Introduction
Neonatal pulmonary hypertension (PH) is a severe
type of PH in children with an incidence rate of 0.43–6.82 per
1,000 live births and a mortality rate of 4–33% according to a
study from the National Institute of Child Health and Human
Development Neonatal Research Network (1). The etiology of neonatal PH includes
hypoxia, parenchymal lung diseases, abnormal pulmonary vascular
development, among which hypoxia is the primary cause (2,3). In
the early stages of PH, which is characterized by pulmonary
vasoconstriction (2), symptomatic
treatments, such as vasodilation, are effective to a limited extent
(2). However, once the disease
progresses towards pulmonary vascular remodeling and right
ventricular hypertrophy, the effectiveness of treatments is
reduced, leading to a high mortality rate (4,5).
Therefore, developing effective strategies for preventing PH
disease progression is essential.
Pulmonary microvascular endothelial cells (PMVECs)
serve a central role in the pathogenesis of adult PH through
apoptosis, proliferation and their interactions with other
pulmonary vascular cell types (6).
Sakao et al (7) demonstrated
that PMVECs are the early damage sites of PH, and early apoptosis
of PMVECs initiates PH pathogenesis. In the early stages of PH, the
apoptosis of PMVECs results in a decrease in cellular function,
leading to the inactivation of the endothelial nitric oxide lyase,
decreased release of the vasodilator nitric oxide and inhibition of
PMVEC-mediated pulmonary vascular tension, resulting in pulmonary
artery constriction, high pulmonary vascular resistance and high
pulmonary artery pressure (8–10). As
the disease progresses, cell proliferation predominates over PMVEC
apoptosis, resulting in tissue imbalance, which coincides with the
formation of plexiform vascular lesions and vascular remodeling
(4,11,12).
Schultz et al (13)
developed a persistant PH mouse model displaying the newborn
phenotype by downregulating the superoxide dismutase 2 gene. These
mice exhibited high right ventricular systolic pressure, pulmonary
artery endothelial cell apoptosis and pulmonary artery smooth
muscle cell proliferation, indicating that PMVEC apoptosis may be
involved in the pathogenesis of neonatal PH. However, whether
PMVECs exhibit an apoptotic phenotype in early hypoxia has not been
previously reported, and the regulatory mechanism underlying PMVEC
apoptosis remains unclear.
Heat shock proteins (HSPs) are a conserved family of
molecular chaperones that protect cells from damage and maintain
cellular homeostasis (14). HSP70,
which is the most widely studied member of the HSP family, is
expressed at low levels in healthy cells under normal environmental
conditions. However, under the effects of heat shock, ischemia,
hypoxia, nutrient deficiency, irradiation or infection, the
expression levels of HSP70 increase compared with those under
normal conditions (14,15). HSP70 is also an antiapoptotic
protein; high expression levels of HSP70 inhibit apoptosis and
promote cell survival, whereas low levels increase the apoptotic
rate (16,17). Through its antiapoptotic activity,
HSP70 participates in the pathogenesis of various diseases,
including heart disease, lung injury and neurodegeneration
(18). Researchers have
demonstrated that HSP70 is highly expressed in breast cancer and
gastric cancer cells, and inhibits apoptosis by acting on multiple
sites upstream and downstream of the mitochondrial apoptotic
signaling pathway, which may contribute to tumor progression by
affecting chemotherapy resistance (17,19,20).
During myocardial ischemia, HSP70 expression is increased, which
protects cardiac cells by removing or refolding misfolded proteins
and inhibiting the activity of apoptosis-inducing factor (AIF)
(21). In addition, HSP70
upregulation stimulated by geranylgeranylacetone, a HSP inducer,
relieves pneumolysin-induced acute lung injury by reducing human
PMVEC apoptosis (22). Kondrikov
et al (14) reported that
hyperoxia elevates the expression levels of HSP70 in PMVECs,
preventing lung endothelial barrier damage by inhibiting
caspase-dependent and AIF-dependent apoptosis. However, the role of
HSP70 in PMVEC apoptosis in the context of neonatal hypoxic PH
(HPH) remains unclear.
Hypoxia-inducible factor-1 (HIF-1), a key regulator
of hypoxia, is a heterodimer composed of HIF-1α and HIF-1β
(23). The expression levels of
HIF-1α are increased under hypoxia and determine the
transcriptional activation of HIF-1 (24). HIF-1α participates in a variety of
hypoxia-related diseases by promoting transcriptional activation of
its downstream target genes (25).
In our previous study, induction of HIF-1α and its downstream
factors, nitric oxide synthase and endothelin-1, promoted HPH
progression in neonatal rats (26).
In addition, recent studies have demonstrated that HIF-1α serves an
important role in vascular endothelial cell apoptosis (27,28).
Therefore, it has been hypothesized that HIF-1α may be involved in
HSP70-mediated regulation of PMVEC apoptosis.
The present study aimed to assess the apoptotic
phenotypic changes in PMVECs during the early stages of hypoxia,
and elucidate the effects and mechanisms of HSP70 on
hypoxia-induced PMVEC apoptosis in neonatal rats.
Materials and methods
Animals
A total of 87 Sprague-Dawley rats (30 female and 57
male; age, 5–7 days; weight, 10–15 g) were purchased from the
Department of Laboratory Animal Science of Xinjiang Medical
University. All neonatal rats were given adaptive feeding after
birth and were maintained in a 12 h light/dark cycle at 22–25°C and
55–60% humidity. Animal health and behavior were monitored once a
day. Every effort was made to minimize animal numbers and
suffering. The specific criteria used to determine when to
euthanize an animal included lethargy, lack of appetite, lost organ
function and rapid weight loss (>10% body weight). No rats were
euthanized or found dead during the course of the present study.
Neonatal rats were anesthetized with an intraperitoneal injection
of 100 mg/kg ketamine and 10 mg/kg xylazine followed by sacrifice
via cervical dislocation. In the present study, the criteria for
verifying animal death were as follows: No spontaneous respiration,
no heartbeat, no corneal reflex and gray skin. All animal
experiments were approved by the Animal Ethics Committee of The
First Affiliated Hospital of Xinjiang Medical University (approval
no. IACUC-20170214029; Urumqi, China).
Cell isolation, culture and
identification
PMVECs were extracted from lung tissue samples of
newborn rats by the tissue adhesion method (29). Lungs were extracted from neonatal
rats and placed in DMEM (Thermo Fisher Scientific, Inc.). The
pleura and lung tissue edges (2 mm) were removed and then 1
mm3 sections were excised from the marginal lung tissue
and incubated at 37°C for 30 min. The tissue blocks were uniformly
placed in a 6-well plate (30 pieces/well) and coated with
poly-D-lysine (Sigma-Aldrich; Merck KGaA). Following tissue block
solidification and attachment to the culture plate at 37°C with 5%
CO2 for 90 min, 750 µl PMVEC primary culture medium
containing DMEM (Gibco; Thermo Fisher Scientific, Inc.), 20% FBS
(Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (Beyotime Institute of Biotechnology) was
added to each well. Cell morphology was examined using an optical
microscope (Nikon Instruments Inc.). PMVECs were digested with
0.25% trypsin (Beyotime Institute of Biotechnology) at 37°C for 5
min and 3–5×105 cells were collected after
centrifugation at 1,000 × g for 5 min. In the present study, three
batches of cell culture were used. Primary cells were cultured on
glass cover slips and fixed with 4% paraformaldehyde (Sangon
Biotech Co., Ltd.) at room temperature for 15 min. Subsequently,
the cell samples were blocked with 6% normal goat serum (HyClone;
Cytiva) at room temperature for 30 min. Following incubation with a
rabbit monoclonal antibody targeting CD31 (1:100; cat. no.
ab222783; Abcam) overnight at 4°C (the control cells were incubated
with PBS instead of the CD31 antibody), the samples were washed and
incubated with an Alexa Fluor 488-conjugated goat anti-rabbit
secondary antibody (1:800; cat. no. ab150077; Abcam) at 37°C for 30
min. The samples were rinsed three times with 1X PBS and incubated
with DAPI (Beyotime Institute of Biotechnology) for 5 min in the
dark at room temperature. Antifade Mounting Medium (Beyotime
Institute of Biotechnology) was subsequently added, and images were
acquired using a TCS SP8 confocal laser scanning microscope (Leica
Microsystems, Inc.).
Overexpression and inhibition of
HSP70
PMVECs were assigned into four groups: i) Hypoxia
(HX), PMVECs transduced with an empty lentivirus (LV-Null) and
cultured under hypoxic conditions; ii) normoxia (NX), PMVECs
transduced with LV-Null and cultured under normoxic conditions;
iii) HX + HSP70, PMVECs transduced with a lentivirus with
HSP70-overexpression lentivirus (LV-HSP70) and cultured under
hypoxic conditions; and iv) HX + HSP70 +
N-formyl-3,4-methylenedioxy-benzylidene-g-butyrolactam (KNK437;
Sigma-Aldrich; Merck KGaA), PMVECs transduced with LV-HSP70 and
treated with KNK437 prior to exposure to hypoxia.
LV-HSP70 and LV-Null were provided by Western
Biotechnology, Inc. The HSP70 coding sequence was amplified using
whole genome synthesis of DNA (NCBI Gene ID: 266759, derived from
rat tissues; chemically synthesized by Genscript, Inc.) as a
template with Premix Taq™ (LA Taq™ Version 2.0; Takara
Biotechnology, Co., Ltd.) and verified via sequencing on the Ion
Proton™ Sequencer (Invitrogen; Thermo Fisher Scientific, Inc.). The
forward (F) and reverse (R) primer sequences used for the
amplification of HSP70 were 5′-ACTAGTGCCACCATGTCGGTGGATGGG-3′ and
5′-GCTCTAGATCAATCAATGTCCATCTC-3′, respectively. The thermocycling
conditions were as follows: Initial denaturation at 98°C for 3 min;
30 cycles of denaturation cycles at 98°C for 10 sec and annealing
and elongation at 55°C for 30 sec and 72°C for 2 min respectively;
followed by a final extension step at 72°C for 10 min. The HSP70
coding sequence was inserted into the pLVX–IRES-Puro vector
(Guangzhou FitGene Biotechnology Co., Ltd.), and namely
pLVX-HSP70-IRES-Puro. Using 56.7 µl NDE™ 3000 (Western Technology,
Inc.), the empty pLVX–IRES-Puro or pLVX-HSP70-IRES-Puro (8.04 µg)
vectors were transduced into the 293T viral packaging cell line
(2–3×106; The Cell Bank of Type Culture Collection of
The Chinese Academy of Sciences) together with 8.34 µg psPAX2
(Genscript, Inc.) and 2.52 µg pMD2.G plasmids (Genscript, Inc.).
The 3rd generation vector system was used. Following incubation at
37°C, lentivirus particle-rich supernatants were collected at 72
and 96 h post-transduction. The lentivirus was concentrated at
5,000 × g for 15 min at 4°C via ultrafiltration using Amicon
Ultra-15 (cat. no. UFC910024; EMD Millipore).
Primary PMVECs cultured to the third generation were
seeded in 24-well culture plates (3–5×104 cells/well).
At ~70% confluency, cells were infected with LV-HSP70 or LV-Null at
a multiplicity of infection of 100 and incubated at 37°C with 5%
CO2 for 72 h. The infected cells were selected using 2
µg/ml puromycin. Subsequently, the cells were cultured in hypoxic
(5% O2) or normoxic (21% O2) environments for
24, 48 or 72 h. In order to ensure the consistency and reliability
of the present study, these three time points were selected to
clarify the apoptotic trend of PMVECs in early hypoxia with
reference to relevant previous studies conducted in adults
(30,31). KNK437, which is a synthetic reagent
that specifically inhibits the synthesis of HSP70 at the
transcriptional level (32–34), was dissolved in DMSO and added to
the cell medium at a final concentration of 100 µM. Following
treatment with KNK437 at room temperature for 3 h, the PMVECs were
subjected to hypoxia as aforementioned.
Apoptotic rate analysis
The apoptotic rate was detected using an Annexin
V-PE/7-AAD kit (Tianjin Sungene Biotech Co., Ltd.) according to the
manufacturer's protocol. Following treatment, PMVECs were washed
twice with 1X PBS and 1–5×105 cells were harvested. The
cells were incubated with a mixture of 50 µl binding buffer and 5
µl 7-Aminoactinomycin D at room temperature in the dark for 15 min.
Following the addition of a further 450 µl binding buffer, cells
were incubated with 10 µl Annexin V-PE for 15 min at room
temperature in the dark. After washing with 1X PBS, the apoptotic
rate (early + late apoptosis) was detected and analyzed using
CytoFLEX flow cytometer and CytExpert software v2.4.0.28 (Beckman
Coulter, Inc.) within 1 h.
Mitochondrial membrane potential (MMP)
analysis
PMVECs (1–5×105) were harvested and
changes in the MMP were detected using a JC-1 MMP Assay kit (cat.
no. C2006; Beyotime Institute of Biotechnology) according to the
manufacturer's instructions and analyzed using CytoFLEX flow
cytometer and CytExpert software v2.4.0.28 (Beckman Coulter, Inc.).
In healthy mitochondria, JC-1 aggregates in the mitochondrial
matrix to form a polymer that emits strong red fluorescence
(excitation wavelength, 585 nm; emission wavelength, 590 nm)
(35). When the MMP is low or
absent, JC-1 is only present in the cytoplasm as a monomer and
emits green fluorescence (excitation wavelength, 514 nm; emission
wavelength, 529 nm) (36). The MMP
was measured as the ratio of red-to-green fluorescence
intensity.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from PMVECs using an RNA
extraction kit (Takara Bio, Inc.) according to the manufacturer's
instructions, and reverse transcribed with random primers. RT was
performed using the RT kit (Takara Bio, Inc.) following the
manufacturer's protocol. Subsequently, qPCR was performed in a 10
µl reaction system comprising 1 µl DNA template, 5 µl 2X PowerUp
SYBR® Green Master Mix (Applied Biosystems; Thermo
Fisher Scientific, Inc.) and 0.5 µl each of forward and reverse
primers. Furthermore, the thermocycling conditions used were as
follows: Initial denaturation, 50°C for 2 min and 95°C for 2 min;
40 cycles of denaturation, 95°C for 15 sec, and annealing and
extension, 60°C for 1 min. mRNA expression levels of the target
genes quantified using the 2−ΔΔCq method (37) and normalized to GADPH mRNA
expression levels. In the present study, the mRNA expression levels
of HSP70, HIF-1α, Bcl-2, cytochrome c (cyt C) and caspase-3,
which are involved in the mitochondrial apoptosis pathway (38,39)
were examined. The following primers were used: HSP70 F,
5′-CTGTGCTCTGCAGTGTGCAAT-3′ and R, 5′-GGCCTCCAGAGTGAACGGT-3′;
HIF-1α F, 5′-ATACATGGGGTTGACTCAGTTTG-3′ and R,
5′-GCTTCGCTGCGTGTTTTGT-3′; Bcl-2 F, 5′-GAGGGGCTACGAGTGGGATAC-3′ and
R, 5′-TCAGGCTGGAAGGAGAAGATG-3′; cyt C F, 5′-ACAAAGGCATCATCTGGGGA-3′
and R, 5′-TAAGTCTGCCCTTTCTTCCTTCT-3′; caspase-3 F,
5′-CCATAAAAGCACTGGAATGTCAG-3′ and R, 5′-CAAAACTGCTCCTTTTGCTGTG-3′;
and GAPDH F, 5′-GGCAAGTTCAACGGCACAG-3′ and R,
5′-CGCCAGTAGACTCCACGACAT-3′.
Western blotting
PMVECs were added to radioimmunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology) to extract total
protein, and the amount of protein was determined using the BCA
method. The proteins (20 µg) were separated via SDS-PAGE on 8, 10
or 12% gels and electroblotted onto PVDF membranes (Bio-Rad
Laboratories, Inc.). After blocking with TBS with 0.05% Tween-20
(TBST) buffer containing 5% skimmed milk at room temperature for 2
h, the membranes were incubated at 4°C overnight with primary
rabbit antibodies targeting HSP70 (cat. no. ab181606), HIF-1α (cat.
no. ab216842), Bcl-2 (cat. no. ab196495), caspase-3 (cat. no.
ab13847) and GAPDH (cat. no. ab181602), and primary mouse
antibodies targeted against cyt C (cat. no. ab13575). All primary
antibodies were purchased from Abcam and used at a final dilution
of 1:1,000. Following three washes with TBST, the membranes were
incubated at room temperature with HRP-labeled secondary antibodies
(goat anti-rabbit and goat anti-mouse; cat. nos. A0545-1ML and
A4416-1ML; 1:1,000; Sigma-Aldrich; Merck KGaA) for 1.5 h. The
membranes were evenly covered with an ECL reagent (Pierce; Thermo
Fisher Scientific, Inc.) for 2 min at room temperature and placed
into an exposure instrument for detection. Protein band densities
were semi-quantified by the Quantity One software (version 4.6.2;
Bio-Rad Laboratories, Inc.), with GAPDH as the loading control.
Statistical analysis
SPSS (version 22.0; IBM Corp.) statistical software
was used for all data analyses. Differences among multiple groups
were analyzed by one-way or two-way ANOVA followed by Bonferroni's
post hoc test. All experiments were conducted with three
independent repeats. Data are presented as the mean ± SD. P<0.05
was considered to indicate a statistically significant
difference.
Results
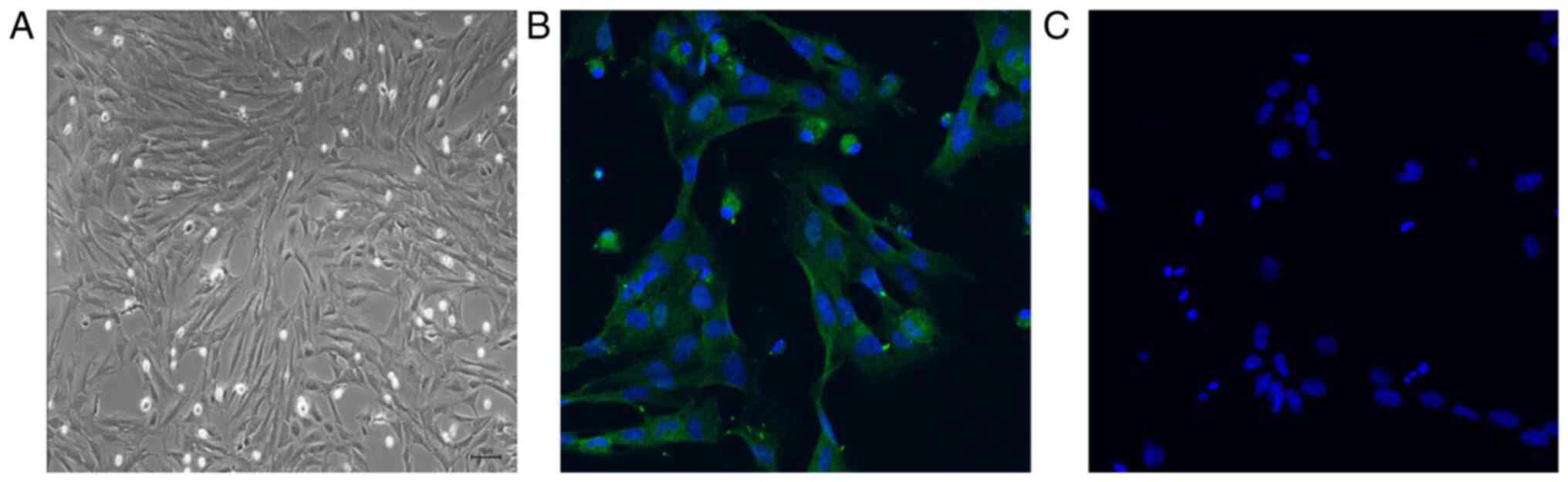
Identification of PMVECs isolated from
neonatal rats
To determine the effects of HSP70 on apoptosis in
PMVECs, PMVECs were extracted from the lung tissues of newborn
rats. PMVECs exited the lung tissue of normal newborn rats 72 h
following lung tissue attachment to the culture plate. Once fused
into a monolayer during culture, PMVECs were analyzed using a light
microscope. The results demonstrated that PMVECs displayed a
polygonal or fusiform morphology with a typical cobblestone-like
arrangement (Fig. 1A). An
endothelial cell-specific antibody, anti-CD31, was used to assess
the cell via immunofluorescence. The results revealed that the
cells expressed CD31, confirming that these cells were PMVECs and
could be used to assess the function of HSP70 under hypoxic
conditions (Fig. 1B and C).
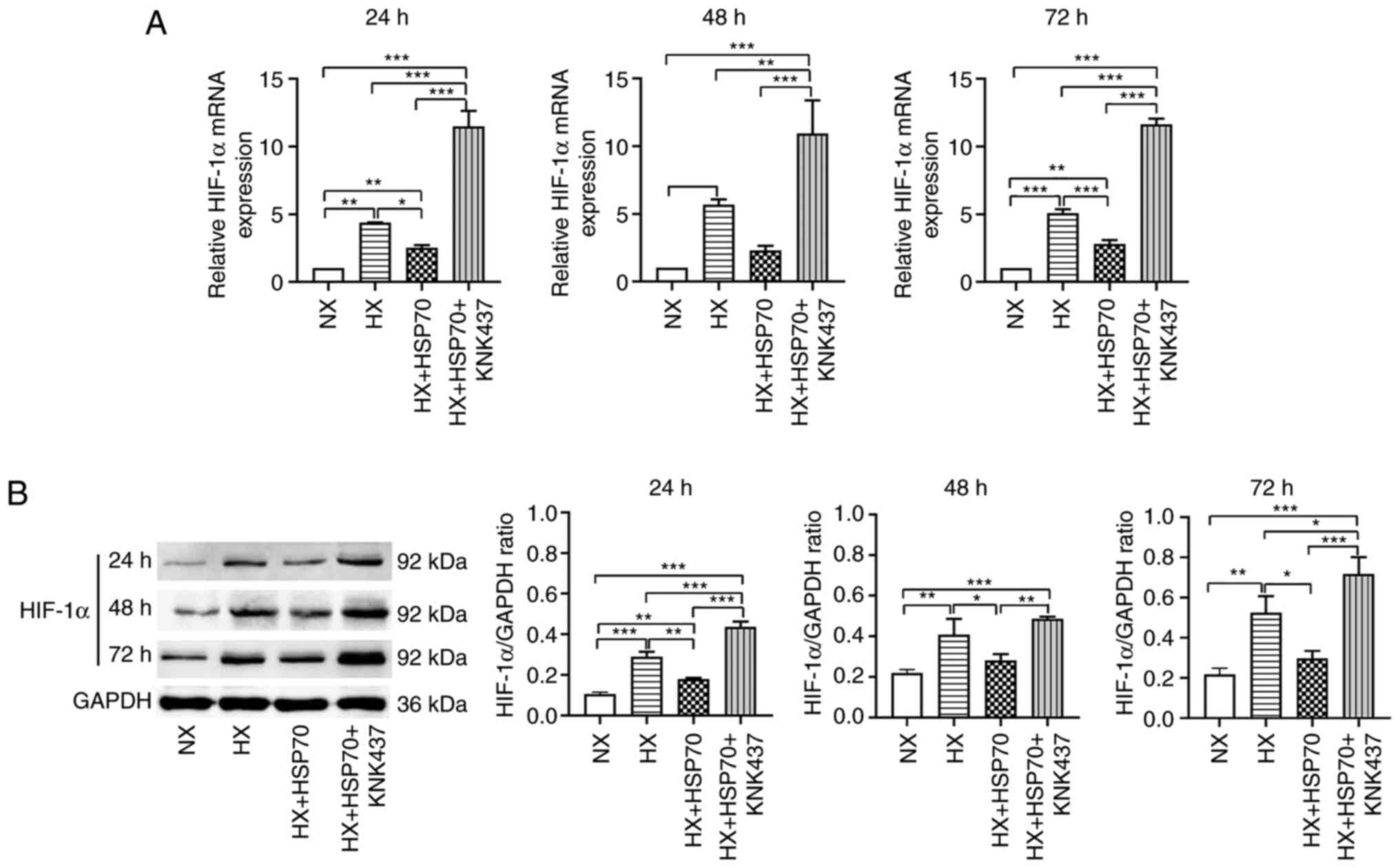
Hypoxia upregulates the expression
levels of HSP70 in PMVECs isolated from neonatal rats
Primary PMVECs from neonatal rats were cultured in a
hypoxic environment for 24, 48 or 72 h. Subsequently, RT-qPCR was
performed to evaluate the mRNA expression levels of HSP70 (Fig. 2A). The results revealed that the
relative HSP70 mRNA expression levels in the HX group were
significantly higher compared with the NX group. Similarly, HSP70
mRNA expression levels were significantly higher in the HX + HSP70
group compared with those in the HX group. By contrast, HSP70 mRNA
expression levels in the HX + HSP70 + KNK437 group were
significantly lower compared with those in the HX + HSP70 group,
suggesting that treatment with KNK437 inhibited HSP70
overexpression at the mRNA level. In addition, western blotting was
performed to assess the protein expression levels of HSP70 in
PMVECs at 24, 48 and 72 h time points during exposure to hypoxia
(Fig. 2B). The results were
consistent with the mRNA expression levels; hypoxia significantly
increased the protein expression levels of HSP70 in PMVECs compared
with those in the NX group. HSP70 overexpression further increased
HSP70 expression in the HX + HSP70 group compared with the HX
group. By contrast, KNK437 significantly reduced the protein
expression levels of HSP70 compared with those in the HX + HSP70
group. Taken together, these results suggested that hypoxia induced
the upregulation of HSP70 mRNA and protein expression levels in the
PMVECs of neonatal rats.
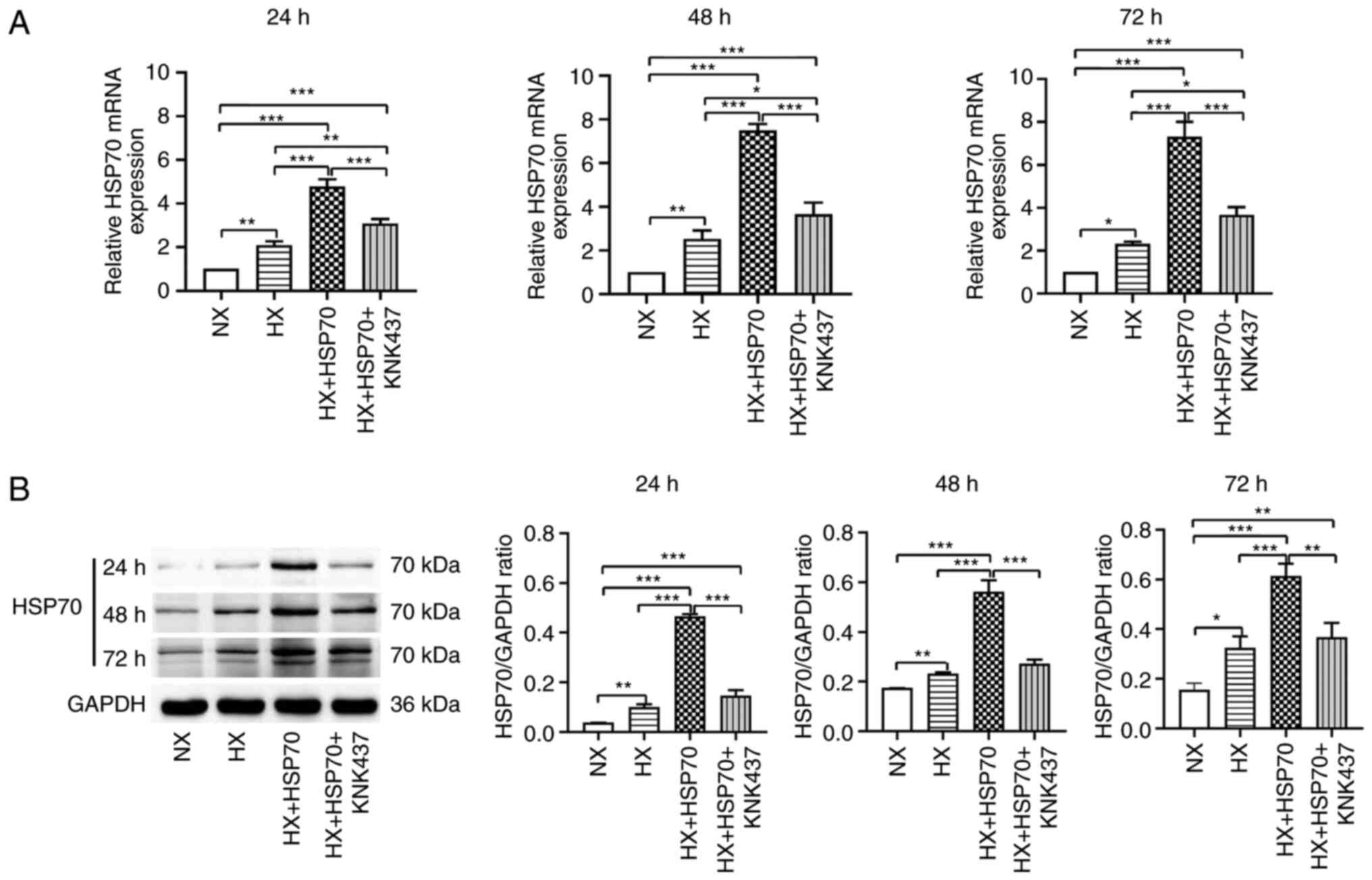 | Figure 2.Hypoxia upregulates HSP70 in PMVECs
isolated from neonatal rats. Following 24, 48 and 72 h hypoxia
exposure, relative HSP70 (A) mRNA and (B) protein expression levels
were assessed in the HX, NX, HX + HSP70 and HX + HSP70 + KNK437
groups. GAPDH was used as the loading control. Data are presented
as the mean ± SD from at least three separate experiments, and were
analyzed by one-way ANOVA followed by Bonferroni's post hoc test.
*P<0.05, **P<0.01 and ***P<0.001. HSP, heat shock protein;
PMVECs, pulmonary microvascular endothelial cells; HX, hypoxia; NX,
normoxia; KNK437,
N-formyl-3,4-methylenedioxy-benzylidene-g-butyrolactam. |
HSP70 inhibits hypoxia-induced
apoptosis in PMVECs isolated from neonatal rats
In the present study, the effects of HSP70 on the
apoptotic rate of hypoxia-treated PMVECs isolated from neonatal
rats were evaluated by flow cytometry. The results demonstrated
that the apoptotic rate of the HX cell group at 24, 48 and 72 h was
significantly higher compared with the NX group at the same time
point, with the increase becoming more notable with longer
incubation times (Fig. 3). The
apoptotic rate in the HX + HSP70 group was significantly lower
compared with that in the HX group, whereas the apoptotic rate was
significantly higher in the HX + HSP70 + KNK437 group compared with
that in the HX + HSP70 group. These results suggested that HSP70
inhibited apoptosis in PMVECs isolated from neonatal rats under
hypoxic conditions.
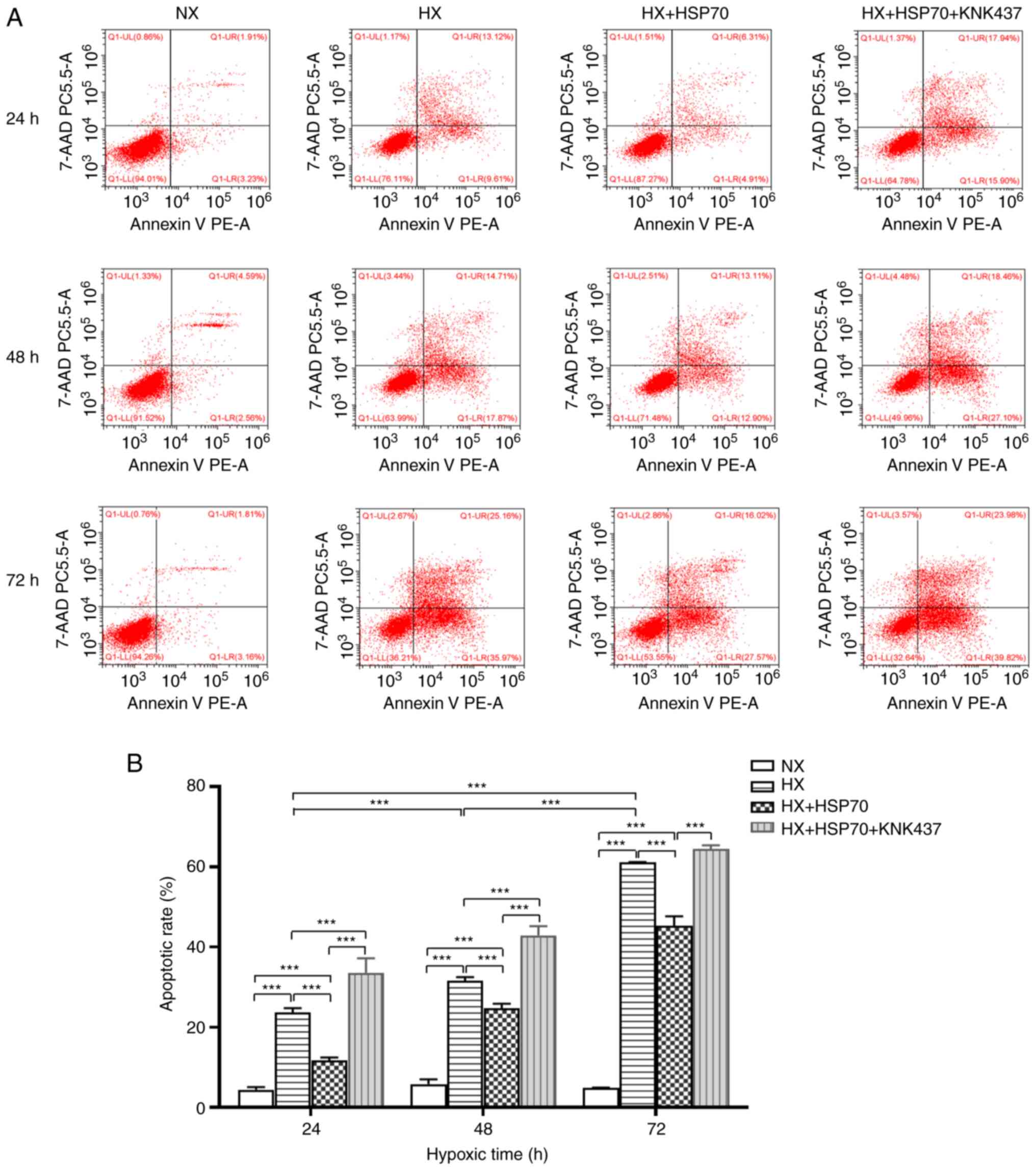 | Figure 3.HSP70 inhibits hypoxia-induced
apoptosis in PMVECs isolated from neonatal rats. (A) Flow
cytometric analysis of the apoptotic rate of PMVECs. (B) Apoptotic
rates of PMVECs were compared in the HX, NX, HX + HSP70 and HX +
HSP70 + KNK437 groups. Apoptotic rates of PMVECs exposed to hypoxia
for 24, 48 and 72 h were significantly higher compared with those
of PMVECs in the NX group, and increased gradually with increasing
hypoxia duration. At all tested time points during hypoxia, the
PMVEC apoptotic rates in the HX + HSP70 group were significantly
lower compared with those of the corresponding HX and HX + HSP70 +
KNK437 groups. Data are presented as the mean ± SD from at least
three separate experiments, and were analyzed by two-way ANOVA
followed by Bonferroni's post hoc test. ***P<0.001. HSP, heat
shock protein; PMVECs, pulmonary microvascular endothelial cells;
HX, hypoxia; NX, normoxia; KNK437,
N-formyl-3,4-methylenedioxy-benzylidene-g-butyrolactam; PE,
phycoerythrin; PC5.5, phycoerythrin-cyanin 5.5. |
HSP70 is associated with the
mitochondrial apoptotic pathway in PMVECs isolated from neonatal
rats
To investigate the association between HSP70 and the
mitochondrial apoptotic pathway, changes in the MMP and expression
levels of Bcl-2, cyt C and caspase-3 were examined. The results
demonstrated that the MMP was significantly lower in PMVECs exposed
to hypoxia for 24, 48 and 72 h compared with that in the
corresponding NX group, especially after 72 h (Fig. 4). For all time points, the MMP in
the HX + HSP70 group was significantly higher compared with that in
the HX group, but significantly lower compared with the NX group.
In addition, the MMP was significantly lower in the HX + HSP70 +
KNK437 group compared with that in the HX + HSP70 group, and
markedly lower compared with the NX group.
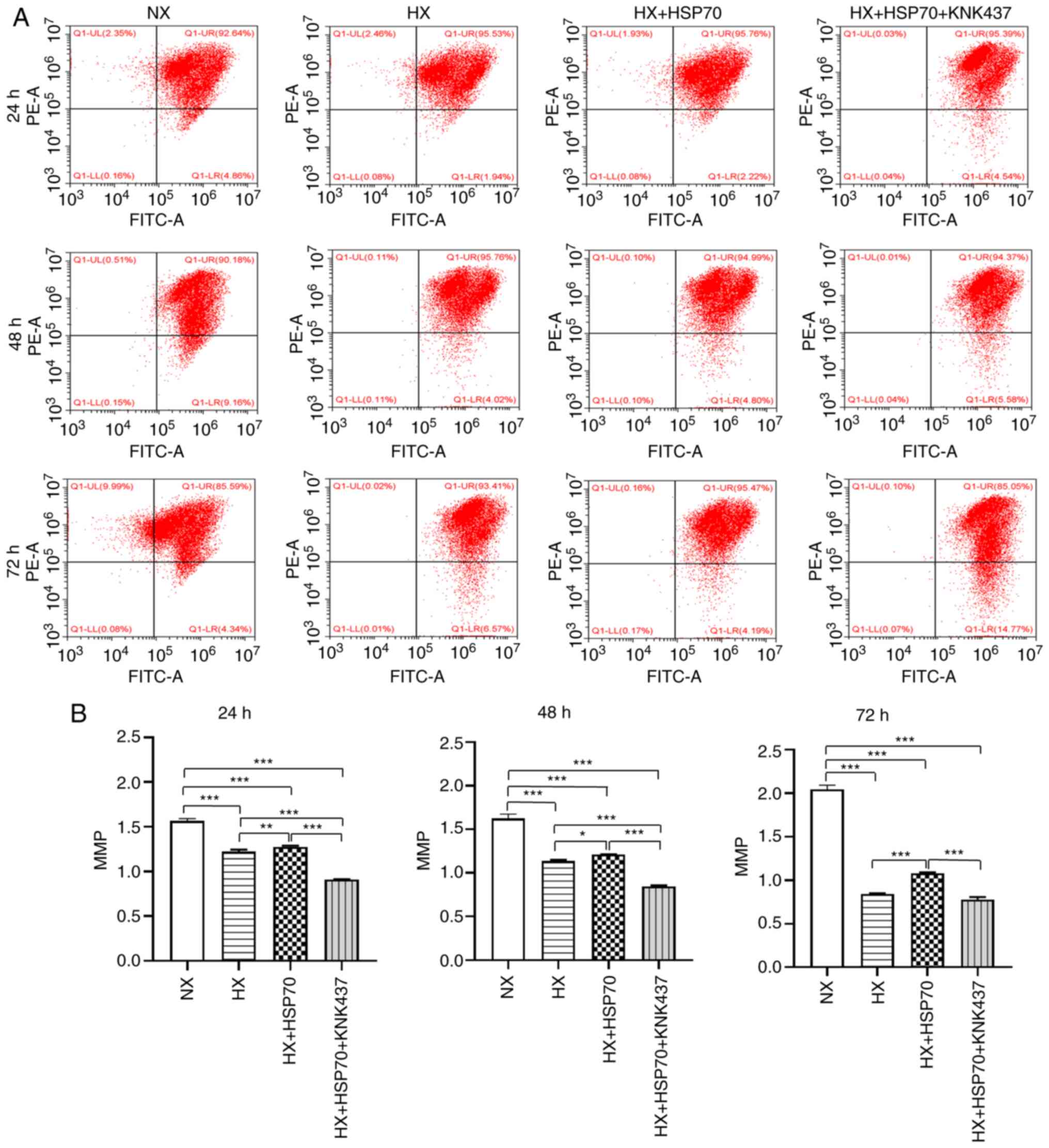 | Figure 4.HSP70 inhibits hypoxia-induced MMP
decrease in PMVECS isolated from neonatal rats. (A) Flow cytometric
analysis of the MMP of PMVECs by JC-1. (B) MMP of PMVECs was
compared in the HX, NX, HX + HSP70 and HX + HSP70 + KNK437 groups.
The MMP was significantly lower in PMVECs exposed to hypoxia for
24, 48 and 72 h compared with normoxic cells. At all tested time
points during hypoxia exposure, the MMP was significantly higher in
the HX + HSP70 group compared with the HX group, but lower compared
with the NX group. The MMP was markedly lower in the HX + HSP70 +
KNK437 group compared with that in the other two HX groups and the
NX group. Data are presented as the mean ± SD from at least three
separate experiments, and were analyzed by one-way ANOVA followed
by Bonferroni's post hoc test. *P<0.05, **P<0.01 and
***P<0.001. HSP, heat shock protein; MMP, mitochondrial membrane
potential; PMVECs, pulmonary microvascular endothelial cells; HX,
hypoxia; NX, normoxia; KNK437,
N-formyl-3,4-methylenedioxy-benzylidene-g-butyrolactam; PE,
phycoerythrin. |
RT-qPCR and western blotting were performed to
assess the expression levels of the antiapoptotic factor Bcl-2 and
the proapoptotic factors cyt C and caspase-3. The results
demonstrated that PMVECs exposed to hypoxia exhibited markedly
decreased protein and mRNA expression levels of Bcl-2, and
increased protein and mRNA expression levels of cyt C and caspase-3
compared with those in the NX group, which was significant for the
HX and HX + HSP70 groups (Fig. 5).
However, overexpression of HSP70 partly recovered the changes in
the mRNA and protein expression levels of Bcl-2, cyt C and
caspase-3 in PMVECs exposed to hypoxia for 24, 48 and 72 h.
Furthermore, treatment with KNK437 significantly decreased the
expression levels of Bcl-2, and significantly increased the protein
and mRNA expression levels of cyt C and caspase-3 in PMVECs
compared with those in the HX + HSP70 group. The observed effects
of HSP70 on the MMP, as well as Bcl-2, cyt C and caspase-3
expression levels suggested that HSP70 exerted its effects on
apoptosis via the mitochondrial pathway.
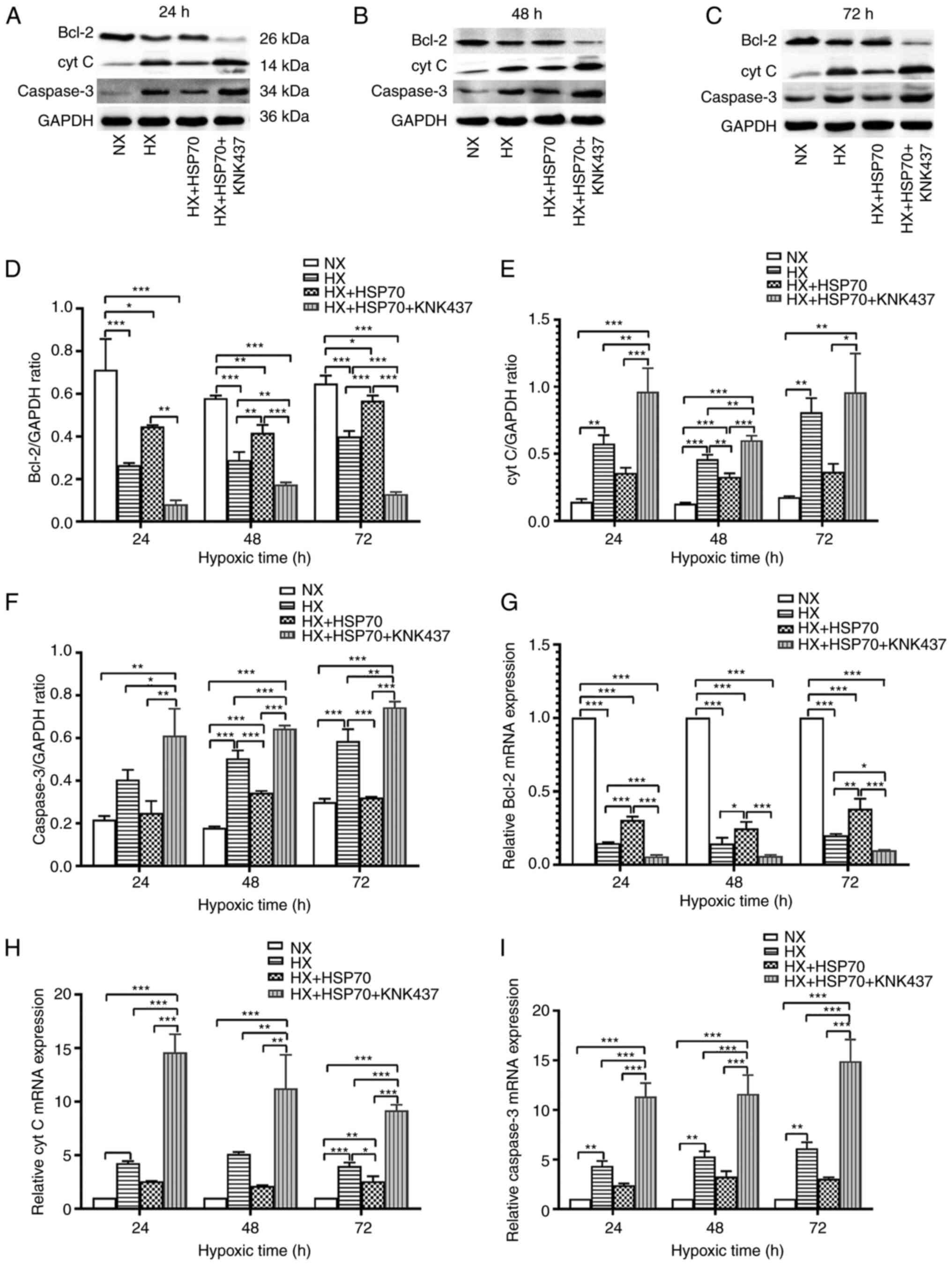 | Figure 5.HSP70 regulates the protein
expression levels of molecules involved in mitochondrial
pathway-mediated apoptosis. At 24, 48 and 72 h of hypoxia, (A-F)
protein and (G-I) mRNA expression levels of the apoptotic inhibitor
Bcl-2 and the proapoptotic factors cyt C and caspase-3 were
detected. Data are presented as the mean ± SD from at least three
separate experiments, and were analyzed by one-way ANOVA followed
by Bonferroni's post hoc test. *P<0.05, **P<0.01 and
***P<0.001. HSP, heat shock protein; cyt C, cytochrome c;
HX, hypoxia; NX, normoxia; KNK437,
N-formyl-3,4-methylenedioxy-benzylidene-g-butyrolactam. |
HSP70 downregulates HIF-1α in
hypoxia-exposed PMVECs
The mRNA and protein expression levels of HIF-1α
were assessed by performing RT-qPCR and western blotting,
respectively, and the results were consistent with the
aforementioned changes in apoptotic rates. HIF-1α mRNA and protein
expression levels were significantly upregulated in the HX groups
compared with those in the NX groups, and HSP70 overexpression
partly alleviated this increase (Fig.
6). Additionally, HIF-1α mRNA and protein expression levels
were significantly increased in the HX + HSP70 + KNK437 group
compared with those in the HX + HSP70 group, which was consistent
with the effects of HSP70 on PMVEC apoptosis. Therefore, the
protective mechanism of HSP70 against apoptosis may be associated
with HIF-1α expression levels.
Discussion
Endothelial cell apoptosis and dysfunction caused by
uncoordinated stimulation and uncontrolled cell responses are
implicated in the pathogenesis of various diseases, including PH,
atherosclerosis and sepsis (40).
Studies on PH have reported that a higher-than-average apoptotic
rate of PMVECs is the initial step in PH pathogenesis (7,41).
Neonatal PH is a unique form of the disease that results primarily
from hypoxia, leading to difficulties in decreasing pulmonary
vascular resistance, achieving continuous fetal circulation and
increasing pulmonary arterial pressure (2). To explore the role of PMVEC apoptosis
in the pathogenesis of PH, the present study investigated apoptotic
changes in the early stages of hypoxia in primary PMVECs derived
from neonatal SD rats. The results demonstrated that the apoptotic
rate of PMVECs exposed to hypoxic conditions for 24 h was
significantly higher compared with that in normoxic cells; the
apoptotic rate gradually increased with hypoxia duration. Following
72 h of hypoxia exposure, the apoptotic rate was >60%. These
results indicated that hypoxia-induced apoptosis of PMVECs in
neonatal rats gradually increased with prolonged exposure to
hypoxic conditions. The present study demonstrated that changes
observed in neonatal rat PMVECs in the context of PH were similar
to early changes in adults, where PMVECs undergo apoptosis in the
early stages of hypoxia (12). This
transition may be associated with the proliferation and repair of
endothelial cells following apoptosis, activation of STAT-3
promoting cell survival, and dysregulation of the expression of
proapoptotic and antiapoptotic factors, such as Bcl-2 and caspase
family members (42,43).
Our previous study revealed that HSP70 expression
was increased in the lung tissues of neonatal rats with HPH,
decreasing pulmonary arterial pressure and pulmonary vascular
remodeling, suggesting that HSP70 may exert a protective effect
against pulmonary vascular damage in this experimental system
(44). However, the mechanism by
which HSP70 alleviates pulmonary vascular damage remains unclear.
In the present study, PMVECs isolated from neonatal rats were
cultured in vitro and subjected to hypoxia, whereby PMVEC
apoptosis increased with prolonged hypoxia exposure. Furthermore,
HSP70 expression levels were significantly upregulated in hypoxic
PMVECs compared with those in normoxic conditions, indicating that
HSP70 may be involved in the apoptotic response of PMVECs to
hypoxia. Lentivirus-mediated overexpression of HSP70 in PMVECs
significantly decreased the apoptotic rate of the cells in the
early stages of hypoxia. On the other hand, KNK437-treated PMVECs
displayed significantly attenuated overexpression of exogenous
HSP70 and significant inhibition of the aforementioned effects of
HSP70 on hypoxia-induced PMVEC apoptosis. Although KNK437 did not
decrease the expression levels of HSP70 in HSP70-overexpression
PMVECs under hypoxic conditions compared with cells exposed to
hypoxia alone, it significantly increased the PMVEC apoptotic rate.
This outcome may be a result of the exacerbation of abnormal
synthesis and release of apoptotic inhibitors and proapoptotic
factors following KNK437-mediated downregulation of HSP70. The
changes detected in apoptosis-related factors also supported this
hypothesis. Moreover, these results suggested that HSP70 may serve
a protective role in the structure and function of PMVECs in
neonatal rats by inhibiting PMVEC apoptosis under hypoxia. This
role is similar to that of HSP70 in inhibiting apoptosis of tumor
cells (17), ischemic
cardiomyocytes (21) and infected
pulmonary vascular endothelial cells (22). Considering these findings and the
results of our previous in vivo study (44), it could be speculated that HSP70 may
reduce pulmonary vascular damage and delay the progression of
neonatal HPH by inhibiting PMVEC apoptosis in the early stages of
the disease.
The mitochondrial apoptotic pathway is important for
normal cell function; when cells encounter foreign stimuli, the
levels of the antiapoptotic protein Bcl-2 are downregulated,
whereas the proapoptotic proteins Bax and BAD are activated and
upregulated. Furthermore, the Bax/Bcl-2 ratio is increased, leading
to a decrease in the MMP, an increase in mitochondrial permeability
and the release of large amounts of cyt C from the mitochondria
(14,45). These changes activate the caspase
family of proteins and induce mitochondrial pathway-mediated
apoptosis. Researchers have reported that overexpression of HSP70
affects mitochondrial apoptotic pathway-mediated inhibition of
ovarian cancer cell apoptosis by increasing the MMP and interfering
with the release of cyt C and AIF, caspase activation and misfolded
protein accumulation (19,46). In addition, Kondrikov et al
(14) demonstrated that HSP70
alleviates hyperoxia-induced damage to the pulmonary endothelial
barrier by inhibiting PMVEC apoptosis via the mitochondrial caspase
and AIF pathway. In the present study, the associations between
HSP70, MMP, Bcl-2, cyt C and caspase-3 were analyzed to determine
the effects of HSP70 on mitochondrial pathway-mediated apoptosis of
PMVECs. The results indicated that hypoxia decreased the MMP (more
noticeably after 72 h of hypoxia) and Bcl-2 mRNA and protein
expression levels, but increased the mRNA and protein expression
levels of the proapoptotic proteins cyt C and caspase-3 compared
with those in the normoxia group, inferring that hypoxia induced
PMVEC apoptosis via the mitochondrial pathway. Under hypoxic
conditions, overexpression of HSP70 in PMVECs resulted in increases
in the MMP and Bcl-2 expression levels, and decreased expression of
cyt C and caspase-3 after 24, 48 and 72 h of hypoxia, whereas HSP70
inhibition exerted the opposite effects, suggesting that HSP70 may
inhibit PMVEC apoptosis via the mitochondrial pathway.
HIF-1α, a major factor in the regulation of cellular
oxygen homeostasis, is involved in the pathogenesis of cancer, PH,
ischemic disease and infections (26,47–49).
Our previous study has demonstrated that exogenous HSP70 reduces
pulmonary artery pressure, weakens pulmonary vascular remodeling
and blocks pulmonary vascular damage by inhibiting the expression
of HIF-1α in the lung tissues of neonatal rats with HPH (42). The present study further
investigated the involvement of HIF-1α in the process of
HSP70-mediated inhibition of apoptosis in neonatal rat PMVEC
cultures exposed to hypoxia. The results demonstrated that HIF-1α
mRNA and protein expression levels were significantly increased in
PMVECs exposed to hypoxia compared with those in cells maintained
under normoxic conditions. HIF-1α upregulation was accompanied by
an increased PMVEC apoptotic rate. In addition, in hypoxic
conditions, HIF-1α expression levels and apoptotic rate were
reduced in HSP70-overexpressing PMVECs; however, these factors were
significantly increased in PMVECs treated with an HSP70 inhibitor.
Therefore, it was hypothesized that HSP70 inhibited apoptosis in
PMVECs under hypoxia, which may be associated with HSP70-mediated
downregulation of HIF-1α. The association between HIF-1α and
apoptosis varies between different cell types; in cervical and
thyroid studies, HIF-1α has been reported to participate in the
mechanisms of tumor cells protection and chemotherapeutic
resistance through antiapoptotic properties and thus, HIF-1α may
serve as an effective therapeutic target for tumors (50,51).
In a recent study assessing myocardial damage caused by
hyperglycemia, HIF-1α expression in the rat myoblast cell line H9c2
was demonstrated to significantly increase following apoptotic
induction by high glucose (52). In
the same study, silencing HIF-1α with small interfering RNAs or
reducing HIF-1α expression levels using propofol significantly
attenuated apoptosis, indicating that HIF-1α may promote apoptosis
in H9c2 cells exposed to high concentrations of glucose (52). The results of the present study
suggested that HIF-1α may promote apoptosis in neonatal rat PMVECs
exposed to hypoxia, however, the exact underlying molecular
mechanism requires further investigation.
The present study had several limitations that
warrant further explanation and should be investigated in future
studies. Firstly, the impact of HSP70 on PMVEC function was
inferred but not confirmed, thus further research is required.
Secondly, the effects of HSP70 on the apoptosis of neonatal rat
PMVECs under hypoxia require further validation in vivo.
Lastly, the association between HIF-1α and PMVEC apoptosis should
be further elucidated.
In summary, the results of the present study
demonstrated that exposure of neonatal rat PMVECs to hypoxia in
vitro gradually elevated the apoptotic rate with increasing
exposure time. HSP70 and HIF-1α expression levels were also
upregulated under hypoxic conditions compared with cells maintained
under normoxic conditions. Overexpression of HSP70 attenuated PMVEC
apoptosis via the mitochondrial pathway under hypoxia, whereas its
inhibition increased the apoptotic rate. The mechanism by which
HSP70 regulates hypoxia-induced apoptosis in PMVECs isolated from
neonatal rats may involve HSP70-mediated downregulation of HIF-1α.
Based on these results, we hypothesize that HSP70 reduces pulmonary
vascular damage in neonatal HPH by inhibiting PMVEC apoptosis,
providing a novel target for the treatment of this disease.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81760278).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YM and ML designed and supervised the study, and
revised the manuscript. JC and LY performed the experiments and
drafted the manuscript. LW supervised the experiments, analyzed the
data and revised the manuscript critically for important
intellectual content. DW and QZ performed the experiments and
analyzed the data. JC and LW confirm the authenticity of all the
raw data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study followed internationally
recognized guidelines on animal welfare, as well as local and
national regulations, in accordance with the UK Animals (Scientific
Procedures) Act and associated guidelines (53), the EU Directive 2010/63/EU for
animal experiments (54) and the
National Institutes of Health guide for the care and use of
laboratory animals (55). The study
complied with the Animal Research: Reporting of in vivo
Experiments guidelines (56) and
the American Veterinary Medical Association euthanasia guidelines
2013 (57). All animal experiments
were approved by the Animal Ethics Committee of the First
Affiliated Hospital of Xinjiang Medical University (Ürümqi, China;
approval no. IACUC-20170214029).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PMVEC
|
pulmonary microvascular endothelial
cell
|
|
PH
|
pulmonary hypertension
|
|
HPH
|
hypoxic pulmonary hypertension
|
|
MMP
|
mitochondrial membrane potential
|
|
HSP
|
heat shock protein
|
|
AIF
|
apoptosis-inducing factor
|
|
HIF-1
|
hypoxia-inducible factor-1
|
|
cyt C
|
cytochrome c
|
References
|
1
|
Walsh-Sukys MC, Tyson JE, Wright LL, Bauer
CR, Korones SB, Stevenson DK, Verter J, Stoll BJ, Lemons JA, Papile
LA, et al: Persistent pulmonary hypertension of the newborn in the
era before nitric oxide: Practice variation and outcomes.
Pediatrics. 105((1 Pt 1)): 14–20. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Distefano G and Sciacca P: Molecular
physiopathogenetic mechanisms and development of new potential
therapeutic strategies in persistent pulmonary hypertension of the
newborn. Ital J Pediatr. 41:62015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Du Y, Fu J, Yao L, Qiao L, Liu N, Xing Y
and Xue X: Altered expression of PPAR-ү and TRPC in neonatal rats
with persistent pulmonary hypertension. Mol Med Rep. 16:1117–1124.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dabral S, Tian X, Kojonazarov B, Savai R,
Ghofrani HA, Weissmann N, Florio M, Sun J, Jonigk D, Maegel L, et
al: Notch1 signalling regulates endothelial proliferation and
apoptosis in pulmonary arterial hypertension. Eur Respir J.
48:1137–1149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hudalla H, Michael Z, Christodoulou N,
Willis GR, Fernandez-Gonzalez A, Filatava EJ, Dieffenbach P,
Fredenburgh LE, Stearman RS, Geraci MW, et al: Carbonic anhydrase
inhibition ameliorates inflammation and experimental pulmonary
hypertension. Am J Respir Cell Mol Biol. 61:512–524. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Majka S, Hagen M, Blackwell T, Harral J,
Johnson JA, Gendron R, Paradis H, Crona D, Loyd JE, Nozik-Grayck E,
et al: Physiologic and molecular consequences of endothelial Bmpr2
mutation. Respir Res. 12:842011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sakao S, Taraseviciene-Stewart L, Lee JD,
Wood K, Cool CD and Voelkel NF: Initial apoptosis is followed by
increased proliferation of apoptosis-resistant endothelial cells.
FASEB J. 19:1178–1180. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rhodes CJ, Im H, Cao A, Hennigs JK, Wang
L, Sa S, Chen PI, Nickel NP, Miyagawa K, Hopper RK, et al: RNA
sequencing analysis detection of a novel pathway of endothelial
dysfunction in pulmonary arterial hypertension. Am J Respir Crit
Care Med. 192:356–366. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singh N, Singh H, Jagavelu K, Wahajuddin M
and Hanif K: Fatty acid synthase modulates proliferation, metabolic
functions and angiogenesis in hypoxic pulmonary artery endothelial
cells. Eur J Pharmacol. 815:462–469. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guerard P, Rakotoniaina Z, Goirand F,
Rochette L, Dumas M, Lirussi F and Bardou M: The HMG-CoA reductase
inhibitor, pravastatin, prevents the development of
monocrotaline-induced pulmonary hypertension in the rat through
reduction of endothelial cell apoptosis and overexpression of eNOS.
Naunyn Schmiedebergs Arch Pharmacol. 373:401–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yeager ME, Halley GR, Golpon HA, Voelkel
NF and Tuder RM: Microsatellite instability of endothelial cell
growth and apoptosis genes within plexiform lesions in primary
pulmonary hypertension. Circ Res. 88:E2–E11. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Masri FA, Xu W, Comhair SA, Asosingh K,
Koo M, Vasanji A, Drazba J, Anand-Apte B and Erzurum SC:
Hyperproliferative apoptosis-resistant endothelial cells in
idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell
Mol Physiol. 293:L548–L554. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Schultz A, Olorundami OA, Teng RJ,
Jarzembowski J, Shi ZZ, Kumar SN, Pritchard K Jr, Konduri GG and
Afolayan AJ: Decreased OLA1 (Obg-Like ATPase-1) expression drives
ubiquitin-proteasome pathways to downregulate mitochondrial SOD2
(Superoxide Dismutase) in persistent pulmonary hypertension of the
newborn. Hypertension. 74:957–966. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kondrikov D, Fulton D, Dong Z and Su Y:
Heat shock protein 70 prevents hyperoxia-induced disruption of lung
endothelial barrier via caspase-dependent and AIF-dependent
pathways. PLos One. 10:e01293432015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li X, Kanegasaki S, Jin F, Deng Y, Kim JR,
Chang HW and Tsuchiya T: Simultaneous induction of HSP70
expression, and degranulation, in IgE/Ag-stimulated or
extracellular HSP70-stimulated mast cells. Allergy. 73:361–368.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Aghdassi A, Phillips P, Dudeja V,
Dhaulakhandi D, Sharif R, Dawra R, Lerch MM and Saluja A: Heat
shock protein 70 increases tumorigenicity and inhibits apoptosis in
pancreatic adenocarcinoma. Cancer Res. 67:616–625. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kumar S, Stokes J 3rd, Singh UP, Scissum
Gunn K, Acharya A, Manne U and Mishra M: Targeting Hsp70: A
possible therapy for cancer. Cancer Lett. 374:156–166. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van Noort JM, Bugiani M and Amor S: Heat
shock proteins: Old and novel roles in neurodegenerative diseases
in the central nervous system. CNS Neurol Disord Drug Targets.
16:244–256. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Garrido C, Brunet M, Didelot C, Zermati Y,
Schmitt E and Kroemer G: Heat shock proteins 27 and 70:
Anti-apoptotic proteins with tumorigenic properties. Cell Cycle.
5:2592–2601. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ciocca DR and Calderwood SK: Heat shock
proteins in cancer: Diagnostic, prognostic, predictive, and
treatment implications. Cell Stress Chaperones. 10:86–103. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ranek MJ, Stachowski MJ, Kirk JA and
Willis MS: The role of heat shock proteins and co-chaperones in
heart failure. Philos Trans R Soc Lond B Biol Sci.
373:201605302018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Yu Y, Gorshkov B, Haigh S, Bordan Z,
Weintraub D, Rudic RD, Chakraborty T, Barman SA, Verin AD, et al:
Hsp70 suppresses mitochondrial reactive oxygen species and
preserves pulmonary microvascular barrier integrity following
exposure to bacterial toxins. Front Immunol. 9:13092018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Semenza GL: The genomics and genetics of
oxygen homeostasis. Annu Rev Genomics Hum Genet. 21:183–204. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Zhou Y, Li M and Zhu Y: Expression
of hypoxia-inducible factor-1α, endothelin-1 and adrenomedullin in
newborn rats with hypoxia-induced pulmonary hypertension. Exp Ther
Med. 8:335–339. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Veith C, Schermuly RT, Brandes RP and
Weissmann N: Molecular mechanisms of hypoxia-inducible
factor-induced pulmonary arterial smooth muscle cell alterations in
pulmonary hypertension. J Physiol. 594:1167–1177. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen S and Sang N: Hypoxia-inducible
factor-1: A critical player in the survival strategy of stressed
cells. J Cell Biochem. 117:267–278. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao L, Ma R, Zhang L, Yuan X, Wu J, He L,
Liu G and Du R: Inhibition of HIF-1a-mediated TLR4 activation
decreases apoptosis and promotes angiogenesis of placental
microvascular endothelial cells during severe pre-eclampsia
pathogenesis. Placenta. 83:8–16. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xie RY, Fang XL, Zheng XB, Lv WZ, Li YJ,
Ibrahim Rage H, He QL, Zhu WP and Cui TX: Salidroside and FG-4592
ameliorate high glucose-induced glomerular endothelial cells injury
via HIF upregulation. Biomed Pharmacother. 118:1091752019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Leong KH, Zhou LL, Lin QM, Wang P, Yao L
and Huang ZJ: Therapeutic effects of various methods of MSC
transplantation on cerebral resuscitation following cardiac arrest
in rats. Mol Med Rep. 13:3043–3051. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu M, Liu Q, Pei Y, Gong M, Cui X, Pan J,
Zhang Y, Liu Y, Liu Y, Yuan X, et al: Aqp-1 gene knockout
attenuates hypoxic pulmonary hypertension of mice. Arterioscler
Thromb Vasc Biol. 39:48–62. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang YD, Li MM, Xu G, Zhang EL, Chen J,
Sun B, Chen DW and Gao YQ: Targeting mitochondria-associated
membranes as a potential therapy against endothelial injury induced
by hypoxia. J Cell Biochem. 120:18967–18978. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Koishi M, Yokota S, Mae T, Nishimura Y,
Kanamori S, Horii N, Shibuya K, Sasai K and Hiraoka M: The effects
of KNK437, a novel inhibitor of heat shock protein synthesis, on
the acquisition of thermotolerance in a murine transplantable tumor
in vivo. Clin Cancer Res. 7:215–219. 2001.PubMed/NCBI
|
|
33
|
Shiota M, Kusakabe H, Izumi Y, Hikita Y,
Nakao T, Funae Y, Miura K and Iwao H: Heat shock cognate protein 70
is essential for Akt signaling in endothelial function.
Arterioscler Thromb Vasc Biol. 30:491–497. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bozaykut P, Sozen E, Kaga E, Ece A,
Ozaltin E, Bergquist J, Kartal Ozer N and Karademir Yilmaz B: HSP70
inhibition leads to the activation of proteasomal system under mild
hyperthermia conditions in young and senescent fibroblasts. Oxid
Med Cell Longev. 2020:93695242020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Chung JW, Piao ZH, Yoon SR, Kim MS, Jeong
M, Lee SH, Min JK, Kim JW, Cho YH, Kim JC, et al: Pseudomonas
aeruginosa eliminates natural killer cells via phagocytosis-induced
apoptosis. PLoS Pathog. 5:e10005612009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang H, Liu Y, Wang L, Li Z, Zhang H, Wu
J, Rahman N, Guo Y, Li D, Li N, et al: Differential effects of
estrogen/androgen on the prevention of nonalcoholic fatty liver
disease in the male rat. J Lipid Res. 54:345–357. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu YL, Li ZL, Zhang XB and Liu H:
Yinchenhao decoction attenuates obstructive jaundice-induced liver
injury and hepatocyte apoptosis by suppressing protein kinase
RNA-like endoplasmic reticulum kinase-induced pathway. World J
Gastroenterol. 25:6205–6221. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Du ZD, Wei W, Yu S, Song QL, Liu K and
Gong SS: NADPH oxidase 2-mediated insult in the auditory cortex of
zucker diabetic fatty rats. Neural Plast. 2019:35916052019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Sakao S, Tatsumi K and Voelkel NF:
Endothelial cells and pulmonary arterial hypertension: Apoptosis,
proliferation, interaction and transdifferentiation. Respir Res.
10:952009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Teichert-Kuliszewska K, Kutryk MJ,
Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, Granton J and
Stewart DJ: Bone morphogenetic protein receptor-2 signaling
promotes pulmonary arterial endothelial cell survival: Implications
for loss-of-function mutations in the pathogenesis of pulmonary
hypertension. Circ Res. 98:209–217. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Pullamsetti SS, Savai R, Seeger W and
Goncharova EA: Translational advances in the field of pulmonary
hypertension. From cancer biology to new pulmonary arterial
hypertension therapeutics. Targeting cell growth and proliferation
signaling hubs. Am J Respir Crit Care Med. 195:425–437. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Marshall JD, Bazan I, Zhang Y, Fares WH
and Lee PJ: Mitochondrial dysfunction and pulmonary hypertension:
Cause, effect, or both. Am J Physiol Lung Cell Mol Physiol.
314:L782–L796. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang L and Li MX: Roles of heat shock
protein 70 toward hypoxia-inducible factor 1α (HIF-1α) blockade in
newborn rats with hypoxia-induced pulmonary hypertension. Int J
Clin Exp Med. 11:13520–13527. 2018.
|
|
45
|
Pena-Blanco A and Garcia-Saez AJ: Bax, Bak
and beyond-mitochondrial performance in apoptosis. FEBS J.
285:416–431. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Hoter A and Naim HY: Heat shock proteins
and ovarian cancer: Important roles and therapeutic opportunities.
Cancers (Basel). 11:13892019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dabral S, Muecke C, Valasarajan C,
Schmoranzer M, Wietelmann A, Semenza GL, Meister M, Muley T,
Seeger-Nukpezah T, Samakovlis C, et al: A RASSF1A-HIF1α loop drives
Warburg effect in cancer and pulmonary hypertension. Nat Commun.
10:21302019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Devraj G, Beerlage C, Brune B and Kempf
VA: Hypoxia and HIF-1 activation in bacterial infections. Microbes
Infect. 19:144–156. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ham PB 3rd and Raju R: Mitochondrial
function in hypoxic ischemic injury and influence of aging. Prog
Neurobiol. 157:92–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Mylonis I, Kourti M, Samiotaki M,
Panayotou G and Simos G: Mortalin-mediated and ERK-controlled
targeting of HIF-1α to mitochondria confers resistance to apoptosis
under hypoxia. J Cell Sci. 130:466–479. 2017.PubMed/NCBI
|
|
51
|
Zhou L, Cha G, Chen L, Yang C, Xu D and Ge
M: HIF1α/PD-L1 axis mediates hypoxia-induced cell apoptosis and
tumor progression in follicular thyroid carcinoma. Onco Targets
Ther. 12:6461–6470. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Pu J, Zhu S, Zhou D, Zhao L, Yin M, Wang Z
and Hong J: Propofol alleviates apoptosis induced by chronic high
glucose exposure via regulation of HIF-1α in H9c2 cells. Oxid Med
Cell Longev. 2019:48240352019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hollands C: The animals (Scientific
Procedures) ACT 1986. Lancet. 2:32–33. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
European Parliament: Directive 2010/63/EU
of the European Parliament and of the Council of 22 September 2010
on the protection of animals used for scientific purposes. Official
J Eur Union. 276:33–79. 2010.
|
|
55
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals, . Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington, DC: 2011
|
|
56
|
McGrath JC, Drummond GB, McLachlan EM,
Kilkenny C and Wainwright CL: Guidelines for reporting experiments
involving animals: The ARRIVE guidelines. Br J Pharmacol.
160:1573–1576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
American Veterinary Medical Association
(AVMA), . AVMA Guidelines for the Euthanasia of Animals: 2013
Edition. AVMA; Schaumburg, IL: pp. 67–73. 2013
|