Introduction
Injury to vascular endothelial cells was found to
alter the biological functions of these cells via the activation of
endothelial-mesenchymal transition (EndMT) (1). EndMT is an intricate cellular
differentiation process, whereby endothelial cells lose their
original endothelial properties and, to varying extents, acquire
mesenchymal features (2), which has
been discovered to be an important pathological process in the
progression and evolution of atherosclerosis (AS) (3). The decreased activity of endothelial
nitric oxide (NO) synthase (eNOS) and decreased NO bioavailability
have been established as significant features of endothelial
function injury (4). eNOS is a key
enzyme required by endothelial cells to synthesize NO and the
expression level of eNOS has been shown to serve an important role
in EndMT-mediated AS-related diseases (5). Therefore, the eNOS/NO signaling axis
has been suggested to be involved in the regulation of vascular
function during EndMT.
Transcription factors (TFs) regulate a large number
of cellular activities by binding to specific target DNA sequences.
Kruppel-like factors (KLFs) are zinc finger TFs, which serve
crucial roles in regulating important biological processes, such as
cell proliferation, the differential expression of phenotypes and
cytoskeletal remodeling (6,7). Therefore, the present study
hypothesized that the KLF4/eNOS signaling pathway may be involved
in the EndMT process.
Salidroside (SAL) is the main active ingredient of
Rhodiola rosea, which belongs to the Crassulaceae family.
Previous studies have reported that SAL could inhibit oxidative
stress, inflammation and endoplasmic reticulum stress in vascular
endothelial cells, as well as delay endothelial cell senescence,
thereby exerting a strong cardioprotective pharmacological effect
(8–13). A number of studies have also
revealed that SAL treatment upregulated the expression levels of
the homocysteine (Hcy)-induced endothelial cell antioxidant
signaling protein, nuclear factor, erythroid 2 like 2 (Nrf2), and
improved endothelial cell oxidative stress injury, suggesting that
SAL may have the potential to protect endothelial cells (12–14).
Therefore, the present study aimed to investigate whether the
effect of SAL on Hcy-induced EndMT occurred via the KLF4/eNOS
signaling pathway, which may provide experimental evidence of the
vascular pharmacological activity of SAL.
Materials and methods
Reagents
SAL was purchased from Shanghai Aladdin Biochemical
Technology Co., Ltd. (purity ≥98%; cat. no. B19208). Endothelial
cell culture medium (cat. no. 26219) and cell cryopreservation
solution (cat. no. 19671) were purchased from ScienCell Research
Laboratories, Inc. VE-cadherin (1:1,000; cat. no. 2500), α-smooth
muscle actin (1:1,000; α-SMA; cat. no. 19245), KLF4 (1:1,000; cat.
no. 4038) and eNOS (1:1,000; cat. no. 9572) primary antibodies were
purchased from Cell Signaling Technology, Inc. The GAPDH primary
antibody (cat. no. MB001) was purchased from Bioworld Technology,
Inc. The BCA protein quantitative kit (cat. no. P0010) was
purchased from Beyotime Institute of Biotechnology, while the ECL
detection kit (cat. no. 1829501) was purchased from MilliporeSigma
and small interfering RNA (siRNA/si) was purchased from Shanghai
Gema Gene Technology Co., Ltd.
Cell lines, culture and experimental
groupings
HUVECs (cat. no. 19195) were purchased from
ScienCell Research Laboratories, Inc. HUVECs were cultured in DMEM
(cat. no. 56499C; Sigma-Aldrich; Merck KGaA) supplemented with 5%
FBS (cat. no. F8687; Sigma-Aldrich; Merck KGaA), 1%
penicillin-streptomycin and 1% EGF, and maintained at a constant
temperature of 37°C with 5% CO2 in an incubator until
the cell confluence reached 80–90%.
For the first experiment (experiment 1), cells were
divided into the following groups: i) Control group; ii) Hcy
(model) group [1 mmol/l Hcy (Sigma-Aldrich; Merck KGaA)]; iii) SAL
low-dose group (1 mmol/l Hcy + 10 µmol/l SAL); and iv) SAL
high-dose group (1 mmol/l Hcy + 50 µmol/l SAL). Cells were
pretreated with SAL for 2 h, then treated with Hcy for 48 h, both
at 37°C to induce EndMT.
For the second experiment (experiment 2), siRNAs
were used to knock down the expression of KLF4 in HUVECs, and
whether the inhibitory effects of SAL on EndMT occurred via the
KLF4/eNOS signaling pathway were investigated. Cells were divided
into the following groups: i) Control group; ii) Hcy group (1
mmol/l Hcy); iii) SAL group (1 mmol/l Hcy + 50 µmol/l SAL); iv)
siKLF4 group [1 mmol/l Hcy + 5 µl siKLF4 (20 µM)]; and iv) siKLF4 +
SAL co-administration group [1 mmol/l Hcy + 5 µl siKLF4 (20 µM) +
50 µmol/l SAL]. To induce EndMT, all groups were preincubated with
SAL for 2 h and then treated with Hcy for 48 h at 37°C.
siRNA transfection
Control siRNA (sense, 5′-UUCUCCGAACGUGUCACGUTT-3′
and antisense, 5′-ACGUGACACGUUCGGAGAATT-3′) and siKLF4 (sense,
5′-GGACUUUAUUCUCUCCAAUTT-3′ and antisense,
5′-AUUGGAGAGAAUAAAGUCCTT-3′) were transfected into HUVECs. Briefly,
HUVECs were cultured to 60% confluence and transfected with 5 µl
siRNA at 37°C using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) diluted in serum-free RPMI-1640
medium (Wisent Biotechnology). Following 6 h of transfection, the
cell culture medium was replaced with antibiotic-free endothelial
cell culture medium supplemented with 5% FBS and cultured in normal
conditions for 24 h, after which cells were used for subsequent
experiments. The transfection efficiency was analyzed using western
blotting.
MTT assay
Cells from at least six wells/experimental group
were used. Briefly, after a 24 h exposure to Hcy (1 mmol/l) at
37°C, the medium was removed, and the cells were gently washed with
PBS. Then, 80 µl fresh culture medium and 20 µl MTT (5 mg/ml) were
added to the cells. After 4 h of incubation at 37°C, 200 µl DMSO
was added to dissolve the formazan crystals, and the absorbance was
measured using a microplate reader (Tecan Group, Ltd.) at a
wavelength of 570 nm. Inhibition of cell viability (%) was
calculated with the following formula: [Optical density
(OD)treated group-ODox-LDL
group)/(ODcontrol group-ODox-LDL
group)] ×100.
Lactate dehydrogenase (LDH) assay
To evaluate cell injury, LDH released from the
cytosol into the culture medium was measured. After treatment with
SAL (10 or 50 µmol/l) for 1 h, followed by incubation with Hcy (1
mmol/l) for 24 h at 37°C, the medium was collected from each well.
Supernatants were obtained by centrifugation at 12,000 × g at 4°C
for 10 min. LDH release was determined using an LDH assay kit
(Nanjing Jiancheng Bioengineering Institute), according to the
manufacturer's protocol. The absorbance was measured at a
wavelength of 440 nm using a microplate reader (SpectraMax 190;
Molecular Devices, LLC).
Western blotting
Total protein was extracted from HUVECs using a
protein lysate buffer (cat. no. P0013; Beyotime Institute of
Biotechnology) supplemented with 1% PMSF (cat. no. 36978; Thermo
Fisher Scientific, Inc.). Total protein was quantified using a BCA
protein assay kit (Abcam) and 40 µg protein/lane was separated via
10 or 12% SDS-PAGE. The separated proteins were subsequently
transferred onto PVDF membranes at room temperature and blocked
with 10% non-fat dried milk diluted in TBS-0.1% Tween-20 (TBST) for
15 min at room temperature. The membranes were then incubated with
the primary antibodies overnight at 4°C. Following the primary
antibody incubation, the membrane was washed with TBST and
incubated with a HRP-conjugated secondary (1:10,000; cat. no.
BM2020; Wuhan Boster Biological Technology, Ltd.) at room
temperature for 2 h. The membranes were washed with 3X TBS-Tween-20
(0.1%) and total protein was visualized using an ECL reagent
(Amersham; Cytiva). Densitometric analysis was performed using
ImageJ version 1.43 software (National Institutes of Health).
Detection of changes in KLF4 levels
using reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from HUVECs using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) and quantified using a NanoDrop ND-2000 system (Thermo Fisher
Scientific, Inc.). cDNA was synthesized from the purified RNA (200
ng/sample) using a reverse transcription kit (cat. no. DRR037A;
Takara Bio, Inc.), according to the manufacturer's protocol.
Subsequently, qPCR was performed using the CFX96 real-time PCR
detection system (Bio-Rad Laboratories, Inc.) and Premix Ex Taq kit
PCR reagents (cat. no. RR390A; Takara Bio, Inc.) with primers
specific for KLF4 and GADPH. The following thermocycling conditions
were used for the qPCR: Initial denaturation at 94°C for 30 sec;
followed by 39 cycles of amplification at 94°C for 5 sec and 56°C
for 30 sec. The following primer sequences were used for the qPCR:
KLF4 forward, 5′-GCATGTGCCCCAAGATTAAG-3′ and reverse,
5′-GTGACAGTCCCTGCTGTTCA-3′; and GAPDH forward,
5′-TGTGAACGGATTTGGCCGTA-3′ and reverse, 5′-GATGGTGATGGGTTTCCCGT-3′.
GAPDH was used as the loading control. Expression levels were
quantified using the 2−∆∆Cq method (15) and target gene expression was
normalized to GAPDH expression.
Wound healing assay
Cells were plated into a 6-well plate and cultured
overnight to confluence, then treated according to the experimental
groups described as experiment 1. Subsequently, an artificial wound
was created by vertically scratching the cell monolayer using a
10-µl pipette tip. The non-adherent cells were removed by washing
with PBS and cells were incubated for 10 min at 37°C with
serum-free medium. Cells were visualized in five randomly selected
fields of view at 0 and 12 h to determine the influence of SAL on
cell migration. The following equation was used to calculate the
migration using images captured with a fluorescence microscope
(Leica DM3000k magnification, ×100): Migration (%) = [(0 h average
scratch distance-12 h average scratch distance)/0 h average scratch
distance] ×100.
Determination of NO content using a
nitrate reductase assay
Following centrifugation at 3,000 × g for 10 min at
4°C, the cell culture supernatant was collected and analyzed using
a commercially available kit (cat. no. S0021S; Beyotime Institute
of Biotechnology), according to the manufacturer's protocol.
Immunofluorescence assay
Immunofluorescence was used to analyze the
expression levels of KLF4. Briefly, the cells were treated
according to the groups described as experiment 2. The cells were
washed with PBS, fixed with 4% paraformaldehyde for 15 min at room
temperature, washed with PBS and permeabilized with Triton X-100
for 10 min. Then, cells were washed with PBS and blocked with 4%
goat serum (Abcam) at room temperature for 1 h. The cells were then
incubated with a primary anti-KLF4 antibody (1:1,000; cat. no.
12173S; Cell Signaling Technology, Inc.) overnight at 4°C.
Following the primary antibody incubation, the cells were washed
with PBS and incubated with the corresponding
fluorescent-conjugated secondary antibody (1:1,000; cat. no.
ab150077; Abcam) for 1 h. After washing with PBS, nuclei were
stained with DAPI at room temperature for 10 min. Finally, the
cells were washed with PBS for 5 min, the fluorescence was
quenched, and stained cells were visualized using an inverted
fluorescence microscope (magnification, ×200) to analyze the
expression of KLF4.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism-6.0 software (GraphPad Software, Inc.) and all data are
presented as the mean ± SD of at least three experimental repeats.
Statistical differences between groups were determined using
one-way ANOVA followed by Tukey's post-hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of SAL on the expression levels
of Hcy-induced EndMT phenotypic markers
The first series of experiments investigated
endothelial cell viability and cell damage. Endothelial cell
viability was detected using an MTT assay. Hcy caused a decrease in
cell viability in a concentration-dependent manner, especially at
48 h, and the extent of cellular injury at the concentration of 1
mM Hcy was the most severe (Fig. S1A
and B). Cell damage was determined by the release of LDH, which
increases with necrosis in cultured cells. LDH leakage
significantly increased with Hcy exposure, which was in line with
the results of cell viability (Fig.
S1D). Moreover, SAL improved the cell viability, with a
significant decrease of the release of LDH compared with Hcy
exposure (Fig. S1C and D).
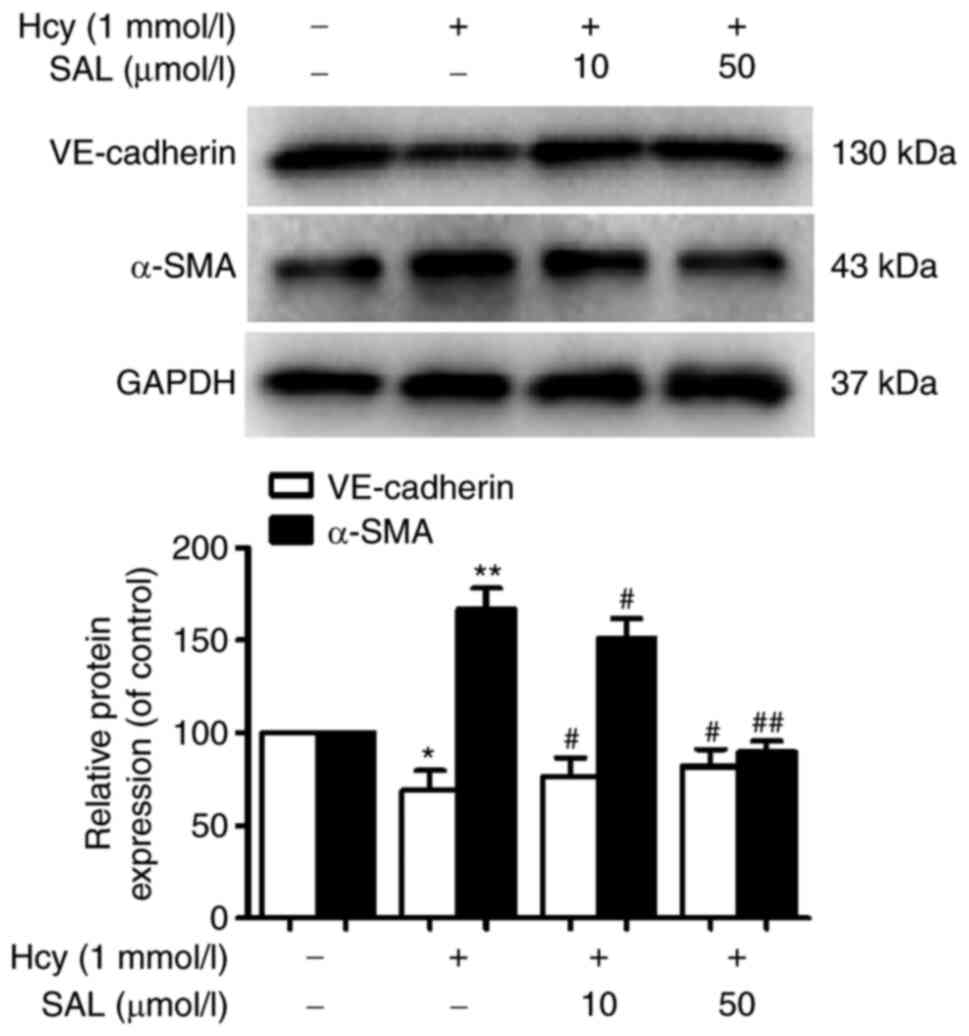
To further investigate the effects of SAL on the
expression levels of Hcy-induced EndMT phenotypic markers, western
blotting was performed. As shown in Fig. 1, Hcy significantly downregulated the
expression levels of the endothelial marker VE-cadherin and
significantly upregulated the expression levels of the mesenchymal
marker α-SMA in HUVECs. Conversely, compared with the Hcy group,
pretreatment with 10 or 50 µmol/l SAL upregulated the expression
levels of VE-cadherin and downregulated α-SMA expression in HUVECs.
These results suggested that SAL pretreatment may inhibit
Hcy-induced changes in the expression levels of endothelial cell
phenotypic markers.
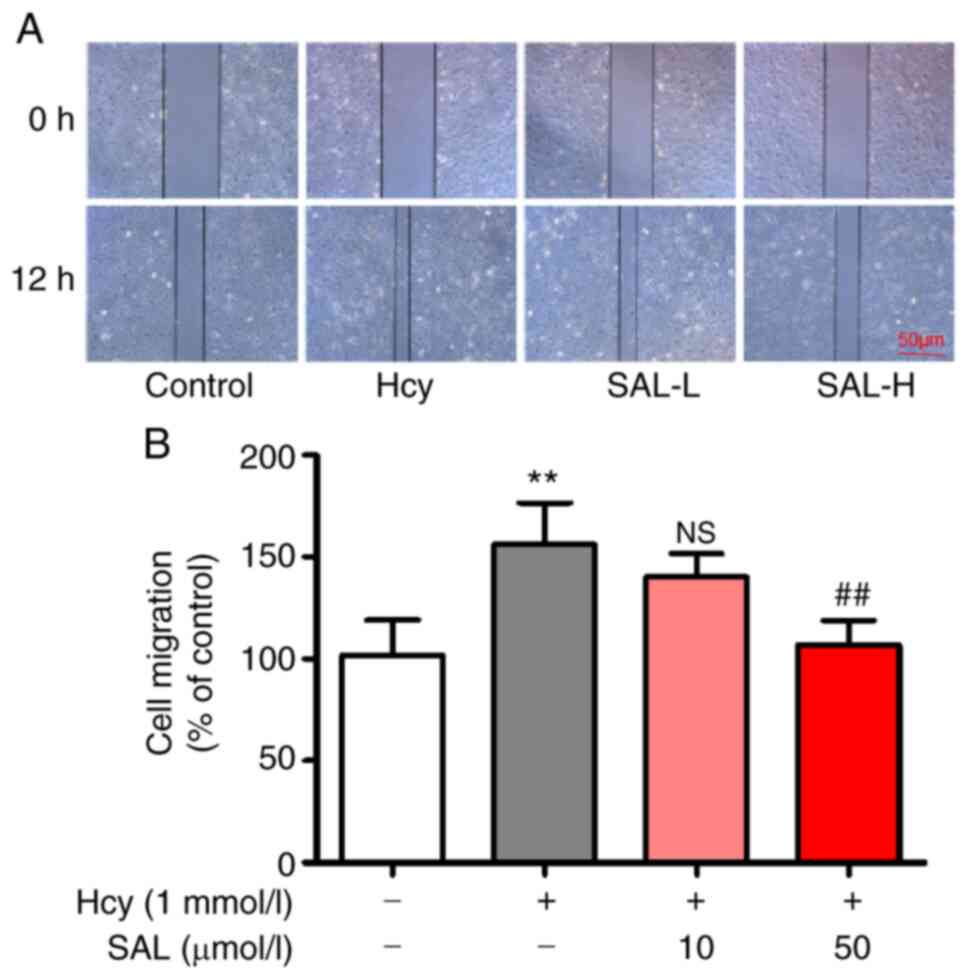
Effect of SAL on Hcy-induced cell
migration
As presented in Fig.
2, compared with the control group, the results of the wound
healing assay revealed that Hcy significantly increased the cell
migration rate (150.69±21.97%), while compared with Hcy group, 10
or 50 µmol/l SAL pretreatment decreased the cell migratory rates to
137.79±11.42 and 105.58±10.44%, respectively, suggesting that SAL
treatment may inhibit the Hcy-induced cell migratory ability.
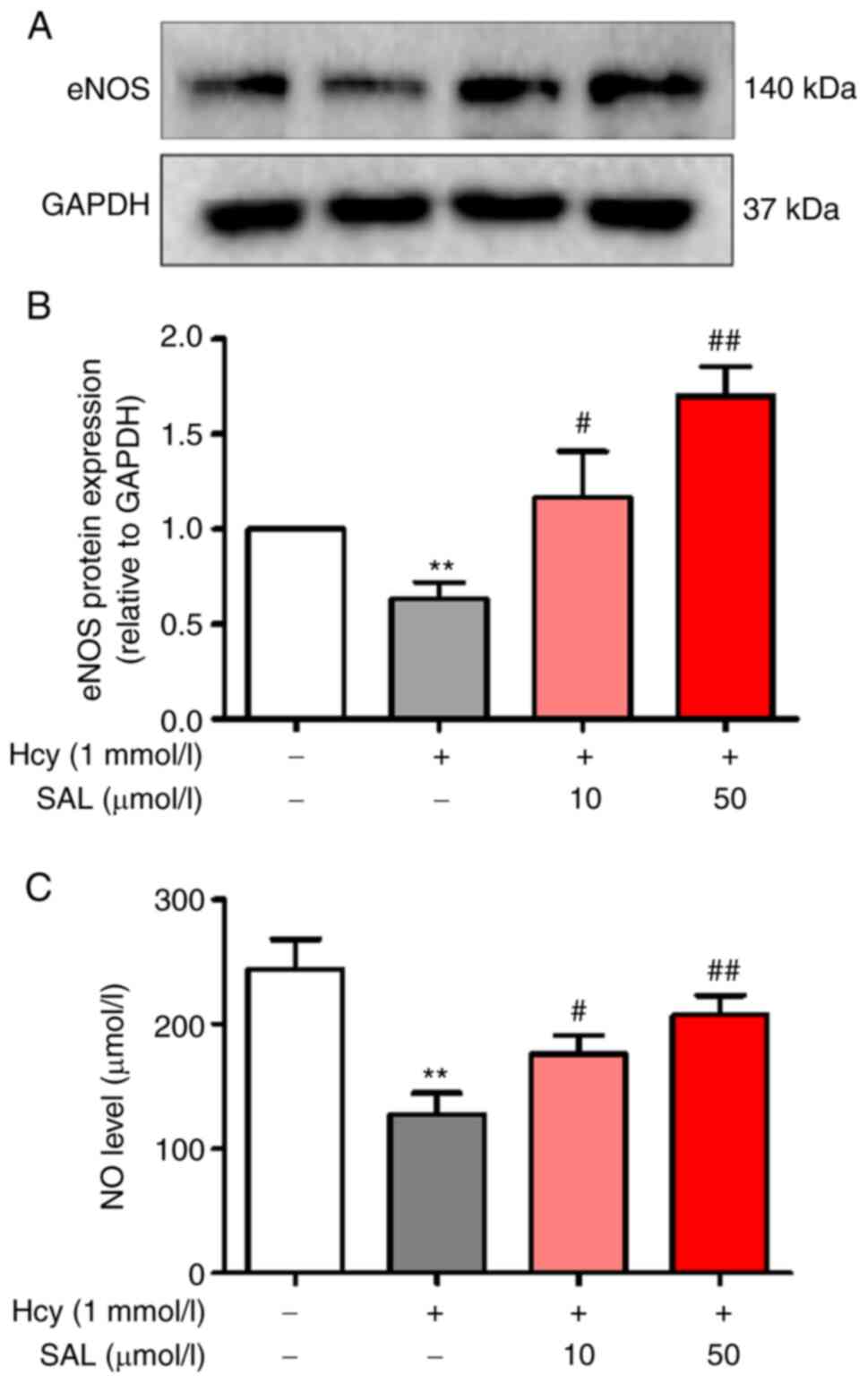
SAL regulates the eNOS/NO signaling
axis in Hcy-induced EndMT
To investigate the role of eNOS in Hcy-induced
EndMT, the expression levels of eNOS were analyzed using western
blotting. The results demonstrated that Hcy downregulated the
expression levels of eNOS, while SAL pretreatment upregulated the
expression levels of eNOS in HUVECs (Fig. 3A and B). To further confirm the role
of the eNOS/NO signaling axis in Hcy-induced EndMT, the NO content
in the cell culture supernatant was measured (Fig. 3C), and similar trends to the eNOS
expression levels were observed. Collectively, it was shown that
Hcy downregulated the protein expression level of eNOS and the
levels of NO. Moreover, these findings indicated that SAL treatment
may upregulate the Hcy-induced downregulated eNOS/NO signaling
axis, which suggested that SAL may promote the recovery of
endothelial dysfunction.
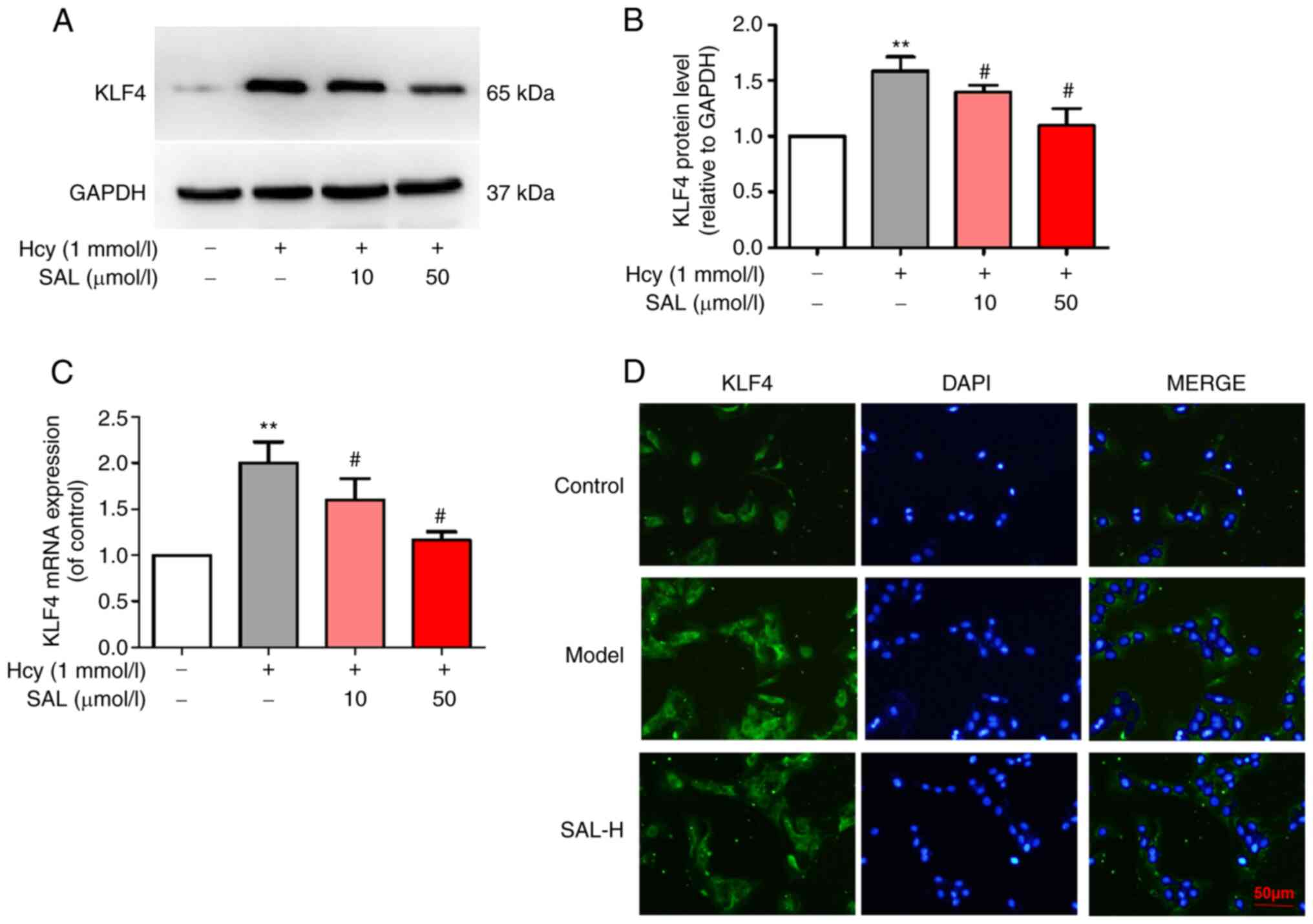
Effect of SAL on the expression levels
of KLF4 in HUVECs
Western blotting and RT-qPCR analysis revealed that
pretreatment with low and high doses of SAL significantly
downregulated the Hcy-induced upregulation of KLF4 expression in
HUVECs (Fig. 4A-C). The results of
the immunofluorescence analysis further demonstrated that, compared
with the control group, Hcy induced the translocation of the
nuclear TF KLF4 from the cytoplasm to the nucleus, while 50 µmol/l
SAL pretreatment decreased the fluorescence intensity of KLF4 and
inhibited KLF4 nuclear translocation (Fig. 4D). These results suggested that SAL
may effectively downregulate the expression levels of the nuclear
TF KLF4 and inhibit the nuclear translocation of KLF4.
Effect of SAL and siKLF4 on the
expression levels of Hcy-induced EndMT phenotypic markers, KLF4 and
eNOS
To determine whether KLF4 could act as an upstream
signaling factor to regulate the expression levels of eNOS and
exert a role in the regulation of Hcy-induced EndMT, siKLF4 was
used to knock down KLF4 expression. The results revealed that
siKLF4 significantly downregulated the expression levels of KLF4,
confirming that the transfection of siKLF4 into HUVECs was
successful (Fig. 5A). The results
of the western blot analysis demonstrated that Hcy upregulated the
expression levels of KLF4 and α-SMA in HUVECs, and downregulated
the expression levels of eNOS and the endothelial marker,
VE-cadherin. Conversely, SAL treatment and KLF4-knockdown inhibited
Hcy-induced EndMT by upregulating the expression levels of eNOS and
VE-cadherin and downregulating the expression levels of KLF4 and
α-SMA (Fig. 5B and C). Compared
with the SAL + siKLF4 co-administration group, no significant
differences were observed in the expression levels of the
phenotypic makers in the SAL or siKLF4 groups. These results
suggested that KLF4 may be an important signaling factor involved
in the inhibitory mechanism of SAL on Hcy-induced EndMT, and that
KLF4 may represent a potential upstream signaling protein to target
to regulate eNOS expression. These results indicated that SAL could
inhibit Hcy-induced EndMT by downregulating the KLF4/eNOS signaling
pathway.
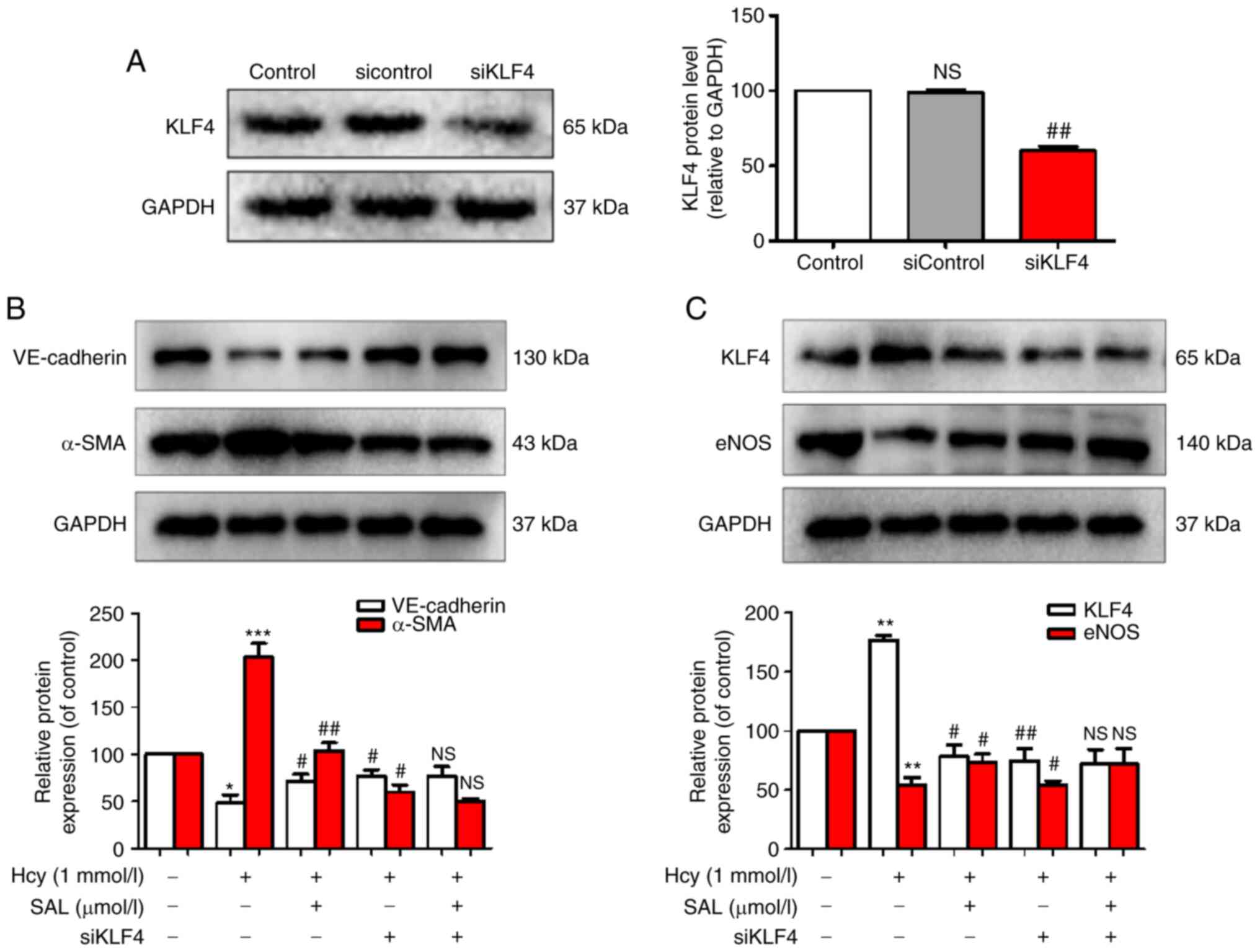 | Figure 5.Effects of SAL and siKLF4 on the
expression levels of Hcy-induced endothelial-mesenchymal
transition-related markers, KLF4 and eNOS. (A) KLF4, (B)
VE-cadherin and α-SMA and (C) KLF4 and eNOS expression levels were
analyzed using western blotting. Data are presented as the mean ±
SD of three independent experiments. *P<0.05, **P<0.01,
***P<0.001 vs. control; #P<0.05,
##P<0.01 vs. Hcy. Hcy, homocysteine; SAL,
salidroside; KLF4, Kruppel-like factor 4; α-SMA, α-smooth muscle
actin; eNOS, endothelial nitric oxide synthase; si, small
interfering RNA; NS, not significant. |
Discussion
EndMT is the mechanism via which endothelial cells
undergo transdifferentiation into a mesenchymal phenotype under the
action of internal and external damage factors (16). EndMT can damage the endothelial cell
microenvironment by destroying the cell surface and secretory
function of endothelial cells, which thereby aggravates endothelial
dysfunction (17). Thus,
determining key molecules involved in the process of EndMT remains
an urgent priority. The findings of the present study demonstrated
that Hcy upregulated the expression levels of the mesenchymal cell
marker α-SMA, KLF4 and the eNOS/NO signaling axis, and
downregulated VE-cadherin expression, as well as promoted cell
migration and KLF4 translocation to the nucleus. Moreover, the
knockdown of KLF4 reversed the aberrant expression of the
aforementioned proteins induced by Hcy treatment. SAL treatment
could also reverse the Hcy-induced effects. These findings
suggested that SAL may provide vascular protection by inhibiting
EndMT via the KLF4/eNOS signaling pathway.
Endothelial cell dysfunction is an early factor of
AS; therefore, devising innovative drugs that protect vascular
endothelial cell function and reverse the biological dysfunction of
endothelial cells is of great significance for the prevention and
treatment of AS (18). Previous
studies have reported that endothelial dysfunction was accompanied
by changes in the expression levels of proteins in the eNOS/NO
signaling axis; this mechanism was discovered to serve a key role
in Hcy-induced endothelial dysfunction (19,20).
NO is widely accepted to be a potent endothelial relaxing factor,
which is mainly produced by the eNOS-induced catalysis of
L-arginine. NO produced by the endothelial cells diffuses to smooth
muscle cells of the blood vessel and is activated by guanylate
cyclization (21). Enzymes mediate
the relaxation effect of the vascular smooth muscle, thereby
regulating the vasodilation function (22). The eNOS/NO signaling axis is known
to help maintain vascular homeostasis and has therefore attracted
significant interest in the field of vascular pharmacology
(23,24). Accumulating evidence has suggested
that eNOS/NO signaling axis was involved in vessel relaxation,
vascular smooth muscle cell proliferation and ischemia/reperfusion
injury (25). Moreover, our
previous study revealed that KLF4 knockdown in endothelial cells
could inhibit TGF-β-induced cell proliferation, migration and
phenotypic changes, and effectively inhibited EndMT, suggesting
that KLF4 may serve a role in EndMT (26). However, to the best of our
knowledge, few studies have investigated the regulatory effect of
the KLF4/eNOS signaling pathway in vascular damage-associated
disorders. Our previous study reported that SAL upregulated the
expression levels of the antioxidant signaling factor, Nrf2, which
improved the Hcy-induced oxidative stress damage of endothelial
cells, thereby suggesting its potential vascular pharmacological
activity (13).
Based on the aforementioned findings, it was
hypothesized that the KLF4/eNOS signaling pathway may serve a role
in EndMT. Therefore, the present study aimed to investigate the
effect and underlying molecular mechanisms of SAL in Hcy-induced
EndMT. The current results revealed that Hcy treatment for 48 h
significantly downregulated the expression levels of the
endothelial specific marker, VE-cadherin, upregulated the
expression levels of the mesenchymal cell marker, α-SMA, and
increased cell migration, indicating that Hcy may induce EndMT.
Pretreatment with SAL inhibited these changes in the cell
phenotypic markers and reduced cell migration, suggesting that SAL
could effectively inhibit EndMT, and that its effect was associated
with the upregulation of the eNOS/NO signaling axis and
downregulation of KLF4 expression. The findings of the
immunofluorescence assay demonstrated that Hcy induced the
translocation of KLF4 from the cytoplasm to the nucleus, while the
pretreatment with SAL reversed these effects, indicating that SAL
intervention in EndMT may be association with the downregulation of
the expression level of the TF KLF4. To further investigate whether
KLF4 affected the eNOS/NO signaling axis, siRNA interference
technology was used to knockdown KLF4 expression in HUVECs. The
present findings showed that KLF4-knockdown increased the levels of
cellular eNOS/NO signaling, suggesting that KLF4 may regulate the
upstream molecular signaling of eNOS and participate in the
regulation of vasodilation. However, no significant differences
were observed between the SAL, siKLF4 and SAL + siKLF4
co-administration groups in the expression levels of the cell
phenotypic markers, VE-cadherin and α-SMA, indicating that KLF4 may
represent a key molecular target for SAL to inhibit EndMT.
In conclusion, to the best of our knowledge, the
results of the present study demonstrated, for the first time, that
SAL may inhibit the effect of Hcy-induced EndMT by regulating the
KLF4/eNOS signaling pathway; however, its precise underlying
mechanism remains to be determined.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was funded by Scientific Research Project
of Hunan Provincial Department of Education (grant no. 19B043), the
Yan'an University Science Foundation Project (grant no. YDQ2020-06)
and the Hunan Provincial Administration of Traditional Chinese
Medicine Research Project Fund (grant no. 202083).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YH and XH designed and undertook experiments, and
analyzed, interpreted and presented results for group discussions.
YH, XH and JT performed some of the experiments and confirm the
authenticity of all the raw data. XL provided methods, and was
involved in the interpretation of results for the manuscript. XW
made substantial contributions to the conception and design of the
study. All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu X, Friehs I, Hu TZ, Melnychenko I,
Tampe B, Alnour F, Iascone M, Kalluri R, Zeisberg M, Del Nido PJ
and Zeisberg EM: Endocardial fibroelastosis is caused by aberrant
endothelial to mesenchymal transition. Circ Res. 116:857–866. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bischoff J: Endothelial-to-mesenchymal
transition. Circ Res. 124:1163–1165. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qin W, Zhang L, Li Z, Xiao D, Zhang Y,
Zhang H, Mokembo JN, Monayo SM, Jha NK, Kopylov P, et al:
Endothelial to mesenchymal transition contributes to
nicotine-induced atherosclerosis. Theranostics. 10:5276–5289. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wadhera RK, Steen DL, Khan I, Giugliano RP
and Foody JM: A review of low-density lipoprotein cholesterol,
treatment strategies, and its impact on cardiovascular disease
morbidity and mortality. J Clin Lipid. 10:472–489. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo Y, Li P, Bledsoe G, Yang ZR, Chao L
and Chao J: Kallistatin inhibits TGF-β-induced
endothelial-mesenchymal transition by differential regulation of
microRNA-21 and eNOS expression. Exp Cell Res. 337:103–110. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yoshida T, Yamashita M and Hayashi M:
Kruppel-like factor 4 contributes to high phosphate-induced
phenotypic switching of vascular smooth muscle cells into
osteogenic cells. J Biol Chem. 287:25706–25714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sun Q, Gong L, Qi R, Qing W, Zou M, Ke Q,
Zhang L, Tang X, Nie Q, Yang Y, et al: Oxidative stress-induced
KLF4 activates inflammatory response through IL17RA and its
downstream targets in retinal pigment epithelial cells. Free Rad
Biol Med. 147:271–281. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhong Z, Han J, Zhang J, Xiao Q, Hu J and
Chen L: Pharmacological activities, mechanisms of action, and
safety of salidroside in the central nervous system. Drug Des Devel
Ther. 12:1479–1489. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zheng L, Su J, Zhang Z, Jiang L, Wei J, Xu
X and Lv S: Salidroside regulates inflammatory pathway of alveolar
macrophages by influencing the secretion of miRNA-146a exosomes by
lung epithelial cells. Sci Rep. 10:207502020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lai W, Xie X, Zhang X, Wang Y, Chu K,
Brown J, Chen L and Hong G: Inhibition of complement drives
increase in early growth response proteins and neuroprotection
mediated by salidroside after cerebral ischemia. Inflammation.
41:449–463. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xing SS, Yang J, Li WJ, Li J, Chen L, Yang
YT, Lei X, Li J, Wang K and Liu X: Salidroside decreases
atherosclerosis plaque formation via inhibiting endothelial cell
pyroptosis. Inflammation. 43:433–440. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhu L, Jia F, Wei J, Yu Y, Yu T, Wang Y,
Sun J and Luo G: Salidroside protects against homocysteine-induced
injury in human umbilical vein endothelial cells via the regulation
of endoplasmic reticulum stress. Cardiovasc Ther. 35:33–39. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Yanyan, He Li, Huang Mei, Jiang
Feng, Fu Lingyun and Li Jiachuan: Salidroside inhibits homocysteine
induced oxidative stress in human umbilical vein endothelial cells.
Journal of Southwest Minzu University (Natural Science Edition).
2020.46((04)): 349–353, (In Chinese).
|
|
14
|
Xing SS, Li J, Chen L, Yang YF, He PL, Li
J and Yang J: Salidroside attenuates endothelial cellular
senescence via decreasing the expression of inflammatory cytokines
and increasing the expression of SIRT3. Mech Ageing Dev. 175:1–6.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Song S, Zhang R, Cao W, Fang G, Yu Y, Wan
Y, Wang C, Li Y and Wang Q: Foxm1 is a critical driver of
TGF-β-induced EndMT in endothelial cells through Smad2/3 and binds
to the snail promoter. J Cell Physiol. 234:9052–9064. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Putten S, Shafieyan Y and Hinz B:
Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol.
93:133–142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 19:620–636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song CL, Liu B, Shi YF, Liu N, Yan YY,
Zhang JC, Xue X, Wang JP, Zhao Z, Liu JG, et al: MicroRNA-130a
alleviates human coronary artery endothelial cell injury and
inflammatory responses by targeting PTEN via activating
PI3K/Akt/eNOS signaling pathway. Oncotarget. 7:71922–71936. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang XJ, Tian DC, Wang FW, Zhang MH, Fan
CD, Chen W, Wang MH, Fu XY and Ma JK: Astaxanthin inhibits
homocysteine-induced endothelial cell dysfunction via the
regulation of the reactive oxygen species-dependent VEGF-VEGFR2-FAK
signaling pathway. Mol Med Rep. 19:4753–4760. 2019.PubMed/NCBI
|
|
21
|
Rajendran S, Shen X, Glawe J, Gopi K,
Kolluru GK and Kevil CG: Nitric oxide and hydrogen sulfide
regulation of ischemic vascular growth and remodeling. Compr
Physiol. 12:1213–1247. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tan X, Feng L, Huang X, Yang Y, Yang C and
Gao Y: Histone deacetylase inhibitors promote eNOS expression in
vascular smooth muscle cells and suppress hypoxia-induced cell
growth. J Cell Mol Med. 21:2022–2035. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu S, Sun Z, Chu P, Li H, Ahsan A, Zhou
Z, Zhang Z, Sun B, Wu J, Xi Y, et al: EGCG protects against
homocysteine-induced human umbilical vein endothelial cells
apoptosis by modulating mitochondrial-dependent apoptotic signaling
and PI3K/Akt/eNOS signaling pathways. Apoptosis. 22:672–680. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Arab HH, Salama SA and Maghrabi IA: Camel
milk attenuates methotrexate-induced kidney injury via activation
of PI3K/Akt/eNOS signaling and intervention with oxidative
aberrations. Food Funct. 9:2661–2672. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Xiang X, Xu H and Shi Y: Cilostazol
promotes angiogenesis and increases cell proliferation after
myocardial ischemia-reperfusion injury through a cAMP-dependent
mechanism. Cardiovasc Eng Technol. 10:638–647. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Y, Li C, Huang Y, Zhao S, Xu Y, Chen
Y, Jiang F, Tao L and Shen X: EOFAZ inhibits
endothelial-to-mesenchymal transition through downregulation of
KLF4. Int J Mol Med. 46:300–310. 2020.PubMed/NCBI
|