Introduction
Hydrogen sulfide (H2S) was initially
considered a toxic gas; however, with the continuation of research,
it has been revealed to serve an important role in living
organisms, becoming another important gas transmitter, alongside
carbon monoxide (CO) and nitric oxide (NO) (1,2). Since
H2S has been confirmed to be present in mammalian
tissues, a large number of studies have suggested that
H2S can exert anti-inflammatory, anti-oxidative stress
and anti-fibrotic effects in the body (3,4).
Previous studies have confirmed that H2S serves a
physiological and pathological role in the cardiovascular system,
brain and nervous system (5–7).
However, due to the uneven distribution of
H2S-generating enzymes in various organs and tissues,
the concentration of H2S differs widely in different
organs (8). The study of the
underlying mechanisms of H2S in physiological and
pathological processes in the kidney may assist in systematically
understanding its molecular biological mechanisms, particularly
with regards to it renoprotective role.
General physicochemical properties of
H2S
H2S is a colorless gas that smells
similar to rotten eggs; the smell of H2S can be picked
up by the human olfactory system when the concentration in the air
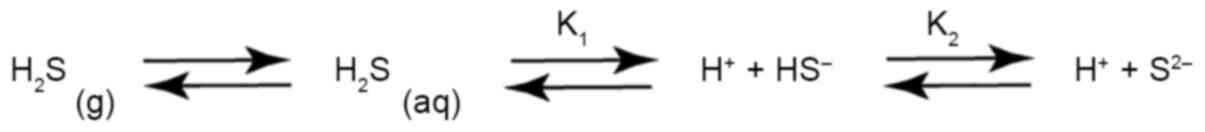
reaches 1/400 of its toxic level (9). As a weak acid, H2S
dissociates in water to reach equilibrium at room temperature
(25°C) with a pKa1 of 6.97–7.06 and pKa2 of
12.35–15.0. Moreover, H2S in aqueous solution is
volatile, and its mutual conversion between the liquid phase and
the gas phase reaches equilibrium, as shown in Fig. 1; this balance is affected by ambient
temperature, pressure and other solutes in the aqueous solution
(10). In addition, H2S
is highly lipophilic, which not only allows it to have a higher
concentration under fat-abundant conditions, but also allows it to
freely penetrate lipid biofilms without relying on membrane
channels to exert its biological activity (11). Since H2S and
HS− coexist in solution, it is difficult to make a clear
distinction between which of them has a role in biological
mechanisms or whether they both have biological effects.
Generation and metabolism of
H2S
Generation of H2S
The synthesis of H2S in mammals primarily
relies on enzymatic pathways. Three traditional enzyme systems that
catalyze H2S production include the synergistic action
of cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and
cysteine transaminase (CAT) with 3-mercaptopyruvate (3-MP)
sulphotransferase (3-MST) (12,13).
With pyridoxal phosphate (also known as vitamin B6) as a cofactor,
CSE and CBS are responsible for the majority of endogenous
H2S generated, as shown in Fig. 2. L-cysteine is catalyzed by CSE or
CBS to produce H2S and L-serine, or by CBS to produce
pyruvate, NH3 and H2S. CSE can polymerize two
L-cysteine residues into L-cystine, and then CSE uses L-cystine as
a substrate to decompose it into thiocysteine, pyruvate and
NH3. The generated thiocysteine reacts with other thiols
to generate H2S through a nonenzymatic reaction. In
addition, L-cysteine polymerizes with L-homocysteine as substrates
for CSE or CBS to produce L-cystathionine and H2S.
L-cystathionine is further decomposed by CSE into L-cysteine,
α-ketobutyrate and NH3, and L-cysteine circulation is
achieved (12,13). It has been reported that in the
reaction in which L-cysteine is metabolized to H2S via
CBS, the amount of H2S produced by β-replacement is 50X
that of β-elimination (14). During
the production of H2S by CSE, the α, β-elimination of
cysteine is the primary source of H2S, accounting for
70% of H2S production (15).
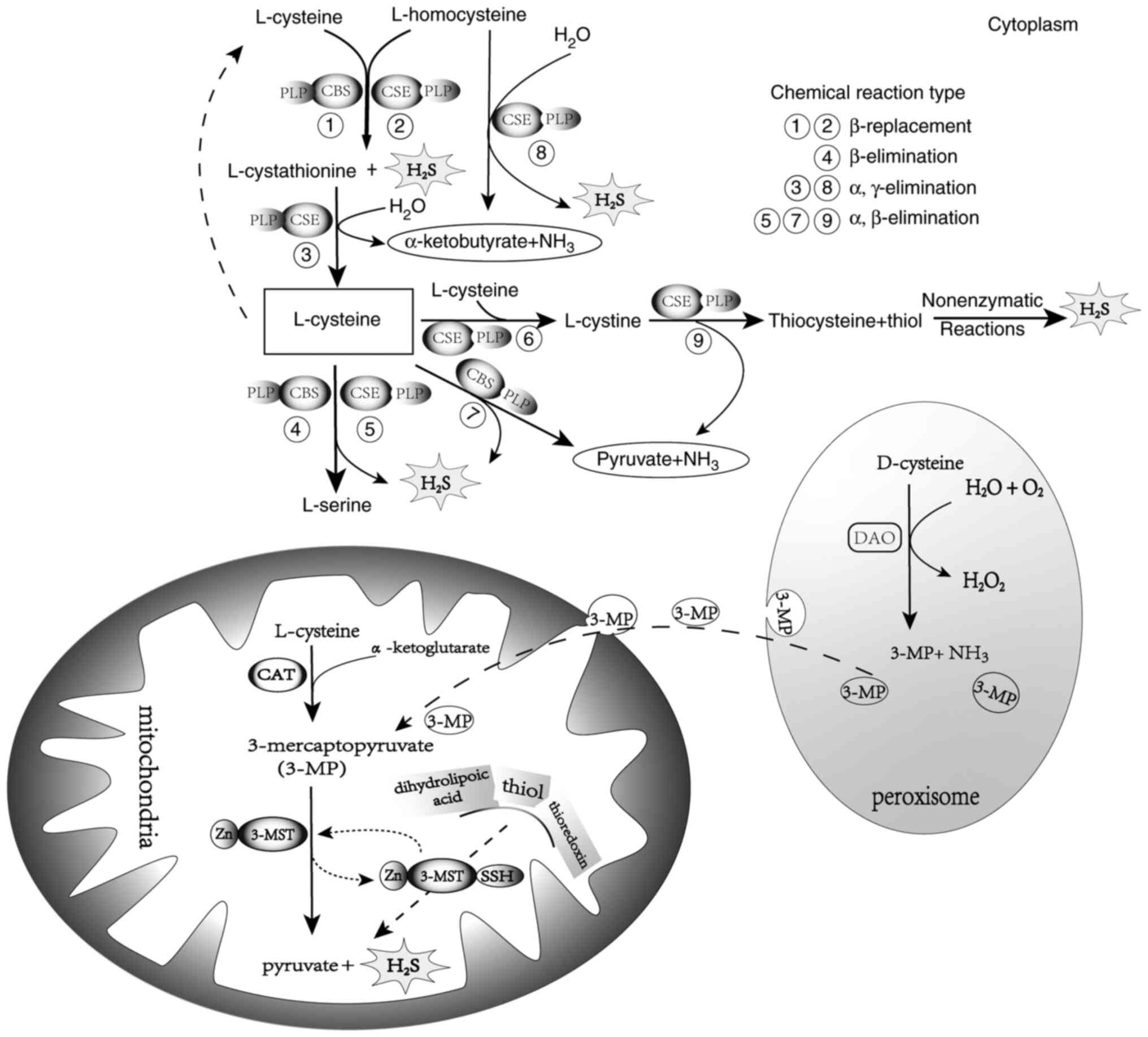 | Figure 2.In the cytoplasm, 1–3: CSE or CBS
catalyzes the β-replacement reaction of L-cysteine and
L-homocysteine to polymerize and form L-cystathionine and
H2S. L-cystathionine is decomposed by CSE into
L-cysteine, α-ketobutyrate and NH3 by means of α,
γ-elimination. L-cysteine continues to participate in the reaction.
4 and 5: L-cysteine is catalyzed to produce L-serine and
H2S via CBS β-elimination or CSE α, β-elimination. 7:
CBS catalyzes L-cysteine to produce pyruvate, NH3 and
H2S through α, β-elimination. 6 and 9: CSE first
polymerizes two L-cysteines into L-cystine, then CSE uses L-cystine
as a substrate to decompose it into thiocysteine (mercaptocysteine,
Cyc-SSH), pyruvate and NH3, resulting in thiocysteine
generating H2S via nonenzymatic reactions with other
thiols. 8: L-homocysteine generates α-ketobutyrate, NH3
and H2S through CSE α, γ-elimination. In the
mitochondria, CAT catalyzes L-cysteine and α-ketoglutarate to
produce 3-MP, which is then catalyzed by 3-MST to produce pyruvate
and H2S. In peroxisomes, 3-MP produced by DAO and
catalyzed by D-cysteine is transported to the mitochondria in
vesicles. H2S, hydrogen sulfide; CBS,
cystathionine-β-synthase; CSE, cystathionine-γ-lyase; 3-MP,
3-mercaptopyruvate; CAT, cysteine transaminase; 3-MST, 3-MP
sulphotransferase; DAO, D-amino acid oxidase. |
Unlike CSE and CBS, 3-MST uses metallic zinc as a
cofactor (14). Moreover,
L-cysteine must be converted into 3-MP and L-glutamic acid through
the reaction of CAT with α-ketoglutarate, and 3-MP is then
desulfurized by 3-MST as a direct substrate to produce
H2S and pyruvate (16,17).
In peroxisomes, D-amino acid oxidase catalyzes D-cysteine, instead
of L-cysteine, to produce 3-MP, NH3 and
H2O2 in the presence of water and oxygen, and
the resulting 3-MP is transferred to mitochondria for 3-MST
utilization to generate H2S (18). The entry of 3-MP in peroxidase into
mitochondria is generally in the form of vesicles, as shown in
Fig. 2. Clinical observations have
reported that the synthesis of CSE and CBS in patients with chronic
kidney disease is reduced, whereas the expression of 3-MST and
hemorrhagic homocysteine is increased (19). This may be explained by the specific
mechanism of action used by the aforementioned enzymes to generate
H2S. When the production of H2S by CBS and
CSE via the L-homocysteine/L-cystathionine pathway is reduced, the
utilization of L-homocysteine is restricted, and the patient may
present with hyperhomocysteinemia.
Metabolism of H2S
H2S in the body is primarily metabolized
by mitochondria (20). Sulfoquinone
oxidoreductase (SQOR) in the mitochondria can utilize
H2S and metabolize it into thiosulfate with the
assistance of thiosulfate sulfur transferase (TST) and
thiodioxygenase (ETHE1). During this process, reduced glutathione
serves an important role, and thiosulfate is further oxidized under
the action of thiosulfate reductase and sulfite oxidase (SUOX), and
finally excreted in the form of sulfate through the kidneys, as
shown in Fig. 3. The role of
O2 in this process is irreplaceable (21,22).
Notably, coenzyme Q (CoQ) is closely related to the aforementioned
enzymes. A previous study revealed that the absence of CoQ may
induce downregulation of the expression levels of thioquinone
oxidoreductase, TST, ETHE1 and SUOX (23). During the early stages of CoQ
deficiency, SQOR levels are significantly decreased, affecting
H2S oxidation, and CoQ supplementation can save
H2S metabolism without affecting its production
(24). While SQOR activity and
protein levels decrease, protein levels of the other mitochondrial
enzymes (TST, ETHE1 and SUOX) in the H2S oxidation
pathway increase in fibroblasts; however, it is not clear whether
the increase in the levels of several enzymes is a temporary
increase in compensation or inversely proportional to the decrease
in SQOR levels (23). Therefore, it
is important to explore the effect of CoQ deficiency on
H2S metabolic enzymes, which may assist in studying the
regulation of H2S concentration through H2S
metabolic pathways to affect several signaling pathways in the
body.
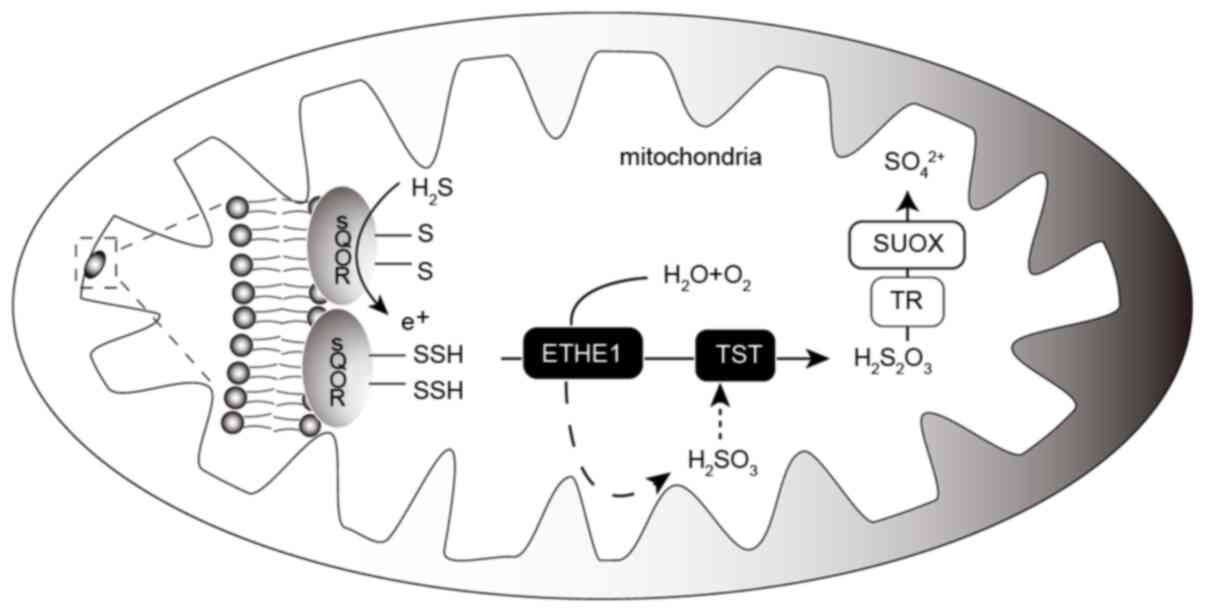 | Figure 3.Oxidative metabolism of
H2S in the mitochondria. H2S in the
mitochondria is activated by SQOR, which receives an-SH group to
form an-SSH group. In the presence of O2 and
H2O, -SSH is used by ETHE1 to generate
H2SO3, which is further converted into
thiosulfate by TST using the-SSH group. Finally, thiosulfate is
oxidized by TR and SUOX, and is eventually excreted in the kidney
as sulfate. H2S, hydrogen sulfide; SQOR, sulfoquinone
oxidoreductase; ETHE1, thiodioxygenase; TST, thiosulfate sulfur
transferase; TR, thiosulfate reductase; SUOX, sulfite oxidase. |
Under normal physiological conditions, when
H2S production in tissues exceeds utilization
metabolism, another metabolic pathway, cytoplasmic
methyltransferase methylation, is required. To date, the known
methyltransferases in the human body are thiopurine
methyltransferase (TPMT) and thiol methyltransferase (TMT). TPMT
selectively methylates thiopurine compounds, whereas TMT
selectively methylates aliphatic mercaptan substrates. Using mass
spectrometry to directly measure the formation of methyl sulfide,
the methylation of H2S and the obtained kinetic curves
have previously been assessed; the Km of methylation of
H2S was 146.2±29.2 µmol (25). It has also been demonstrated that
human methyltransferase-like protein 7B can catalyze the transfer
of a methyl group from S-adenosine 1-methionine to H2S
and other exogenous mercaptan small molecules, thereby metabolizing
H2S (25). In addition,
H2S can be removed by methemoglobin or
metallic/nonmetallic molecules, such as oxidized glutathione
(26).
Physiological role of H2S in the
kidney
Renal excretory function
Clinical studies have confirmed that plasma
H2S levels are positively correlated with glomerular
filtration rate in patients with chronic kidney disease (CKD). In
addition, serum homocysteine content in patients with advanced CKD
(CKD3-5) has been reported to be significantly higher than that in
patients with early CKD (CKD1-2), and increases in serum
homocysteine levels are associated with decreased renal function
(19). Hyperhomocysteinemia has
been shown to aggravate the deposition of extracellular matrix
(ECM) proteins and the destruction of connexin, and lead to the
phosphorylation of endothelial NO synthase (eNOS) in renal vascular
endothelial cells, thereby reducing the bioavailability of NO to
induce vasoconstriction and decrease renal blood flow, which is
manifested by a decrease in plasma H2S levels and
glomerular filtration rate (GFR) (27). H2S can increase urinary
sodium and potassium excretion by inhibiting Na-K-2Cl
co-transporters and Na-K-ATPase. In vivo experiments have
shown that intra-renal artery infusion of the H2S donor
NaHS may increase renal blood flow, GFR and excretion of urinary
sodium [U (Na) × volume] and potassium [U (K) × volume), and the
infusion of L-cysteine via the renal artery to increase the
concentration of H2S substrate could simulate this
effect (28). In addition,
H2S may block the opening of phosphatidylinositol
3,4,5-triphosphate-dependent distal renal epithelial sodium
channels induced by H2O2, reduce the
reabsorption of sodium by nephrons and increase urinary sodium
excretion (29). In addition, the
use of CSE and CBS enzyme inhibitors propargylglycine and
amino-oxoacetate has been shown to increase urine volume and
decrease urine osmotic pressure in mice; this is related to the
H2S-induced decrease in the expression of aquaporin
(AQP)-2 in the renal medulla. Following treatment with GYY4137, a
H2S donor sustained release agent, expression levels of
AQP-2 were significantly upregulated (30).
H2S can directly target some
H2S-sensitive disulfide bonds in the epidermal growth
factor receptor (EGFR), which can induce endocytosis and inhibition
of Na-K-ATPase in renal tubular epithelial cells by regulating the
EGFR/GAB1/PI3K/Akt pathway, thus reducing sodium and potassium ion
exchange of renal tubular epithelial cells, and promoting sodium
excretion (31). However, how the
EGFR/GAB1/PI3K/Akt pathway acts on Na-K-ATPase remains to be
determined. EGFR is known to possess tyrosine kinase activity, and
its family members can bind to a variety of ligands to form
homodimers or heterodimers, leading to the phosphorylation of
specific tyrosine residues in intracellular domains. In renal
vascular endothelial cells, inhibition of EGFR has been reported to
dilate renal vessels and improve renal blood flow; in podocytes,
inhibition of EGFR may reduce podocyte damage and loss induced by
high glucose levels, and reduce proteinuria, whereas in renal
tubular epithelial cells, inhibition of EGFR was shown to alleviate
renal tubular injury and epithelial-mesenchymal transition (EMT)
(32,33). However, studies on inhibitors of
EGFR tyrosine kinase activity have shown that inhibition of EGFR
can also lead to renal tubular damage and electrolyte disturbance
(34). Therefore, more in-depth
studies are required, particularly with regard to the advantages
and disadvantages of H2S in regulating EGFR pathway
activity.
Thus, these aforementioned previous studies
indicated that H2S has a role in the metabolism of water
and electrolytes via a variety of methods. In general, it has been
suggested that the increased concentration of H2S is
conducive to regulating the excretion of electrolytes by the
kidney, whereas the inhibition of its production can preserve
sodium drainage. Therefore, H2S-generating enzyme CBS
and CSE inhibitors may be potential diuretics.
Oxygen sensing
H2S-mediated O2 sensing has
been detected in various O2-sensing tissues in the
cardiovascular and respiratory systems of vertebrates (35,36).
The effect of H2S on downstream signaling events is
consistent with that of hypoxia activation (37,38).
In normal kidneys, due to the intrarenal arteriovenous oxygen
shunt, the kidney is in a state of low oxygen partial pressure
compared with other organs, and the renal medulla oxygen partial
pressure is lower than that of the renal parenchyma (39,40).
Therefore, H2S is regarded as an oxygen sensor in the
kidney, particularly in the medulla (41). As an oxygen sensor, H2S
is inseparable from its generation and oxidative metabolic balance.
H2S generation is not dependent on O2, but
its oxidative metabolism in mitochondria is dependent on oxygen, as
aforementioned; therefore, hypoxia can lead to an increase in
H2S concentration and an inverse relationship exists
between the two (37). The
mitochondrial oxidative respiratory electron transport chain is the
primary means of energy generation; thus it is necessary and
significant to prove that H2S participates in energy
generation under physiological conditions in the renal medulla
under normal hypoxia. As an oxygen sensor, H2S can
affect the blood flow supply and regulate the oxygen balance in the
heart and lungs. Whether H2S also regulates the
distribution of oxygen supply in the renal cortex and medulla under
physiological conditions through this mechanism or via other means
remains to be determined. Investigating the location and molecular
mechanism of H2S as an oxygen sensor affecting the
occurrence of downstream signaling events will further enrich our
understanding of H2S as an oxygen sensor.
Role of H2S in renal disease
Renal injury
Our previous study revealed that the expression
levels of CBS and CSE, two enzymes that produce H2S,
were decreased in renal tissues following urinary tract obstruction
(42). In vivo studies also
demonstrated that supplementation with an H2S donor, to
provide sufficient H2S, improved renal injury (42); the mechanisms and molecular pathways
involved are relevant to the disease model studied. Kidney injury
can be divided into two categories: Acute kidney injury (AKI) and
CKD. AKI may occur as a result of ischemia-reperfusion (hemorrhagic
or septic shock) or after exposure to toxic substances (such as
iodized contrast agents, aminoglycosides and cisplatin). CKD occurs
in glomerular and tubular interstitial lesions, such as diabetic
nephropathy (DN) and hypertensive nephropathy, amongst other causes
(43).
Ischemia-reperfusion injury (IRI)
In the process of kidney transplantation, the
temporary cessation of renal blood flow leads to acute ischemic
injury, and reperfusion further enhances the functional and
structural damage to human kidneys, namely renal
ischemia-reperfusion injury (IRI). Animal experiments have shown
that following renal ischemia-reperfusion, serum and tissues
exhibit markedly increased levels of IL and tumor necrosis factor-α
(TNF-α), alongside other inflammatory indicators, significantly
elevated malondialdehyde (MDA) concentrations, significantly
reduced superoxide dismutase (SOD) activity and renal tubular
necrosis; conversely, the H2S donor Na2S has
been shown to significantly reduce inflammation, oxidative stress
and kidney damage, as shown in Fig.
4 (44). Increased levels of
MDA and reduced activity of SOD have been shown to promote lipid
peroxidation and upregulate nuclear factor-κB (NF-κB), IL-2 and
Toll-like receptor-4 (TLR-4), which can stimulate an inflammatory
response, thereby increasing renal cell apoptosis (45). The CSE inhibitor, propargyl glycine,
or the CBS inhibitor, hydroxylamine, have been shown to aggravate
AKI and apoptosis, presenting with higher levels of
pro-inflammatory factors, significantly increased levels of NF-κB
(P65), and phosphorylated (p)-apoptosis signal-regulating kinase 1
and p-TNFR-associated factor 2. These changes were accompanied by
the increased expression levels of TLR-2 and TLR-4, indicating that
a TLR-mediated inflammatory response and apoptosis are also
involved in renal IRI (46).
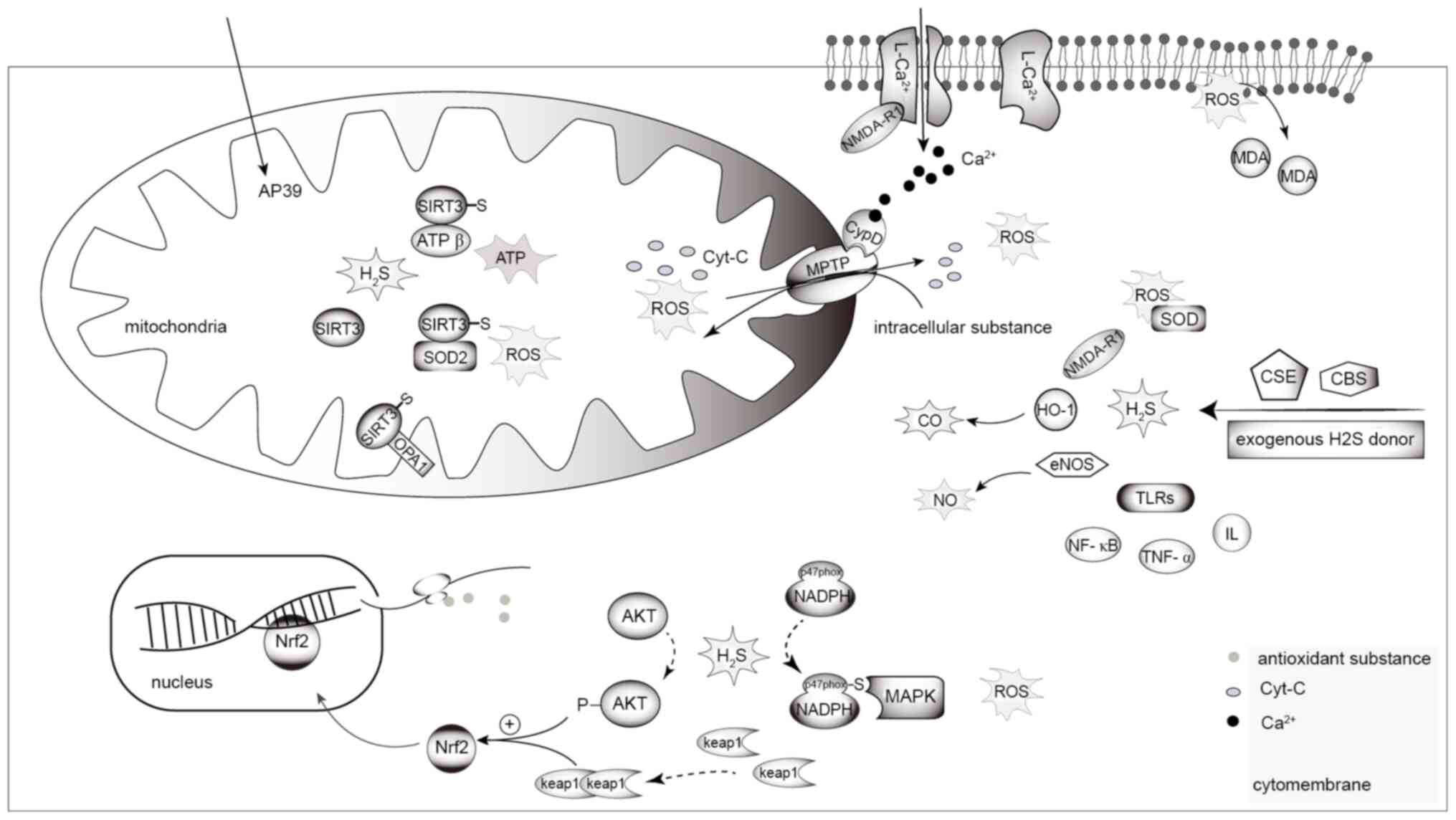 | Figure 4.H2S and renal injury. In
the mitochondria, H2S increases S-mercaptoylation of the
four cysteine residues of SIRT3, and induces deacetylation of its
target proteins, OPA1, ATP synthase (depicted as ATP in the figure)
and SOD2, thus reducing mitochondrial division and increasing ATP
production. H2S reduces MPTP opening and loss of
mitochondrial membrane potential via Ca2+-dependent CypD
activation by inhibiting NMDA-R1 mediated Ca2+ influx,
thus avoiding mitochondrial morphological and functional damage,
leading to ROS accumulation. ROS leads to the peroxidation of
membrane lipids to MDA, causing damage to cells and organelles. The
prevulcanization of H2S on NADPH oxidase subunit P47PHOx
inhibits the activity of NADPH oxidase, thereby inhibiting the
generation of MAPKs and intracellular ROS. H2S leads to
the phosphorylation of AKT and dimerization of Keap1, and induces
nuclear translocation of Nrf2 to promote the expression of
antioxidant substances, thereby inhibiting ROS in cells. NaHS
downregulates the overexpression of renal iNOS, upregulates eNOS
and HO-1, and regulates the T-AOC and IL-10 via the CO/NO pathway
to exert an anti-inflammatory and antioxidant effect.
H2S, hydrogen sulfide; OPA1, dynamin-like 120 kDa
protein; SIRT3, NAD-dependent deacetylase sirtuin-3; SOD2,
superoxide dismutase 2; MPTP, mitochondrial permeability transition
pore; iNOS, inducible nitric oxide synthase; eNOS, endothelial
nitric oxide synthase; HO-1, heme oxygenase-1; T-AOC, total
antioxidant capacity; Nrf2, nuclear factor erythroid 2-related
factor 2; Keap1, Kelch-like ECH-related protein 1; ROS, reactive
oxygen species; TLR, Toll-like receptor; CBS,
cystathionine-β-synthase; CSE, cystathionine-γ-lyase; p-,
phosphorylated; MDA, malondialdehyde; CO, carbon monoxide; NO,
nitric oxide. |
The mitochondrial targeted H2S donor,
AP39, has been reported to significantly improve the survival and
function of donor kidney transplantation, and reduce cell apoptosis
and necrosis (47,48). H2S has been shown to
attenuate apoptosis and necrosis during cryopreservation of a donor
kidney, and may increase the survival rate and function of
transplanted kidneys by regulating the mitochondrial membrane
potential and reducing reactive oxygen species (ROS) production
(47). Oxidative stress induced by
glucose oxidase can lead to mitochondrial dysfunction, which
reduces the levels of ATP in renal epithelial cells, increases the
formation of cellular ROS at a relatively high concentration and
promotes cell necrosis. A previous study using both in vitro
and in vivo experiments found that AP39 pretreatment has a
concentration-dependent protective effect on renal IRI, with the
most significant effect observed at a concentration of 300
nm·l−1 (48). The
H2S protection of AP39 was 1,000X higher than that of
GYY4137, a nonspecific exogenous H2S donor (47). In addition, H2S may
reduce the inflammatory response by inhibiting activation of the
Nod2 signaling pathway and suppressing the type A macrophage
scavenger receptor signaling pathway to upregulate endoplasmic
reticulum stress-induced autophagy to protect the kidney from IRI
(49). However, how H2S
acts on these targets is unclear.
Studies on renal transplantation storage also
demonstrated that long-term static storage of donation after
cardiac death (DCD) kidneys at 21°C in UW solution supplemented
with AP39 may increase the activity of renal tubular epithelial
cells and reduce tissue necrosis compared with long-term static
storage at 4°C in UW solution. However, the experimental results
also revealed that the UW solution supplemented with AP39 exhibited
improved cell-protective effects at 4°C compared with that at 21°C
(50). This is consistent with
static cryogenic storage (SCS) and continuous cryogenic machine
perfusion commonly used in our clinic. However, it is worth noting
that organ preservation at a physiological temperature (37°C), such
as normal temperature machine perfusion, may be worthy of study to
better prevent the damage of transplanted organs caused by low
temperatures (51). Renal function
was revealed to be improved in transplanted kidneys stored at a
normal physiological temperature compared with those stored in the
cold state (52). In a previous
study, kidneys from expanded criteria donors (ECD) were normally
perfused in vitro for 63±16 min with a plasma-free red
cell-based solution at an average temperature of 34.6°C and
compared with 47 ECD kidneys with CSC in a control group; the
results showed that all donor kidneys were successfully
transplanted with good renal function (53). In addition, subnormothermic machine
perfusion of DCD porcine grafts at 20°C has been shown to improve
graft prognosis compared with hypothermic machine perfusion and SCS
(54). Therefore, the effects of
H2S and the storage temperature of transplanted kidneys
should be studied further to determine the ideal storage
conditions.
Drug nephrotoxicity
Cisplatin is a common chemotherapeutic drug that is
widely used in the clinic. Cisplatin, by downregulating the
expression levels of CSE, is known to disrupt H2S
generation and lead to the death of proximal tubular cells, thereby
causing renal toxicity. The H2S donors, NaHS and
GYY4137, have been reported to reduce cisplatin-induced cell death
and renal toxicity (55). A
previous study revealed that H2S can increase
S-sulfhydrylation of the Cys256, Cys259, Cys280 and Cys283 residues
of NAD-dependent deacetylase sirtuin-3 (SIRT3), which induces the
deacetylation of its target proteins, dynamin-like 120 kDa protein
(OPA1), ATP synthase and SOD2, thus reducing mitochondrial division
and increasing ATP production, and thereby reducing oxidative
damage (56). In addition,
H2S may inhibit the generation of intracellular ROS and
MAPKs by inhibiting the activity of NADPH oxidase, which is related
to the vulcanization effect of H2S on the NADPH oxidase
subunit P47PHOx (55). Whilst
reducing NADPH oxidase activity, H2S may also induce
nuclear translocation of the nuclear factor erythroid 2-related
factor 2 (Nrf2) to inhibit the production of ROS in cells. Further
experiments have revealed that exogenous H2S donors lead
to the phosphorylation of Akt and dimerization of Kelch-like
ECH-related protein 1 (Keap1); the inhibition of Akt activation has
been reported to not only weaken the nuclear translocation of Nrf2,
but also reduce the protective effects of exogenous H2S
donors (57). H2S can
activate Nrf2 translocation to the nucleus by dimerizing Keap1,
thus promoting the expression of antioxidant genes (58). Therefore, H2S is
hypothesized to inhibit ROS production in cells via Akt/Keap1 and
the activation of MAPKs, thereby mediating the nuclear
translocation of Nrf2. It may also inhibit the production of ROS in
cells by reducing the activity of NADPH oxidase. Recent studies
have shown that exogenous H2S serves a renoprotective
role in cyclophosphamide-induced nephrotoxicity, which is
associated with increased expression of Nrf2 and downstream
antioxidant proteins, such as heme oxygenase-1 (HO-1), and reduced
glutathione and SOD in renal tissues (57,59),
as shown in Fig. 4.
The histopathological results of a previous study
revealed that the renal tissues of a cisplatin group were positive
for desmin protein expression, with notable podocyte injury,
increased quantities of mesangial matrix and increased
proliferation of mesangial cells. Notably, NaHS therapy could
improve podocyte injury and increase nephrin protein levels
(60). These findings suggested
that H2S may improve cisplatin-induced renal injury by
protecting renal podocyte cells. In gentamicin-induced kidney
injury in rats, NaHS significantly reduced renal NO and TNF-α
levels, whilst increasing total antioxidant capacity (T-AOC), HO-1
and IL-10 levels, and reduced the increase in renal inducible NOS
(iNOS), whilst upregulating eNOS levels. Zinc proporphyrin (a
selective HO-1 inhibitor) could reverse these changes, and block
the anti-inflammatory and antioxidant effects of H2S
(61). Therefore, H2S
may serve an anti-inflammatory and antioxidant role in protecting
AKI, partly by relying on the CO/NO pathway, and this mechanism may
function to primarily downregulate NO levels, or to downregulate
the effects of NO by increasing CO levels (61).
DN
Streptomycin-induced DN rats have been shown to
exhibit notable inflammation and oxidative stress, with obvious
renal decline and insufficiency, decreased activities of SIRT1 and
SOD, and increased relative expression of caspase-3, p53 and MDA;
however, NaHS may improve renal function, manifested as
significantly reduced serum urea and creatinine levels, and markers
of renal injury, and reversal of the aforementioned indicators of
DN (62,63). ATP-sensitive potassium
(KATP) channels and L-type calcium channels have been
shown to be related to the increase in ROS levels and oxidative
stress in DN renal cells. NaHS may increase T-AOC and reduce the
total NO levels in a rat model of DN, and the use of
KATP inhibitors may further increase T-AOC and reduce NO
levels (62). Therefore, the
renoprotective mechanism of H2S on DN may be partly
dependent on KATP channel activation-mediated effects on
renal tissue antioxidants and NO.
In a previous study, renal injury was simulated in
C57BL/6J and Akita (C57BL/6JIns2Akita) mice under a high-glucose
environment, and the experiment showed increased cytoplasmic
Ca2+ influx, activation of the mitochondrial matrix
protein cyclophilin D (CypD), increased mitochondrial permeability
transition opening, loss of mitochondrial membrane potential and
oxidative burst. The H2S donor GYY4137 could reduce the
aforementioned effects following treatment. Similar results were
also observed with the N-methyl-D-aspartate receptor-R1 (NMDA-R1)
blocker MK-801, which further confirmed that H2S
function may involve NMDA-R1 (64).
H2S has been reported to reduce intracellular
Ca2+ by inhibiting NMDA-R1-mediated inflow of
Ca2+ ions, and thus reducing Ca2+-dependent
CypD activation to result in mitochondrial permeability transition
pore opening and loss of mitochondrial membrane potential. This
effects may avoid damage to the mitochondrial morphology and
function, and could cause outbreaks of active oxygen substances and
protect diabetic kidney cells from oxidative stress injury, as
shown in Fig. 4. In dose-response
experiments assessing the protective effects of H2S on
DN kidney, it was found that at a dose of 100 mol/kg/day, the
activity/expression of SIRT1 returned to normal, and the kidney
function of DN rats was improved (63). However, this previous study did not
elaborate on the relationship between SIRT1 and oxidative stress
and inflammation, and the molecular mechanism of action between
them still remains to be further explored.
Renal fibrosis
Long-term damage to the kidney by various factors
can lead to the occurrence of renal fibrosis. In the kidney of
diabetic rats, NaHS therapy downregulated the expression of
transforming growth factor-β1 (TGF-β1), extracellular signal
regulated kinase 1/2 (ERK1/2), tissue inhibitors of
metalloproteinases (TIMPs) and matrix metalloproteinases (MMPs),
leading to the improvement of renal fibrosis (65,66).
Renal fibrosis is associated with TGF-β/Smad signaling,
AMP-activated protein kinase (AMPK) activation, ERK1/2 expression
and MMP/TIMP dysregulation (65,66),
as shown in Fig. 5.
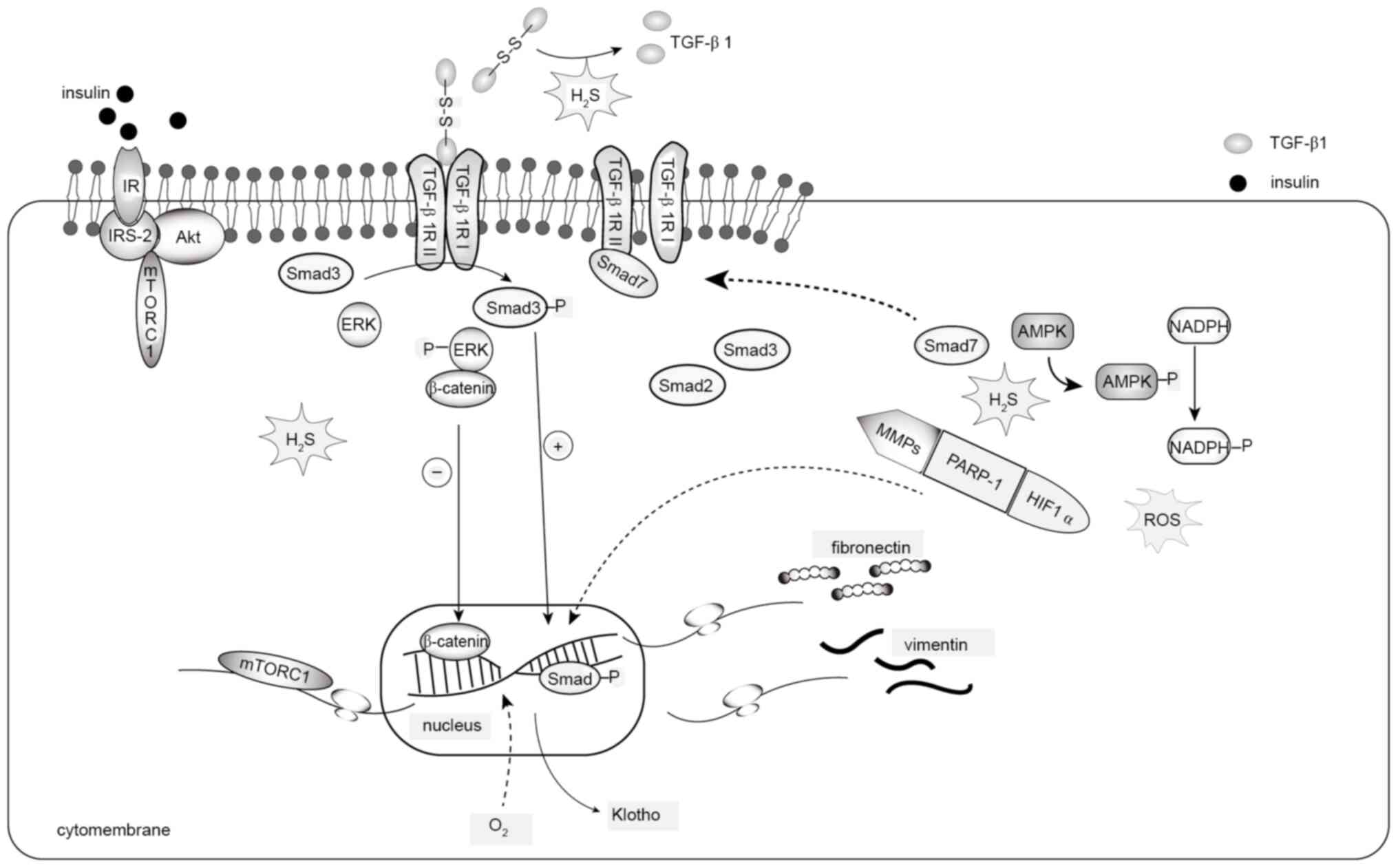 | Figure 5.H2S and renal fibrosis.
TGF-β1 binds to TβR and promotes downstream Smad protein
activation, leading to the overexpression of fibronectin and
vimentin. Activated by TβR, ERK promotes the conversion of
β-catenin in the nucleus, leading to the increased expression of
fibronectin. H2S promotes Smad7 expression to reduce the
combination of TβRII and TβRI, preventing this process. At the same
time, H2S lyses the disulfide bond in the active TGF-β1
dimer, promoting the formation of inactive TGF-β1 monomers. In
addition, increases in the expression of matrix-associated proteins
are associated with the activation of the IR/IRS-2/Akt-mTORC1/mRNA
transcriptional signaling axis. H2S reduces ROS and
collagen cross-linking by regulating MMPs/PARP-1/HIF-1. Hypoxia is
associated with methylation and expression silencing of the Klotho
promoter. H2S can significantly improve hypoxia, reverse
Klotho promoter methylation and increase Klotho expression. TGF-β1,
transforming growth factor-β1; TβR, TGF-β receptor; ERK,
extracellular signal-regulated kinase; ROS, reactive oxygen
species; H2S, hydrogen sulfide; IR, insulin receptor;
IRS, IR substrate; mTORC, mammalian target of rapamycin complex 1;
MMP, matrix metalloproteinase; PARP, poly ADP-ribose-polymerase;
HIF-1, hypoxia-inducible factor-1; p-, phosphorylated. |
The novel H2S-releasing compound,
S-propylcysteine, was revealed to inhibit the mRNA expression
levels of hyperglycemic fibulin and type IV collagen, as well as
the over-proliferation and hypertrophy of mesangial cells. Further
experiments confirmed that this was related to the inhibition of
TGF-β1- and Smad3-related signaling pathways (67). Following unilateral ureteral
obstruction (UUO) in male Lewis rats, H2S treatment was
shown to reduce serum creatinine and urine protein/creatinine
excretion rate, and tissues exhibited reduced expression of
EMT-related proteins, including fibronectin, vimentin, Smad2,
TGF-β1 and TGF-β1 receptor (TβR)II. Pathological analysis also
showed that H2S alleviated cortical loss, inflammatory
damage and renal tubulointerstitial fibrosis (68). Previous studies have shown that
H2S-mediated Smad7 expression may reduce TβRII
expression, and improve UUO renal fibrosis in a rat model via the
upregulation of cadherin expression and downregulation of vimentin
expression in endothelial cells (69–71).
In this mechanism, TβRII binds to and activates TβRI, which can
increase the activation of downstream Smad expression, leading to
the upregulation of vimentin expression and downregulation of
cadherin expression in endothelial cells; Smad7 can interact with
TβRI/TβRII to prevent this process (70,71).
In vitro experiments using human recombinant active TGF-β1
to induce EMT also found that H2S cleaved the disulfide
bonds in the active dimer of TGF-β1, and promoted the formation of
inactive TGF-β1 monomers (72). In
addition, NaHS reduced the increase in expression of β-catenin
induced by TGF-β1, increased the phosphorylation of ERK and
inhibited the nuclear translocation of β-catenin induced by TGF-β1.
Using the ERK inhibitor U0126 or β-catenin small interfering RNA
(siRNA) agent XAV939 abrogated the effects of NaHS on fibronectin,
E-cadherin and TGF-βRI. These findings indicated that
H2S may block TGF-β-induced EMT by inhibiting ERK
activation and β-catenin translocation, thus preventing renal
fibrosis (73).
In diabetic Akita mice, the levels of plasma
H2S, ROS and its regulator ROS modulator 1, and the
expression of collagen cross-linking proteins (prolyl 4-hydroxylase
subunit α 1 and procollagen-lysine, 2-oxoglutarate 5-dioxynenase 2)
were increased, and the activity and the expression levels of poly
ADP-ribose-polymerase-1 (PARP-1), hypoxia-inducible factor-1
(HIF-1), and MMP-9, −13 and −14 were increased. These findings may
be related to the downregulation of microRNA (miR)-194. Notably,
GYY4137 was shown to restore expression of miR-194. In addition,
in vivo and in vitro experiments revealed that cells
transfected with miR-194 mimic exhibited alleviation of high
glucose-induced ROS production (74). A high-glucose environment mat
increase ROS levels and lead to PARP activation, whereas PARP-1
deficiency may alleviate DN (75).
Furthermore, blocking HIF-1 may reduce glomerular hypertrophy, ECM
deposition and urinary albumin excretion in diabetic kidneys
(76). These results suggested that
H2S may alleviate diabetic renal ECM deposition and
thereby reduce renal fibrosis by regulating MMPs/PARP-1/HIF-1
expression to reduce ROS levels and the increase in collagen
cross-linking.
The increase in matrix protein content involved in
renal fibrosis has been reported to be associated with AMPK
activity and activation of the insulin receptor (IR)/IR substrate
(IRS)-2/Akt/mammalian target of rapamycin complex 1 (mTORC1)/mRNA
transcriptional signaling axis (77). In proximal renal tubular epithelial
cells, high glucose levels inhibited AMPK phosphorylation and
activity, increased NADPH oxidase 4 (NOX4) expression and activity,
and the production of ROS and matrix protein synthesis, which was
reversed by NaHS. In further experiments, an AMPK inhibitor
prevented NaHS from reducing the expression of NOX4 induced by high
glucose (78). In addition, it was
revealed that N (ω)-nitro-L-arginine methyl ester (a NOS inhibitor)
could abolish NaHS inhibition of NOX4 expression induced by high
glucose. NaHS enhanced the expression of iNOS instead of eNOS.
Further experiments showed that iNOS siRNA and 1400W (a selective
iNOS inhibitor) eliminated the favorable effects of NaHS on the
expression of high glucose-induced NOX4, ROS and matrix laminin
expression (78). Therefore, NaHS
may regulate oxidative stress and the expression of renal
interstitial matrix protein by inducing NO production and mediating
the AMPK pathway to inhibit hyperglycemic renal fibrosis and
protect diabetic renal function. Two gas transmitters,
H2S and NO, and their interactions can be used as
therapeutic targets for DN (78).
Hypoxia and inflammation can lead to renal
fibrosis, and renal hypoxia is associated with methylation and
silencing of the Klotho promoter. Notably, NaHS treatment has been
reported to significantly reduce hypoxia, reverse Klotho promoter
methylation to increase Klotho expression, and thereby improve
renal tubular interstitial fibrosis in mice (79). Inhibition of M1/M2 macrophage
infiltration and NLRP3 inflammasome activation, and subsequent
inactivation of the NF-κB and IL-4/STAT6 signaling pathways can
also exert anti-inflammatory and anti-fibrotic roles in the
protection against renal fibrosis and renal injury following
obstruction (80). In renal tubular
epithelial cells, H2S has been shown to sulfurize the
two conserved domains of SIRT1 (Cys371/374 and Cys395/398), and
induce the dephosphorylation and deacetylation of its target
proteins NF-κB (p65) and STAT3, thereby reducing oxidative stress,
inflammation and EMT caused by high glucose (81). Renal fibrosis is also associated
with aging and obesity. Notably, NaHS restored AMPK activity,
inhibited activation of the IR/IRS-2/Akt/mTORC1/mRNA translation
axis, and improved renal function in aged mice (77). In addition, miR-21 has been shown to
be associated with renal injury in the elderly. After inhibition of
miR-21 expression, the expression levels of
H2S-generating enzymes, CBS and CSE, in mouse
endothelial cells were upregulated, and the expression levels of
MMP-9 and type IV collagen were downregulated (82). In a high-fat diet (HFD)-induced
model of kidney injury, H2S reduced the phosphorylation
levels of IR and Akt in the renal cortex of male mice, which may
suggest that obesity-related kidney injury is related to the IR/Akt
pathway; however, this association was not observed in female mice,
and whether it is related to sex-related factors remains to be
studied (83). Furthermore,
H2S significantly reduced lipid accumulation in the
kidneys of HFD-induced obese mice, and studies have shown that
H2S may downregulate NF-κB (P65) expression to reduce
renal inflammation and alleviate HFD-induced renal injury in obese
mice (84). These findings
suggested that obesity may aggravate inflammatory-mediated renal
fibrosis and renal injury.
Conclusions
H2S deficiency is a potential risk
factor for the development and progression of renal diseases. A
variety of renal injuries, including IRI, drug nephrotoxicity and
DN, exhibit metabolic imbalances of H2S during their
pathological development. Supplementing exogenous H2S
can alleviate renal injury caused by these diseases, delay the
progression of renal fibrosis and improve renal function. The
signaling pathways and molecules in which H2S serves an
antioxidant, anti-inflammatory, anti-apoptotic and anti-fibrotic
role in renal protection are being increasingly better understood.
In addition, organelles serve a notable role in the progression of
AKI and CKD. At present, studies on the damage to organelles, such
as mitochondrial homeostasis, mitochondrial autophagy and
endoplasmic reticulum oxidative stress, are relatively limited with
regard to the pathological mechanism of AKI and CKD, and more
in-depth studies are required. In terms of exogenous H2S
donors, research into drugs targeting the mitochondria and inducing
the controlled release of agents may form an indispensable means of
treatment for AKI and CKD.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
NG conceived the idea of the present review, and
was responsible for reviewing the images and main text. HuZ and HaZ
were major contributors to the writing of the manuscript, and
designed the figures. All authors read and approved the final
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang R: Gasotransmitters: Growing pains
and joys. Trends Biochem Sci. 39:227–232. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Szabo C: A timeline of hydrogen sulfide
(H2S) research: From environmental toxin to biological
mediator. Biochem Pharmacol. 149:5–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ngowi EE, Sarfraz M, Afzal A, Khan NH,
Khattak S, Zhang X, Li T, Duan SF, Ji XY and Wu DD: Roles of
hydrogen sulfide donors in common kidney diseases. Front Pharmacol.
11:5642812020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mao YG, Chen X, Zhang Y and Chen G:
Hydrogen sulfide therapy: A narrative overview of current research
and possible therapeutic implications in future. Med Gas Res.
10:185–188. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Paul BD and Snyder SH: Gasotransmitter
hydrogen sulfide signaling in neuronal health and disease. Biochem
Pharmacol. 149:101–109. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wen YD, Wang H and Zhu YZ: The drug
developments of hydrogen sulfide on cardiovascular disease. Oxid
Med Cell Longev. 2018:40103952018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kumar M and Sandhir R: Hydrogen sulfide in
physiological and pathological mechanisms in brain. CNS Neurol
Disord Drug Targets. 17:654–670. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yadav PK, Vitvitsky V, Carballal S,
Seravalli J and Banerjee R: Thioredoxin regulates human
mercaptopyruvate sulfurtransferase at physiologically-relevant
concentrations. J Biol Chem. 295:6299–6311. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang R: Two's company, three's a crowd:
Can H2S be the third endogenous gaseous transmitter? FASEB J.
16:1792–1798. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Q and Lancaster JR Jr: Chemical
foundations of hydrogen sulfide biology. Nitric Oxide. 35:21–34.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mathai JC, Missner A, Kügler P, Saparov
SM, Zeidel ML, Lee JK and Pohl P: No facilitator required for
membrane transport of hydrogen sulfide. Proc Natl Acad Sci USA.
106:16633–16638. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singh S, Padovani D, Leslie RA, Chiku T
and Banerjee R: Relative contributions of cystathionine
beta-synthase and gamma-cystathionase to H2S biogenesis via
alternative trans-sulfuration reactions. J Biol Chem.
284:22457–22466. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Módis K, Coletta C, Erdélyi K,
Papapetropoulos A and Szabo C: Intramitochondrial hydrogen sulfide
production by 3-mercaptopyruvate sulfurtransferase maintains
mitochondrial electron flow and supports cellular bioenergetics.
FASEB J. 27:601–611. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen X, Jhee KH and Kruger WD: Production
of the neuromodulator H2S by cystathionine beta-synthase via the
condensation of cysteine and homocysteine. J Biol Chem.
279:52082–52086. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chiku T, Padovani D, Zhu W, Singh S,
Vitvitsky V and Banerjee R: H2S biogenesis by human cystathionine
gamma-lyase leads to the novel sulfur metabolites lanthionine and
homolanthionine and is responsive to the grade of
hyperhomocysteinemia. J Biol Chem. 284:11601–11612. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li L, Hsu A and Moore PK: Actions and
interactions of nitric oxide, carbon monoxide and hydrogen sulphide
in the cardiovascular system and in inflammation-a tale of three
gases! Pharmacol Ther. 123:386–400. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Whiteman M, Le Trionnaire S, Chopra M, Fox
B and Whatmore J: Emerging role of hydrogen sulfide in health and
disease: Critical appraisal of biomarkers and pharmacological
tools. Clin Sci (Lond). 121:459–488. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schumann U and Subramani S: Special
delivery from mitochondria to peroxisomes. Trends Cell Biol.
18:253–256. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuang Q, Xue N, Chen J, Shen Z, Cui X,
Fang Y and Ding X: Low plasma hydrogen sulfide is associated with
impaired renal function and cardiac dysfunction. Am J Nephrol.
47:361–371. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lagoutte E, Mimoun S, Andriamihaja M,
Chaumontet C, Blachier F and Bouillaud F: Oxidation of hydrogen
sulfide remains a priority in mammalian cells and causes reverse
electron transfer in colonocytes. Biochim Biophys Acta.
1797:1500–1511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hildebrandt TM and Grieshaber MK: Three
enzymatic activities catalyze the oxidation of sulfide to
thiosulfate in mammalian and invertebrate mitochondria. FEBS J.
275:3352–3361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Libiad M, Yadav PK, Vitvitsky V, Martinov
M and Banerjee R: Organization of the human mitochondrial hydrogen
sulfide oxidation pathway. J Biol Chem. 289:30901–30910. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ziosi M, Di Meo I, Kleiner G, Gao XH,
Barca E, Sanchez-Quintero MJ, Tadesse S, Jiang H, Qiao C, Rodenburg
RJ, et al: Coenzyme Q deficiency causes impairment of the sulfide
oxidation pathway. EMBO Mol Med. 9:96–111. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kleiner G, Barca E, Ziosi M, Emmanuele V,
Xu Y, Hidalgo-Gutierrez A, Qiao C, Tadesse S, Area-Gomez E, Lopez
LC and Quinzii CM: CoQ10 supplementation rescues
nephrotic syndrome through normalization of H2S
oxidation pathway. Biochim Biophys Acta Mol Basis Dis.
1864:3708–3722. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Maldonato BJ, Russell DA and Totah RA:
Human METTL7B is an alkyl thiol methyltransferase that metabolizes
hydrogen sulfide and captopril. Sci Rep. 11:48572021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kabil O and Banerjee R: Redox biochemistry
of hydrogen sulfide. J Biol Chem. 285:21903–21907. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pushpakumar S, Kundu S and Sen U: Hydrogen
sulfide protects hyperhomocysteinemia-induced renal damage by
modulation of caveolin and eNOS interaction. Sci Rep. 9:22232019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xia M, Chen L, Muh RW, Li PL and Li N:
Production and actions of hydrogen sulfide, a novel gaseous
bioactive substance, in the kidneys. J Pharmacol Exp Ther.
329:1056–1062. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang J, Chen S, Liu H, Zhang B, Zhao Y,
Ma K, Zhao D, Wang Q, Ma H and Zhang Z: Hydrogen sulfide prevents
hydrogen peroxide-induced activation of epithelial sodium channel
through a PTEN/PI(3,4,5)P3 dependent pathway. PLoS One.
8:e643042013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo R, Hu S, Liu Q, Han M, Wang F, Qiu M,
Li S, Li X, Yang T, Fu X, et al: Hydrogen sulfide upregulates renal
AQP-2 protein expression and promotes urine concentration. FASEB J.
33:469–483. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ge SN, Zhao MM, Wu DD, Chen Y, Wang Y, Zhu
JH, Cai WJ, Zhu YZ and Zhu YC: Hydrogen sulfide targets EGFR
Cys797/Cys798 residues to induce Na(+)/K(+)-ATPase endocytosis and
inhibition in renal tubular epithelial cells and increase sodium
excretion in chronic salt-loaded rats. Antioxid Redox Signal.
21:2061–2082. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Z, Li Y, Overstreet JM, Chung S, Niu A,
Fan X, Wang S, Wang Y, Zhang MZ and Harris RC: Inhibition of
epidermal growth factor receptor activation is associated with
improved diabetic nephropathy and insulin resistance in type 2
diabetes. Diabetes. 67:1847–1857. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang ZJ, Chang LL, Wu J, Pan HM, Zhang QY,
Wang MJ, Xin XM, Luo SS, Chen JA, Gu XF, et al: A novel
rhynchophylline analog, Y396, inhibits endothelial dysfunction
induced by oxidative stress in diabetes through epidermal growth
factor receptor. Antioxid Redox Signal. 32:743–765. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Izzedine H and Perazella MA: Adverse
kidney effects of epidermal growth factor receptor inhibitors.
Nephrol Dial Transplant. 32:1089–1097. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Prieto-Lloret J and Aaronson PI: Hydrogen
sulfide as an O2 sensor: A critical analysis. Adv Exp
Med Biol. 967:261–276. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Olson KR: Hydrogen sulfide is an oxygen
sensor in the carotid body. Respir Physiol Neurobiol. 179:103–110.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Olson KR: Hydrogen sulfide as an oxygen
sensor. Antioxid Redox Signal. 22:377–397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Olson KR: Hydrogen sulfide as an oxygen
sensor. Clin Chem Lab Med. 51:623–632. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Evans RG, Smith DW, Khan Z, Ngo JP and
Gardiner BS: Letter to the editor: ‘The plausibility of
arterial-to-venous oxygen shunting in the kidney: It all depends on
radial geometry’. Am J Physiol Renal Physiol. 309:F179–F180. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hirakawa Y, Tanaka T and Nangaku M: Renal
hypoxia in CKD; pathophysiology and detecting methods. Front
Physiol. 8:992017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koning AM, Frenay AR, Leuvenink HG and van
Goor H: Hydrogen sulfide in renal physiology, disease and
transplantation-the smell of renal protection. Nitric Oxide.
46:37–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen Q, Yu S, Zhang K, Zhang Z, Li C, Gao
B, Zhang W and Wang Y: Exogenous H2S inhibits autophagy in
unilateral ureteral obstruction mouse renal tubule cells by
regulating the ROS-AMPK signaling pathway. Cell Physiol Biochem.
49:2200–2213. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lobb I, Sonke E, Aboalsamh G and Sener A:
Hydrogen sulphide and the kidney: Important roles in renal
physiology and pathogenesis and treatment of kidney injury and
disease. Nitric Oxide. 46:55–65. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sekijima M, Sahara H, Miki K, Villani V,
Ariyoshi Y, Iwanaga T, Tomita Y and Yamada K: Hydrogen sulfide
prevents renal ischemia-reperfusion injury in CLAWN miniature
swine. J Surg Res. 219:165–172. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Du Y, Liu XH, Zhu HC, Wang L, Wang ZS,
Ning JZ and Xiao CC: Hydrogen sulfide treatment protects against
renal ischemia-reperfusion injury via induction of heat shock
proteins in rats. Iran J Basic Med Sci. 22:99–105. 2019.PubMed/NCBI
|
|
46
|
Tan Z, Shi Y, Yan Y, Liu W, Li G and Li R:
Impact of endogenous hydrogen sulfide on toll-like receptor pathway
in renal ischemia/reperfusion injury in rats. Ren Fail. 37:727–733.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lobb I, Jiang J, Lian D, Liu W, Haig A,
Saha MN, Torregrossa R, Wood ME, Whiteman M and Sener A: Hydrogen
sulfide protects renal grafts against prolonged cold
ischemia-reperfusion injury via specific mitochondrial actions. Am
J Transplant. 17:341–352. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ahmad A, Olah G, Szczesny B, Wood ME,
Whiteman M and Szabo C: AP39, a mitochondrially targeted hydrogen
sulfide donor, exerts protective effects in renal epithelial cells
subjected to oxidative stress in vitro and in acute renal injury in
vivo. Shock. 45:88–97. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ling Q, Yu X, Wang T, Wang SG, Ye ZQ and
Liu JH: Roles of the exogenous H2S-mediated SR-A signaling pathway
in renal ischemia/reperfusion injury in regulating endoplasmic
reticulum stress-induced autophagy in a rat model. Cell Physiol
Biochem. 41:2461–2474. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Juriasingani S, Akbari M, Chan JY,
Whiteman M and Sener A: H2S supplementation: A novel
method for successful organ preservation at subnormothermic
temperatures. Nitric Oxide. 81:57–66. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hosgood SA, van Heurn E and Nicholson ML:
Normothermic machine perfusion of the kidney: Better conditioning
and repair? Transpl Int. 28:657–664. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Bagul A, Hosgood SA, Kaushik M, Kay MD,
Waller HL and Nicholson ML: Experimental renal preservation by
normothermic resuscitation perfusion with autologous blood. Br J
Surg. 95:111–118. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nicholson ML and Hosgood SA: Renal
transplantation after ex vivo normothermic perfusion: The first
clinical study. Am J Transplant. 13:1246–1252. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hoyer DP, Gallinat A, Swoboda S,
Wohlschläger J, Rauen U, Paul A and Minor T: Subnormothermic
machine perfusion for preservation of porcine kidneys in a donation
after circulatory death model. Transpl Int. 27:1097–1106. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Cao X, Xiong S, Zhou Y, Wu Z, Ding L, Zhu
Y, Wood ME, Whiteman M, Moore PK and Bian JS: Renal protective
effect of hydrogen sulfide in cisplatin-induced nephrotoxicity.
Antioxid Redox Signal. 29:455–470. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yuan Y, Zhu L, Li L, Liu J, Chen Y, Cheng
J, Peng T and Lu Y: S-sulfhydration of SIRT3 by hydrogen sulfide
attenuates mitochondrial dysfunction in cisplatin-induced acute
kidney injury. Antioxid Redox Signal. 31:1302–1319. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao X, Nie X, Xiong S, Cao L, Wu Z, Moore
PK and Bian JS: Renal protective effect of polysulfide in
cisplatin-induced nephrotoxicity. Redox Biol. 15:513–521. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Koike S, Ogasawara Y, Shibuya N, Kimura H
and Ishii K: Polysulfide exerts a protective effect against
cytotoxicity caused by t-buthylhydroperoxide through Nrf2 signaling
in neuroblastoma cells. FEBS Lett. 587:3548–3555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Waz S, Heeba GH, Hassanin SO and
Abdel-Latif RG: Nephroprotective effect of exogenous hydrogen
sulfide donor against cyclophosphamide-induced toxicity is mediated
by Nrf2/HO-1/NF-κB signaling pathway. Life Sci. 264:1186302021.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Karimi A, Absalan F, Khorsandi L,
Valizadeh A and Mansouri E: Sodium hydrogen sulfide (NaHS)
ameliorates alterations caused by cisplatin in filtration slit
diaphragm and podocyte cytoskeletal in rat kidney. J Nephropathol.
6:150–156. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Aziz NM, Elbassuoni EA, Kamel MY and Ahmed
SM: Hydrogen sulfide renal protective effects: Possible link
between hydrogen sulfide and endogenous carbon monoxide in a rat
model of renal injury. Cell Stress Chaperones. 25:211–221. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Elbassuoni EA, Аziz NM and Habeeb WN: The
role of activation of KАTP channels on hydrogen sulfide
induced renoprotective effect on diabetic nephropathy. J Cell
Physiol. 235:5223–5228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ahmed HH, Taha FM, Omar HS, Elwi HM and
Abdelnasser M: Hydrogen sulfide modulates SIRT1 and suppresses
oxidative stress in diabetic nephropathy. Mol Cell Biochem.
457:1–9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Papu John AS, Kundu S, Pushpakumar S, Amin
M, Tyagi SC and Sen U: Hydrogen sulfide inhibits
Ca2+-induced mitochondrial permeability transition pore
opening in type-1 diabetes. Am J Physiol Endocrinol Metab.
317:E269–E283. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Li L, Xiao T, Li F, Li Y, Zeng O, Liu M,
Liang B, Li Z, Chu C and Yang J: Hydrogen sulfide reduced renal
tissue fibrosis by regulating autophagy in diabetic rats. Mol Med
Rep. 16:1715–1722. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li Y, Li L, Zeng O, Liu JM and Yang J:
H2S improves renal fibrosis in STZ-induced diabetic rats
by ameliorating TGF-β1 expression. Ren Fail. 39:265–272. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Qian X, Li X, Ma F, Luo S, Ge R and Zhu Y:
Novel hydrogen sulfide-releasing compound, S-propargyl-cysteine,
prevents STZ-induced diabetic nephropathy. Biochem Biophys Res
Commun. 473:931–938. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lin S, Visram F, Liu W, Haig A, Jiang J,
Mok A, Lian D, Wood ME, Torregrossa R, Whiteman M, et al: GYY4137,
a slow-releasing hydrogen sulfide donor, ameliorates renal damage
associated with chronic obstructive uropathy. J Urol.
196:1778–1787. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lin S, Lian D, Liu W, Haig A, Lobb I,
Hijazi A, Razvi H, Burton J, Whiteman M and Sener A: Daily therapy
with a slow-releasing H2S donor GYY4137 enables early
functional recovery and ameliorates renal injury associated with
urinary obstruction. Nitric Oxide. 76:16–28. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yan X, Liao H, Cheng M, Shi X, Lin X, Feng
XH and Chen YG: Smad7 protein interacts with receptor-regulated
Smads (R-Smads) to inhibit transforming growth factor-β
(TGF-β)/Smad signaling. J Biol Chem. 291:382–392. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Yan X and Chen YG: Smad7: Not only a
regulator, but also a cross-talk mediator of TGF-β signalling.
Biochem J. 434:1–10. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Huang Y, Zhang Z, Huang Y, Mao Z, Yang X,
Nakamura Y, Sawada N, Mitsui T, Takeda M and Yao J: Induction of
inactive TGF-β1 monomer formation by hydrogen sulfide contributes
to its suppressive effects on Ang II- and TGF-β1-induced EMT in
renal tubular epithelial cells. Biochem Biophys Res Commun.
501:534–540. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Guo L, Peng W, Tao J, Lan Z, Hei H, Tian
L, Pan W, Wang L and Zhang X: Hydrogen sulfide inhibits
transforming growth factor-β1-induced EMT via Wnt/catenin pathway.
PLoS One. 11:e01470182016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
John AMSP, Kundu S, Pushpakumar S, Fordham
M, Weber G, Mukhopadhyay M and Sen U: GYY4137, a hydrogen sulfide
donor modulates miR194-dependent collagen realignment in diabetic
kidney. Sci Rep. 7:109242017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Shevalye H, Maksimchyk Y, Watcho P and
Obrosova IG: Poly(ADP-ribose) polymerase-1 (PARP-1) gene deficiency
alleviates diabetic kidney disease. Biochim Biophys Acta.
1802:1020–1027. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Nayak BK, Shanmugasundaram K, Friedrichs
WE, Cavaglierii RC, Patel M, Barnes J and Block K: HIF-1 mediates
renal fibrosis in OVE26 type 1 diabetic mice. Diabetes.
65:1387–1397. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Lee HJ, Feliers D, Barnes JL, Oh S,
Choudhury GG, Diaz V, Galvan V, Strong R, Nelson J, Salmon A, et
al: Hydrogen sulfide ameliorates aging-associated changes in the
kidney. Geroscience. 40:163–176. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lee HJ, Lee DY, Mariappan MM, Feliers D,
Ghosh-Choudhury G, Abboud HE, Gorin Y and Kasinath BS: Hydrogen
sulfide inhibits high glucose-induced NADPH oxidase 4 expression
and matrix increase by recruiting inducible nitric oxide synthase
in kidney proximal tubular epithelial cells. J Biol Chem.
292:5665–5675. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gu Y, Chen J, Zhang H, Shen Z, Liu H, Lv
S, Yu X, Zhang D, Ding X and Zhang X: Hydrogen sulfide attenuates
renal fibrosis by inducing TET-dependent DNA demethylation on
Klotho promoter. FASEB J. 34:11474–11487. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhou Y, Zhu X, Wang X, Peng Y, Du J, Yin
H, Yang H, Ni X and Zhang W: H2S alleviates renal injury
and fibrosis in response to unilateral ureteral obstruction by
regulating macrophage infiltration via inhibition of NLRP3
signaling. Exp Cell Res. 387:1117792020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Sun HJ, Xiong SP, Cao X, Cao L, Zhu MY, Wu
ZY and Bian JS: Polysulfide-mediated sulfhydration of SIRT1
prevents diabetic nephropathy by suppressing phosphorylation and
acetylation of p65 NF-κB and STAT3. Redox Biol. 38:1018132021.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Pushpakumar S, Kundu S, Weber G and Sen U:
Exogenous hydrogen sulfide and miR-21 antagonism attenuates
macrophage-mediated inflammation in ischemia reperfusion injury of
the aged kidney. Geroscience. 43:1349–1367. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lee HJ, Mariappan MM, Norton L, Bakewell
T, Feliers D, Oh SB, Donati A, Rubannelsonkumar CS, Venkatachalam
MA, Harris SE, et al: Proximal tubular epithelial insulin receptor
mediates high-fat diet-induced kidney injury. JCI Insight.
6:e1436192021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Wu D, Gao B, Li M, Yao L, Wang S, Chen M,
Li H, Ma C, Ji A and Li Y: Hydrogen sulfide mitigates kidney injury
in high fat diet-induced obese mice. Oxid Med Cell Longev.
2016:27157182016. View Article : Google Scholar : PubMed/NCBI
|