergy and autophagy. The study
showed that mouse cardiomyocytes and endothelial cells lacking
MCUR1 severely impaired mitochondrial Ca2+ absorption
and current flow through the MCU. The loss of MCU or MCUR1 will
interfere with the MCU hetero-oligomeric complex, damage
mitochondrial bioenergy, cell proliferation and migration, and
trigger AMPK-dependent autophagy. In addition to ITPR, some other
calcium-permeable channels have also been shown to participate in
autophagy regulation. For example, the role of intracellular
voltage-gated calcium channels in autophagy has been revealed. Tian
et al (37) reported that
the P/Q-type calcium channel CACNA1A/Cav2.1 (calcium voltage-gated
channel subunit α1A) is located in the lysosome, and the loss of
lysosome CACNA1A in the cerebellar cultured neurons leads to
lysosome unable to merge with the endosome. It was shown that
lysosomal fusion requires CACNA1A, instead of CACNA1A residing on
the plasma membrane (37). Current
evidence also supports the view that ryanodine receptor, TPCN, TRP
channels and SOCE channels are also involved in the regulation of
autophagy (15,16,38–40).
Other ion channels regulate
autophagy
The accumulated data indicates that the complex
regulation of other ion channels and autophagy opens up a new way
to target autophagy in disease treatment. For example, the
acid-sensitive ion channel 3 (ASIC3) in the sodium channel can
regulate the apoptosis of chondrocytes in articular cartilage, and
the overexpression of ASIC3 and hypoxia-inducible factor-1α
(HIF-1α) can inhibit the cells in intervertebral disc degeneration
by preventing the G1 cell cycle transition and activating the MAPK
pathway of autophagy to promote cell apoptosis (41,42).
In addition, non-selective cation channels (such as members of the
TRP channel family) can also permeate Na+ ions,
suggesting that these channels may regulate autophagy through
Na+-dependent mechanisms. There are also studies that
have proposed several potassium channels to regulate autophagy.
According to reports, the opening of the ATP-sensitive
K+ (K+ + ATP) channel induces autophagy in
different cell types. For example, according to Williams et
al (43), minoxidil is a
K+ + ATP channel agonist that can enhance autophagy in
PC12 cells, while K+ + ATP channel blockers such as
quinine sulfate and tosylamide can slow down autophagic substrate
removal. One study suggested that KCNH7/Kv11.3/HERG3 (member 7 of
potassium voltage-gated channel subfamily H) plasma membrane
potassium channels are involved in the regulation of melanoma
autophagy and aging (44). In
detail, by stimulating the KCNH7 channel with the small molecule
activator NS1643, AMPK-dependent signaling pathways in melanoma
cell lines are activated to induce autophagy (44). There are few data on the role of
chloride channels in the regulation of autophagy. In fact, the
Cl-channel is very low in selectivity among anions and allows
various anions to penetrate. A previous study showed that small
interference RNA-mediated knockdown of CLIC4/mtCLIC (chloride
intracellular channel 4) can enhance BECN1-dependent autophagy and
apoptosis in human glioma U251 cells induced by starvation
(45). In addition, Wang et
al (46) reported that
volume-sensitive outward rectification (LRRC8/VSOR) chloride
channels can promote high glucose-induced cardiomyocyte apoptosis
by inhibiting autophagy. The authors demonstrate that LRRC8/VSOR
channel blockers induce autophagy through MTOR inhibition (46). In summary, the aforementioned data
indicate the diversity and importance of ion channels in the
regulation of autophagy. Today's limited data proves that ion
channel dysfunction and autophagy dysregulation are associated with
several diseases, and it is possible to consider ion channels and
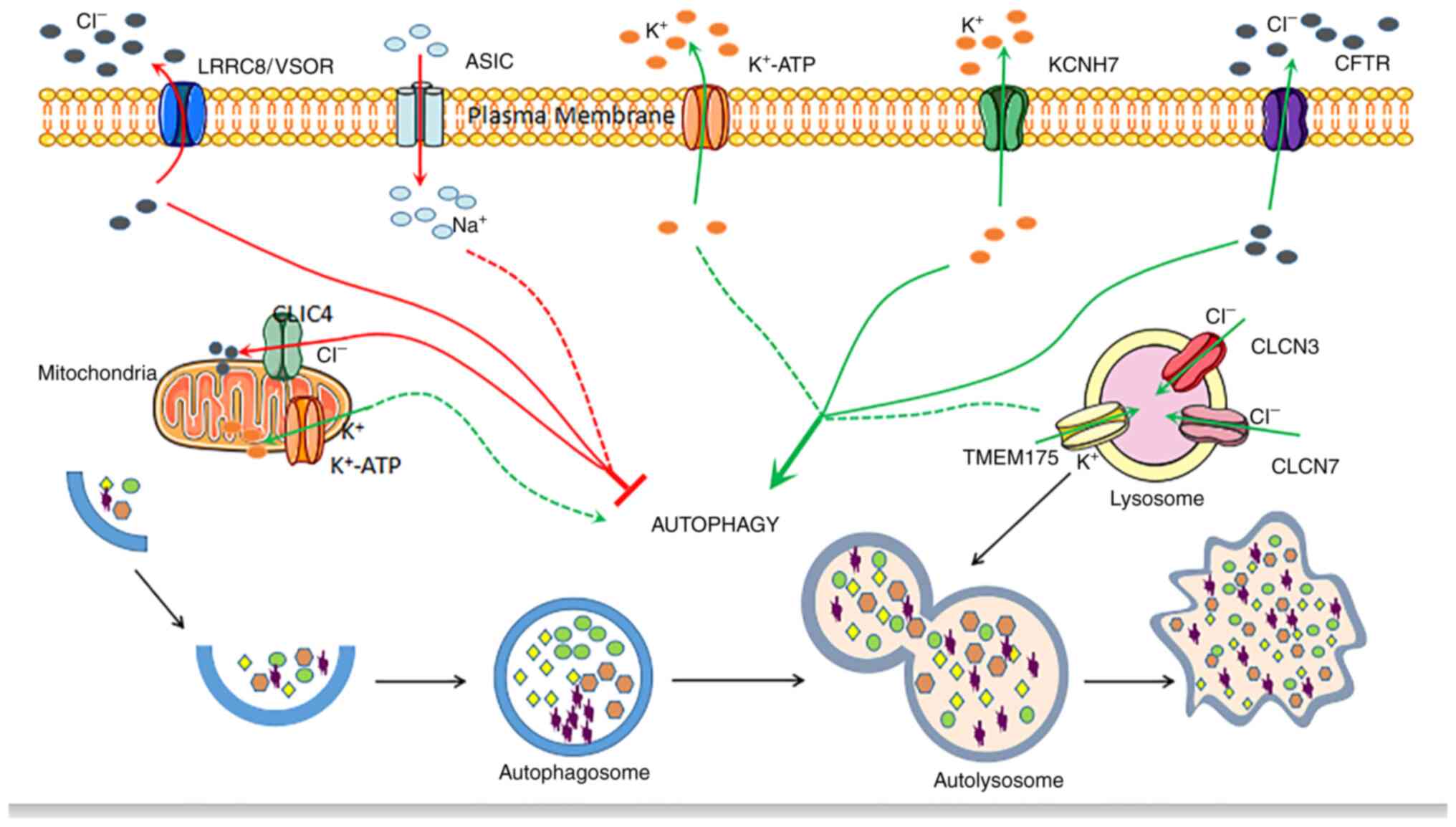
autophagy as potential therapeutic targets. A graphic overview of
the mechanisms associated with other ion channels on autophagy
regulation is presented in Fig.
3.
In recent years, cytoplasmic and extracellular ion
concentrations and ion channel types, which mediate ion flux across
cell membranes, have become important regulators of basic autophagy
and induced autophagy. The dysfunction of ion channels may cause
the dysregulation of autophagy, which in turn leads to a series of
diseases. Therefore, ion channels and autophagy can be regarded as
potential therapeutic targets. However, there is still limited
information about the molecular nature of autophagy-regulated
channels and their regulatory mechanisms, and the most important
ones are reports of calcium ions and calcium channels regulating
autophagy. The existing data on ion channels participating in the
autophagy regulatory pathway was discussed in the present
review.
Interaction between diseases, ion channels
and autophagy
Calcium and calcium channels in liver
disease regulate autophagy
The liver can provide the nutrients used by the
body's organs. When the body is under starvation conditions, it
will induce autophagy in the liver to help maintain the homeostasis
of the whole body. One study showed that during starvation,
glucagon stimulates the InsP3R1-CaMKII pathway, which mediates the
phosphorylation of O-linked β-N-acetylglucosamine (O-GlcNAc)
transferase (OGT) at a post-translational level (47). ULK protein is a target to fine-tune
liver autophagy. During starvation, glucagon stimulates the
InsP3R1-CaMKII pathway, thereby mediating OGT phosphorylation. This
approach uses post-translational levels of ULK protein as a target
to fine-tune liver autophagy and provide substrates for
gluconeogenesis and ketogenic to maintain systemic glucose
homeostasis (47). In addition,
autophagy caused by nutrient deficiency or starvation can also be
inhibited by BAPTA-AM (BAPTA is a selective Ca2+
chelator, and its acetyl methyl ester derivative is BAPTA-AM),
which indicates that other autophagy-inducing conditions, such as
starvation or rapamycin, may also activate autophagy through the
Ca2+ signaling pathway. In this study, to demonstrate
whether calreticulin was stimulated by ER stress, the authors
generated an ER stress murine model via intraperitoneal
administration of an ER stress inducer, tunicamycin, and determined
the hepatic expression of calreticulin. Researchers found that the
overexpression of calreticulin stimulates the formation of
autophagosomes and increases autophagic flux. These findings
indicate that a calreticulin-based mechanism couples endoplasmic
reticulum stress with autophagy activation, thereby reducing
cellular stress. This may be achieved by reducing the formation of
abnormally folded proteins (48).
Under ER stress conditions, calreticulin induces autophagy by
interacting with microtubule-associated protein 1A/1B-light chain 3
(LC3). Further studies have shown that calreticulin-mediated
autophagy activation and decrease in ER stress require an area that
interacts with the LC3 in calreticulin (48).
Non-alcoholic fatty liver disease (NAFLD) is the
most common cause of chronic liver disease. NAFLD involves the
accumulation of excess lipids in hepatocyte cytoplasmic lipid
droplets, which can develop into nonalcoholic steatosis, fibrosis
and HCC. Furthermore, elevated free fatty acids, especially
saturated fatty acids, such as palmitic acid, may play an important
role in the lipotoxic mechanism of NAFLD (49). Saturated fatty acids can induce
autophagy dysfunction and ER stress, increase intracellular
calcium, and ultimately lead to hepatocyte apoptosis (50,51).
Studies on the mechanism of autophagy impairment in fatty liver
disease have determined that the increase in intracellular calcium
is a mediator of autophagy dysfunction. Park et al (52) demonstrated that saturated free fatty
acids (FFA) induce increased cytosolic calcium in liver cells,
thereby inhibiting autophagy. It was determined that obesity and
lipotoxicity can induce a chronic increase in cytosolic calcium
levels in liver cells, thereby interfering with the fusion between
autophagosomes and lysosomes, which can weaken liver autophagy
flux. Subsequently, Czaja (53)
found that in obese mice, the decrease in autophagy can be reversed
by calcium channel blockers, resulting in a decrease in
steatohepatitis. In addition, zinc (Zn) deficiency is the most
commonly found nutritional manifestation of fatty liver disease. Zn
is known to stimulate liver lipid oxidation, but research by Wei
et al (54) demonstrated
that zinc is an effective promoter of fat phagocytosis, which can
significantly decrease the accumulation of lipid in hepatocytes and
activate fat phagocytosis through the Zn2+/MTF-1/PPARα
and Ca2+/CaMKKβ/AMPK pathways. The aforementioned
results provide new insights into Zn nutrition and its potential
beneficial role in preventing fatty liver disease. In addition,
calcium can also be used to control autophagy in liver injury to
protect the liver from inflammation. LPS is known to induce liver
injury and promote hepatocyte autophagy (55). CD38 is an enzyme that produces
NAADP, which is the most effective intracellular Ca2+
mobilization signal molecule (56).
In a study by Rah et al (57), it was found that administration of
NAADP to mice can alleviate LPS/GalN-induced liver injury by
enhancing the autophagy process, which revealed that CD38 and
Ca2+-activated messenger NAADP are important regulators
of hepatocyte autophagy. Although the downstream targets of
Ca2+ signaling pathway mediated by NAADP-mediated
autophagy have not yet been found, the Ca2+ signaling
pathway mediated by CD38/NAADP triggered the autophagy process,
induced the expression of autophagy-associated genes and protects
the liver from injury caused by LPS (57).
In recent years, many researchers have worked hard
to find new therapeutic targets to successfully treat
hepatocellular carcinoma (HCC), and there are also therapeutic
strategies that study the interaction mechanism between calcium
signaling and autophagy. The earliest research was conducted by Shi
et al (58), who found that
Trichokonin VI (TKVI), a peptaibol from Trichoderma pseudokoningii
SMF2, induced growth inhibition of HCC cells in a dose-dependent
manner. TK VI induces the influx of Ca2+ across the cell
membrane or the release of intracellular Ca2+ stores,
resulting in increased cytoplasmic calcium activation of Bak and
calpain, which induces two types of cell death, including
calcium-calpain-Bax in HepG2 mediated apoptosis and
calcium-Bak-mediated autophagy (58). It is suggested that the increase in
intracellular calcium level in HCC mediates the autophagy of HCC
cells, thereby inducing cell apoptosis. 5-fluorouracil (5-FU) is
the most widely used chemotherapeutic agent for the
gastrointestinal tract, breast cancer, head and neck cancer and
ovarian cancer. Accumulating evidence suggests that various cancer
cells including colon cancer (59),
pancreatic cancer (60) and gastric
cancer cells (61) can induce
autophagy through 5-FU. It is known that increased SOCE associated
with upregulation of Stim1, Orai1 or TRPC1 expression has been
observed in several different types of tumors (62–64),
indicating that SOC is a potential therapeutic target for the
treatment of cancer. In liver cancer, recent studies have shown
that inhibition of TRPC1 by regulating SOCE can inhibit cell
proliferation (65,66). A study showed that with increased
expression of Orai1 in liver cancer tissues, 5-FU can inhibit
Ca2+ entry by down-regulating Orai1-mediated SOCE, and
by inhibiting the activation of PI3K/AKT/mTOR signaling pathway,
causes 5-FU in HepG2 cells to induce autophagic cell death and
enhances the chemical sensitivity of liver cancer cells to 5-FU
(67). Based on the fact that
changes in mitochondrial dynamics and cytosolic calcium may occur
at the same time, the study by Huang et al (68) provides a new idea. The study by
Huang et al found that in HCC cells, mitochondrial fission
through STIM1-mediated storage-operated Ca2+ entry
(SOCE) significantly enhances cytoplasmic Ca2+
signaling, and increased cytoplasmic calcium signaling promotes
mitochondrial fission, forming a positive feedback loop. It is
mitochondrial fission and the cytoplasmic calcium signaling pathway
that form a positive feedback loop, and promote the autophagy of
liver cancer cells through the Ca2+/CAMKK/AMPK signaling
pathway (68).
Calcium and calcium channels in
pancreatic diseases regulate autophagy
Acute pancreatitis (AP) is the most common
pancreatic disease, which is mainly caused by a variety of factors.
It is usually accompanied by inflammation, edema, bleeding and even
necrosis of its own tissues or remote organs (69–71).
During the onset of AP, under the stimulation of cholecystokinin
(CCK), bile acids, alcohol metabolites or some other reason, the
influx of Ca2+ was significantly increased (71–74).
It was determined that storage-operated Ca2+ entry
(SOCE) is a key pathogenic step in AP development and can lead to
trypsin activation, inflammation and vacuoles (73,74).
Recent studies have shown that Orai1 is the main SOCE channel in
pancreatic acinar cells. After the ER Ca2+ reservoir is
emptied, its opening interacts with stromal interaction molecule 1
(STIM1) (75–78). Research by Zhu et al
(79) found that caerulein (CCK
receptor agonist) triggers SOCE by inducing the interaction between
STIM1 and Orai1, thereby activating calcineurin (CaN), which
activates activated T cells nuclear translocation and
dephosphorylation of nuclear factor and transcription factor EB
(TFEB), thereby promoting the transcriptional activation of
multiple chemokine genes and autophagy-associated genes (79). These findings suggest that TFEB may
play a vital role in the long-term effect of SOCE/CaN on autophagy
and vacuoles in AP development. In addition, the inhibition of SOCE
or CaN decreases the formation of autophagosomes and the severity
of vacuoles, edema and inflammation, which supports the hypothesis
that CaN is a key regulator of autophagy and inflammation in AP. In
summary, CaN-mediated TFEB activation regulates the autophagy of
SOCE in AP. Moreover, there is another statement about the
signaling pathway that triggers and promotes acute pancreatitis
(AP), namely, the pathogenesis of AP has been associated with
abnormal increases in cytosolic Ca2+, mitochondrial
dysfunction, impaired autophagy and ER stress (80).
Accumulated evidence indicates that interleukin-1β
(IL-1β) is important in AP (81,82).
IL-1β can stimulate the autophagy of macrophages and induce ER
stress (83–86). Among them, ER is an important
organelle for calcium storage and can regulate intracellular
Ca2+ homeostasis. ER stress may cause the ER to release
calcium into the cytoplasm (87).
Additionally, it has been thought that Ca2+ plays an
important role in many aspects of the cellular processes involved
in pancreatitis (88). Studies have
also shown that there is a connection between intracellular
Ca2+ signaling and autophagy, indicating that elevated
free cytoplasmic Ca2+ can lead to autophagy activation
(89,90). Based on the aforementioned analysis,
Xu et al (91) hypothesized
that IL-1β caused ER stress, resulting in the release of
Ca2+ into the cytoplasm, and subsequent activation of
trypsinogen by AP autophagy damage. The data indicate that IL-1β
can induce autophagy damage to acinar cells, and this effect is
time-dependent. Besides, studies have shown that ER can release
Ca2+ into the cytoplasm in response to IL-1β
stimulation, and cause a transient oscillation signal of
Ca2+ mediated by the InsP3 signaling pathway, which
increases the cytoplasmic Ca2+ in acinar cells. After
inhibiting Ca2+ signal, the expression of LC3-II and
trypsinogen activation were both decreased. In addition, capsaicin
is an effective stimulant for TRPV1. Díaz-Laviada and
Rodríguez-Henche (92) demonstrated
that capsaicin increases intracellular calcium, produces reactive
oxygen species, disrupts mitochondrial membrane transition
potential, and activates transcription factors such as NFκB and
STATS, and triggers AMP-dependent kinases in cell death (AMPK) and
autophagy pathways trigger apoptosis in human pancreatic cancer
cells (92).
Calcium and calcium channels in
gastric diseases regulate autophagy
Gastric cancer (GC) is the fifth most common cancer
in the world and ranks third in the cause of death (93). Although the diagnosis and treatment
of early GC have been developed, the long-term survival rate of
patients with advanced GC is still low (94). Therefore, understanding the basic
mechanisms of GC will help elucidate specific molecular targets and
develop more effective therapies for the disease. Currently, there
is increasing evidence that TRPM2 function is particularly critical
in many cellular events by inducing several intracellular pathways
(such as oxidative stress signaling, MAPK and autophagy events),
including insulin secretion, cytokine production and cell
metabolism, temperature-steady state and cell death. In the study
by Almasi et al (95) it was
demonstrated that TRPM2 channel-mediated autophagy regulation
maintains mitochondrial function through JNK signaling pathway and
promotes the survival of GC cells. The results indicate that TRPM2
regulates autophagy through mechanistic targets of c-Jun N-terminal
kinase (JNK)-dependent and rapamycin-independent pathways. In the
absence of TRPM2, the downregulation of the JNK signaling pathway
impairs autophagy, eventually leading to the accumulation of
damaged mitochondria and the death of gastric cancer cells.
Acid-sensitive ion channels (ASICs) are insensitive cation channels
on the media of the epithelial Na+ channel/phenylephrine
superfamily and are activated by extracellular protons (96,97).
According to reports, ASICs can activate autophagy (98,99).
In a study by Zhang et al (100), it was confirmed that ASIC1 and
autophagy were activated in the gastric cancer tissues and cells of
patients. ASIC1 regulates autophagy through ATG5 activation. At the
same time, in the mouse xenograft model established, ASIC1 or ATG5
knockdown inhibited the growth of cancer cells, while ASIC1 shRNA
inhibited tumor volume and prolong the survival time of animals.
Therefore, the downregulation of ASIC1 can inhibit GC by inhibiting
autophagy (100).
Helicobacter pylori (H. pylori) is a
common pathogenic bacterium that causes stomach diseases and
exhibits severe stomach diseases, including gastritis and gastric
malignancies (101). The secreted
vacuolar cytotoxin A (VacA) is a key bacterial virulence factor
that is inserted into the host cell membrane and essentially acts
as a chloride channel (102). It
can target mitochondria and may penetrate VacA, resulting in loss
of mitochondrial membrane potential, thereby causing mitochondrial
autophagy (103). Inhibiting the
autophagy of the host's macrophages is one of the strategies used
by several pathogen cells (including H. pylori) to escape
killing. Studies have pointed out that VacA will inhibit the
lysosomal calcium channel MCOLN1/TRPML1, resulting in increased
lysosomal calcium levels, thereby disrupting the transport events
between lysosomes and autophagosomes, late endosomes and plasma
membranes, leading to large enzyme-like organelles and autophagic
vesicles aggregate, and the autophagic flux is severely damaged,
which ultimately promotes the survival of bacteria in the cell
(104); the latest research has
also confirmed such results (105). For the treatment of H.
pylori, recent research suggests that vitamin D3 has an ideal
anti-H. pylori effect, and can even resist
antibiotic-resistant strains (106). Cells treated with vitamin D3
activate the PDIA3 receptor to restore the lysosomal degradation
function and promote the nuclear translocation of the PDIA3-STAT3
protein complex, and subsequently upregulate the MCOLN3 channel,
resulting in enhanced release of lysosomal Ca2+ and
lysozyme. The body acidification is normal, and the restored
lysosomal degradation function eliminates H. pylori through
the autolysosome pathway.
Calcium and calcium channels in
intestinal diseases regulate autophagy
Despite many treatment and screening attempts,
colorectal cancer (CRC) remains the main life-threatening malignant
tumor (107). Autophagy is known
to be associated with various clinical diseases, such as CRC.
Emerging research shows that in human cancer, STIM1/Orai1-mediated
SOCE is crucial in regulating autophagy-associated mechanisms. A
recent study reported that in colorectal cancer cells, the SOCE
inhibitor SKF-96365 induces cytoprotective autophagy (108). Calcium antagonist SKF-96365
decreases the concentration of Ca2+ by inhibiting SOCE
in human colorectal cancer cells, while decreasing the colon cancer
cell line through the calcium/calmodulin-dependent protein kinase
IIγ (CaMKIIγ)/AKT signaling cascade Akt activity. The present study
highlights the unique role of STIM1/Orai1-mediated SOCE in
regulating autophagy. In addition, studies have shown that once
activated by integrin-mediated adhesion, the hERG potassium channel
will form a macromolecular complex with the p85 regulatory subunit
of PI3K and regulate the PI3K-Akt pathway, resulting in Akt
phosphorylation (109). The latest
research on ion channels regulating autophagy affects CRC indicates
that clarithromycin (Cla) exerts antiproliferative activity and
regulates autophagy through hERG1-dependent PI3K/Akt pathway and
p53 regulation, thereby enhancing chemotherapeutic drugs' antitumor
effect (110).
Discussion
Autophagy has been studied in detail as a key factor
in cell physiology and pathology, which has revealed many
intracellular signal transduction pathways and the strict
regulation of autophagy by molecules. Among them, the field of
interaction between ion channels and autophagy needs further
development. Thus far, accumulated data prove that calcium,
calcium-permeable ion channel dysfunction and autophagy are
associated with several diseases. Thus, calcium, calcium-permeable
ion channel and autophagy are potential therapeutic targets.
Calcium and calcium-permeable ion channels are considered as
autophagy regulators, opening up a new way for targeting autophagy
in the treatment of digestive tract diseases. In cancer, changes in
ion channel expression are associated with several
cancer-associated processes, including autophagy. For example, the
TRPV1 channel that targets tumor suppressor (overexpressed in
pancreatic cancer and actively regulates autophagy) may be used to
regulate autophagy and tumor cell apoptosis. In different digestive
tract diseases, the regulation of autophagy by calcium and the
calcium permeable channel is also different. For example, saturated
FFA in NAFLD induce increased cytosolic calcium in liver cells,
thereby inhibiting autophagy, and the use of calcium channel
blockers can reverse this phenomenon, leading to a decrease in
steatohepatitis. In HCC, inhibiting the entry of Ca2+
mediated by Orai1 can enhance the chemical sensitivity of HepG2
liver cancer cells to 5-FU. For different digestive tract diseases,
a deeper understanding of the interaction between calcium,
calcium-permeable ion channels and autophagy will be of great value
for the diagnosis and treatment of digestive system diseases in the
future.
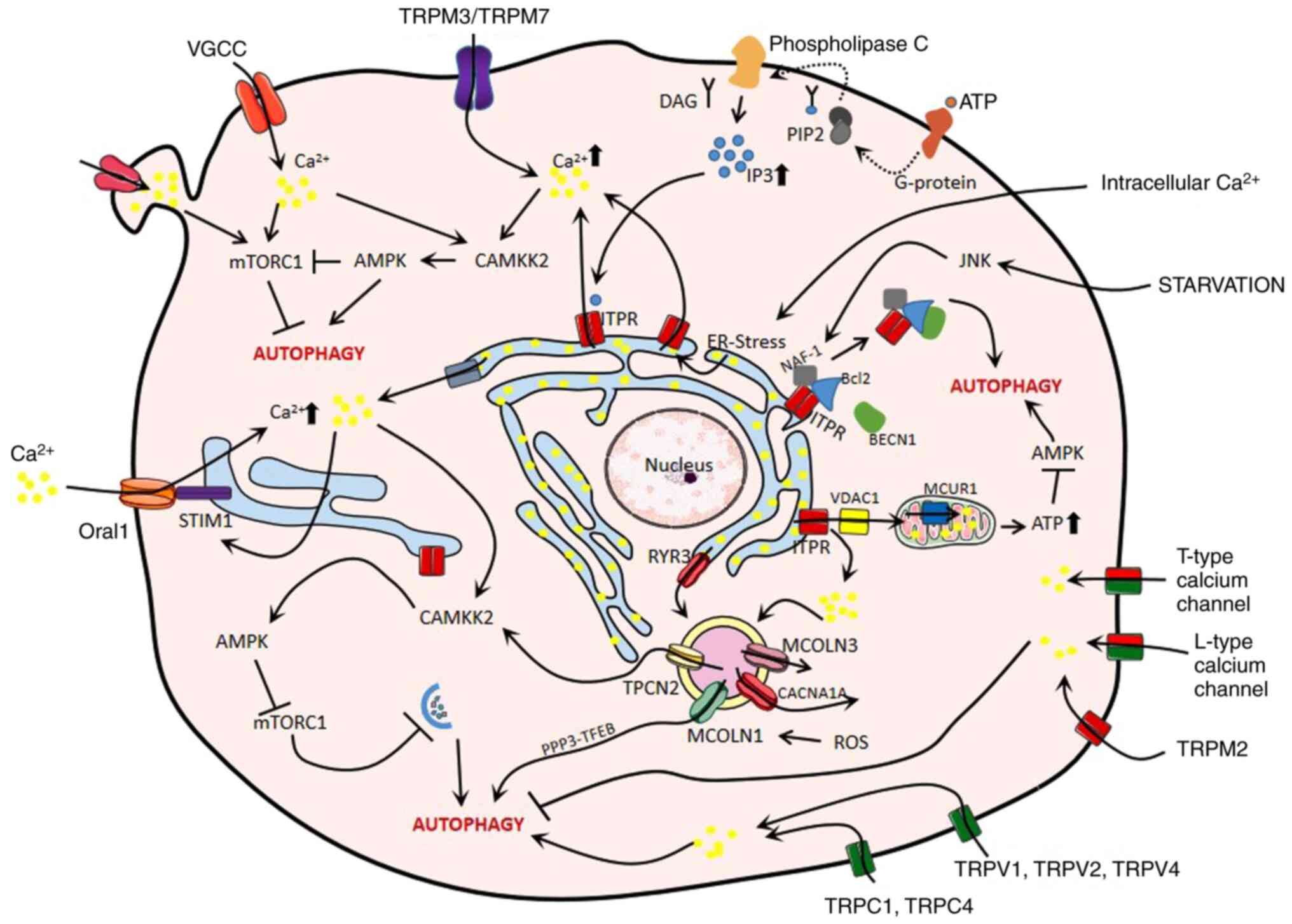
Overall, the data in the present review clearly
demonstrate the diversity and particular importance of calcium ion
channels in the regulation of autophagy. However, little is known
about the mechanism of action of autophagy through calcium channel
in the occurrence and development of digestive system diseases and
its application in disease treatment. A graphic overview of the
calcium and calcium channel-associated mechanisms of autophagy
regulation in digestive diseases is presented in Fig. 4. The present review discussed the
association between digestive system diseases and autophagy, and
reveals the role of autophagy in digestive system diseases, that
is, the regulatory mechanism, in order to provide more theoretical
basis for the diagnosis and treatment of digestive system
diseases.
Acknowledgements
The authors would like to thank Professor Biguang
Tuo (Department of Gastroenterology, Affiliated Hospital to Zunyi
Medical University) for his highly professional knowledge
explanation and scientific research services.
Funding
This study was supported by research grants from the
National Natural Science Foundation of China (grant no. 81970541 to
JY); and the Graduate Research Fund Project of Guizhou Province
[grant no. YJSCXJH (2019) 088 to XXY].
Availability of data and materials
Not applicable.
Authors' contributions
JL and XXY wrote the manuscript. WXS, ZJ, JHD, YXH,
QD and QSL collected the literature. JYX primarily revised and
finalized the manuscript. RX revised the manuscript for clarity and
style. All authors read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ER
|
endoplasmic reticulum
|
|
CMA
|
chaperone-mediated autophagy
|
|
MTORC1
|
mechanistic target of rapamycin
complex 1
|
|
AMPK
|
AMP-activated protein kinase
|
|
PI3K
|
Class III phosphatidylinositol
3-kinase
|
|
AMBRA-1
|
Beclin 1 modulator 1
|
|
TRP
|
transient receptor potential
|
|
SOC
|
store-operated calcium
|
|
TPCN
|
2-pore segment channel
|
|
MPTP
|
mitochondrial permeability transition
pores
|
|
MCU
|
mitochondrial calcium unidirectional
transporters
|
|
CAMKK2/CaMKKb
|
calcium/calmodulin dependent kinase
2
|
|
MTOR
|
mechanistic target of rapamycin
|
|
InsP3
|
inositol 1,4,5-trisphosphate
|
|
ITPR
|
inositol 1,4,5-trisphosphate
receptor
|
|
VDAC1
|
voltage-dependent anion channel 1
|
|
MCUR1
|
mitochondrial calcium uniporter
regulator1
|
|
ULK
|
unc-51 like autophagy activating
kinase
|
|
SOCE
|
store-operated calcium entry
|
|
ASIC
|
acid sensing ion channel
|
|
KCNH7/HERG3
|
potassium voltage-gated channel
subfamily H member 7
|
|
CLIC4
|
chloride intracellular channel 4
|
|
OGT
|
O-linked β-N-acetylglucosamine
(O-GlcNAc) transferase
|
|
NAFLD
|
nonalcoholic fatty liver disease
|
|
HCC
|
hepatocellular carcinoma
|
|
FFA
|
free fatty acids
|
|
LPS
|
lipopolysaccharide
|
|
NAADP
|
nicotinic acid adenine dinucleotide
phosphate
|
|
5-FU
|
5-fluorouracil
|
|
AP
|
acute pancreatitis
|
|
CCK
|
cholecystokinin
|
|
STIM1
|
stromal interaction molecule 1
|
|
TFEB
|
transcription factor EB
|
|
CaN
|
calcineurin
|
|
IL-1β
|
interleukin-1β
|
|
VacA
|
vacuolar cytotoxin A
|
References
|
1
|
Klionsky DJ and Codogno P: The mechanism
and physiological function of macroautophagy. J Innate Immun.
5:427–433. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kroemer G, Mariño G and Levine B:
Autophagy and the integrated stress response. Mol Cell. 40:280–293.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li WW, Li J and Bao JK: Microautophagy:
Lesser-known self-eating. Cell Mol Life Sci. 69:1125–1136. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cuervo AM and Wong E: Chaperone-mediated
autophagy: Roles in disease and aging. Cell Res. 24:92–104. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yin Z, Pascual C and Klionsky DJ:
Autophagy: Machinery and regulation. Microb Cell. 3:588–596. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mizushima N, Yoshimori T and Ohsumi Y: The
role of Atg proteins in autophagosome formation. Annu Rev Cell Dev
Biol. 27:107–132. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Behrends C, Sowa ME, Gygi SP and Harper
JW: Network organization of the human autophagy system. Nature.
466:68–76. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alexander SP, Kelly E, Marrion N, Peters
JA, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Southan C,
Buneman OP, et al: The concise guide to PHARMACOLOGY 2015/16:
Overview. Br J Pharmacol. 172:5729–5743. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rizzuto R, De Stefani D, Raffaello A and
Mammucari C: Mitochondria as sensors and regulators of calcium
signalling. Nat Rev Mol Cell Biol. 13:566–578. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berridge MJ, Bootman MD and Roderick HL:
Calcium signalling: Dynamics, homeostasis and remodelling. Nat Rev
Mol Cell Biol. 4:517–529. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ramsey IS, Delling M and Clapham DE: An
introduction to TRP channels. Annual Rev Physiol. 68:619–647. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Pedersen SF, Owsianik G and Nilius B: TRP
channels: An overview. Cell Calcium. 38:233–252. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Catterall WA: Voltage-gated calcium
channels. Cold Spring Harb Perspect Biol. 3:a0039472011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bernardi P and von Stockum S: The
permeability transition pore as a Ca(2+) release channel: New
answers to an old question. Cell Calcium. 52:22–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu FH and Catterall WA: Overview of the
voltage-gated sodium channel family. Genome Biol. 4:2072003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kellenberger S and Schild L: Epithelial
sodium channel/degenerin family of ion channels: A variety of
functions for a shared structure. Physiol Rev. 82:735–767. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kuang Q, Purhonen P and Hebert H:
Structure of potassium channels. Cell Mol Sci. 72:3677–3693. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nilius B and Droogmans G: Amazing chloride
channels: An overview. Acta Physiol Scand. 177:119–147. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kondratskyi A, Yassine M, Kondratska K,
Skryma R, Slomianny C and Prevarskaya N: Calcium-permeable ion
channels in control of autophagy and cancer. Front Physiol.
4:2722013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Berridge MJ, Lipp P and Bootman MD: The
versatility and universality of calcium signaling. Nature reviews.
Mol Cell Biol. 1:11–21. 2000.
|
|
25
|
Gordon PB, Holen I, Fosse M, Røtnes JS and
Seglen PO: Dependence of hepatocytic autophagy on intracellularly
sequestered calcium. J Biol Chem. 268:26107–26112. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Høyer-Hansen M, Bastholm L, Szyniarowski
P, Campanella M, Szabadkai G, Farkas T, Bianchi K, Fehrenbacher N,
Elling F, Rizzuto R, et al: Control of macroautophagy by calcium,
calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell.
25:193–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Grotemeier A, Alers S, Pfisterer SG,
Paasch F, Daubrawa M, Dieterle A, Viollet B, Wesselborg S,
Proikas-Cezanne T and Stork B: AMPK-independent induction of
autophagy by cytosolic Ca2+ increase. Cell Signal.
22:914–925. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gao W, Ding WX, Stolz DB and Yin XM:
Induction of macroautophagy by exogenously introduced calcium.
Autophagy. 4:754–761. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Decuypere JP, Bultynck G and Parys JB: A
dual role for Ca(2+) in autophagy regulation. Cell Calcium.
50:242–250. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
East DA and Campanella M: Ca2+
in quality control: An unresolved riddle critical to autophagy and
mitophagy. Autophagy. 9:1710–1719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Parys JB, Decuypere JP and Bultynck G:
Role of the inositol 1,4,5-trisphosphate
receptor/Ca2+-release channel in autophagy. Cell Commun
Signal. 10:172012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Decuypere JP, Parys JB and Bultynck G:
ITPRs/inositol 1,4,5-trisphosphate receptors in autophagy: From
enemy to ally. Autophagy. 11:1944–1948. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kania E, Roest G, Vervliet T, Parys JB and
Bultynck G: IP3 receptor-mediated calcium signaling and
its role in autophagy in cancer. Front Oncol. 7:1402017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khan MT and Joseph SK: Role of inositol
trisphosphate receptors in autophagy in DT40 cells. J Biol Chem.
285:16912–16920. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cárdenas C, Miller RA, Smith I, Bui T,
Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, et al:
Essential regulation of cell bioenergetics by constitutive InsP3
receptor Ca2+ transfer to mitochondria. Cell.
142:270–283. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tomar D, Dong Z, Shanmughapriya S, Koch
DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL,
Corbally DP, et al: MCUR1 Is a scaffold factor for the MCU complex
function and promotes mitochondrial bioenergetics. Cell Rep.
15:1673–1685. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tian X, Gala U, Zhang Y, Shang W, Nagarkar
Jaiswal S, di Ronza A, Jaiswal M, Yamamoto S, Sandoval H, Duraine
L, et al: A voltage-gated calcium channel regulates lysosomal
fusion with endosomes and autophagosomes and is required for
neuronal homeostasis. PLoS Biol. 13:e10021032015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bround MJ, Wambolt R, Luciani DS, Kulpa
JE, Rodrigues B, Brownsey RW, Allard MF and Johnson JD:
Cardiomyocyte ATP production, metabolic flexibility, and survival
require calcium flux through cardiac ryanodine receptors in vivo. J
Biol Chem. 288:18975–18986. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pereira GJ, Hirata H, Fimia GM, do Carmo
LG, Bincoletto C, Han SW, Stilhano RS, Ureshino RP, Bloor-Young D,
Churchill G, et al: Nicotinic acid adenine dinucleotide phosphate
(NAADP) regulates autophagy in cultured astrocytes. J Biol Chem.
286:27875–27881. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Abdelmohsen K, Srikantan S, Tominaga K,
Kang MJ, Yaniv Y, Martindale JL, Yang X, Park SS, Becker KG,
Subramanian M, et al: Growth inhibition by miR-519 via multiple
p21-inducing pathways. Mol Cell Biol. 32:2530–2548. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guo Y, Meng Y, Liu H, Wang B, Ding C, Rong
X, Yang Y and Hong Y: Acid-sensing ion channels mediate the
degeneration of intervertebral disc via various pathways-A
systematic review. Channels (Austin). 13:367–373. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wang D, Zhu H, Cheng W, Lin S, Shao R and
Pan H: Effects of hypoxia and ASIC3 on nucleus pulposus cells: From
cell behavior to molecular mechanism. Biomed Pharmacother.
117:1090612019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Williams A, Sarkar S, Cuddon P, Ttofi EK,
Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, et
al: Novel targets for Huntington's disease in an mTOR-independent
autophagy pathway. Nat Chem Biol. 4:295–305. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Perez-Neut M, Haar L, Rao V, Santha S,
Lansu K, Rana B, Jones WK and Gentile S: Activation of hERG3
channel stimulates autophagy and promotes cellular senescence in
melanoma. Oncotarget. 7:21991–22004. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhong J, Kong X, Zhang H, Yu C, Xu Y, Kang
J, Yu H, Yi H, Yang X and Sun L: Inhibition of CLIC4 enhances
autophagy and triggers mitochondrial and ER stress-induced
apoptosis in human glioma U251 cells under starvation. PLoS One.
7:e393782012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang L, Shen M, Guo X, Wang B, Xia Y, Wang
N, Zhang Q, Jia L and Wang X: Volume-sensitive outwardly rectifying
chloride channel blockers protect against high glucose-induced
apoptosis of cardiomyocytes via autophagy activation. Sci Rep.
7:442652017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ruan HB, Ma Y, Torres S, Zhang B, Feriod
C, Heck RM, Qian K, Fu M, Li X, Nathanson MH, et al:
Calcium-dependent O-GlcNAc signaling drives liver autophagy in
adaptation to starvation. Genes Dev. 31:1655–1665. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang Y, Ma F, Liu Z, Su Q, Liu Y, Liu Z
and Li Y: The ER-localized Ca2+-binding protein
calreticulin couples ER stress to autophagy by associating with
microtubule-associated protein 1A/1B light chain 3. J Biol Chem.
294:772–782. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cusi K: Role of obesity and lipotoxicity
in the development of nonalcoholic steatohepatitis: Pathophysiology
and clinical implications. Gastroenterology. 142:711–725.e6. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
González-Rodríguez A, Mayoral R, Agra N,
Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillón J,
Lo Iacono O, Corazzari M, Fimia GM, et al: Impaired autophagic flux
is associated with increased endoplasmic reticulum stress during
the development of NAFLD. Cell Death Dis. 5:e11792014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Miyagawa K, Oe S, Honma Y, Izumi H, Baba R
and Harada M: Lipid-induced endoplasmic reticulum stress impairs
selective autophagy at the step of autophagosome-lysosome fusion in
hepatocytes. Am J Pathol. 186:1861–1873. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Park HW, Park H, Semple IA, Jang I, Ro SH,
Kim M, Cazares VA, Stuenkel EL, Kim JJ, Kim JS and Lee JH:
Pharmacological correction of obesity-induced autophagy arrest
using calcium channel blockers. Nat Commun. 5:48342014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Czaja MJ: A new mechanism of lipotoxicity:
Calcium channel blockers as a treatment for nonalcoholic
steatohepatitis? Hepatology. 62:312–314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wei CC, Luo Z, Hogstrand C, Xu YH, Wu LX,
Chen GH, Pan YX and Song YF: Zinc reduces hepatic lipid deposition
and activates lipophagy via Zn2+/MTF-1/PPARα and
Ca2+/CaMKKβ/AMPK pathways. FASEB J. fj2018004632018.doi:
10.1096/fj.201800463. PubMed/NCBI
|
|
55
|
Lalazar G, Ilyas G, Malik SA, Liu K, Zhao
E, Amir M, Lin Y, Tanaka KE and Czaja MJ: Autophagy confers
resistance to lipopolysaccharide-induced mouse hepatocyte injury.
Am J Physiol Gastrointest Liver Physiol. 311:G377–G386. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Guse AH and Lee HC: NAADP: A universal
Ca2+ trigger. Sci Signal. 1:re102008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Rah SY, Lee YH and Kim UH: NAADP-mediated
Ca2+ signaling promotes autophagy and protects against
LPS-induced liver injury. FASEB J. 31:3126–3137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shi M, Wang HN, Xie ST, Luo Y, Sun CY,
Chen XL and Zhang YZ: Antimicrobial peptaibols, novel suppressors
of tumor cells, targeted calcium-mediated apoptosis and autophagy
in human hepatocellular carcinoma cells. Mol Cancer. 9:262010.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sui X, Kong N, Wang X, Fang Y, Hu X, Xu Y,
Chen W, Wang K, Li D, Jin W, et al: JNK confers 5-fluorouracil
resistance in p53-deficient and mutant p53-expressing colon cancer
cells by inducing survival autophagy. Sci Rep. 4:46942014.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Suzuki R, Kang Y, Li X, Roife D, Zhang R
and Fleming JB: Genistein potentiates the antitumor effect of
5-Fluorouracil by inducing apoptosis and autophagy in human
pancreatic cancer cells. Anticancer Res. 34:4685–4692.
2014.PubMed/NCBI
|
|
61
|
Li LQ, Xie WJ, Pan D, Chen H and Zhang L:
Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity
of gastric cancer cells. Tumour Biol. 37:653–659. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yang N, Tang Y, Wang F, Zhang H, Xu D,
Shen Y, Sun S and Yang G: Blockade of store-operated Ca(2+) entry
inhibits hepatocarcinoma cell migration and invasion by regulating
focal adhesion turnover. Cancer Lett. 330:163–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kondratska K, Kondratskyi A, Yassine M,
Lemonnier L, Lepage G, Morabito A, Skryma R and Prevarskaya N:
Orai1 and STIM1 mediate SOCE and contribute to apoptotic resistance
of pancreatic adenocarcinoma. Biochim Biophys Acta. 1843:2263–2269.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Guéguinou M, Harnois T, Crottes D, Uguen
A, Deliot N, Gambade A, Chantôme A, Haelters JP, Jaffrès PA,
Jourdan ML, et al: SK3/TRPC1/Orai1 complex regulates SOCE-dependent
colon cancer cell migration: A novel opportunity to modulate
anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget.
7:36168–36184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Selli C, Erac Y, Kosova B, Erdal ES and
Tosun M: Silencing of TRPC1 regulates store-operated calcium entry
and proliferation in Huh7 hepatocellular carcinoma cells. Biomed
Pharmacother. 71:194–200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Selli C, Pearce DA, Sims AH and Tosun M:
Differential expression of store-operated calcium- and
proliferation-related genes in hepatocellular carcinoma cells
following TRPC1 ion channel silencing. Mol Cell Biochem.
420:129–140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tang BD, Xia X, Lv XF, Yu BX, Yuan JN, Mai
XY, Shang JY, Zhou JG, Liang SJ and Pang RP: Inhibition of
Orai1-mediated Ca2+ entry enhances chemosensitivity of
HepG2 hepatocarcinoma cells to 5-fluorouracil. J Cell Mol Med.
21:904–915. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Huang Q, Cao H, Zhan L, Sun X, Wang G, Li
J, Guo X, Ren T, Wang Z, Lyu Y, et al: Mitochondrial fission forms
a positive feedback loop with cytosolic calcium signaling pathway
to promote autophagy in hepatocellular carcinoma cells. Cancer
Lett. 403:108–118. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Pandol SJ, Saluja AK, Imrie CW and Banks
PA: Acute pancreatitis: Bench to the bedside. Gastroenterology.
132:1127–1151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Lankisch PG, Apte M and Banks PA: Acute
pancreatitis. Lancet. 386:85–96. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Banks PA, Bollen TL, Dervenis C, Gooszen
HG, Johnson CD, Sarr MG, Tsiotos GG and Vege SS; Acute Pancreatitis
Classification Working Group, : Classification of acute
pancreatitis-2012: Revision of the Atlanta classification and
definitions by international consensus. Gut. 62:102–111. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Ward JB, Petersen OH, Jenkins SA and
Sutton R: Is an elevated concentration of acinar cytosolic free
ionised calcium the trigger for acute pancreatitis? Lancet.
346:1016–1019. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Petersen OH and Tepikin AV: Polarized
calcium signaling in exocrine gland cells. Ann Rev Physiol.
70:273–299. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Petersen OH and Sutton R: Ca2+
signalling and pancreatitis: Effects of alcohol, bile and coffee.
Trends Pharmacol Scie. 27:113–120. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Gerasimenko JV, Gerasimenko OV and
Petersen OH: The role of Ca2+ in the pathophysiology of
pancreatitis. J Physiol. 592:269–280. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Lur G, Haynes LP, Prior IA, Gerasimenko
OV, Feske S, Petersen OH, Burgoyne RD and Tepikin AV: Ribosome-free
terminals of rough ER allow formation of STIM1 puncta and
segregation of STIM1 from IP(3) receptors. Curr Biol. 19:1648–1653.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Gerasimenko JV, Gryshchenko O, Ferdek PE,
Stapleton E, Hébert TO, Bychkova S, Peng S, Begg M, Gerasimenko OV
and Petersen OH: Ca2+ release-activated Ca2+
channel blockade as a potential tool in antipancreatitis therapy.
Proc Natl Acad Sci USA. 110:13186–13191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Petersen OH, Courjaret R and Machaca K:
Ca2+ tunnelling through the ER lumen as a mechanism for
delivering Ca2+ entering via store-operated
Ca2+ channels to specific target sites. J Physiol.
595:2999–3014. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhu ZD, Yu T, Liu HJ, Jin J and He J: SOCE
induced calcium overload regulates autophagy in acute pancreatitis
via calcineurin activation. Cell Death Dis. 9:502018. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Biczo G, Vegh ET, Shalbueva N, Mareninova
OA, Elperin J, Lotshaw E, Gretler S, Lugea A, Malla SR, Dawson D,
et al: Mitochondrial dysfunction, through impaired autophagy, leads
to endoplasmic reticulum stress, deregulated lipid metabolism, and
pancreatitis in animal models. Gastroenterology. 154:689–703. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Marrache F, Tu SP, Bhagat G, Pendyala S,
Osterreicher CH, Gordon S, Ramanathan V, Penz-Osterreicher M, Betz
KS, Song Z and Wang TC: Overexpression of interleukin-1beta in the
murine pancreas results in chronic pancreatitis. Gastroenterology.
135:1277–1287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Gukovsky I, Li N, Todoric J, Gukovskaya A
and Karin M: Inflammation, autophagy, and obesity: Common features
in the pathogenesis of pancreatitis and pancreatic cancer.
Gastroenterology. 144:1199–1209.e1194. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Harris J: Autophagy and cytokines.
Cytokine. 56:140–144. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Shi CS and Kehrl JH: MyD88 and Trif target
Beclin 1 to trigger autophagy in macrophages. J Biol Chemistry.
283:33175–33182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
O'Neill CM, Lu C, Corbin KL, Sharma PR,
Dula SB, Carter JD, Ramadan JW, Xin W, Lee JK and Nunemaker CS:
Circulating levels of IL-1B + IL-6 cause ER stress and dysfunction
in islets from prediabetic male mice. Endocrinology. 154:3077–3088.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Berridge MJ: The endoplasmic reticulum: A
multifunctional signaling organelle. Cell Calcium. 32:235–249.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Frick TW: The role of calcium in acute
pancreatitis. Surgery. 152 (Suppl 1):S157–S163. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chen X, Li M, Chen D, Gao W, Guan JL,
Komatsu M and Yin XM: Autophagy induced by calcium phosphate
precipitates involves endoplasmic reticulum membranes in
autophagosome biogenesis. PLoS One. 7:e523472012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Smaili SS, Pereira GJ, Costa MM, Rocha KK,
Rodrigues L, do Carmo LG, Hirata H and Hsu YT: The role of calcium
stores in apoptosis and autophagy. Curr Mol Med. 13:252–265. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Xu B, Bai B, Sha S, Yu P, An Y, Wang S,
Kong X, Liu C, Wei N, Feng Q and Zhao Q: Interleukin-1β induces
autophagy by affecting calcium homeostasis and trypsinogen
activation in pancreatic acinar cells. Int J Clin Exp Pathol.
7:3620–3631. 2014.PubMed/NCBI
|
|
92
|
Díaz-Laviada I and Rodríguez-Henche N: The
potential antitumor effects of capsaicin. Prog Drug Res.
68:181–208. 2014.PubMed/NCBI
|
|
93
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: Sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Röcken C: Molecular classification of
gastric cancer. Expert Rev Mol Diagn. 17:293–301. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Almasi S, Kennedy BE, El-Aghil M, Sterea
AM, Gujar S, Partida-Sánchez S and El Hiani Y: TRPM2
channel-mediated regulation of autophagy maintains mitochondrial
function and promotes gastric cancer cell survival via the
JNK-signaling pathway. J Biol Chem. 293:3637–3650. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wemmie JA, Taugher RJ and Kreple CJ:
Acid-sensing ion channels in pain and disease. Nat Rev
Neuroscience. 14:461–471. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Du J, Reznikov LR, Price MP, Zha XM, Lu Y,
Moninger TO, Wemmie JA and Welsh MJ: Protons are a neurotransmitter
that regulates synaptic plasticity in the lateral amygdala. Proc
Natl Acad Sci USA. 111:8961–8966. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sun X, Cao YB, Hu LF, Yang YP, Li J, Wang
F and Liu CF: ASICs mediate the modulatory effect by paeoniflorin
on α-synuclein autophagic degradation. Brain Res. 1396:77–87. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Zhou RP, Wu XS, Wang ZS, Xie YY, Ge JF and
Chen FH: Novel insights into acid-sensing ion channels:
Implications for degenerative diseases. Aging Dis. 7:491–501. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Zhang Q, Wu S, Zhu J, Chai D and Gan H:
Down-regulation of ASIC1 suppressed gastric cancer via inhibiting
autophagy. Gene. 608:79–85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Fukase K, Kato M, Kikuchi S, Inoue K,
Uemura N, Okamoto S, Terao S, Amagai K, Hayashi S and Asaka M;
Japan Gast Study Group, : Effect of eradication of Helicobacter
pylori on incidence of metachronous gastric carcinoma after
endoscopic resection of early gastric cancer: An open-label,
randomised controlled trial. Lancet. 372:392–397. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Foegeding NJ, Caston RR, McClain MS, Ohi
MD and Cover TL: An overview of Helicobacter pylori VacA
Toxin Biology. Toxins. 8:1732016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Rassow J and Meinecke M: Helicobacter
pylori VacA: A new perspective on an invasive chloride channel.
Microbes Infection. 14:1026–1033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Capurro MI, Greenfield LK, Prashar A, Xia
S, Abdullah M, Wong H, Zhong XZ, Bertaux-Skeirik N, Chakrabarti J,
Siddiqui I, et al: VacA generates a protective intracellular
reservoir for Helicobacter pylori that is eliminated by
activation of the lysosomal calcium channel TRPML1. Nat Microbiol.
4:1411–1423. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Capurro MI, Prashar A and Jones NL:
MCOLN1/TRPML1 inhibition-a novel strategy used by Helicobacter
pylori to escape autophagic killing and antibiotic eradication
therapy in vivo. Autophagy. 16:169–170. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Hu W, Zhang L, Li MX, Shen J, Liu XD, Xiao
ZG, Wu DL, Ho IHT, Wu JCY, Cheung CKY, et al: Vitamin D3 activates
the autolysosomal degradation function against Helicobacter
pylori through the PDIA3 receptor in gastric epithelial cells.
Autophagy. 15:707–725. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Arnold M, Sierra MS, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global patterns and trends in
colorectal cancer incidence and mortality. Gut. 66:683–691. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Jing Z, Sui X, Yao J, Xie J, Jiang L, Zhou
Y, Pan H and Han W: SKF-96365 activates cytoprotective autophagy to
delay apoptosis in colorectal cancer cells through inhibition of
the calcium/CaMKIIγ/AKT-mediated pathway. Cancer Lett. 372:226–238.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Crociani O, Zanieri F, Pillozzi S,
Lastraioli E, Stefanini M, Fiore A, Fortunato A, D'Amico M,
Masselli M, De Lorenzo E, et al: hERG1 channels modulate integrin
signaling to trigger angiogenesis and tumor progression in
colorectal cancer. Sci Rep. 3:33082013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Petroni G, Bagni G, Iorio J, Duranti C,
Lottini T, Stefanini M, Kragol G, Becchetti A and Arcangeli A:
Clarithromycin inhibits autophagy in colorectal cancer by
regulating the hERG1 potassium channel interaction with PI3K. Cell
Death Dis. 11:1612020. View Article : Google Scholar : PubMed/NCBI
|