Introduction
Long-term hyperuricemia leads to uric acid (UA)
stone formation and irreversible kidney injury (1); however, the precise mechanism
responsible for the development of UA-related kidney disease is
still largely unknown. Serum uric acid is commonly elevated in
subjects with chronic kidney disease (CKD). Recent evidence implied
that an elevated UA level predicts the development of CKD and
end-stage kidney disease (2).
Therefore, it is necessary to determine the mechanism of UA-related
kidney injury and develop novel and effective preventative
strategies. Notably, apoptosis is widely considered as an important
pathological process of UA-induced renal tubular epithelial cell
injury (3). Therefore,
understanding the underlying mechanism of apoptosis in UA-induced
renal tubular cell injury is necessary. In recent years, there has
been increased research on antiapoptotic therapy (4). Uric acid crystals and soluble UA can
activate the immune system and increase the production of the
reactive oxygen species (ROS), which can stimulate neutrophil
chemotaxis and activate NLR family pyrin domain containing 3
(NLRP3) inflammasomes (5,6). Never in mitosis gene A (NIMA)-related
kinase 7 (NEK7) is a protein kinase that acts as a selective
upstream regulator of NLRP3 inflammasome activation (7). Previous studies have revealed that
NEK7 can connect the adjacent NLRP3 subunits and mediate NLRP3
inflammasome activation through mitotic interaction (8,9).
Currently, mitochondrial ROS is the most studied aspect of NLRP3
inflammasome activation and apoptosis (10,11).
Our previous study confirmed that UA could stimulate excess ROS
production, which could further trigger the assembly of the NLRP3
inflammasome and induce renal tubular injury in hyperuricemic rats
(12). NEK7 is required for NLRP3
activation via the direct NEK7-NLRP3 interaction (13). However, whether UA can induce renal
tubular epithelial cell apoptosis by regulating ROS release and
activating the NEK7-NLRP3 pathway remains to be elucidated.
Therefore, the aim of the present study was to determine whether UA
can induce ROS release and activation of NEK7-NLRP3 pathways to
increase renal tubular epithelial cell apoptosis.
Materials and methods
Soluble uric acid preparation
In experiments requiring soluble uric acid, an
appropriate amount of monosodium urate (MSU) crystals (cat. no.
U2875 Sigma-Aldrich; Merck KGaA) were added into 1 M NaOH to a
final concentration of 10 mg/ml. The MSU crystals were then crushed
using ultrasound and the solution was sterilized through 0.20 µm
filters. Crystals were not detected under polarizing microscopy
(magnification, ×200), nor did they develop during the experiments.
The soluble uric acid solution was pre-warmed for 30 min at 37°C
before use.
Cell culture and treatment
The normal renal tubular epithelial NRK-52E cell
line was purchased from The Cell Bank of Type Culture Collection of
The Chinese Academy of Sciences. Cells were cultured in
high-glucose Dulbecco's modified Eagle's medium (DMEM; cat. no.
C11995500BT; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (cat. no. 10099-141C; Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin and streptomycin (cat.
no. C0222; Beyotime Institute of Biotechnology) at 37°C in a
humidified incubator containing 5% CO2 and 95%
O2.
When the adherent cells reached 80–90% confluence,
they were passaged using 0.25% trypsin and 0.02%
ethylenediaminetetraacetic acid (cat. no. C0201; Beyotime Institute
of Biotechnology). The NRK-52E cells were cultured in 6-well
polystyrene plates at ~4×105 cells per well and treated
using different concentrations of UA (0, 50 and 100 µg/ml) for 24 h
to establish the cell injury model. Subsequently, cultured cells
were divided into three groups: The control group, cells exposed to
100 µg/ml UA (model group) and cells pretreated with 20 mM
N-acetyl-l-cysteine (NAC; cat. no. HY-B0215; MedChemExpress) and
then treated with UA (the model plus NAC group). Apoptosis was
detected using a terminal deoxynucleotidyl transferase
nick-end-labeling (TUNEL) assay, western blotting and flow
cytometry analysis. Triplicate wells per group were cultured for 24
h and the experiments were repeated three times.
Cell morphology
The NRK-52E cells were seeded in equal amounts
(4×105 cells per well) into 6-well plates and then
exposed to UA (0, 50 and 100 µg/ml) for 24 h. After the cell-drug
interaction, the growth of NRK-52E cells in each group was observed
under an inverted phase contrast microscope (Nikon Corporation) to
investigate the morphological changes (magnification, ×400).
Flow cytometry detection of cell
apoptosis
Following treatment, the cells were counted and then
resuspended in 100 µl 1X binding buffer at a concentration of
1×106 cells/ml. Subsequently, the cells in binding
buffer were added to a 5 ml culture tube, followed by gentle
vortexing and incubation with 5 µl Annexin V-fluorescein
isothiocyanate (FITC) and 5 µl propidium iodide (PI) solution (cat.
no. 556547; BD Pharmingen; BD Biosciences) for 15 min at room
temperature in the dark. After adding 400 µl 1X binding buffer to
each tube, the apoptotic cell ratio was detected on a BD
FACSCalibur flow cytometer (BD Biosciences) using FlowJo software
(version 7.6.1; FlowJo LLC). Early apoptotic cells were
differentiated by their unique characteristics of being Annexin
V-FITC+ and PI−, and late apoptotic cells
were counted as Annexin V-FITC+ and PI+
cells.
TUNEL assay
Apoptotic cells were measured using a One-step TUNEL
Apoptosis Detection kit (cat. no. KGA7072; Nanjing KeyGen Biotech
Co., Ltd.). Briefly, cultured cells were fixed with 4% formaldehyde
at room temperature for 30 min. Then, the cells were incubated with
1% Triton X-100. Subsequently, the cells were mixed with 50 µl
TUNEL reaction mixture containing biotin-11-dUTP and TdT Enzyme and
then with streptavidin fluorescein at 37°C for 30 min. The nucleus
was counterstained using 4′,6-diamidino-2-phenylindole at room
temperature for 10 min in the dark. Slides were mounted using
glycerol. Finally, apoptotic cells were examined using a
fluorescence microscope under three fields of vision
(magnification, ×200; Nikon Eclipse 80i; Nikon Corporation).
Western blotting
The cells were grown to 80–90% confluence,
homogenized in ice-cold phenylmethylsulfonyl fluoride in
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology) and incubated at 4°C for 30 min. After
centrifugation at 7,992 × g for 15 min at 4°C, the protein
concentration of supernatant was measured using a bicinchoninic
acid protein assay kit (Beyotime Institute of Biotechnology). The
proteins (40 µg) were electrophoresed on 6–10% SDS-PAGE gels and
then electrotransferred to a polyvinylidene fluoride membrane (EMD
Millipore). The membranes were blocked for 1 h at room temperature
using 5% skimmed milk powder (Beyotime Institute of Biotechnology)
in phosphate buffered saline containing 0.1% Tween-20 (PBST),
followed by probing with primary antibodies at 4°C overnight.
Thereafter, the membranes were washed with PBST three times for 10
min each, followed by incubation with respective horseradish
peroxidase-linked secondary antibodies at room temperature for 60
min. Finally, ECL solution (cat. no. 180-5001W; Tanon Science and
Technology Co., Ltd.) was applied to the membrane evenly before
detection of the signals (T1600; Tanon Science and Technology Co.,
Ltd.). Quantitative analysis was performed using ImageJ software
(v1.48; National Institutes of Health) The primary antibodies used
were as follows: Rabbit anti-cleaved caspase-3 (cat. no. 9661;
1:1,000; Cell Signaling Technology, Inc.), rabbit anti-Bcl-xl (cat.
no. ab32370; 1:1,000; Abcam), rabbit anti-Bax (cat. no. ab32503;
1:5,000; Abcam), rabbit anti-NEK7 (cat. no. ab133514; 1:5,000;
Abcam), rabbit anti-pro-Caspase-1+ p10 + p12 (cat. no. ab179515;
1:1,000; Abcam), rabbit anti-NLRP3 (cat. no. 19771-1-AP; 1:1,000;
ProteinTech Group, Inc.), mouse anti-β-actin (cat. no. 66009-1-lg;
1:5,000; ProteinTech Group, Inc.) and rabbit
anti-apoptosis-associated speck-like (ASC; cat. no. YT0365;
1:1,000; ImmunoWay Biotechnology Company). Horseradish
peroxidase-labeled goat anti-mouse IgG (H+L; cat. no. A0216;
1:1,000;) and horseradish peroxidase-labeled goat anti-rabbit IgG
(H+L; cat. no. A0208; 1:1,000;) were purchased from Beyotime
Institute of Biotechnology.
Flow cytometry detection of ROS
Detection of intracellular ROS in NRK-52E cells:
Following treatment, cells were washed twice in PBS and incubated
with 0.1% 2,7-dichlorofluorescin diacetate (DCFH-DA; cat. no.
E004-1-1; Nanjing Jiancheng Bioengineering Institute) as a probe at
37°C for 60 min. Subsequently, the cells were digested with 0.25%
trypsin. After being centrifuged at 55.5 × g for 5 min, the cells
were resuspended in 400 µl of 1X binding buffer and then subjected
to flow cytometry using a BD FACSCalibur flow cytometer (BD
Biosciences). The excitation wavelength was 500 (500±15 nm) and the
emission wavelength was 525 (530±20 nm). The results were expressed
as fluorescence values. FlowJo software was used for analysis of
data (v10; FlowJo LLC).
Statistical analysis
Data are expressed as the mean ± standard error of
the mean. All statistical tests were performed using SPSS 21
software (IBM Corp.). Data were analyzed using an unpaired
Student's t test; comparisons between groups involved one-way
analysis of variance with Tukey-Kramer post-hoc test. Data were
presented using GraphPad Prism 8 (GraphPad Software, Inc.). All the
experiments were repeated three times for each experimental
condition and the representative results are presented. P<0.05
was considered to indicate a statistically significant
difference.
Results
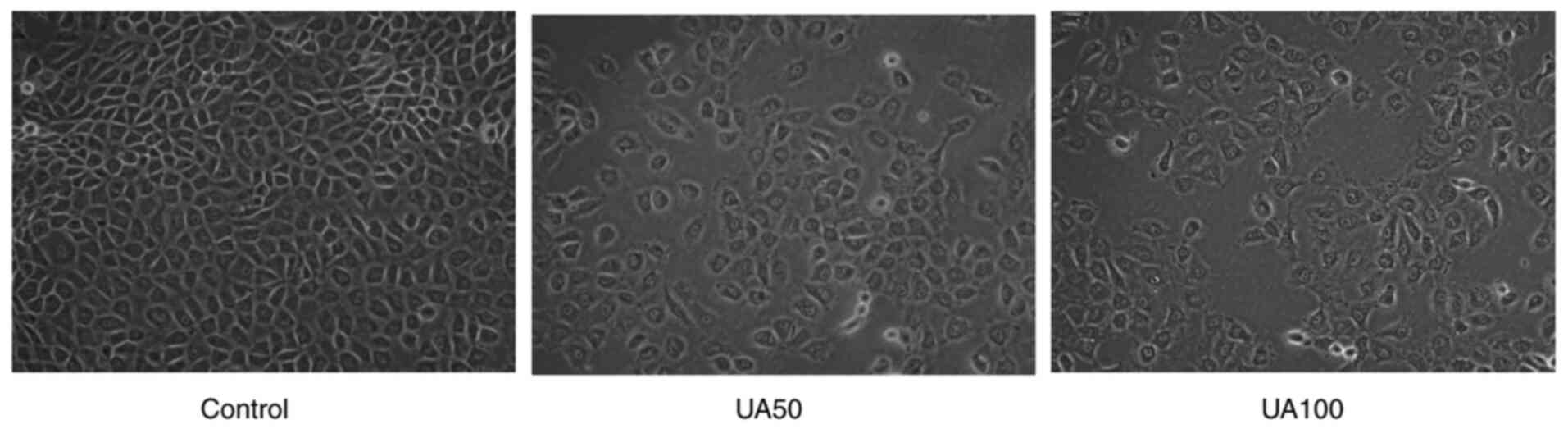
Effect of UA on morphological changes
of NRK-52E cells
Initially, the CCK8 assay was performed to show that
uric acid could inhibit the viability of NRK-52E cells when the
concentration was 100 µg/ml UA for 24, 48 and 72 h (Fig. S1,Fig.
S2,Fig. S3). After statistical
analysis, there was significant difference between the cells
stimulated with 100 µg/ml uric acid for 24 and 48 h and normal
cells, the results of 72 h showed that the cell survival rate of
100 µg/ml uric acid decreased significantly, which could not meet
the requirements of the experiment (data not shown). Combined with
this study, the effect of early uric acid levels on NRK-52E cells
was mainly observed, hence, 100 µg/ml UA was selected to stimulate
NRK-52E cells 24 h in the following assay (data not shown). The
morphological changes of NRK-52E cells were observed after 24 h of
treatment with UA. NRK-52E cells appeared pebble shaped in the
control group (Fig. 1); however,
following treatment with 50 and 100 µg/ml UA for 24 h, the cells
became progressively swollen, the membrane ruptured and became
detached (Fig. 1). In addition, the
cell adhesion ability was weakened and the distribution density of
the cells was reduced significantly.
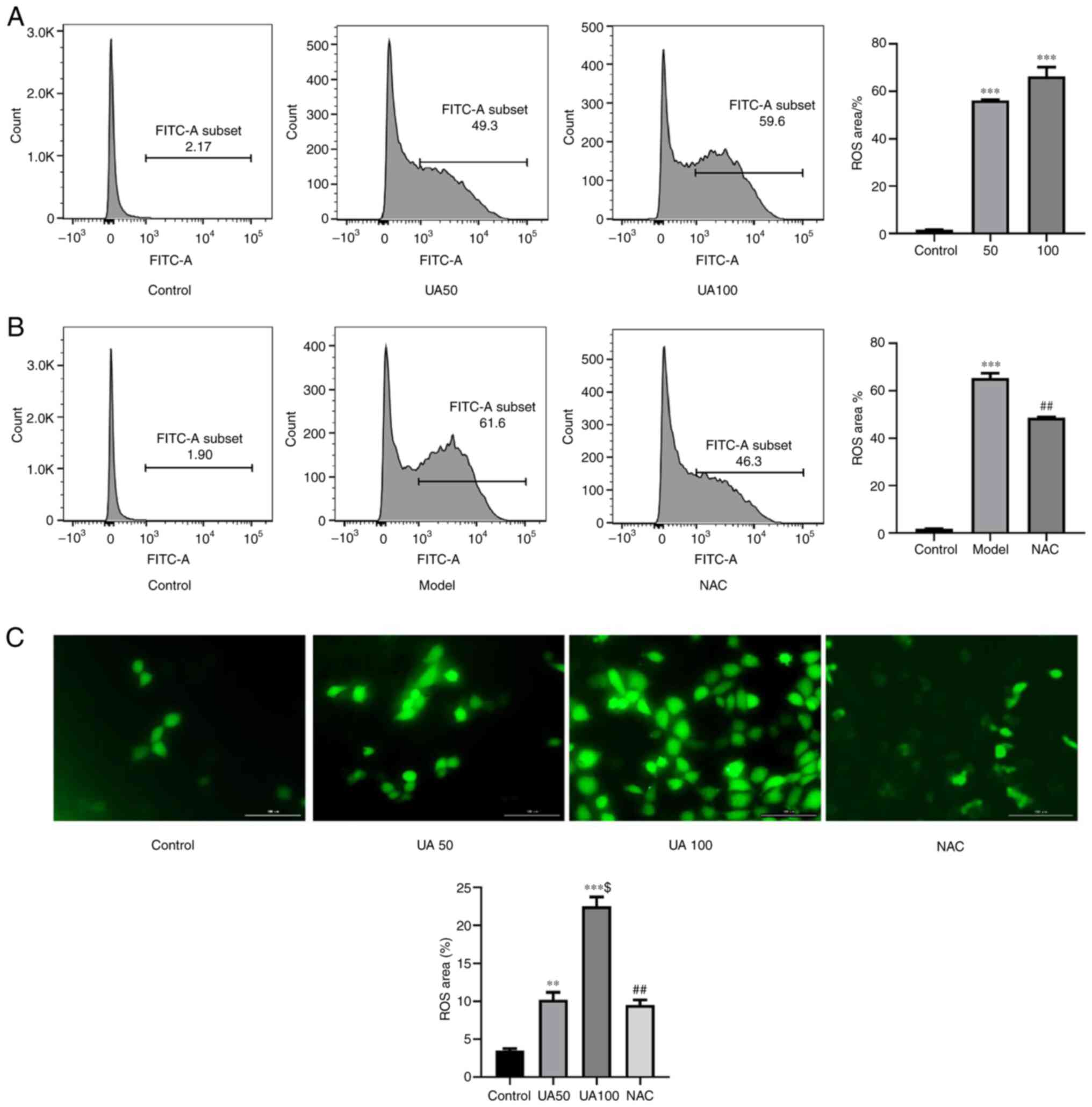
Effect of UA on ROS production in
NRK-52E cells
As shown in Fig. 2A and
C, treating NRK-52E cells with 50 and 100 µg/ml UA for 24 h
significantly increased ROS levels compared with those in the
control group. To further assess whether NAC inhibited the
UA-induced increase in ROS levels, NRK-52E cells were pretreated
with NAC for 1 h; and then the cells were co-cultured with UA for
24 h. As expected, the number of DCFH-DA-positive cells was
increased by UA treatment compared with that in the NAC group
(Fig. 2B and C). Thus, pretreatment
with NAC significantly inhibited the UA-induced increase in ROS
levels.
Effect of UA on the NEK7-NLRP3
signaling pathway activation
The present study evaluated the effects of 50 and
100 µg/ml UA on the NEK7-NLRP3 signaling pathway. Fig. 3A shows that UA upregulated the
levels of NEK7, NLRP3, ASC and Cleaved-caspase-1 in a
dose-dependent manner. Compared with that in the 100 µg/ml UA
group, NAC attenuated the effect of UA on the levels of NEK7,
NLRP3, ASC and Cleaved-caspase-1 (Fig.
3B).
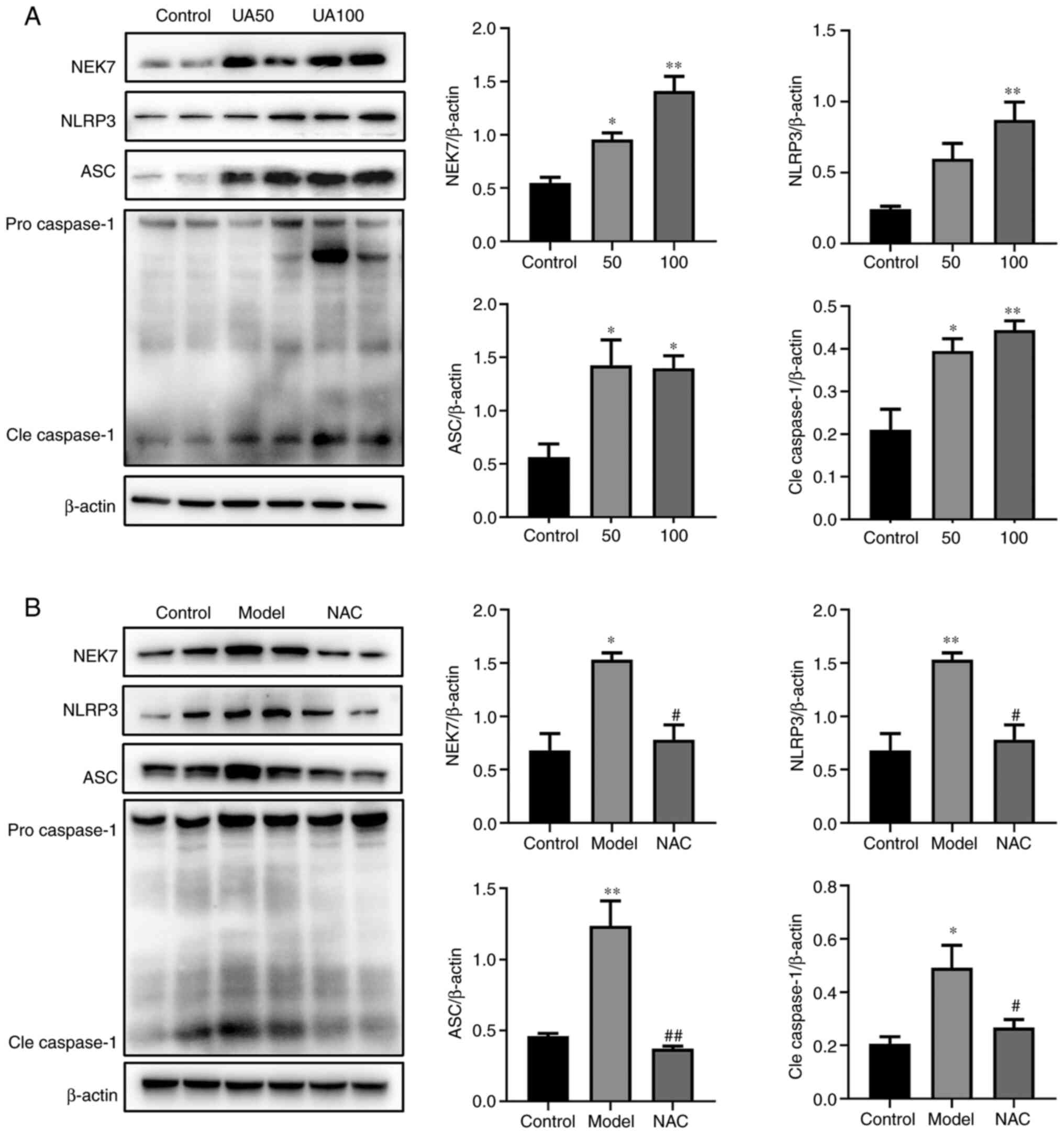 | Figure 3.Effects of UA on the apoptotic
pathway in injured NRK-52E cells. (A and B) Levels of apoptotic
pathway proteins (NEK7, NLRP3, ASC and caspase-1) in response to UA
(0, 50 and 100 µg/ml) treatment of NRK-52E cells, as assessed using
western blotting. NRK-52E cells were pretreated with 20 mM NAC for
1 h and then treated with 100 µg/ml UA for an additional 24 h and
the levels of NEK7, NLRP3, ASC and caspase-1 were determined. Data
represent the mean ± standard error of the mean; n=4 independent
samples repeated three times. *P<0.05, **P<0.01 vs. the
control group. #P<0.05, ##P<0.01 vs.
the model group. UA, uric acid; NAC, N-acetyl-l-cysteine; NEK7,
Never in mitosis gene A-related kinase 7; NLRP3, NLR family pyrin
domain containing 3; ASC, apoptosis-associated speck-like; Cle,
cleaved. |
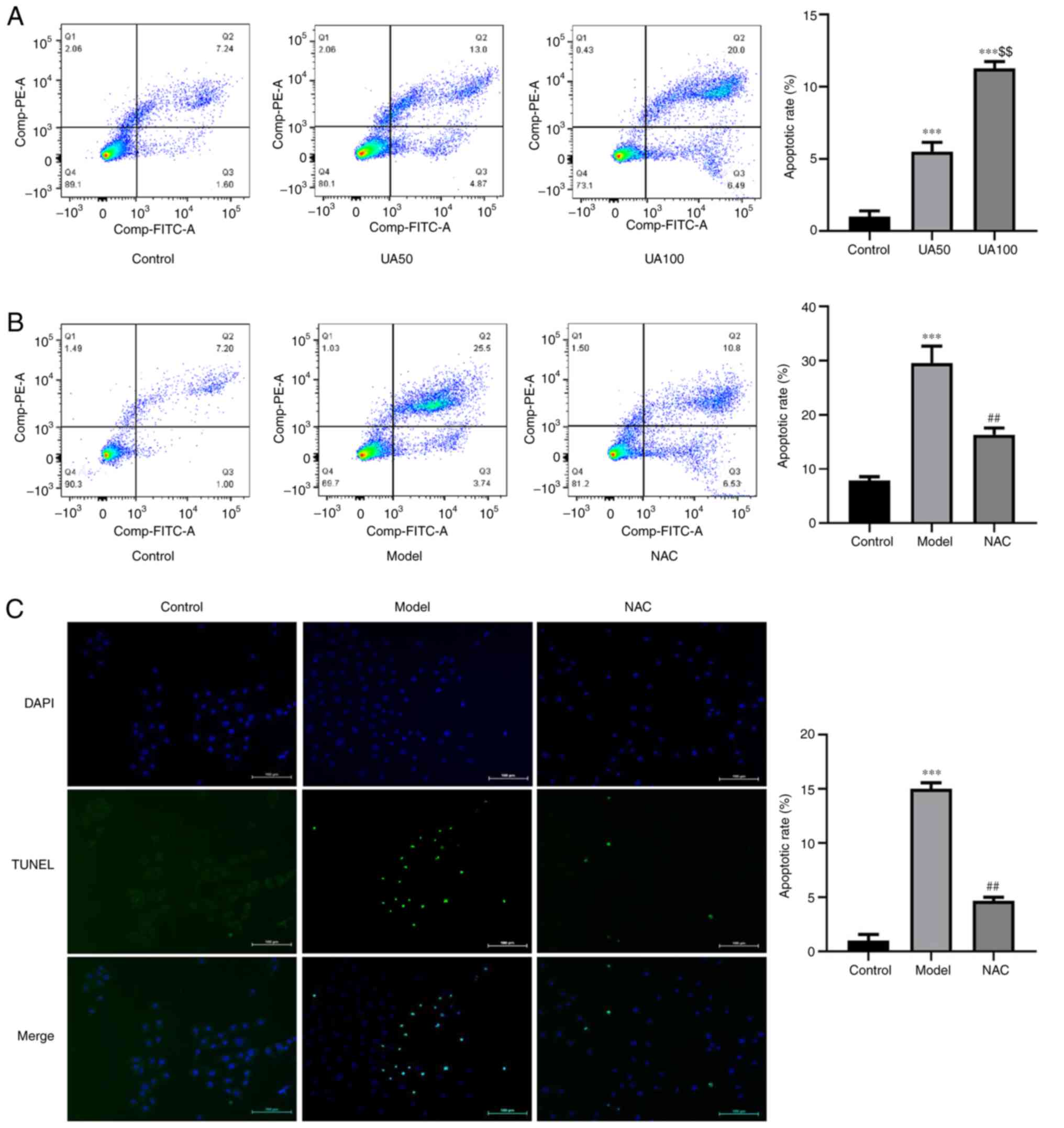
Effect of UA on NRK-52E cell
apoptosis
To evaluate UA-induced apoptosis of NRK-52E cells
following treatment with UA, the apoptosis rate was detected using
flow cytometry and TUNEL assays. The results showed that UA
significantly promoted apoptosis of NRK-52E cells compared with the
non-treated control group (Fig.
4A-C). Flow cytometry analysis showed that the number of
FITC+ and PI− cells increased following UA
treatment. The ratio of FITC+ and PI+ cells
was higher compared with the control group, indicating that the
cells exhibited advanced apoptosis or necrosis. Treatment with NAC
abrogated the increases in cell apoptosis induced by UA compared
with that in cells treated with UA alone (Fig. 4B and C). In addition, the levels of
apoptosis markers (Cleaved-caspase-3, Bcl-xl and Bax) were also
assessed via western blotting. As shown in Fig. 5A, UA decreased the expression of
Bcl-xl and increased the expression levels of Bax and
Cleaved-caspase-3 in a dose-dependent manner in NRK-52E cells. NAC
attenuated the effect of UA on the levels of these proteins
(Fig. 5B).
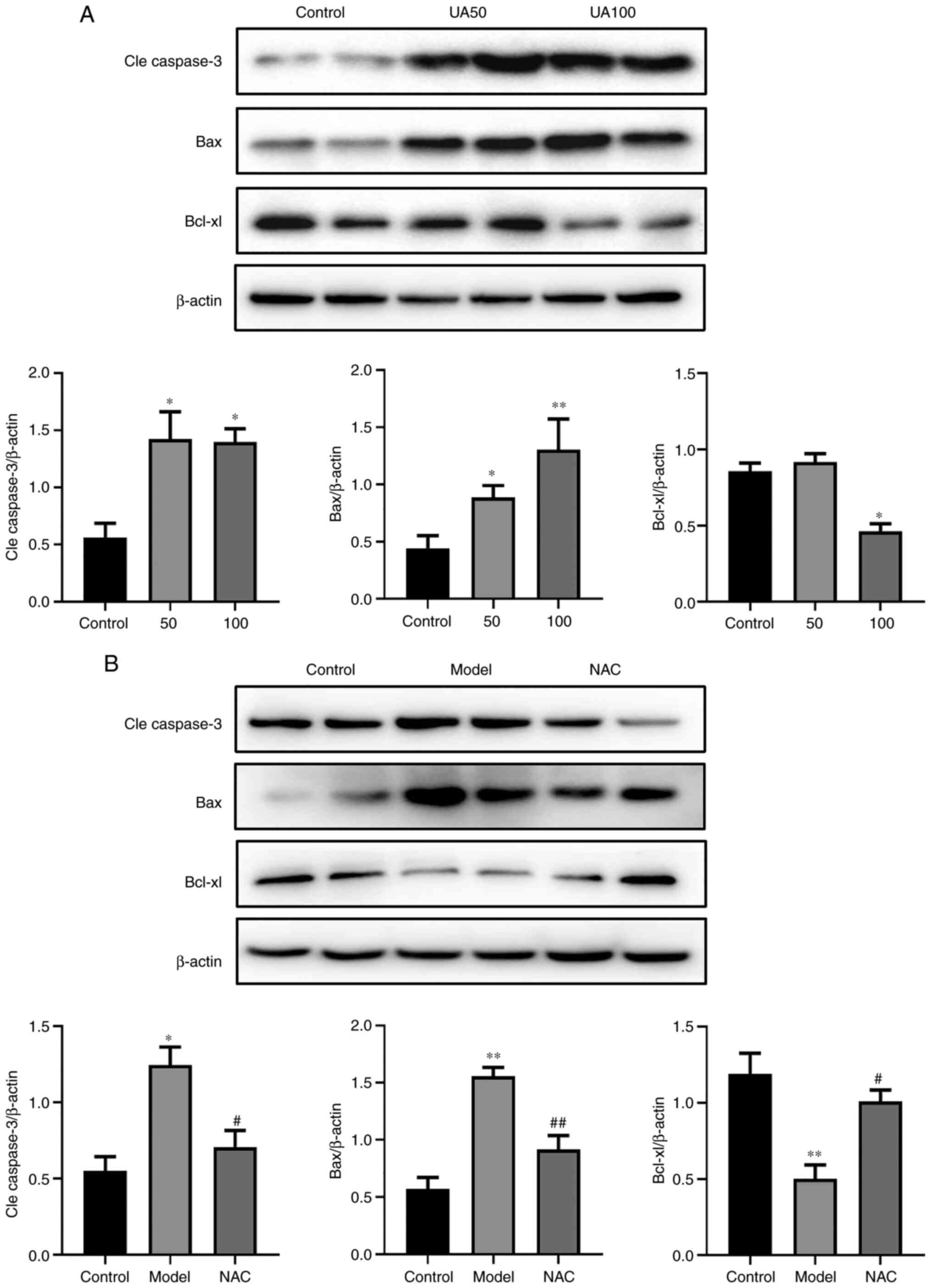 | Figure 5.Effects of UA on the apoptotic
proteins of NRK-52E cells during injury. (A) Levels of apoptotic
proteins (Cle caspase-3, Bax and Bcl-xl) in response to UA (0, 50,
100 µg/ml) in NRK-52E cells subjected to injury, as assessed using
western blotting. (B) NRK-52E cells were pretreated with 20 mM NAC
for 1 h and then treated with 100 µg/ml UA for an additional 24 h
and the levels of Cle caspase-3, Bax and Bcl-xl were examined using
western blotting. Data represent the mean ± standard error of the
mean; n=4 independent samples and repeated three times. *P<0.05,
**P<0.01 vs. the control group; #P<0.05,
##P<0.05 vs. the model group. UA, uric acid; NAC,
N-acetyl-l-cysteine; Cle, cleaved. |
Discussion
Hyperuricemia has been widely reported as a risk
factor for a variety of kidney diseases (14,15).
The observation that hyperuricemia frequently precedes the
development of CKD suggests that factors other than renal
insufficiency are likely involved in the pathogenesis of the
elevation in uric acid (16).
Studies have confirmed that ~2/3 of UA in the human body is
reabsorbed and secreted by the renal tubules (17,18).
Therefore, renal tubular epithelial cell injury might be an
important factor in the renal damage caused by UA (19). High levels of UA, as a dangerous
molecular model, are ingested into the cell lysosome, increase the
permeability of lysosomal membrane. Lysosomal membrane
permeabilization results in translocation to the cytoplasm of the
intraluminal contents, followed by the release of cathepsins into
the cytosol. Specific cathepsins are released from the
permeabilized lysosomes resulting in mitochondrial damage,
mitochondria is an important place for cells to produce ROS
(20,21). Abnormal function and structure of
mitochondrial respiratory chain can cause electron leakage and
produce a large amount of ROS. Oxidative stress plays an important
role in renal apoptosis; ROS generated by mitochondria after the
binding of UA crystals to TLR is a pivotal mediator for activation
of the NLRP3 inflammasome in the development of MSU-crystal-induced
renal inflammation (22). The
current study revealed that oxidative stress generated by UA
crystals induced renal apoptosis through activation of a
caspase-dependent apoptosis pathway (23). A previous study demonstrated that UA
at a concentration of 10 mg/dl activates NALP3 and increases
cleaved caspase-1 level and caspase-1 activity. Glyburide, a NALP3
inhibitor, prevents the above almost completely (24). In the present study, when the
stimulation time of UA was fixed, the cell survival rate gradually
decreased with the increase of the stimulation concentration and
the damage of UA to cells increased with the increase of the
stimulation concentration. In addition, the relevant literature was
consulted, so 100 µg/ml UA for 24 h, and not 48 or 72 h, was used
as the timepoint (23). It was
found that UA promoted dose-dependent apoptotic damage in NRK-52E
cells over 24 h. The results of the present study demonstrated a
dose-dependent increase in cell apoptosis upon UA treatment. It
showed that NEK7/NLRP3 signaling might be involved in the mechanism
of tubular epithelial cells apoptosis in a high UA environment.
More significantly, ROS, which is a key factor in the process of
apoptosis, was associated closely with tubular epithelial cells
injury and thus might represent a direct mechanism of UA-induced
tubular epithelial cell apoptosis.
Cell death mechanisms have been proven to be
associated with the development of UA-induced kidney disease
(25). High apoptosis rates have
been observed in the renal tubular epithelial cells of rats with
UA-induced kidney disease (26).
Increasing evidence shows that apoptosis could serve as a
therapeutic paradigm in hyperuricemia (27). Yang et al (28) found that renal tubular epithelial
cell apoptosis occurs in rats with UA-induced kidney disease and
the use of the antioxidant glutathione can inhibit cell apoptosis
significantly and reduce kidney damage. The results of the present
study also indicated that NAC inhibited UA-induced apoptosis. In
addition, the levels of the proapoptotic factors cleaved caspase-3
and Bax were upregulated significantly and the level of
antiapoptotic factor Bcl-xl was downregulated after stimulation
with UA. NAC attenuated the effect of UA on the levels of these
proteins.
Previous studies demonstrated that various stress
stimuli, including the oxidative stress caused by ROS, might induce
the activation of NLRP3 inflammasome signaling pathways (29,30).
In addition, NLRP3 inflammasome activation contributes to
stress-induced apoptosis (31).
NLRP3 inflammasomes can also activate caspase-8 and activated
caspase-8 promoted caspase-3 cleavage in response to nigericin,
which initiates apoptosis (32).
Tsuchiya et al (33)
reported that caspase-1 induction of apoptosis involves the
Bid-dependent mitochondrial apoptosis pathway. NEK7 is a key
protein in the assembly and activation of NLRP3 inflammasomes
(34). In the downstream pathway of
mitochondrial ROS production, NEK7 binds to the NLRP3 leucine rich
repeat region in a kinase-independent manner. NEK7 mediates the
activation of NLRP3 inflammasomes by connecting adjacent NLRP3
subunits through a biphasic interaction (13). In a Nek7 knockout mouse
model, the absence of NEK7 inhibits NLRP3 activation, which
verifies the key role of NEK7 in the assembly and activation of
NLRP3 (35). The results of the
present study demonstrated that ROS upregulated the levels of NEK7,
NLRP3, ASC and caspase-1. NAC was also observed to inhibit
NEK7/NLRP3 activation, suggesting that NAC attenuated renal tubular
epithelial cell apoptosis by reducing UA-induced oxidative stress
via the NEK7/NLRP3 signaling pathway. ROS-mediated activation of
NLRP3 inflammatory bodies appeared to be related to NEK7. A
previous study demonstrated that artemisinin can regulate
NEK7-mediated NLRP3 inflammasome activation by suppressing the
interaction between NEK7 and NLRP3 in lipopolysaccharide and
UA-induced inflammation in human U937 macrophages and in UA-induced
arthritic mice (36). Thus,
inhibition of NEK7/NLRP3 activation reduced UA-induced renal
tubular cell damage.
Oxidative stress in tubular cells has emerged as a
major cause of renal damage in different pathophysiological
processes, such as UA-induced kidney disease and diabetic
nephropathy (23,37). ROS are considered to be important
mediators of several biological responses, such as inflammation and
apoptosis (38,39). ROS is a normal metabolite of redox
reactions. The normal balance of ROS can bidirectionally regulate
cell apoptosis and proliferation and activates a series of signal
transduction pathways essential for transcription (40). Excessive ROS can damage cell
integrity and lead to tissue dysfunction through lipid, protein,
mitochondria and cellular DNA peroxidation (22). The present study demonstrated that
oxidative stress generated by UA activated a specific proteases
termed the executioner caspases (e.g., cleaved caspase-3) and
downregulated the expression of the anti-apoptotic molecule Bcl-xl
in the caspase-dependent apoptosis pathway. Recent studies indicate
that UA triggers the generation of oxidant stress in several
different cell types (41–43). In early chronic kidney disease, UA
can increase the production of ROS, regulate the activation of the
NLRP3 inflammasome and further induce early vascular endothelial
cell injury of chronic kidney disease (5). Oxidative stress injury is also
reported to be involved in UA-induced pancreatic cell apoptosis
(44). The present study showed
that UA increased the production of ROS in a dose-dependent manner.
Subsequently, it was confirmed that the apoptotic events in NRK-52E
cells cultivated under high UA levels were dependent on ROS
generation. Inhibition of ROS has been demonstrated as protective
in several experimental cell models, such as hypoxia reoxygenation,
unilateral ureteral obstruction (UUO), chronic renal failure and
diabetic nephropathy (45–48). The findings of the present study
offered an explanation for the renal cell protective effects of ROS
scavengers. It was observed that UA-induced ROS production was
inhibited by the antioxidant NAC, suggesting a major role of UA in
inducing oxidative stress in renal tubular epithelial cell
injury.
Accumulating evidence demonstrates that the intake
of toxic agents (such as uric acid, oxalic acid and heavy metals)
might lead to cell antioxidant defense dysfunction and apoptosis
(49–51); however, the exact mechanism of
apoptosis caused by UA-induced antioxidant imbalance requires
further study. There have been a number of studies on the mechanism
of UA-mediated ROS-induced apoptosis. It is reported that the
ROS-p53 signaling pathway is involved in the pathogenesis of UA
kidney injury. Li et al (11) observe that UA promotes ROS-induced
apoptosis in HK-2 cells. The mechanism involves the activation of
P38 and ERK1/2. The present study verified the role of NEK7 in
UA-induced apoptosis of renal tubular cells. However, it only
verified the role of NEK7 in ROS-activated, NLRP3-induced apoptosis
and did not use NLRP3 inhibitors and NEK7 knockdown; this is the
limitation of the present study. Thus, further research is required
to understand the mechanism by which UA stimulates ROS to influence
NEK7-induced NLRP3 inflammasome activation and apoptosis. Hence,
the exact mechanisms and inhibitors of NEK7-licensed NLRP3
inflammasome activation should be investigated further, NLRP3
inhibitors and NEK7 knockout should be further investigated.
Therefore, the results of the present study suggested that the
ROS-NEK7-NLRP3 signaling pathway might be one of the main
mechanisms through which UA causes renal tubular epithelial cell
apoptosis. However, there might be other mechanisms by which UA
induces apoptosis and a study reports that inhibition of EZH2
attenuates renal tubular cell apoptosis in hyperuricemic mice
(52), thus further experiments are
needed.
In summary, ROS can lead to the apoptosis of NRK-52E
cells, which might be an important pathogenic characteristic of
renal tubular damage caused by UA. The upregulation of cleaved
caspase-3 and Bax, the downregulation of Bcl-xl levels and the
activation of the NEK7-NLRP3 signaling pathway might serve key
roles in the apoptosis of NRK-52E cells induced by ROS.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81874437 and
81904126), the Three Year Action Plan Project of Shanghai
Accelerating Development of Traditional Chinese Medicine [grant no.
ZY (2018-2020)-CCCX-2003-08] and the Key Disciplines Group
Construction Project of Pudong Health Bureau of Shanghai (grant no.
PWZxq2017-07).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DL and JG conceived and designed the study. DL wrote
the manuscript. LW, JO, LL, CW and JZ performed the experiments; LL
and YW analyzed the data; JG reviewed and revised the manuscript.
DL and JG confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
He L, Fan Y, Xiao W, Chen T, Wen J, Dong
Y, Wang Y, Li S, Xue R, Zheng L, et al: Febuxostat attenuates ER
stress mediated kidney injury in a rat model of hyperuricemic
nephropathy. Oncotarget. 8:111295–111308. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kang DH: Hyperuricemia and progression of
chronic kidney disease: Role of phenotype transition of renal
tubular and endothelial cells. Contrib Nephrol. 192:48–55. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tao M, Shi Y, Tang L, Wang Y, Fang L,
Jiang W, Lin T, Qiu A, Zhuang S and Liu N: Blockade of ERK1/2 by
U0126 alleviates uric acid-induced EMT and tubular cell injury in
rats with hyperuricemic nephropathy. Am J Physiol Renal Physiol.
316:F660–F673. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Priante G, Gianesello L, Ceol M, Del Prete
D and Anglani F: Cell death in the kidney. Int J Mol Sci.
20:35982019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yin W, Zhou QL, OuYang SX, Chen Y, Gong YT
and Liang YM: Uric acid regulates NLRP3/IL-1β signaling pathway and
further induces vascular endothelial cells injury in early CKD
through ROS activation and K+ efflux. BMC Nephrol.
20:3192019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Moreno-Irusta A, Dominguez EM,
Marín-Briggiler CI, Matamoros-Volante A, Lucchesi O, Tomes CN,
Treviño CL, Mariano GB, Lascano R, Losinno L and Giojalas LC:
Reactive oxygen species are involved in the signaling of equine
sperm chemotaxis. Reproduction. 159:423–436. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu G, Chen X, Wang Q and Yuan L: NEK7: A
potential therapy target for NLRP3-related diseases. Biosci Trends.
14:74–82. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shi H, Wang Y, Li X, Zhan X, Tang M, Fina
M, Su L, Pratt D, Bu CH, Hildebrand S, et al: NLRP3 activation and
mitosis are mutually exclusive events coordinated by NEK7, a new
inflammasome component. Nat Immunol. 17:250–258. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma ZZ, Sun HS, Lv JC, Guo L and Yang QR:
Expression and clinical significance of the NEK7-NLRP3 inflammasome
signaling pathway in patients with systemic lupus erythematosus. J
Inflamm (Lond). 15:162018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Braga TT, Forni MF, Correa-Costa M, Ramos
RN, Barbuto JA, Branco P, Castoldi A, Hiyane ML, Davanso MR, Latz
E, et al: Soluble uric acid activates the NLRP3 inflammasome. Sci
Rep. 7:398842017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Z, Sheng Y, Liu C, Li K, Huang X, Huang
J and Xu K: Nox4 has a crucial role in uric acid-induced oxidative
stress and apoptosis in renal tubular cells. Mol Med Rep.
13:4343–4348. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu Y, He F, Li Y, Wang H, Shi L, Wang Q,
Ou J, Zhang X, Huang D, Xu L, et al: Effects of Shizhifang on NLRP3
inflammasome activation and renal tubular injury in hyperuricemic
rats. Evid Based Complement Alternat Med. 2017:76742402017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sharif H, Wang L, Wang WL, Magupalli VG,
Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Núñez G, Mao Y and Wu H:
Structural mechanism for NEK7-licensed activation of NLRP3
inflammasome. Nature. 570:338–343. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Oh TR, Choi HS, Kim CS, Bae EH, Ma SK,
Sung SA, Kim YS, Oh KH, Ahn C and Kim SW: Hyperuricemia has
increased the risk of progression of chronic kidney disease:
Propensity score matching analysis from the KNOW-CKD study. Sci
Rep. 9:66812019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han Y, Xu X, Tang C, Gao P, Chen X, Xiong
X, Yang M, Yang S, Zhu X, Yuan S, et al: Reactive oxygen species
promote tubular injury in diabetic nephropathy: The role of the
mitochondrial ros-txnip-nlrp3 biological axis. Redox Biol.
16:32–46. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu YY, Qiu XH, Ye Y, Gao C, Wu F and Xia
G: Risk factors analysis for hyperuricemic nephropathy among CKD
stages 3–4 patients: An epidemiological study of hyperuricemia in
CKD stages 3–4 patients in Ningbo, China. Ren Fail. 40:666–671.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hyndman D, Liu S and Miner JN: Urate
handling in the human body. Curr Rheumatol Rep. 18:342016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jung SW, Kim SM, Kim YG, Lee SH and Moon
JY: Uric acid and inflammation in kidney disease. Am J Physiol
Renal Physiol. 318:F1327–F1340. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xiao J, Zhang X, Fu C, Yang Q, Xie Y,
Zhang Z and Ye Z: Impaired Na+-K+-ATPase
signaling in renal proximal tubule contributes to
hyperuricemia-induced renal tubular injury. Exp Mol Med.
50:e4522018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Isaka Y, Takabatake Y, Takahashi A, Saitoh
T and Yoshimori T: Hyperuricemia-induced inflammasome and kidney
diseases. Nephrol Dial Transplant. 31:890–896. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang F, Gomez-Sintes R and Boya P:
Lysosomal membrane permeabilization and cell death. Traffic.
19:918–931. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou R, Yazdi AS, Menu P and Tschopp J: A
role for mitochondria in NLRP3 inflammasome activation. Nature.
469:221–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choe JY, Park KY and Kim SK: Oxidative
stress by monosodium urate crystals promotes renal cell apoptosis
through mitochondrial caspase-dependent pathway in human embryonic
kidney 293 cells: Mechanism for urate-induced nephropathy.
Apoptosis. 20:38–49. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eleftheriadis T, Pissas G, Antoniadi G,
Makri P, Liakopoulos V and Stefanidis I: Urate crystals induce
NLRP3 inflammasome-dependent IL-1β secretion and proliferation in
isolated primary human T-cells. Hippokratia. 19:41–46.
2015.PubMed/NCBI
|
|
25
|
Shi Y, Tao M, Ma X, Hu Y, Huang G, Qiu A,
Zhuang S and Liu N: Delayed treatment with an autophagy inhibitor
3-MA alleviates the progression of hyperuricemic nephropathy. Cell
Death Dis. 11:4672020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu J, Yang Z, Wu H and Wang D: Rhein
attenuates renal inflammatory injury of uric acid nephropathy via
lincRNA-Cox2/miR-150-5p/STAT1 axis. Int Immunopharmacol.
85:1066202020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
He Y, Qu Q, Luo T, Gong Y, Hou Z, Deng J,
Xu Y, Wang B and Hao S: Human hair keratin hydrogels alleviate
rebleeding after intracerebral hemorrhage in a rat model. ACS
Biomater Sci Eng. 5:1113–1122. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang L, Chang B, Guo Y, Wu X and Liu L:
The role of oxidative stress-mediated apoptosis in the pathogenesis
of uric acid nephropathy. Ren Fail. 41:616–622. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang Y, Liu L, Sun D, He Y, Jiang Y,
Cheng KW and Chen F: DHA protects against monosodium urate-induced
inflammation through modulation of oxidative stress. Food Funct.
10:4010–4021. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang M, Zhao J, Zhang N and Chen J:
Astilbin improves potassium oxonate-induced hyperuricemia and
kidney injury through regulating oxidative stress and inflammation
response in mice. Biomed Pharmacother. 83:975–988. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun L, Ma W, Gao W, Xing Y, Chen L, Xia Z,
Zhang Z and Dai Z: Propofol directly induces caspase-1-dependent
macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell
Death Dis. 10:5422019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gaidt MM, Ebert TS, Chauhan D, Ramshorn K,
Pinci F, Zuber S, O'Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et
al: The DNA inflammasome in human myeloid cells is initiated by a
STING-cell death program upstream of NLRP3. Cell.
171:1110–1124.e18. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tsuchiya K, Nakajima S, Hosojima S, Thi
Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y,
Miura M, et al: Caspase-1 initiates apoptosis in the absence of
gasdermin D. Nat Commun. 10:20912019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chiu HW, Li LH, Hsieh CY, Rao YK, Chen FH,
Chen A, Ka SM and Hua KF: Glucosamine inhibits IL-1β expression by
preserving mitochondrial integrity and disrupting assembly of the
NLRP3 inflammasome. Sci Rep. 9:56032019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zeng Q, Deng H, Li Y, Fan T, Liu Y, Tang
S, Wei W, Liu X, Guo X, Jiang J, et al: Berberine directly targets
the NEK7 protein to block the NEK7-NLRP3 interaction and exert
anti-inflammatory activity. J Med Chem. 64:768–781. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kim SK, Choe JY and Park KY:
Anti-inflammatory effect of artemisinin on uric acid-induced NLRP3
inflammasome activation through blocking interaction between NLRP3
and NEK7. Biochem Biophys Res Commun. 517:338–345. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Huang C, Zhang Y, Kelly DJ, Tan CY, Gill
A, Cheng D, Braet F, Park JS, Sue CM, Pollock CA and Chen XM:
Thioredoxin interacting protein (TXNIP) regulates tubular autophagy
and mitophagy in diabetic nephropathy through the mTOR signaling
pathway. Sci Rep. 6:291962016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Giribabu N, Karim K, Kilari EK and Salleh
N: Phyllanthus niruri leaves aqueous extract improves kidney
functions, ameliorates kidney oxidative stress, inflammation,
fibrosis and apoptosis and enhances kidney cell proliferation in
adult male rats with diabetes mellitus. J Ethnopharmacol.
205:123–137. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang Y, Lv X, Hu Z, Ye X, Zheng X, Ding
Y, Xie P and Liu Q: Protection of Mcc950 against
high-glucose-induced human retinal endothelial cell dysfunction.
Cell Death Dis. 8:e2941. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhou X, Bai C, Sun X, Gong X, Yang Y, Chen
C, Shan G and Yao Q: Puerarin attenuates renal fibrosis by reducing
oxidative stress induced-epithelial cell apoptosis via MAPK signal
pathways in vivo and in vitro. Ren Fail. 39:423–431. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cai W, Duan XM, Liu Y, Yu J, Tang YL, Liu
ZL, Jiang S, Zhang CP, Liu JY and Xu JX: Uric acid induces
endothelial dysfunction by activating the HMGB1/RAGE signaling
pathway. Biomed Res Int. 2017:43919202017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hong Q, Wang L, Huang Z, Feng Z, Cui S, Fu
B, Cai G, Chen X and Wu D: High concentrations of uric acid and
angiotensin II act additively to produce endothelial injury.
Mediators Inflamm. 2020:83876542020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang H, Ma Y, Cao R, Wang G, Li S, Cao Y,
Zhang H, Liu M, Liu G, Zhang J, et al: Soluble uric acid induces
myocardial damage through activating the NLRP3 inflammasome. J Cell
Mol Med. 24:8849–8861. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xin Y, Wang K, Jia Z, Xu T, Xu Q, Zahng C,
Liu J, Chen R, Du Z and Sun J: Zurampic protects pancreatic β-cells
from high uric acid induced-damage by inhibiting URAT1 and
inactivating the ROS/AMPK/ERK pathways. Cell Physiol Biochem.
47:1074–1083. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang H, Peng X, Huang Y, Xiao Y, Wang Z
and Zhan L: Propofol attenuates hypoxia/reoxygenation-induced
apoptosis and autophagy in HK-2 cells by inhibiting JNK activation.
Yonsei Med J. 60:1195–1202. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Sun L, Xu H, Wang Y, Ma X, Xu Y and Sun F:
The mitochondrial-targeted peptide SBT-20 ameliorates inflammation
and oxidative stress in chronic renal failure. Aging (Albany NY).
12:18238–18250. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Jin JZ, Li HY, Jin J, Piao SG, Shen XH, Wu
YL, Xu JC, Zhang LY, Jiang YJ, Zheng HL, et al: Exogenous
pancreatic kininogenase protects against renal fibrosis in rat
model of unilateral ureteral obstruction. Acta Pharmacol Sin.
41:1597–1608. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shopit A, Niu M, Wang H and Tang Z, Li X,
Tesfaldet T, Ai J, Ahmad N, Al-Azab M and Tang Z: Protection of
diabetes-induced kidney injury by phosphocreatine via the
regulation of ERK/Nrf2/HO-1 signaling pathway. Life Sci.
242:1172482020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Han B, Li S, Lv Y, Yang D, Li J, Yang Q,
Wu P, Lv Z and Zhang Z: Dietary melatonin attenuates
chromium-induced lung injury via activating the Sirt1/Pgc-1α/Nrf2
pathway. Food Funct. 10:5555–5565. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yang D, Yang Q, Fu N, Li S, Han B, Liu Y,
Tang Y, Guo XY, Lv Z and Zhang Z: Hexavalent chromium induced heart
dysfunction via Sesn2-mediated impairment of mitochondrial function
and energy supply. Chemosphere 264 (Pt 2). 1285472021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu B, Yu H, Baiyun R, Lu J, Li S, Bing Q,
Zhang X and Zhang Z: Protective effects of dietary luteolin against
mercuric chloride-induced lung injury in mice: Involvement of
AKT/Nrf2 and NF-κB pathways. Food Chem Toxicol. 113:296–302. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Shi Y, Xu L, Tao M, Fang L, Lu J, Gu H, Ma
S, Lin T, Wang Y, Bao W, et al: Blockade of enhancer of zeste
homolog 2 alleviates renal injury associated with hyperuricemia. Am
J Physiol Renal Physiol. 316:F488–F505. 2019. View Article : Google Scholar : PubMed/NCBI
|