Introduction
Microglia are glial cells that are located
throughout the brain and spinal cord. Microglia act as resident
macrophages and serve a major role in immune defense and
homeostasis of the central nerve system (CNS) (1). Microglia scavenge the CNS, and
activated microglia phagocytose pathogens, plaques, and damaged or
unnecessary neurons (2). However,
over-activation of microglia can result in excessive production of
proinflammatory molecules, including nitric oxide (NO) radical,
reactive oxygen species (ROS), cytokines and chemokines, which may
cause neuronal cell death and brain injury (3,4).
Previous studies have demonstrated that microglial activation
contributes to neuronal damage and the progression of
neurodegenerative diseases, such as Alzheimer's disease,
Parkinson's disease, multiple sclerosis and amyotrophic lateral
sclerosis (ALS) (5,6).
Lipopolysaccharide (LPS) is a highly conserved outer
membrane component of Gram-negative bacteria, which promotes the
activation of macrophages and microglial cells. In response to LPS,
microglia produce a variety of inflammatory modulators, such as
IL-1β, TNF-α, IL-6, NO, ROS and prostaglandins (2). A previous demonstrated that LPS binds
to target cells through CD14 and Toll-like receptors 4 (TLR4)
(7). Ligation of TLR4 induces
recruitment of adaptor proteins and activates subsequent downstream
signaling, including mitogen-activated protein kinases (MAPKs),
Ca2+/calmodulin-dependent protein kinases (CAMKs) and
NF-κB. In unstimulated cells, NF-κB is present in the cytosol bound
to IκB; however, when stimulated by factors, such as LPS, IκB is
phosphorylated by IκB kinases. Subsequently, phosphorylated (p)-IκB
is rapidly ubiquitinated and degraded by the 26S proteasome
complex, and the free NF-κB translocates to the nucleus leading to
the expression of proinflammatory molecules (7). Moreover, MAPKs and CAMKs have been
reported to be associated with inflammation in the brain and glial
cells (8–11).
Panax ginseng C.A. Meyer has been used as a
traditional and herbal medicine in Asia and Western countries.
Ginsenosides are pharmacologically active natural constituents of
ginseng (12). Ginsenoside Re
(G-Re) is a panaxatriol saponin and is one of the most extensively
studied ginsenosides. G-Re exhibits diverse effects, including
antioxidant (13–16), anti-inflammatory (17–19)
and angiogenic activities (20).
Furthermore, G-Re has been reported to improve cardiac function
(21,22) and exert antidiabetic effects
(23–26). However, to the best of our
knowledge, the effects of G-Re on neuroinflammation-associated
neurotoxicity have not been fully investigated. The present study
investigated the effects of G-Re on the neuroinflammatory response
in LPS-stimulated microglia and its protective activities on
hippocampal neurons.
Materials and methods
Materials
G-Re (purity, >98%) was purchased from Ambo
Institute. LPS (phenol extracted from Salmonella
enteritidis), MTT, and poly-L-lysine (PLL) were purchased from
MilliporeSigma. KN93 (CAMK inhibitor) and KN92 (inactive analog of
KN93) were purchased from Cayman Chemical Company. PD98059 (ERK
inhibitor) and SP600125 (JNK inhibitor) were purchased from AG
Scientific, Inc. Antibodies against inducible NO synthase (iNOS;
cat. no. sc-651), cyclooxygenase 2 (COX-2; cat. no. sc-19999),
NF-κB p65 (cat. no. sc-372), IκB-α (cat. no. sc-371), extracellular
signal-regulated kinase (ERK; cat. no. sc-94), c-Jun N-terminal
kinase (JNK; cat. no. sc-571), CAMK2 (cat. no. sc-9035), p-CAMK2
(cat. no. sc-12886-R) and β-actin (cat. no. sc-47778) were
purchased from Santa Cruz Biotechnology, Inc. Antibodies against
p-ERK (cat. no. 4377S), p-JNK (cat. no. 9251S), p-IκB-α (cat. no.
2859S) and CAMK4 (cat. no. 4032S) were purchased from Cell
Signaling Technology, Inc. The antibody against p-CAMK4 (cat. no.
A0831) was purchased from Bioss Antibodies, Inc. The antibody
against TATA-binding protein (TBP; cat. no. PAB703Mu01) was
purchased from Cloud-Clone Corp. Horseradish peroxidase
(HRP)-conjugated goat anti-rabbit (cat. no. ADI-SAB-300-J) and
anti-mouse antibody (cat. no. ADI-SAB-100-J) were purchased from
Enzo Life Sciences. Anti-CD11b-APC antibody (cat. no. 17-0112-81),
CM-H2DCFDA, DMEM and fetal bovine serum (FBS) were
purchased from Thermo Fisher Scientific, Inc. FITC Annexin V
Apoptosis Detection kit I was purchased from BD Pharmingen (BD
Biosciences). Mouse TNF-α (cat. no. SMTA00B) and IL-6 ELISA kits
(cat. no. S6050) were purchased from R&D Systems, Inc.
Isolation of mouse primary microglia
and cell culture
ICR mice (8 weeks; weight, 30–35 g) were purchased
from DBL Co., Ltd. Experimental mice were housed in plastic cages
and maintained at a constant temperature (25±2°C) and humidity
(50±10%) under a 12/12-h light/dark cycle. Mice were provided with
free access to food and water. One male and one female mouse were
mated to obtain neonates. The animal experiments in the present
study were approved by the Pusan National University Institutional
Animal Care and Use Committee (approval no. PNU-2020-2651; Busan,
South Korea) and were conducted in accordance with the principles
in the Pusan National University Institutional Animal Care and Use
Committee guidelines. Mouse primary microglia were isolated as
previously described (27). After
10–14 neonates (postnatal day 2–5) of ICR mice housed in the
aforementioned conditions were euthanized by decapitation, primary
mixed glial cell cultures from the whole brains were prepared in
PLL-coated culture flasks (3.75×105 cells/ml) and
maintained in DMEM containing 10% FBS, 1 mM sodium pyruvate, 2 mM
L-glutamine and 50 mg/ml penicillin/streptomycin at 37°C in 5%
CO2. After 2 weeks, the culture flasks were placed on an
orbital shaker at 200 rpm and 37°C for 5 h. The cells in medium
were seeded in new PLL-coated plates and incubated at 37°C in 5%
CO2. After 2 h, unattached cells were removed and the
remaining microglia were used for further studies. To monitor
purity, cells (1.25×105 cells/0.5 ml) were immunostained
with CD11b-APC antibody (0.06 µg/0.5 ml) for 30 min on ice in the
dark and washed with cold PBS 3 times. Then, cells were resuspended
in 0.5 ml PBS and analyzed by flow cytometry (BD Accuri C6 flow
cytometer; BD Biosciences). >90% of cells were stained
positively (data not shown). BV2 mouse microglial cells and HT22
mouse hippocampal cells were kindly provided by Professor Youn-Chul
Kim (Wonkwang University, Iksan, South Korea). Cells were grown in
DMEM supplemented with 5% heat-inactivated FBS and 0.1%
penicillin/streptomycin (Thermo Fisher Scientific, Inc.) at 37°C in
a humidified atmosphere of 5% CO2 and 95% air.
Neurotoxicity of
microglial-conditioned medium
Mouse primary microglia (2.5×105
cells/ml) and BV2 microglial cells (2.5×105 cells/ml)
were treated with 2.5, 5.5 or 7.5 µg/ml G-Re for 1 h at 37°C and
then incubated with LPS (1 µg/ml) for 24 h at 37°C. After
incubation, cells were centrifuged at 400 × g at 4°C for 20 min to
obtain the cell-free supernatant (conditioned medium). HT22
hippocampal cells (4×104 cells/ml) were serum-starved
for 4 h and then treated with 50% BV2 cell-conditioned medium or
primary microglia-conditioned medium and 50% fresh DMEM at 37°C for
24 h. For controls, HT22 cells (4×104 cells/ml) were
treated with 2.5, 5.5 or 7.5 µg/ml G-Re for 1 h at 37°C and then
incubated with LPS (1 µg/ml) for 24 h at 37°C. The viability of
HT22 hippocampal cells was assessed by MTT assay or Annexin V assay
after incubation.
Cell viability assay
Cell viability was assessed using the MTT-based
colorimetric assay. BV2 (2.5×105 cells/ml) or HT22 cells
(4×104 cells/ml) were treated with MTT (50 µg/ml) for 3
h at 37°C in 5% CO2. After incubation, cell culture
supernatant was removed and the formazan crystals produced in
viable cells were solubilized with dimethyl sulfoxide. The
absorbance of each well was then measured at 570 nm using a
microplate reader (Bio-Rad Laboratories, Inc.).
Cell apoptosis assay
The Annexin V apoptosis assay was conducted using
flow cytometry according to the manufacturer's instructions.
Briefly, following incubation with the conditioned medium or G-Re
and/or LPS, HT22 cells (4×104 cells/ml) were washed with
PBS and resuspended in binding buffer at a density of
1×106 cells/ml. Cells were stained with Annexin V FITC
(2.5 µl) at 4°C for 15 min and propidium iodide (2.5 µl) at 4°C for
5 min in the dark, and then analyzed by flow cytometry (BD Accuri
C6 flow cytometer; BD Biosciences) within 1 h. Data were analyzed
using BD Accuri C6 software (BD Biosciences).
Measurement of nitrite
concentration
To measure nitrite (an indicator of NO levels), 100
µl aliquots were removed from culture supernatant of BV2 cells
(2.5×105 cells/ml) or primary microglia
(2.5×105 cells/ml) and incubated with an equal volume of
Griess reagent [1% sulfanilamide/0.1%
N-(1-naphthyl)-ethylenediamine dihydrochloride/2.5%
H3PO4] at room temperature for 10 min.
Nitrite concentration was determined by measuring the absorbance at
540 nm with a microplate spectrophotometer (Bio-Rad Laboratories,
Inc.). Sodium nitrite was used as a standard.
Measurement of TNF-α and IL-6
concentration
Mouse primary microglia (2.5×105
cells/ml) or BV2 cells (2.5×105 cells/ml) were first
incubated with 2.5, 5.5 or 7.5 µg/ml G-Re for 1 h at 37°C and then
treated with LPS (1 µg/ml) for 24 h at 37°C under 5%
CO2. Subsequently, TNF-α and IL-6 levels in the culture
medium were quantified using ELISA kits according to the
manufacturer's instructions.
Measurement of ROS
To evaluate the levels of intracellular ROS, BV2
cells (2.5×105 cells/ml) were treated with 1 µM
CM-H2DCFDA (general oxidative stress indicator) at 37°C
under 5% CO2 for 30 min. The cells were then harvested
and washed three times with PBS, after which the fluorescence
intensity was measured by fluorescence microscopy using an Axioplan
2 microscope (Zeiss GmbH) or flow cytometry (BD Accuri C6 flow
cytometer; BD Biosciences). Data were analyzed using BD Accuri C6
software (BD Biosciences). DMSO (0.04%) was used as vehicle.
Western blot analysis
Cytosolic extracts were harvested in ice-cold lysis
buffer (1% Triton X-100 and 1% deoxycholate in PBS). Nuclear
extracts were prepared as described previously (28). Briefly, BV2 cells were washed 3
times with cold PBS and the cell pellets were suspended in
hypotonic buffer (10.0 mM HEPES-KOH; pH 7.9; 1.5 mM
MgCl2; 10.0 mM KCl; 0.5 mM dithiothreitol; 0.2 mM PMSF)
and incubated for 15 min on ice. NP-40 (0.1%) was added to the cell
extract, incubated on ice for 1 min and centrifuged at 1,700 × g
for 1 min at 4°C. Following collection of cytosolic proteins from
the supernatant, nuclear proteins were extracted using buffer B
(20.0 mM HEPES-KOH; pH 7.9; 25% glycerol; 420.0 mM NaCl; 1.5 mM
MgCl2; 0.2 mM EDTA; 0.5 mM dithiothreitol; 0.2 mM PMSF)
for 30 min at 4°C with occasional vortexing. Following
centrifugation at 1,700 × g for 5 min at 4°C, supernatant was
collected and stored at −70°C. Protein content in these extracts
was determined using Bradford reagent (Bio-Rad Laboratories, Inc.).
The proteins (20 µg) in each sample were resolved by
SDS-polyacrylamide gel electrophoresis on 10% gels and transferred
to a polyvinylidene difluoride membrane. The blotted membrane was
incubated with 5% skimmed milk in PBS for 1 h at room temperature
and incubated with the appropriate antibodies (1:1,000) at 4°C
overnight. Subsequently, the membrane was incubated with
HRP-conjugated anti-rabbit or anti-mouse secondary antibodies (both
1:5,000) for 1 h at room temperature and the proteins were
visualized using an enhanced chemiluminescence detection system
(Amersham; Cytiva). Anti-β-actin was used as the loading control
for cytosolic proteins and anti-TBP was used as the loading control
for nuclear proteins. Quantitative image analysis was performed
using ImageJ 1.38× (National Institutes of Health) and data are
presented as fold of control.
Statistical analysis
All results are expressed as the mean ± SEM. Each
experiment was conducted in duplicate and repeated over three
times. Statistical analysis was performed using GraphPad Prism
software (version 7; GraphPad Software, Inc.). All data were
statistically analyzed using one-way analysis of variance followed
by Tukey's multiple comparisons test. P<0.05 was considered to
indicate a statistically significant difference.
Results
G-Re suppresses LPS-induced
inflammatory molecules in microglia
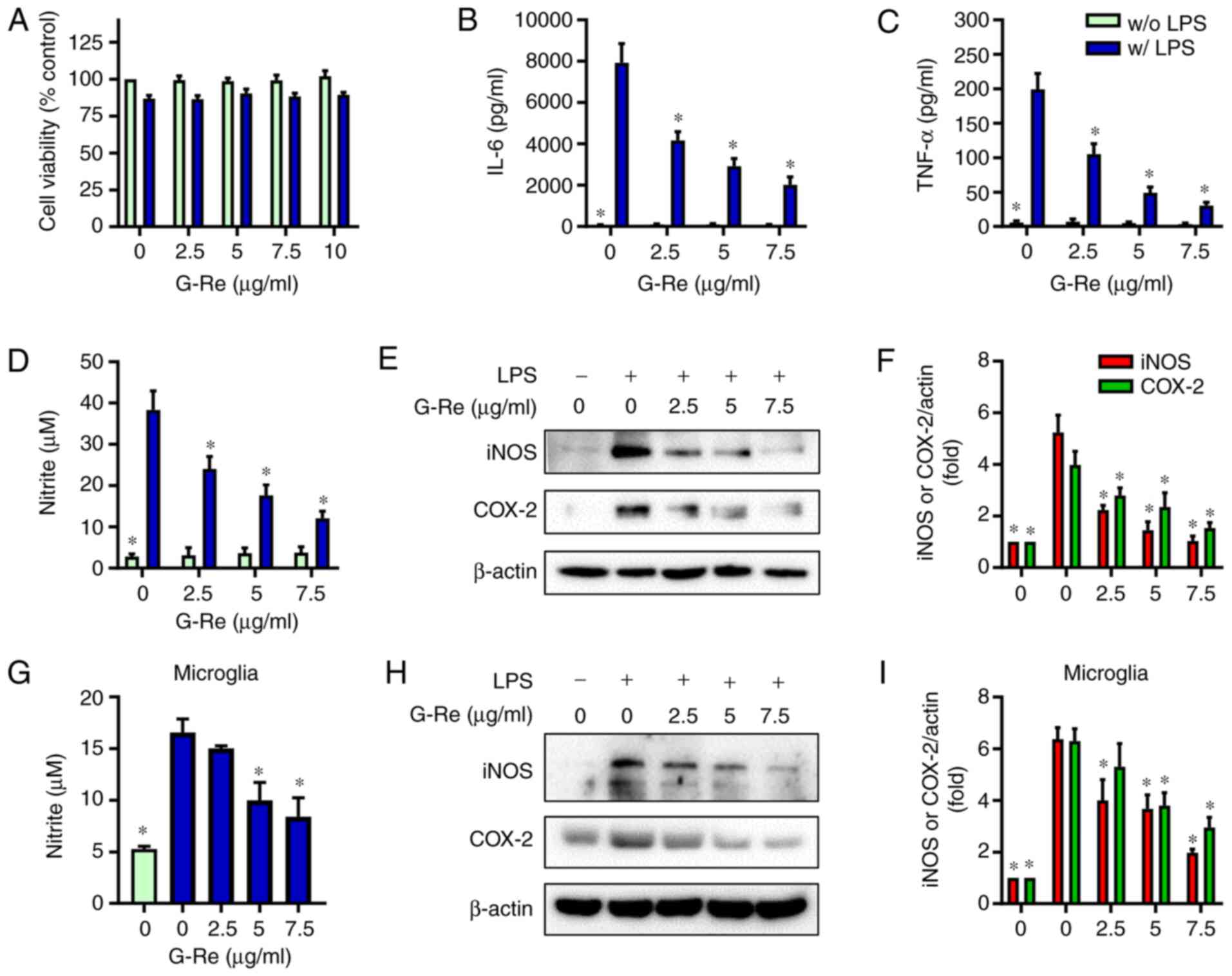
To determine the dose of G-Re, the effect of G-Re on
cell viability was examined using the MTT assay. While LPS induced
a little toxicity in BV2 microglial cells, G-Re at concentrations
up to 10 µg/ml exhibited no cytotoxicity in the presence or absence
of LPS (Fig. 1A). Therefore, cells
were treated with G-Re at concentrations <10 µg/ml in all
subsequent experiments.
In our preliminary study, LPS dose-dependently
induced an inflammatory response in BV2 cells (data not shown) and
1 µg/ml LPS induced a sufficient inflammatory response, which is
consistent with other reports (29–31).
To investigate whether G-Re could ameliorate the LPS-mediated
neuroinflammatory response, the effects of G-Re on cytokine and NO
production were investigated in BV2 cells. G-Re pretreatment
markedly inhibited the LPS-induced secretion of IL-6, TNF-α and NO
in a dose-dependent manner (Fig.
1B-D). Consistent with these results, G-Re dose-dependently
reduced the protein expression levels of iNOS and COX-2 in
LPS-stimulated BV2 cells (Fig. 1E and
F). Moreover, G-Re inhibited NO production, and iNOS and COX-2
expression in LPS-stimulated BV2 cells in a time-dependent manner.
Because preincubation of cells with G-Re for 1 and 2 h exhibited
similar effects (Fig. S1), the
present study pretreated cells with G-Re for 1 h in all
experiments. In addition, G-Re inhibited NO production, and the
expression levels of iNOS and COX-2 in LPS-stimulated mouse primary
microglia in a dose-dependent manner (Fig. 1G-I). These results suggested that
G-Re may inhibit LPS-induced expression of neuroinflammatory
molecules in microglia without damaging the cells.
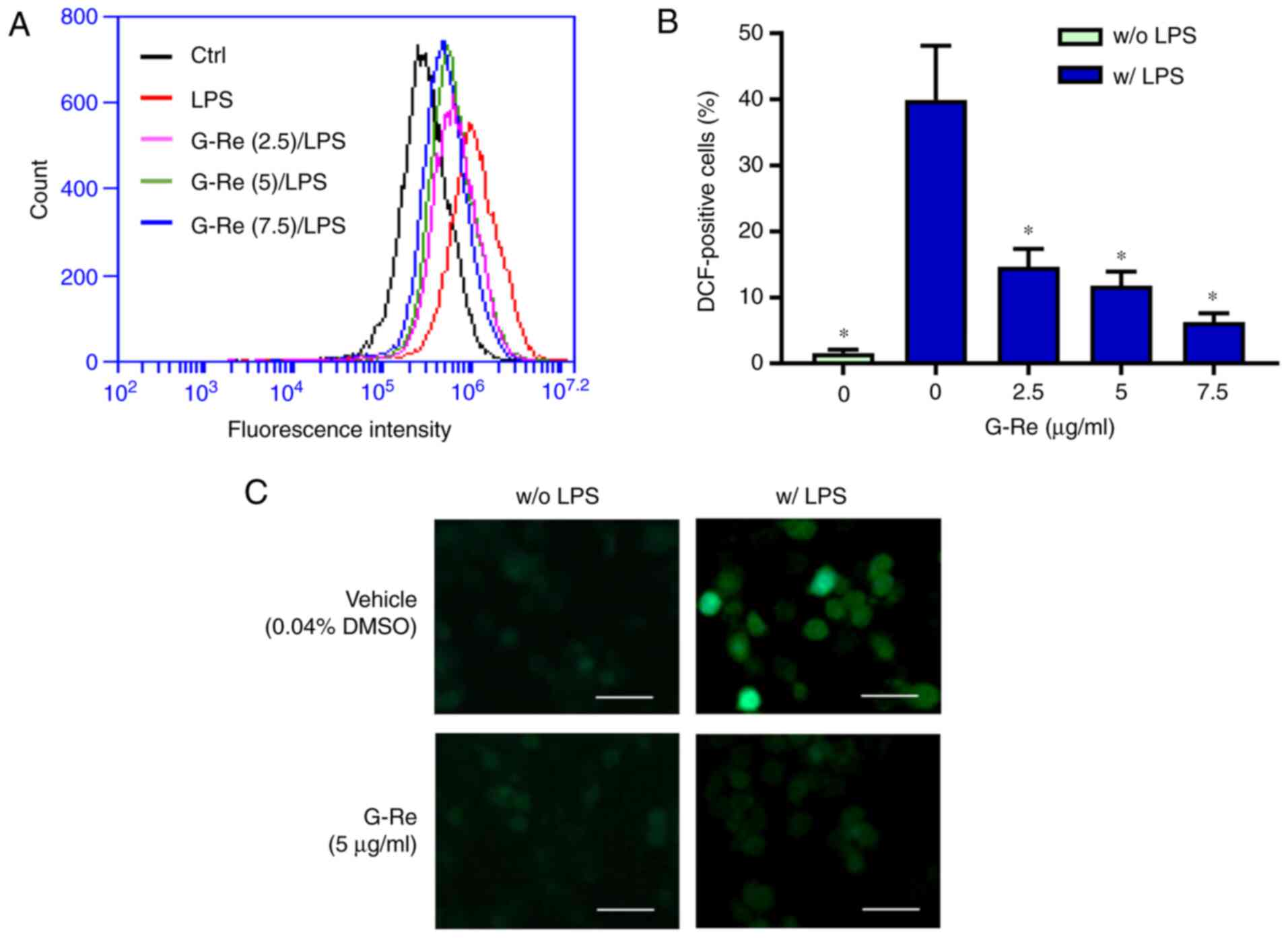
G-Re suppresses ROS production in BV2
microglial cells
To investigate the effect of G-Re on ROS production,
the levels of ROS in BV2 cells were detected using
CM-H2DCFDA. Pre-incubation with G-Re significantly
diminished the levels of ROS in LPS-stimulated cells, as determined
by flow cytometry (Fig. 2A and B)
and fluorescence microscopy (Fig.
2C), whereas G-Re did not affect basal ROS levels. These
results indicated that G-Re may inhibit LPS-induced production of
ROS in microglia.
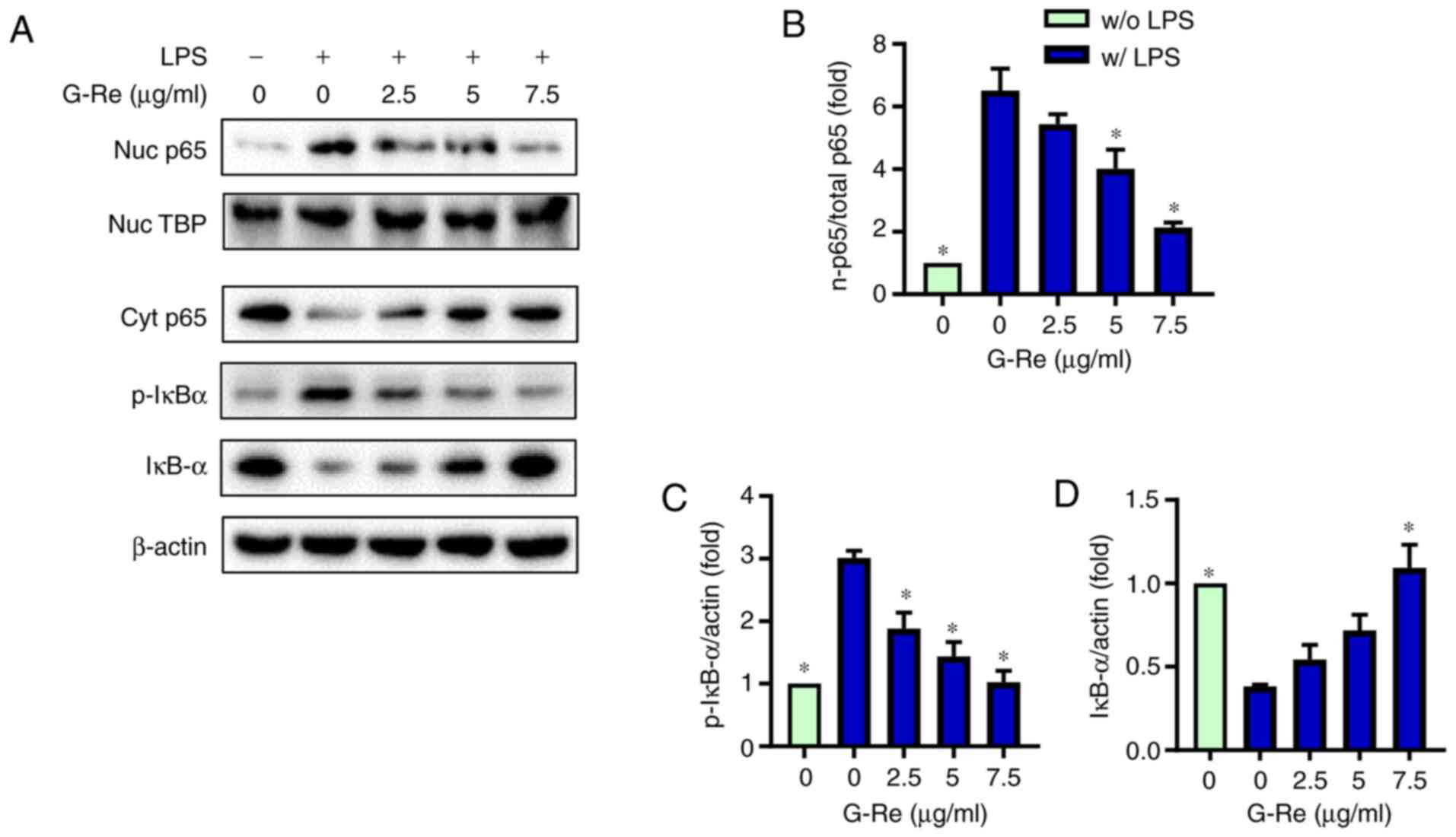
G-Re inhibits LPS-induced activation
of NF-κB
Since NF-κB is a major transcription factor
mediating the expression of numerous proinflammatory genes,
including IL-6, TNF-α, iNOS and COX-2, the present study
investigated the effects of G-Re on NF-κB activation. As shown in
Fig. 3A and B, LPS markedly
increased the nuclear levels of NF-κB p65 whereas it markedly
reduced the cytosolic levels of p65, indicating the nuclear
translocation of p65. However, upon G-Re pretreatment, the nuclear
level of p65 was decreased and cytosolic level of p65 was
simultaneously increased in a dose-dependent manner. Consistent
with this result, G-Re suppressed LPS-induced phosphorylation and
degradation of IκB-α in a dose-dependent manner (Fig. 3A, C and D). These results suggested
that G-Re inhibited LPS-induced nuclear translocation of NF-κB by
preventing phosphorylation and degradation of IκB-α.
G-Re inhibits CAMKs and MAPKs involved
in inflammatory mediator expression
To identify the molecular target of G-Re in the
upstream signaling pathway, the effects of pharmaceutical protein
kinase inhibitors of CAMK (KN93), ERK (PD98059) and JNK (SP600125)
were examined. NO production, and the expression levels of iNOS and
COX-2 were significantly inhibited by these inhibitors, whereas
KN92, an inactive analog of KN93, exhibited no significant effects
compared with those in LPS-treated cells (Fig. 4A-C). Furthermore, co-treatment with
kinase inhibitors and G-Re exhibited a slightly greater inhibition
of iNOS and COX-2 expression compared with treatment with either
G-Re alone or kinase inhibitors alone, but there was no significant
difference in NO production (Fig.
S2). Moreover, G-Re suppressed LPS-induced phosphorylation of
CAMK2, CAMK4, ERK and JNK in a dose-dependent manner (Fig. 4D-H). Because these kinases are
activated when they are phosphorylated (32), these results indicated that G-Re
inhibited activation of these kinases. Therefore, G-Re may suppress
the synthesis of proinflammatory mediators by decreasing CAMK2,
CAMK4, ERK and JNK activities.
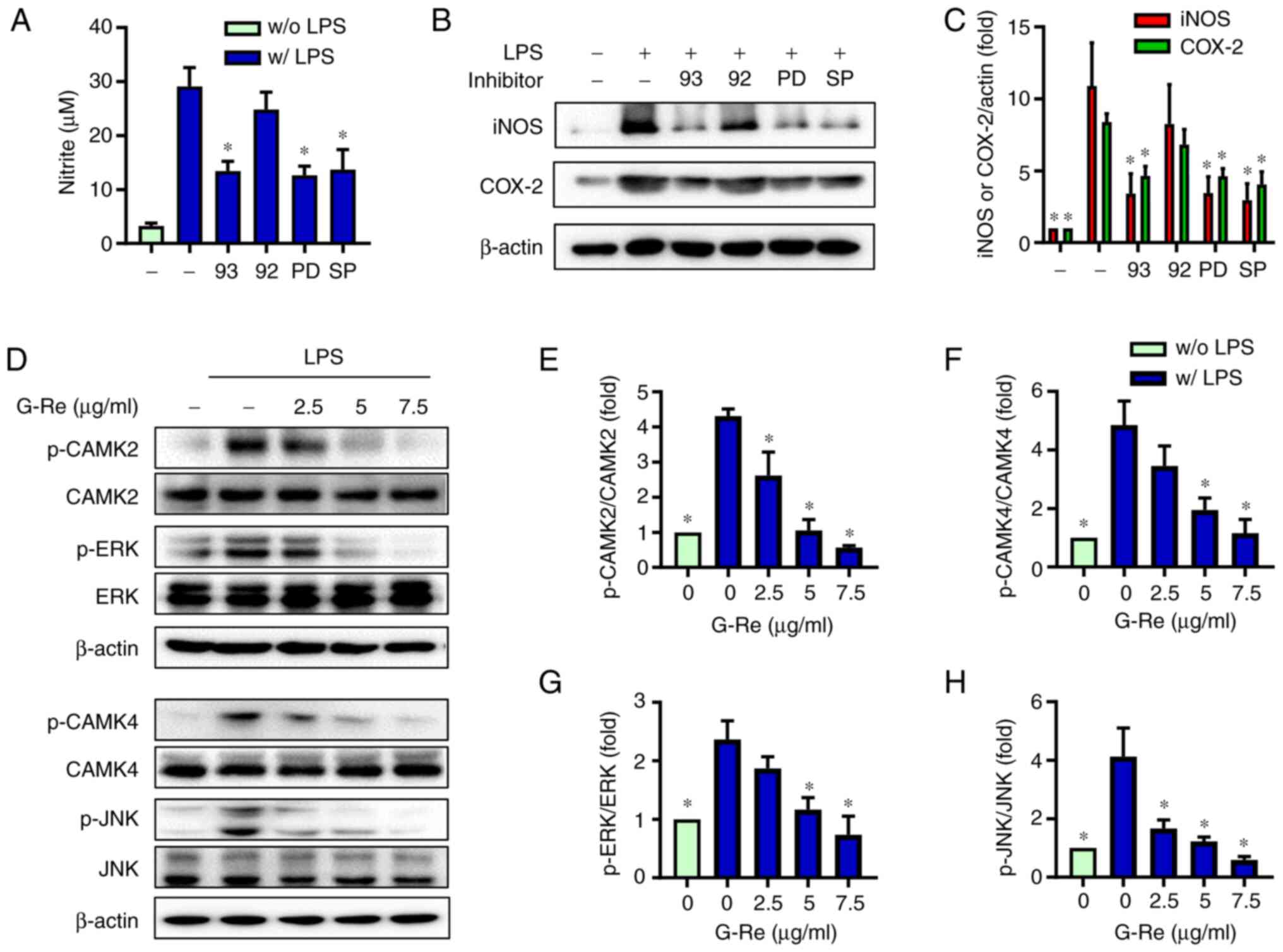 | Figure 4.Effects of G-Re on LPS-induced
activation of CAMKs and mitogen-activated protein kinases. BV2
cells were incubated with KN93 (5 µM), KN92 (5 µM), PD98059 (5 µM)
or SP600125 (5 µM), followed by LPS (1 µg/ml) treatment for 24 h.
(A) Nitric oxide content was measured, and (B) protein expression
levels of iNOS and COX-2 were detected by western blotting. (C)
Relative intensity of each band (normalized to β-actin) was
indicated as a ratio to control. BV2 cells were treated with
various concentrations of G-Re for 1 h, followed by LPS treatment
(1 µg/ml) for 30 min. (D) Expression levels of unphosphorylated
kinases or p-kinases were analyzed by western blotting. (E-H)
Relative intensity of p-kinases (normalized to respective
unphosphorylated kinases) was indicated as a ratio to control.
*P<0.05 vs. the group treated with LPS alone. CAMK,
Ca2+/calmodulin-dependent protein kinase; COX-2,
cyclooxygenase 2; ERK, extracellular signal-regulated kinase; G-Re,
ginsenoside Re; iNOS, inducible nitric oxide synthase; JNK, c-Jun
N-terminal kinases; LPS, lipopolysaccharide; p, phosphorylated. |
Protective effects of G-Re against
microglia-mediated neuronal cell death
Growing evidence has indicated that inflammatory
mediators produced by microglia can induce neuronal cell death
(3–6). Since the present results indicated
that G-Re inhibited the expression of inflammatory mediators in
LPS-induced microglia, the effects of G-Re on indirect toxicity to
HT22 hippocampal neuronal cells were investigated. To accomplish
this, HT22 cells were treated with BV2-conditioned medium and an
MTT assay was performed to assess cell viability and an Annexin V
assay was conducted to detect apoptosis. When HT22 cells were
incubated with the conditioned medium from LPS-stimulated BV2
cells, cell viability was markedly decreased. However, treatment of
HT22 cells with the conditioned medium from BV2 cells incubated
with both LPS and G-Re enhanced HT22 cell viability in a
dose-dependent manner; treatment of the cells with the conditioned
medium from only G-Re-treated BV2 cells exhibited no effect
(Fig. 5A). Consistent with the MTT
assay results, conditioned medium from LPS-treated BV2 cells led to
marked apoptotic cell death [both early (Annexin V-positive and
PI-negative fraction) and late (Annexin V-positive and PI-positive
fraction) apoptosis], whereas application of the conditioned medium
from LPS + G-Re-treated BV2 microglial cells decreased Annexin
V-positive cells (Fig. 5C and D).
Notably, direct stimulation of HT22 neuronal cells with either LPS
alone or both LPS and G-Re had no significant effect on cell
viability (Fig. 5B) and apoptotic
cell death (Fig. 5C and D).
Furthermore, similar results were obtained from the MTT assay
following treatment of cells with the conditioned medium from mouse
primary microglial cells (Fig. 5E).
Overall, these results suggested that factors released from
LPS-treated microglial cells may induce neuronal toxicity and that
G-Re could protect HT22 hippocampal cells by reducing these factors
released from microglial cells.
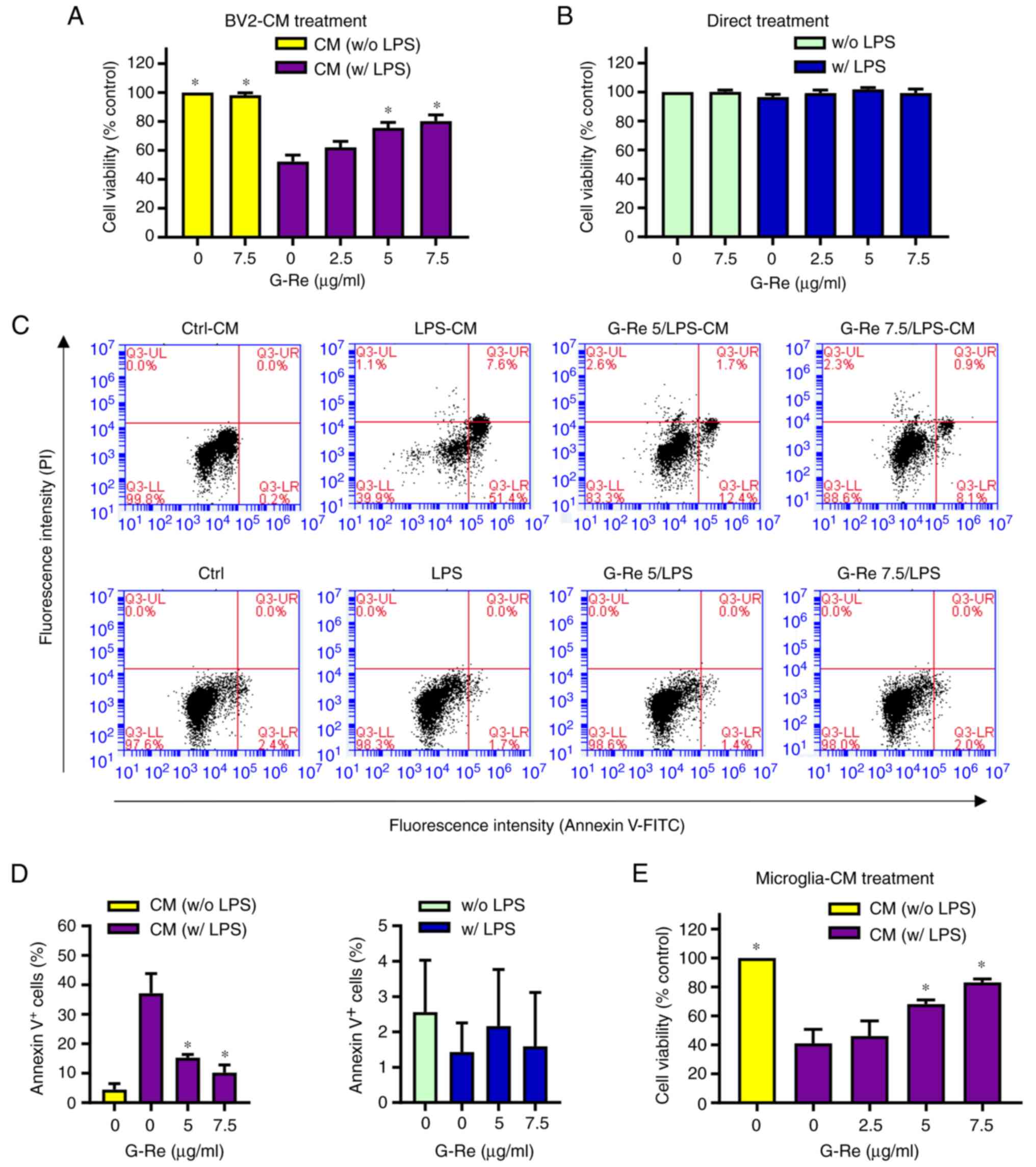 | Figure 5.Effects of G-Re on
inflammation-induced neurotoxicity of HT22 hippocampal cells. (A)
BV2 cells were incubated with LPS (1 µg/ml) in the presence or
absence of G-Re for 24 h, and then HT22 cells were treated with
BV2-CM. After 24 h, the viability of HT22 cells was estimated by
MTT assay. (B) HT22 cells were incubated with LPS (1 µg/ml) in the
presence or absence of G-Re for 24 h, and cell viability was
estimated by MTT assay. (C and D) HT22 cells were treated with
BV2-CM as aforementioned, or with LPS (1 µg/ml) in the presence or
absence of G-Re for 24 h, and an apoptosis assay was performed. (C)
Representative flow cytometry plots and (D) percentage of Annexin
V-positive cells were shown. (E) Mouse primary microglia were
incubated with LPS (1 µg/ml) in the presence or absence of G-Re for
24 h, and then the microglia-CM was added to HT22 cells. After 24
h, the viability of HT22 cells was estimated by MTT assay.
*P<0.05 vs. the group treated with LPS alone. CM, conditioned
medium; G-Re, ginsenoside Re; LPS, lipopolysaccharide; PI,
propidium iodide. |
Discussion
The present study investigated the anti-inflammatory
effects of G-Re on LPS-stimulated microglial cells. The results
revealed that G-Re pretreatment significantly inhibited the
LPS-mediated production of IL-6, TNF-α, NO and ROS, and the
expression levels of iNOS and COX-2. IL-6 and TNF-α are
proinflammatory cytokines, which are known to be involved in the
pathogenesis of various inflammation-related diseases. IL-6
administration has been shown to cause mechanical allodynia and
thermal hyperalgesia (33). In
addition, IL-6 may impair oligodendrocyte regeneration and induce
demyelination (34). Moreover,
chronic microglial activation in GFAP-IL6 mice contributed to the
age-dependent loss of cerebellar volume and impairment in motor
function (35). IL-6 levels have
been reported to be increased in the cerebrospinal fluid of
patients with viral meningitis, encephalitis, systemic lupus
erythematosus and stroke (36–39).
IL-6 may also enhance neuronal damage induced by β-amyloid peptide
in cultured rat cortical neurons (40). Furthermore, the TNF-α protein
synthesis inhibitor has been shown to restore neuronal function and
reverse cognitive deficits induced by chronic neuroinflammation
(41), and it has been demonstrated
that inhibition of TNF-α can lead to favorable outcomes in
Alzheimer's disease (42).
ROS in the brain are involved in the development of
oxidative neuronal damage and progression of neurodegenerative
diseases (43,44). In addition, high amounts of NO
produced by iNOS in activated microglial cells are considered to
cause neuronal cell damage and lead to neurodegeneration (45,46),
because neurons are notably sensitive to NO-induced cell death
(47). Furthermore, NO reacts with
superoxide to produce peroxynitrite, which is a powerful oxidant
and a potent inducer of cell death (48). Thus, suppression of IL-6, TNF-α, NO
and ROS production by G-Re in microglial cells may contribute to
reduced neurodegeneration and neuroinflammation. In agreement with
the present results, previous studies have reported that G-Re
exhibits anti-inflammatory effects (17–19).
G-Re has been shown to inhibit the release of histamine from human
mast cells, and the expression of IL-1α, IL-8, IL-10 and RANTES in
a human alveolar cancer cell line (17).
NF-κB is a vital transcription factor for several
genes, which is involved in regulating immune and inflammatory
responses, such as the expression of cytokines, iNOS and COX-2
(49). Improper regulation of NF-κB
has been shown to be directly involved in a wide range of human
disorders, including neuroinflammatory and neurodegenerative
diseases (50,51); therefore, the development of drugs
that regulate NF-κB is considered a promising strategy for
therapeutic manipulation of inflammatory disease (52). The present study revealed that G-Re
significantly inhibited the nuclear translocation of NF-κB and the
degradation of IκB-α. Moreover, G-Re effectively inhibited the
phosphorylation, and thus the activation, of CAMK2, CAMK4, ERK and
JNK. Thus, these results suggested that G-Re may inhibit the
expression of inflammatory mediators, such as IL-6, TNF-α, iNOS and
COX-2 via blocking CAMK2/CAMK4/ERK/JNK/NF-κB signaling in
microglial cells. To the best of our knowledge, the present study
is the first to demonstrate that G-Re suppressed CAMK2/CAMK4
activities.
Brain inflammation is considered to promote
neurodegenerative diseases and cognitive dysfunction. Notably, an
increasing number of reports have shown that systemic or
intracerebroventricular administration of LPS can lead to β-amyloid
generation and memory deficiency (53–55).
LPS-induced neuroinflammation may also be accompanied by
hippocampal neuronal death and microglia activation. During
neuroinflammation, microglia are activated and release
proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α. The
neuroinflammatory molecules released by activated microglial cells
can induce indirect neuronal toxicity, which may also contribute to
neurodegenerative disorders (56–59).
The results of the present study demonstrated that apoptosis of
HT22 hippocampal neuronal cells was induced following treatment
with the conditioned medium from LPS-stimulated microglia, which is
hypothesized to secrete neurotoxic molecules. By contrast, G-Re
significantly attenuated HT22 cell death, and this protective
effect of G-Re on indirect neuronal toxicity may be due to its
ability to reduce the secretion of these neurotoxic molecules from
microglia. Neither LPS nor G-Re exhibited any direct effects on the
viability of HT22 cells after treatment for 24 h; therefore, it may
be hypothesized that it is not the direct effects of G-Re that
inhibit HT22 cell death, but the effects of G-Re on the microglia
that suppress HT22 cell death. These results suggested that G-Re
could potentially ameliorate various neuroinflammatory and
neurodegenerative diseases. This hypothesis is supported by a study
demonstrating that G-Re attenuated neuroinflammation in a
symptomatic ALS animal model generated using human-superoxide
dismutase 1 transgenic mice (60).
This previous study demonstrated that administration of G-Re
enhanced the number of motor neurons and reduced the number of
microglia.
In conclusion, the present study demonstrated that
G-Re suppressed LPS-induced overproduction of pro-inflammatory
mediators, including IL-6, TNF-α, NO and ROS, in microglial cells.
In addition, G-Re significantly inhibited LPS-induced activation of
NF-κB p65, CAMK2, CAMK4 and MAPKs, such as ERK and JNK.
Furthermore, G-Re attenuated HT22 hippocampal cell death induced by
neurotoxic molecules released from activated microglia. These
findings suggested that G-Re may have therapeutic potential for the
treatment of neuroinflammatory and neurodegenerative diseases.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Education (grant no. 2015R1D1A3A01020633)
and PNU-RENovation (2019–2020).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
IM, JHK and JES performed the experiments. YK and IM
analyzed the data. YK and IM confirm the authenticity of all the
raw data. YK wrote the manuscript and conceptualized the study
design. YK and JHK revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments in the present study were
approved by the Pusan National University Institutional Animal Care
and Use Committee (approval no. PNU-2020-2651).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yang I, Han SJ, Kaur G, Crane C and Parsa
AT: The role of microglia in central nervous system immunity and
glioma immunology. J Clin Neurosci. 17:6–10. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Streit WJ, Conde JR, Fendrick SE, Flanary
BE and Mariani CL: Role of microglia in the central nervous
system's immune response. Neurol Res. 27:685–691. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boje KM and Arora PK: Microglial-produced
nitric oxide and reactive nitrogen oxides mediate neuronal cell
death. Brain Res. 587:250–256. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Block ML, Zecca L and Hong JS:
Microglia-mediated neurotoxicity: Uncovering the molecular
mechanisms. Nat Rev Neurosci. 8:57–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hickman S, Izzy S, Sen P, Morsett L and El
Khoury J: Microglia in neurodegeneration. Nat Neurosci.
21:1359–1369. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Song WM and Colonna M: The identity and
function of microglia in neurodegeneration. Nat Immunol.
19:1048–1058. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Yin H, Zhao M and Lu Q: TLR2 and
TLR4 in autoimmune diseases: A comprehensive review. Clin Rev
Allergy Immunol. 47:136–147. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaminska B, Gozdz A, Zawadzka M,
Ellert-Miklaszewska A and Lipko M: MAPK signal transduction
underlying brain inflammation and gliosis as therapeutic target.
Aanat Rec. 292:1902–1913. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sun J and Nan G: The extracellular
signal-regulated kinase 1/2 pathway in neurological diseases: A
potential therapeutic target (Review). Int J Mol Med. 39:1338–1346.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Song Q, Fan C, Wang P, Li Y, Yang M and Yu
SY: Hippocampal CA1 βCaMKII mediates neuroinflammatory responses
via COX-2/PGE2 signaling pathways in depression. J
Neuroinflammation. 15:3382018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang HH, Hsieh HL and Yang CM: Calmodulin
kinase II-dependent transactivation of PDGF receptors mediates
astrocytic MMP-9 expression and cell motility induced by
lipoteichoic acid. J Neuroinflammation. 7:842010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leung KW and Wong AS: Pharmacology of
ginsenosides: A literature review. Chin Med. 5:202010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang GD, Zhong XF, Deng ZY and Zeng R:
Proteomic analysis of ginsenoside Re attenuates hydrogen
peroxide-induced oxidative stress in human umbilical vein
endothelial cells. Food Funct. 7:2451–2461. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee GH, Lee WJ, Hur J, Kim E, Lee HG and
Seo HG: Ginsenoside Re mitigates 6-hydroxydopamine-induced
oxidative stress through upregulation of GPX4. Molecules.
25:1882020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xie JT, Shao ZH, Vanden Hoek TL, Chang WT,
Li J, Mehendale S, Wang CZ, Hsu CW, Becker LB, Yin JJ and Yuan CS:
Antioxidant effects of ginsenoside Re in cardiomyocytes. Eur J
Pharmacol. 532:201–207. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
López MV, Cuadrado MP, Ruiz-Poveda OM, Del
Fresno AM and Accame ME: Neuroprotective effect of individual
ginsenosides on astrocytes primary culture. Biochim Biophys Acta.
1770:1308–1316. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bae HM, Cho OS, Kim SJ, Im BO, Cho SH, Lee
S, Kim MG, Kim KT, Leem KH and Ko SK: Inhibitory effects of
ginsenozside Re isolated from ginseng berry on histamine and
cytokine release in human mast cells and human alveolar epithelial
cells. J Ginseng Res. 36:369–374. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee IA, Hyam SR, Jang SE, Han MJ and Kim
DH: Ginsenoside Re ameliorates inflammation by inhibiting the
binding of lipopolysaccharide to TLR4 on macrophages. J Agric Food
Chem. 60:9595–9602. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lee KW, Jung SY, Choi SM and Yang EJ:
Effects of ginsenoside Re on LPS-induced inflammatory mediators in
BV2 microglial cells. BMC Complement Altern Med. 12:1962012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang YC, Chen CT, Chen SC, Lai PH, Liang
HC, Chang Y, Yu LC and Sung HW: A natural compound (ginsenoside Re)
isolated from Panax ginseng as a novel angiogenic agent for
tissue regeneration. Pharm Res. 22:636–646. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu Y, Sun J, Liu J, Wang P and Wang C:
Ginsenoside Re preserves cardiac function and ameliorates left
ventricular remodeling in a rat model of myocardial infarction. J
Cardiovasc Pharmacol. 75:91–97. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen RC, Wang J, Yang L, Sun GB and Sun
XB: Protective effects of ginsenoside Re on
lipopolysaccharide-induced cardiac dysfunction in mice. Food Funct.
7:2278–2287. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cho WC, Chung WS, Lee SK, Leung AW, Cheng
CH and Yue KK: Ginsenoside Re of Panax ginseng possesses
significant antioxidant and antihyperlipidemic efficacies in
streptozotocin-induced diabetic rats. Eur J Pharmacol. 550:173–179.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Quan HY, Yuan HD, Jung MS, Ko SK, Park YG
and Chung SH: Ginsenoside Re lowers blood glucose and lipid levels
via activation of AMP-activated protein kinase in HepG2 cells and
high-fat diet fed mice. Int J Mol Med. 29:73–80. 2012.PubMed/NCBI
|
|
25
|
Gao Y, Yang MF, Su YP, Jiang HM, You XJ,
Yang YJ and Zhang HL: Ginsenoside Re reduces insulin resistance
through activation of PPAR-γ pathway and inhibition of TNF-α
production. J Ethnopharmacol. 147:509–516. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi Y, Wan X, Shao N, Ye R, Zhang N and
Zhang Y: Protective and anti-angiopathy effects of ginsenoside Re
against diabetes mellitus via the activation of p38 MAPK, ERK1/2
and JNK signaling. Mol Med Rep. 14:4849–4856. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lian H, Roy E and Zheng H: Protocol for
primary microglial culture preparation. Bio Protoc.
6:e19892016.PubMed/NCBI
|
|
28
|
Kim Y, Moon JS, Lee KS, Park SY, Cheong J,
Kang HS, Lee HY and Kim HD: Ca2+/calmodulin-dependent
protein phosphatase calcineurin mediates the expression of iNOS
through IKK and NF-kappaB activity in LPS-stimulated mouse
peritoneal macrophages and RAW 264.7 cells. Biochem Biophys Res
Commun. 314:695–703. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park SY, Kim YH, Kim Y and Lee SJ:
Aromatic-turmerone's anti-inflammatory effects in microglial cells
are mediated by protein kinase A and heme oxygenase-1 signaling.
Neurochem Int. 61:767–777. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song JS, Shin JE, Kim JH and Kim Y:
Gardenia jasminoides exerts anti-inflammatory activity via
Akt and p38-dependent heme oxygenase-1 upregulation in microglial
cells. J Life Sci. 27:8–14. 2017. View Article : Google Scholar
|
|
31
|
You MM, Chen YF, Pan YM, Liu YC, Tu J,
Wang K and Hu FL: Royal jelly attenuates lps-induced inflammation
in BV-2 microglial cells through modulating NF-κB and p38/JNK
signaling pathways. Mediators Inflamm. 2018:78343812018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ardito F, Giuliani M, Perrone D, Troiano G
and Muzio LL: The crucial role of protein phosphorylation in cell
signaling and its use as targeted therapy (Review). Int J Mol Med.
40:271–280. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou YQ, Liu Z, Liu ZH, Chen SP, Li M,
Shahveranov A, Ye DW and Tian YK: Interleukin-6: An emerging
regulator of pathological pain. J Neuroinflammation. 13:1412016.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Petkovic F and Castellano B: The role of
interleukin-6 in central nervous system demyelination. Neural Regen
Res. 11:19222016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gyengesi E, Rangel A, Ullah F, Liang H,
Niedermayer G, Asgarov R, Venigalla M, Gunawardena D, Karl T and
Münch G: Chronic Microglial activation in the GFAP-IL6 mouse
contributes to age-dependent cerebellar volume loss and impairment
in motor function. Front Neurosci. 13:3032019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hirohata S and Miyamoto T: Elevated levels
of interleukin-6 in cerebrospinal fluid from patients with systemic
lupus erythematosus and central nervous system involvement.
Arthritis Rheum. 33:644–649. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Frei K, Leist TP, Meager A, Gallo P,
Leppert D, Zinkernagel RM and Fontana A: Production of B cell
stimulatory factor-2 and interferon gamma in the central nervous
system during viral meningitis and encephalitis. Evaluation in a
murine model infection and in patients. J Exp Med. 168:449–453.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bodro M, Compta Y, Llansó L, Esteller D,
Doncel-Moriano A, Mesa A, Rodríguez A, Sarto J, Martínez-Hernandez
E, Vlagea A, et al: Increased CSF levels of IL-1β, IL-6, and ACE in
SARS-CoV-2-associated encephalitis. Neurol Neuroimmunol
Neuroinflamm. 7:e8212020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tarkowski E, Rosengren L, Blomstrand C,
Wikkelsö C, Jensen C, Ekholm S and Tarkowski A: Early intrathecal
production of interleukin-6 predicts the size of brain lesion in
stroke. Stroke. 26:1393–1398. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qiu Z and Gruol DL: Interleukin-6,
beta-amyloid peptide and NMDA interactions in rat cortical neurons.
J Neuroimmunol. 139:51–57. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Belarbi K, Jopson T, Tweedie D, Arellano
C, Luo W, Greig NH and Rosi S: TNF-α protein synthesis inhibitor
restores neuronal function and reverses cognitive deficits induced
by chronic neuroinflammation. J Neuroinflammation. 9:232012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shamim D and Laskowski M: Inhibition of
inflammation mediated through the tumor necrosis factor α
biochemical pathway can lead to favorable outcomes in Alzheimer
disease. J Cent Nerv Syst Dis. 9:11795735177225122017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Singh A, Kukreti R, Saso L and Kukreti S:
Oxidative stress: A key modulator in neurodegenerative diseases.
Molecules. 24:15832019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kim GH, Kim JE, Rhie SJ and Yoon S: The
role of oxidative stress in neurodegenerative diseases. Exp
Neurobiol. 24:325–340. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tewari D, Sah AN, Bawari S, Nabavi SF,
Dehpour AR, Shirooie S, Braidy N, Fiebich BL, Vacca RA and Nabavi
SM: Role of Nitric oxide in neurodegeneration: Function,
regulation, and inhibition. Curr Neuropharmacol. 19:114–126. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang L, Hagemann TL, Kalwa H, Michel T,
Messing A and Feany MB: Nitric oxide mediates glial-induced
neurodegeneration in Alexander disease. Nat Commun. 6:89662015.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wei T, Chen C, Hou J, Xin W and Mori A:
Nitric oxide induces oxidative stress and apoptosis in neuronal
cells. Biochim Biophys Acta. 1498:72–79. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Pacher P, Beckman JS and Liaudet L: Nitric
oxide and peroxynitrite in health and disease. Physiol Rev.
87:315–424. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ahn KS and Aggarwal BB: Transcription
factor NF-kappaB: A sensor for smoke and stress signals. Ann N Y
Acad Sci. 1056:218–233. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sivandzade F, Prasad S, Bhalerao A and
Cucullo L: NRF2 and NF-κB interplay in cerebrovascular and
neurodegenerative disorders: Molecular mechanisms and possible
therapeutic approaches. Redox Biol. 21:1010592019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shih RH, Wang CY and Yang CM: NF-kappaB
signaling pathways in neurological inflammation: A mini review.
Front Mol Neurosci. 8:772015. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Giridharan S and Srinivasan M: Mechanisms
of NF-kappaB p65 and strategies for therapeutic manipulation. J
inflamm Res. 11:407–419. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Zhao J, Bi W, Xiao S, Lan X, Cheng X,
Zhang J, Lu D, Wei W, Wang Y, Li H, et al: Neuroinflammation
induced by lipopolysaccharide causes cognitive impairment in mice.
Sci Rep. 9:57902019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB,
Oh KW and Hong JT: Neuro-inflammation induced by lipopolysaccharide
causes cognitive impairment through enhancement of beta-amyloid
generation. J Neuroinflammation. 5:372008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Czerniawski J, Miyashita T, Lewandowski G
and Guzowski JF: Systemic lipopolysaccharide administration impairs
retrieval of context-object discrimination, but not spatial,
memory: Evidence for selective disruption of specific
hippocampus-dependent memory functions during acute
neuroinflammation. Brain Behav Immun. 44:159–166. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dean JM, Wang X, Kaindl AM, Gressens P,
Fleiss B, Hagberg H and Mallard C: Microglial MyD88 signaling
regulates acute neuronal toxicity of LPS-stimulated microglia in
vitro. Brain Behav Immun. 24:776–783. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chao CC, Hu S, Molitor TW, Shaskan EG and
Peterson PK: Activated microglia mediate neuronal cell injury via a
nitric oxide mechanism. J Immunol. 149:2736–2741. 1992.PubMed/NCBI
|
|
58
|
Banati RB, Gehrmann J, Schubert P and
Kreutzberg GW: Cytotoxicity of microglia. Glia. 7:111–118. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vitner EB, Farfel-Becker T, Eilam R, Biton
I and Futerman AH: Contribution of brain inflammation to neuronal
cell death in neuronopathic forms of Gaucher's disease. Brain.
135:1724–1735. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cai M and Yang EJ: Ginsenoside Re
attenuates neuroinflammation in a symptomatic ALS animal model. Am
J Chin Med. 44:401–413. 2016. View Article : Google Scholar : PubMed/NCBI
|