Introduction
Atherosclerotic plaque rupture is the most common
cause of cardiac mortality worldwide (1). Previous studies have reported that
vascular smooth muscle cells (VSMCs) are the primary cell type
involved in all stages of human atherosclerotic plaque development
(2,3). Leukocyte recruitment to the vessel
also directly contributes to the progression of atherosclerosis
(AS), as well as the expression of adhesion molecules on VSMCs
(4).
Following cholesterol loading, VSMCs undergo
significant morphological and functional alterations (5,6); in
particular, adhesion molecule expression is significantly
upregulated (7). Intercellular
adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1
(VCAM-1)-mediated leukocyte recruitment to the vessel intima is a
crucial event in AS (8,9). Since the accumulation of
monocyte-derived cells in atherosclerotic plaques directly
contributes to the progression of AS, the inhibition of
cholesterol-induced adhesion molecule expression on VSMCs may serve
as a potential therapeutic strategy.
Hypercholesterolemia, which is considered to be the
most common and significant risk factor of AS, has been linked to
increased reactive oxygen species (ROS) production (10,11).
Abnormal ROS accumulation is associated with numerous
pathophysiological conditions, such as diabetes, obesity and heart
failure (11). Oxidative stress
mediates endothelial dysfunction (12), modifies the phenotype of VSMCs and
affects extracellular matrix synthesis (13). Due to the involvement in the
phenotypic switching of VSMCs, ROS may also contribute to cellular
morphological and functional alterations. However, the role of ROS
in cholesterol-induced VSMC functional damage is not completely
understood.
Metformin is used extensively as a first-line
medication for type 2 diabetes mellitus (14). In addition to its glucose-lowering
effect, metformin displays cardiovascular protective effects,
including protection against cardiac ischemia-reperfusion injury
(15), and subsequent suppression
of the inflammatory response and development of AS (16). However, the effects of metformin on
cholesterol loading-induced morphological and functional
alterations in VSMCs are not completely understood. The present
study aimed to further the current understanding of the mechanisms
underlying the damaging effects of cholesterol on VSMC function,
and to clarify the protective effect of metformin on VSMCs.
Materials and methods
Materials
Human aortic VSMCs (cat. no. 6110) and smooth muscle
cell medium (SMCM; cat. no. 1101) were obtained from ScienCell
Research Laboratories, Inc. Cholesterol-methyl-β-cyclodextrin
(cholesterol; cat. no. C4951), metformin (cat. no. D150959),
paraformaldehyde and 2′,7′-dichlorofluorescin diacetate (DCFH-DA;
cat. no. D6883) were obtained from Sigma-Aldrich; Merck KGaA.
Compound C (cat. no. HY-13418A), SB203580 (cat. no. HY-10256) and
BAY11-7082 (cat. no. HY-13453) were obtained from MCE.
TRIzol® and CELLTRACE Violet were obtained from
Invitrogen (Thermo Fisher Scientific, Inc.). The Transcriptor First
Strand cDNA Synthesis kit and FastStart Universal SYBR-Green Master
Mix were obtained from Roche Applied Science. Human monocytic THP-1
cells were obtained from National Collection of Authenticated Cell
Center (cat. no. TCHu 57).
Cell culture conditions
Human aortic VSMCs were cultured in SMCM (Science
Cell Research Laboratories, cat. no. 1101) with 2% FBS, 1% smooth
muscle cell growth supplement (SMCGS; both included in the medium),
100 U/ml penicillin and 100 µg/ml streptomycin at 37°C with 5%
CO2. At 80% confluence, VSMCs were synchronized by
replacing the culture media with FBS- and SMCGS-free basic SMCM.
For experiments involving pharmacological reagents, VSMCs were
pretreated which metformin (10 and 100 nm, 1 µm), Compound C (10
µm), SB203580 (10 µm) or BAY11-7082 (10 µm) for 2 h and
subsequently followed by cholesterol (5 µg/ml) for 72 h at 37°C
with 5% CO2. In all experiments, passage 3–6 VSMCs were
used.
Reverse transcription-quantitative PCR
(RT-qPCR)
Following treatment with 5 µg/ml cholesterol at 37°C
with 5% CO2 for 72 h, total RNA was extracted from VSMCs
using TRIzol according to the manufacturer's protocol. Total RNA (4
µg) was reverse transcribed into cDNA using the Transcriptor First
Strand cDNA Synthesis kit. Subsequently, qPCR was performed using
FastStart Universal SYBR-Green Master Mix and a quantitative
fluorescence PCR system (Bio-Rad Laboratories; CFX96 Real-Time PCR
Detection System). The following conditions were used: 95°C for 30
sec, 95°C for 5 sec and 40 cycles at 60°C for 5 sec. The primers
used for qPCR are presented in Table
I. mRNA expression levels were quantified using the
2−∆∆Cq method and normalized to the internal reference
gene β-actin (17). RT-qPCR was
performed in triplicate.
 | Table I.Sequences of primers used for reverse
transcription-quantitative PCR. |
Table I.
Sequences of primers used for reverse
transcription-quantitative PCR.
| Gene | Sequence
(5′→3′) |
|---|
| ICAM-1 | F:
ACCTATGGCAACGACTCCTTC |
|
| R:
CCTTCTGAGACCTCTGGCTTC |
| VCAM-1 | F:
AATGGGAATCTACAGCACCTTTC |
|
| R:
GTCTCCAATCTGAGCAGCAATC |
| β-actin | F:
TTCCTGGGCATGGAGTCCT |
|
| R:
AGGAGGAGCAATGATCTTGATC |
Cell adhesion assay
VSMCs were incubated with 5 µg/ml cholesterol at
37°C with 5% CO2 for 72 h. Following washing three times
with PBS, CELLTRACE Violet-labeled human monocytic THP-1 cells
(1×106 cells/well) were added to the VSMC cultures
(2×106 cells/well) and incubated for 1 h at 10 rpm at
37°C. Cells were then washed twice with PBS to eliminate
non-attached cells. VSMC layers with attached monocytes were fixed
with 4% paraformaldehyde at room temperature for 10 min. Adhered
THP-1 cells were visualized using a DMI4000B fluorescence
microscope (DMI4000B; Leica Microsystems GmbH).
ROS detection
VSMCs were pretreated with cholesterol (5 µg/ml),
metformin (1 µM) or Compound C (10 µM) for 10 h at 37°C.
Subsequently, cells were incubated with diluted fluoroprobe DCFH-DA
for 20 min at 37°C with gentle agitation every 5 min. After washing
with serum-free culture medium, cells were harvested and examined
using the fluorescent microscope at excitation wavelength 502 nm
and emission wavelength 530 nm.
Western blotting
RIPA lysis buffer (Beyotime Institute of
Biotechnology; cat. no. P0013B) was used to lyse the cultured cells
for 30 min at 4°C, after which a bicinchoninic acid (BCA) kit
(Beyotime Institute of Biotechnology; cat. no. P0012) was used to
test the protein concentration. Proteins were denatured at 95°C for
5 min in SDS-PAGE Sample Loading Buffer (Beyotime Institute of
Biotechnology; cat. no. P0015). Equal amounts of protein (20 µg)
were separated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred to PVDF membranes.
Subsequently, the membranes were blocked with 5% non-fat dried milk
(R&D Systems) in tris-buffered saline Tween-20 (0.1%; TBST) for
1 h at room temperature, and then incubated overnight at 4°C with
the following primary antibodies (diluted with TBST in a ratio of
1:1,000): Anti-ICAM-1 (cat. no. ab53013; Abcam), anti-VCAM-1 (cat.
no. ab134047; Abcam), anti-phosphorylated (p)-P38 (cat. no. 4511;
Cell Signaling Technology, Inc.), anti-total-P38 (cat. no. 8690;
Cell Signaling Technology, Inc.), anti-p-P65 (cat. no. 3033; Cell
Signaling Technology, Inc.), anti-total-P65 (cat. no. 8242; Cell
Signaling Technology, Inc.), anti-p-AMPK (cat. no. 2535; Cell
Signaling Technology, Inc.), anti-total AMPK (cat. no. 2532; Cell
Signaling Technology, Inc.) and anti-β-actin (cat. no. sc-8432
Santa Cruz Biotechnology, Inc.). Following TBST washing, the
membranes were incubated with HRP-conjugated goat anti-rabbit IgG
(cat. no. ZB-2301; OriGene Technologies, Inc.) or anti-mouse IgG
(cat. no. ZB-2305; OriGene Technologies, Inc.) secondary antibodies
for 1 h at room temperature. Protein bands were visualized using a
Tanon 6600 Luminescent Imaging Workstation (Tanon Science and
Technology Co., Ltd.; 6600) and BeyoECL Plus (Beyotime Institute of
Biotechnology). Protein levels were semi-quantified using ImageJ
1.8.0 software (National Institutes of Health) with β-actin as the
loading control. Western blotting was performed in triplicate.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism software (version 7.0; GraphPad Software, Inc.). For
comparisons between two groups, the unpaired Student's t-test was
performed. For comparisons among multiple groups, one-way ANOVA
followed by Tukey's post hoc test was performed. Each experiment
was repeated at least three times, and P<0.05 was considered to
indicate a statistically significant difference.
Results
Cholesterol loading induces adhesion
molecule expression and monocyte adhesion on VSMCs
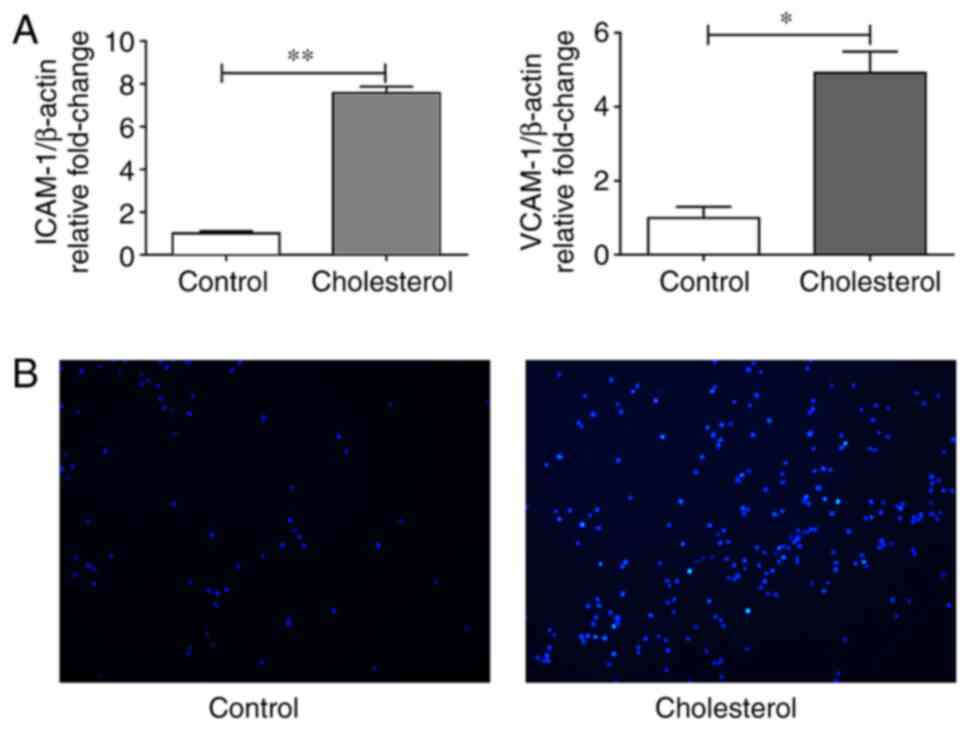
To investigate the effect of cholesterol loading on
the expression of adhesion molecules, VSMCs were incubated with
cholesterol for 72 h and the mRNA expression levels of ICAM-1 and
VCAM-1 were evaluated via RT-qPCR. The incubation of VSMCs with
cholesterol for 72 h significantly upregulated ICAM-1 and VCAM-1
mRNA expression levels compared with control cells (Fig. 1A). Following co-incubation with
CELLTRACE-Violet-labeled THP-1 monocytic cells, a marked
upregulation of monocyte adhesion on VSMCs after cholesterol
treatment for 72 h was observed (Fig.
1B).
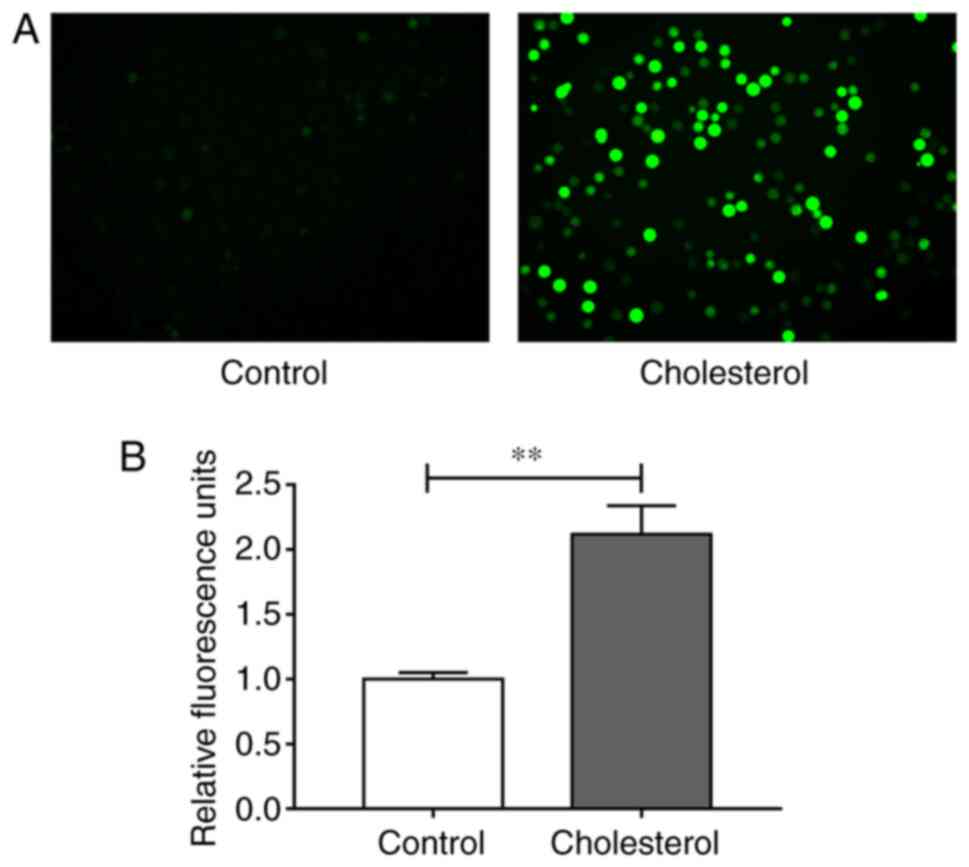
Since oxidative stress is an important factor
regulating cell function (18,19),
the effect of cholesterol on intracellular oxidant levels was
assessed. Intracellular ROS were labeled with DCFH-DA probes and
measured by conducting fluorescence assays. Compared with the
control group, cholesterol significantly increased intracellular
ROS accumulation, which may be closely related to
cholesterol-induced upregulation of adhesion molecule expression
levels (Fig. 2A and B).
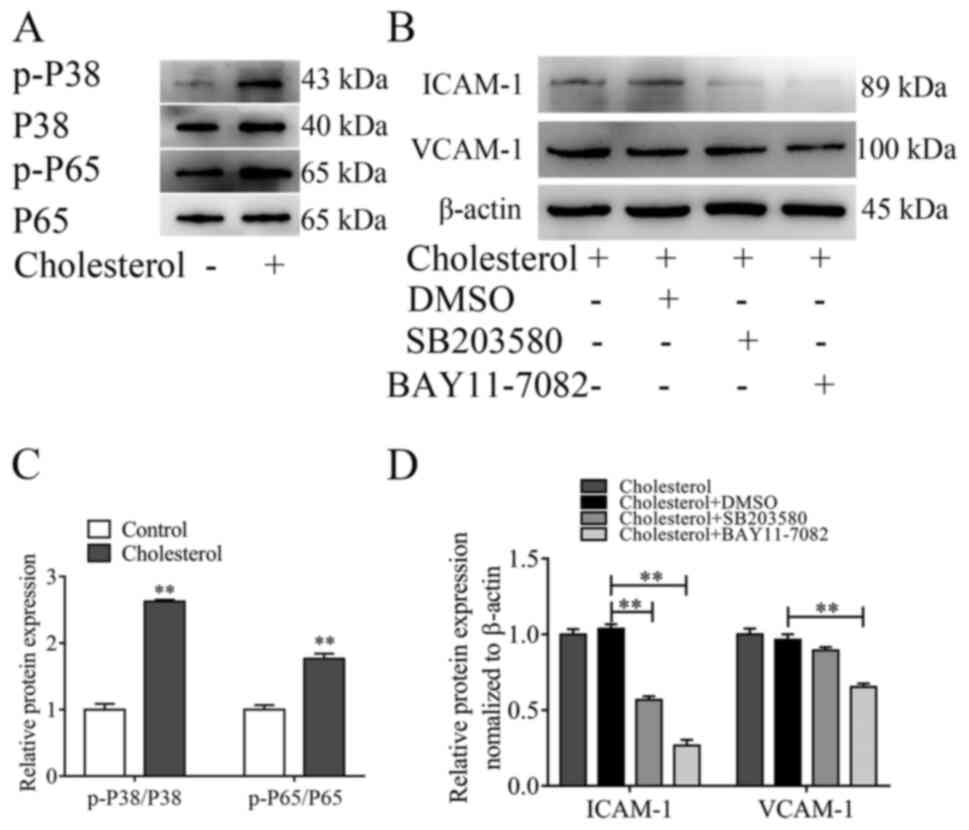
p38 MAPK and NF-κB signaling pathways
are associated with adhesion molecule expression on VSMCs
The redox-sensitive transcription factors p38 MAPK
and NF-κB are involved in regulating the expression of ICAM-1 and
VCAM-1 (20–24). However, the mechanism underlying the
expression of adhesion molecules on VSMCs is not completely
understood. To investigate whether the p38 MAPK and NF-κB signaling
pathways were involved in ICAM-1 and VCAM-1 expression on VSMCs,
the activation of the two signaling pathways was assessed via
western blotting. Cholesterol loading significantly increased p38
and p65 phosphorylation levels compared with the control group
(Fig. 3A and C). To further
evaluate the regulatory effects of p38 MAPK and NF-κB on the
expression of adhesion molecules on VSMCs, the p38 MAPK signaling
pathway inhibitor SB203580 and the NF-κB signaling pathway
inhibitor BAY11-7082 were used to treated VSMCs prior to treatment
with cholesterol. BAY11-7082 significantly inhibited
cholesterol-induced increases in ICAM-1 and VCAM-1 expression
levels, whereas SB203580 only significantly inhibited
cholesterol-induced increases in ICAM-1 expression levels (Fig. 3B and D).
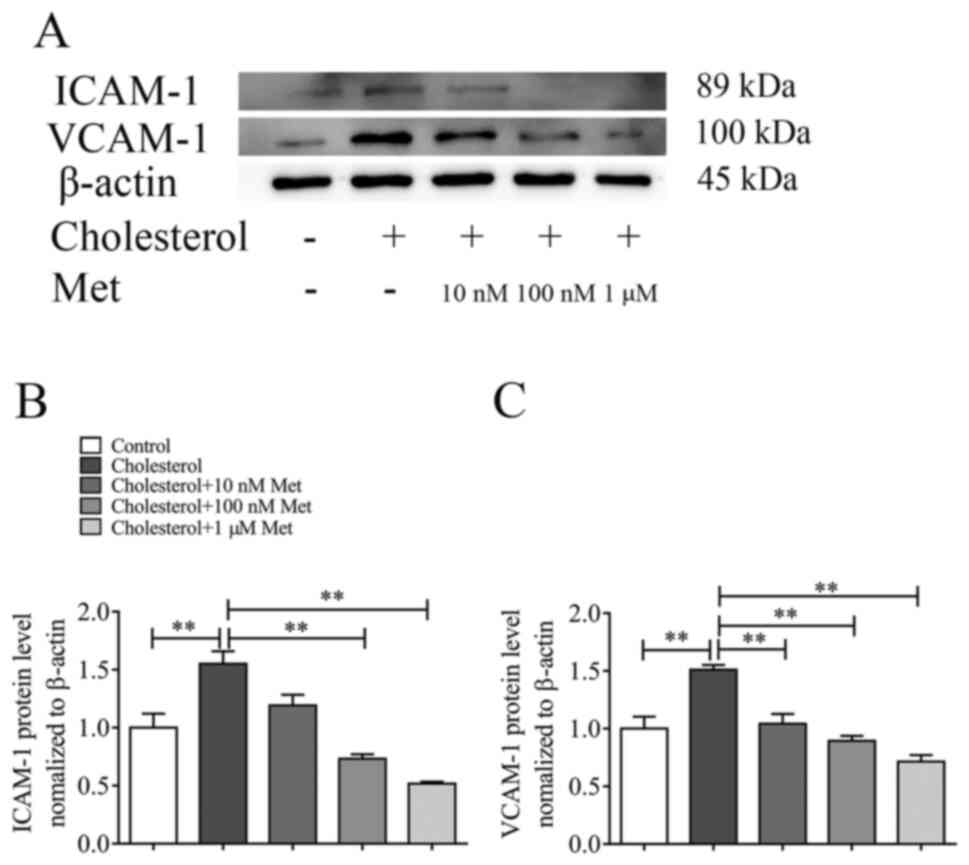
Metformin inhibits cholesterol-induced
adhesion molecule expression on VSMCs in a dose-dependent
manner
To investigate whether metformin inhibited
cholesterol-induced adhesion molecule expression, VSMCs were
incubated with or without metformin at different concentrations (10
and 100 nm or 1 µm) for 30 min, and then co-incubated with
cholesterol for 72 h. The protein expression levels of ICAM-1 and
VCAM-1 were evaluated via western blotting. Metformin treatment
inhibited cholesterol-induced upregulation of adhesion molecule
expression levels in a dose-dependent manner (Fig. 4A). Compared with the cholesterol
group, ICAM-1 and VCAM-1 expression levels were significantly
reduced following treatment with 100 nm (Fig. 4B) and 10 nm (Fig. 4C) metformin, respectively.
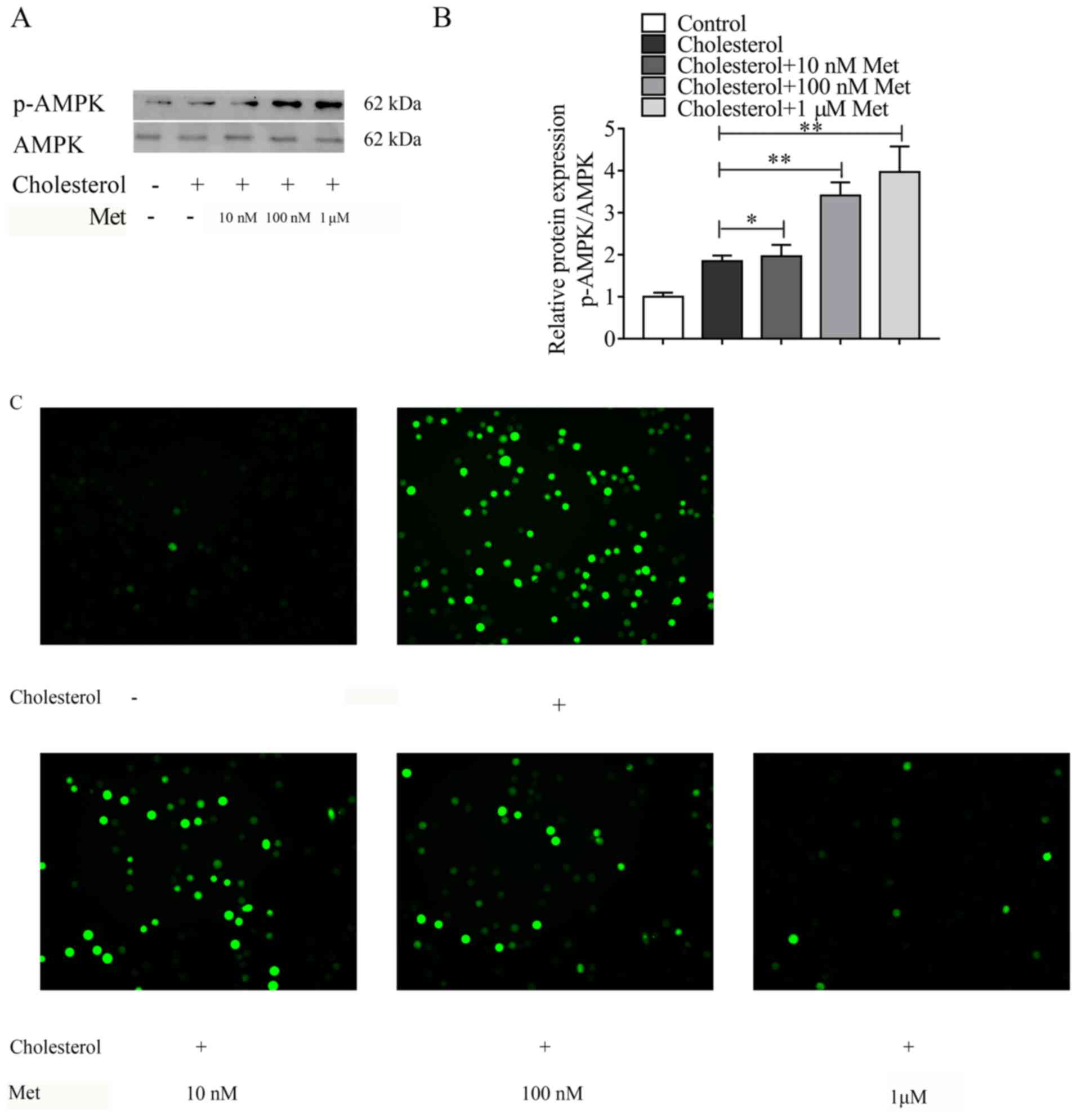
Metformin inhibits cholesterol-induced
ROS accumulation, p38 MAPK and NF-κB activation via the AMPK
signaling pathway
To elucidate the mechanism underlying
metformin-mediated inhibition of cholesterol-induced expression of
adhesion molecules on VSMCs, AMPK signaling pathway-related protein
expression levels were measured. Metformin significantly increased
the expression levels of p-AMPK in cholesterol-treated VSMCs in a
dose-dependent manner, especially at a concentration of 1 µm
(Fig. 5A and B). According to the
above experimental results, when the concentration of metformin is
1 µm, its effect on VSMCs is the most obvious. Subsequently,
whether metformin inhibited cholesterol-induced ROS accumulation in
VSMCs was investigated. Metformin notably decreased
cholesterol-induced ROS accumulation in a concentration-dependent
manner (Fig. 5C), and. To further
explore the role of AMPK in the protective effect of metformin, the
AMPK signaling pathway inhibitor Compound C was used to suppress
AMPK activation. VSMCs were pretreated with Compound C (10 µm) for
30 min, followed by treatment with metformin for 30 min and then
cells were exposed to cholesterol for 72 h. First, the expression
levels of p-AMPK and AMPK were measured via western blotting.
Pre-treatment with Compound C significantly decreased p-AMPK
protein expression levels in cholesterol- and metformin-treated
VSMCs to a similar level to the control group (Fig. 6A and B), confirming the ability of
Compound C to inhibit the phosphorylation of AMPK. Moreover, the
effects of Compound C and metformin on the activation of the p38
MAPK and NF-κB signaling pathways were investigated (Fig. 6C and D). Metformin significantly
decreased the expression levels of p-P38 and p-P65 in
cholesterol-treated VSMCs, whereas Compound C antagonized the
effects of metformin, significantly upregulating the expression
levels of p-P38 and p-P65 in cholesterol- and metformin-treated
VSMCs. In addition, the levels of intracellular ROS were also
measured (Fig. 6E). The results
demonstrated that treatment with Compound C notably increased ROS
levels in cholesterol- and metformin-treated VSMCS to a distinctly
higher level compared with the cholesterol + metformin group.
Furthermore, the adhesion of THP-1 cells on VSMCs and the
expression levels of intracellular adhesion molecules were
assessed. In cholesterol- and metformin-treated VSMCs, Compound C
treatment significantly increased the expression levels of ICAM-1
and VCAM-1, and elevated the ability of THP-1 cells to adhere to
VSMCs, which were both downregulated by metformin (Fig. 6F-H). Collectively, the results
indicated that AMPK signaling may serve an important role in the
protective effects of metformin.
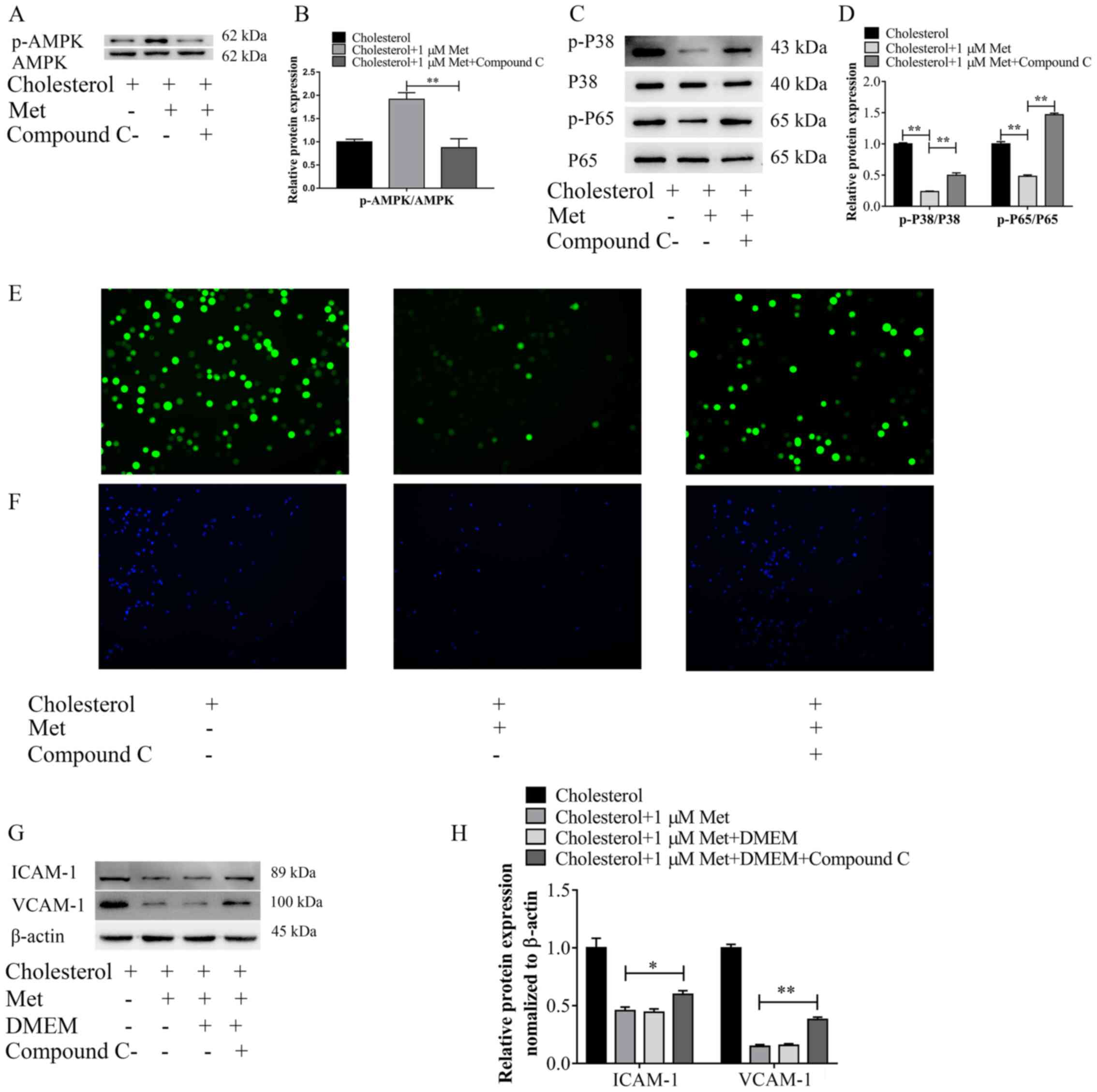 | Figure 6.Metformin inhibits cholesterol
loading-induced adhesion molecule expression, and p38 MAPK and
NF-κB pathway signaling activation via AMPK signaling. VSMCs were
exposed to Compound C (10 µm) for 30 min, followed by treatment
with metformin (1 µm) and cholesterol (5 µg/ml). Protein expression
levels were (A) determined via western blotting and (B) the ratio
of p-AMPK/AMPK was semi-quantified. Protein expression levels were
(C) determined via western blotting and (D) the ratios of p-P38/P38
and p-P65/P65 were semi-quantified. (E) Reactive oxygen species
accumulation in VSMCs was examined via immunofluorescence analysis
(magnification, ×100). (F) Representative images of THP-1 cell
adhesion to VSMCs (magnification, ×50). ICAM-1 and VCAM-1 protein
expression levels were (G) determined via western blotting and (H)
semi-quantified. *P<0.05 and **P<0.01. AMPK, AMP-activated
protein kinase; VSMC, vascular smooth muscle cell; p,
phosphorylated; ICAM-1, intercellular adhesion molecule-1; VCAM-1,
vascular cell adhesion molecule-1; Met, metformin. |
Discussion
The present study indicated that cholesterol loading
led to abnormal ROS accumulation in VSMCs and upregulated the
expression levels of adhesion molecules via activation of the p38
MAPK and NF-κB signaling pathways. Metformin protected VSMCs
against cholesterol loading by decreasing ROS accumulation,
downregulating adhesion molecule expression levels, and blocking
the activation of the p38 MAPK and NF-κB signaling pathways via
AMPK. Collectively, the results indicated that metformin may serve
as a novel modulator of vascular inflammation.
Lymphocyte recruitment to the arterial wall
accelerates AS progression (25).
The adhesion molecules ICAM-1 and VCAM-1 are important functional
mediators of the interactions between leukocytes and the vascular
wall (26,27). Numerous previous studies reported
that ICAM-1 and VCAM-1 are also expressed in VSMCs, especially when
cells are exposed to damaging stimuli, such as Salusin-β or
cholesterol (7,28). In the present study, the results
also demonstrated that, compared with the control group,
cholesterol treatment significantly upregulated expression levels
of adhesion molecules in VMSCs and promoted the adhesion of
monocytes to VSMCs in vitro, which may form the basis of the
initial development of AS.
NADPH oxidase (Nox) family members contribute
substantially to the production of ROS in the cardiovascular system
(29). Enhanced Nox expression and
subsequent ROS formation are directly associated with the severity
of structural-functional alterations at the vascular wall (30). The results of the present study
demonstrated that cholesterol loading significantly increased ROS
accumulation in VSMCs compared with the control group. ROS levels
are a key regulator of the p38 MAPK and NF-κB/p65 signaling
pathways (31,32). The present study examined the
expression levels of key proteins in the p38 MAPK and NF-κB/p65
signaling pathways, and the results suggested that cholesterol
loading in VSMCs activated the inflammatory signaling pathways. Hsu
et al (29) demonstrated
that the expression of adhesion molecules is associated with
NF-κB/p65 and p38 MAPK phosphorylation. Therefore, it was
hypothesized that cholesterol may promote the expression of
adhesion molecules in VSMCs by activating the p38 MAPK and NF-κB
inflammatory signaling pathways. By using inhibitors of the
signaling pathways, the results indicated that ICAM-1 expression in
VSMCs was associated with the activation of both inflammatory
signaling pathways; however, VCAM-1 expression in VSMCs was
exclusively associated with the NF-κB signaling pathway. Further
investigation is required to clarify the specific mechanisms
underlying the molecular interactions.
In recent years, extensive clinical studies have
confirmed the close relationship between diabetes mellitus or
impaired glucose tolerance and AS (33,34).
Metformin, a hypoglycemic drug, has been reported to reduce
cardiovascular events in patients with diabetes (35), which has attracted the attention of
scholars in various fields. However, thus far, the mechanisms
underlying metformin-mediated inhibition of AS formation and
progression have not been previously reported. An oral dose of
metformin is able to induce AMPK activation (36). The results of the present study
demonstrated that metformin displayed a dose-dependent effect on
the activation of the AMPK signaling pathway in cholesterol-treated
VSMCs. AMPK is a highly conserved serine/threonine protein kinase,
which is an energy receptor in eukaryotic cells and regulates
numerous cell functions, including inhibiting oxidative
stress-induced mitochondrial dysfunction (37). In the present study, the results
indicated that metformin decreased ROS accumulation and
downregulated expression levels of adhesion molecules in
cholesterol-treated VSMCs. To investigate whether the
aforementioned effects were related to activation of the AMPK
signaling pathway, the pathway was inhibited using Compound C.
Pre-treatment with Compound C weakened metformin-induced effects on
adhesion molecule expression, ROS accumulation and phosphorylation
of NF-κB/p65 and p38 MAPK in cholesterol-treated VSMCs, suggesting
that metformin exerted anti-inflammatory effects via activation of
the AMPK signaling pathway.
The results of the present study demonstrated that
cholesterol increased ROS accumulation, and p38 MAPK and NF-κB
signaling pathway activation in VSMCs, thereby upregulating the
expression levels of ICAM-1 and VCAM-1. By contrast, metformin
inhibited cholesterol loading-induced VSMC damage, which may serve
as a promising therapeutic strategy for vascular lesions in AS.
Collectively, the present study suggested that
cholesterol upregulated adhesion molecule expression levels via ROS
accumulation and activation of the p38 MAPK and NF-κB signaling
pathways. Moreover, in cholesterol-treated VSMCS, metformin
modulated activation of the p38 MAPK and NF-κB signaling pathways
by activating AMPK, and reduced abnormal ROS accumulation, thus
suppressing adhesion molecule expression.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81671794 and
81801803), the China Postdoctoral Science Foundation (grant nos.
2018M641870 and 2018M640310), the Heilongjiang Postdoctoral Science
Foundation (grant nos. LBH-Z18217 and LBH-Z18141), the Key
Laboratory of Myocardial Ischemia, Chinese Ministry of Education
(Harbin, Heilongjiang, China; grant no. KF201811) and the General
Undergraduate Colleges and Universities Young Innovative Talents
Training Plan (Heilongjiang, China; grant no.
UNPYSCT-2018075.).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
QL, MY, LZ, RZ and XH performed the experiments. QL
and XW performed statistical analyses. WD interpreted the data for
the work and reviewed the final version of the manuscript. QL and
JH designed the study and drafted the manuscript. QL and JH confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman
M, Das SR, Deo R, de Ferranti SD, Floyd J, Fornage M, Gillespie C,
et al: Heart disease and stroke statistics-2017 update: A report
from the American heart association. Circulation. 135:e146–e603.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dubland JA and Francis GA: So much
cholesterol: The unrecognized importance of smooth muscle cells in
atherosclerotic foam cell formation. Curr Opin Lipidol. 27:155–161.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bennett MR, Sinha S and Owens GK: Vascular
smooth muscle cells in atherosclerosis. Circ Res. 118:692–702.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Miano JM, Fisher EA and Majesky MW: Fate
and state of vascular smooth muscle cells in atherosclerosis.
Circulation. 143:2110–2116. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shankman LS, Gomez D, Cherepanova OA,
Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AC, Greene
ES, Straub AC, et al: KLF4-dependent phenotypic modulation of
smooth muscle cells has a key role in atherosclerotic plaque
pathogenesis. Nat Med. 21:628–637. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vengrenyuk Y, Nishi H, Long X, Ouimet M,
Savji N, Martinez FO, Cassella CP, Moore KJ, Ramsey SA, Miano JM
and Fisher EA: Cholesterol loading reprograms the
microRNA-143/145-myocardin axis to convert aortic smooth muscle
cells to a dysfunctional macrophage-like phenotype. Arterioscler
Thromb Vasc Biol. 35:535–546. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Q, Zhang H, Lin J, Zhang R, Chen S,
Liu W, Sun M, Du W, Hou J and Yu B: C1q/TNF-related protein 9
inhibits the cholesterol-induced vascular smooth muscle cell
phenotype switch and cell dysfunction by activating AMP-dependent
kinase. J Cell Mol Med. 21:2823–2836. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Williams JW, Martel C, Potteaux S,
Esaulova E, Ingersoll MA, Elvington A, Saunders BT, Huang LH,
Habenicht AJ, Zinselmeyer BH and Randolph GJ: Limited macrophage
positional dynamics in progressing or regressing murine
atherosclerotic plaques-brief report. Arterioscler Thromb Vasc
Biol. 38:1702–1710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Clemente C, Rius C, Alonso-Herranz L,
Martín-Alonso M, Pollán A, Camafeita E, Martínez F, Mota RA, Núñez
V, Rodríguez C, et al: MT4-MMP deficiency increases patrolling
monocyte recruitment to early lesions and accelerates
atherosclerosis. Nat Commun. 9:9102018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Förstermann U, Xia N and Li H: Roles of
vascular oxidative stress and nitric oxide in the pathogenesis of
atherosclerosis. Circ Res. 120:713–735. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Niemann B, Rohrbach S, Miller MR, Newby
DE, Fuster V and Kovacic JC: Oxidative stress and cardiovascular
risk: Obesity, diabetes, smoking, and pollution: Part 3 of a 3-part
series. J Am Coll Cardiol. 70:230–251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li C, Zhang WJ and Frei B: Quercetin
inhibits LPS-induced adhesion molecule expression and oxidant
production in human aortic endothelial cells by p38-mediated Nrf2
activation and antioxidant enzyme induction. Redox Biol. 9:104–113.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Akoumianakis I and Antoniades C: Impaired
vascular redox signaling in the vascular complications of obesity
and diabetes mellitus. Antioxid Redox Signal. 30:333–353. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Frias JP, Bonora E, Nevárez Ruiz L, Hsia
SH, Jung H, Raha S, Cox DA, Bethel MA and Konig M: Efficacy and
safety of dulaglutide 3.0 and 4.5 mg in patients aged younger than
65 and 65 years or older: Post hoc analysis of the AWARD-11 trial.
Diabetes Obes Metab. Jun 22–2021.(Epub ahead of print). View Article : Google Scholar
|
|
15
|
Chen X, Li X, Zhang W, He J, Xu B, Lei B,
Wang Z, Cates C, Rousselle T and Li J: Activation of AMPK inhibits
inflammatory response during hypoxia and reoxygenation through
modulating JNK-mediated NF-κB pathway. Metabolism. 83:256–270.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Q, Zhang M, Torres G, Wu S, Ouyang C,
Xie Z and Zou MH: Metformin suppresses diabetes-accelerated
atherosclerosis via the inhibition of Drp1-mediated mitochondrial
fission. Diabetes. 66:193–205. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bañuls C, Rovira-Llopis S, de Marañon AM,
Veses S, Jover A, Gomez M, Rocha M, Hernandez-Mijares A and Victor
VM: Metabolic syndrome enhances endoplasmic reticulum, oxidative
stress and leukocyte-endothelium interactions in PCOS. Metabolism.
71:153–162. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yan S, Zhang X, Zheng H, Hu D, Zhang Y,
Guan Q, Liu L, Ding Q and Li Y: Clematichinenoside inhibits VCAM-1
and ICAM-1 expression in TNF-α-treated endothelial cells via NADPH
oxidase-dependent IκB kinase/NF-κB pathway. Free Radic Biol Med.
78:190–201. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Daiber A: Redox signaling (cross-talk)
from and to mitochondria involves mitochondrial pores and reactive
oxygen species. Biochim Biophys Acta. 1797:897–906. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:e124132017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
He H, Guo F, Li Y, Saaoud F, Kimmis BD,
Sandhu J, Fan M, Maulik D, Lessner S, Papasian CJ, et al:
Adiporedoxin suppresses endothelial activation via inhibiting MAPK
and NF-κB signaling. Sci Rep. 6:389752016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Altieri P, Murialdo R, Barisione C,
Lazzarini E, Garibaldi S, Fabbi P, Ruggeri C, Borile S, Carbone F,
Armirotti A, et al: 5-fluorouracil causes endothelial cell
senescence: Potential protective role of glucagon-like peptide 1.
Br J Pharmacol. 174:3713–3726. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang M, Zeng S, Zou Y, Shi M, Qiu Q, Xiao
Y, Chen G, Yang X, Liang L and Xu H: The suppression of bromodomain
and extra-terminal domain inhibits vascular inflammation by
blocking NF-κB and MAPK activation. Br J Pharmacol. 174:101–115.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gerhardt T and Ley K: Monocyte trafficking
across the vessel wall. Cardiovasc Res. 107:321–330. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hsu WY, Chao YW, Tsai YL, Lien CC, Chang
CF, Deng MC, Ho LT, Kwok CF and Juan CC: Resistin induces
monocyte-endothelial cell adhesion by increasing ICAM-1 and VCAM-1
expression in endothelial cells via p38MAPK-dependent pathway. J
Cell Physiol. 226:2181–2188. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Moore KJ, Sheedy FJ and Fisher EA:
Macrophages in atherosclerosis: A dynamic balance. Nat Rev Immunol.
13:709–721. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun HJ, Zhao MX, Liu TY, Ren XS, Chen Q,
Li YH, Kang YM and Zhu GQ: Salusin-β induces foam cell formation
and monocyte adhesion in human vascular smooth muscle cells via
miR155/NOX2/NFκB pathway. Sci Rep. 6:235962016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hsu SY, Liou JW, Cheng TL, Peng SY, Lin
CC, Chu YY, Luo WC, Huang ZK and Jiang SJ: β-Naphthoflavone
protects from peritonitis by reducing TNF-α-induced endothelial
cell activation. Pharmacol Res. 102:192–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lassegue B and Clempus RE: Vascular
NAD(P)H oxidases: Specific features, expression, and regulation. Am
J Physiol Regul Integr Comp Physiol. 285:R277–R297. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rasheduzzaman M, Yin H and Park SY:
Cardiac glycoside sensitized hepatocellular carcinoma cells to
TRAIL via ROS generation, p38MAPK, mitochondrial transition, and
autophagy mediation. Mol Carcinog. 58:2040–2051. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang P, Yin Y, Wang T, Li W, Li C, Zeng
X, Yang W, Zhang R, Tang Y, Shi L, et al: Maresin 1 mitigates
concanavalin A-induced acute liver injury in mice by inhibiting
ROS-mediated activation of NF-κB signaling. Free Radic Biol Med.
147:23–36. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Di Pino A and DeFronzo RA: Insulin
resistance and atherosclerosis: Implications for
insulin-sensitizing agents. Endocr Rev. 40:1447–1467. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang T and Redline S: Cross-sectional and
prospective associations of actigraphy-assessed sleep regularity
with metabolic abnormalities: The multi-ethnic study of
atherosclerosis. Diabetes Care. 42:1422–1429. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Roumie CL, Chipman J, Min JY, Hackstadt
AJ, Hung AM, Greevy RA Jr, Grijalva CG and Elasy T: Association of
treatment with metformin vs sulfonylurea with major adverse
cardiovascular events among patients with diabetes and reduced
kidney function. JAMA. 322:1167–1177. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Foretz M, Guigas B, Bertrand L, Pollak M
and Viollet B: Metformin: From mechanisms of action to therapies.
Cell Metab. 20:953–966. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan Y, Yang Q, Yang Y, Gao Z, Ma Y, Zhang
L, Liang W and Ding G: Sirt6 suppresses high glucose-induced
mitochondrial dysfunction and apoptosis in podocytes through AMPK
activation. Int J Biol Sci. 15:701–713. 2019. View Article : Google Scholar : PubMed/NCBI
|