Introduction
Atrial fibrillation (AF) is a cardiac arrhythmia in
clinical settings with an increasing prevalence and estimated to
affect >15 million individuals, which can result in left
ventricular hypofunction, embolism and infarction (1–3). It is
a progressive disease associated with a high mortality worldwide,
and its onset and development are accompanied by atrial remodeling
(4). Moreover, the influence of AF
on human life is significant, and treating AF is associated with
high expenses, thus exacerbating the socioeconomic burden of this
disease (5). A characteristic of AF
at the early stage is electrical remodeling, with a shortened
atrial effective refractory period (AERP) and reduced physiological
rate adaptation (5). Furthermore,
in the late period, AF is manifested by structural remodeling and
structural alterations such as atrial fibrosis (6). At present, treatment measures
encompassing drug transfer, electric shock cardioversion,
interventional therapy and surgical treatment are capable of
restoring AF to sinus rhythm (2),
but they lack efficacy.
MicroRNA (miRNA/miR), short non-coding RNA,
modulates the translation of protein-coding genes by binding to the
3′untranslated regions (3′UTR) of mRNA (7). In the cardiovascular system, miRNA
serves an important role in regulating a series of physiological
functions in heart diseases, including cellular proliferation,
apoptosis and differentiation (8).
Accumulating evidence has shown the involvement of miRNAs in AF
(9). For instance, miR-27b-3p
overexpression controlled the Wnt/β-catenin signaling pathway by
targeting Wnt3a, thereby relieving atrial fibrosis in rats with AF
(10). Of note, miR-101 was found
to be a regulatory factor in myocardial infarction, myocardial
hypertrophy, rheumatic heart diseases and myocardial fibrosis
(11–14). Previous studies have reported that
overexpression of miR-101 could attenuate cardiac fibrosis and
deterioration of cardiac function in rats after aortic constriction
or coronary artery ligation (14,15).
In addition, miR-101 expression was reported to be significantly
downregulated in atrial samples from a canine model of AF and
patients with AF (5).
EZH2 encodes a member of the polycomb-group family
and exerts important roles in heart development and regeneration
(16). An increase of EZH2
expression has been observed in atrial muscle and atrial
fibroblasts of patients with AF (17). At the same time, inhibition of EZH2
mitigated the atrial enlargement and fibrosis induced by
angiotensin II, and reduced the susceptibility of AF (17). Nevertheless, the detailed function
of miR-101a-3p in atrial fibrosis triggered by AF has not been
fully clarified.
In the current study, the AF model in rats was
established as described in previous studies (10,18,19).
The present study preliminarily investigated the effect of
miR-101a-3p overexpression on the occurrence and development of AF
via targeted regulation of EZH2. Given that both miR-101a-3p and
EZH2 are closely associated with cardiac fibrosis (15,17),
the role of miR-101a-3p overexpression in AF-mediated fibrosis was
further examined. These results demonstrated that miR-101a-3p
overexpression reduced atrial fibrosis by lowering the levels of
collagen I, collagen III, TGF-β1, connective tissue growth factor
(CTGF), fibronectin and α-smooth muscle actin (α-SMA). Moreover, it
was verified that EZH2 was a target gene of miR-101a-3p. These
findings provided insights for the prevention and treatment of
AF.
Materials and methods
Experiment design
The animal experimental protocol was reviewed and
approved by the Ethics Committee of The First Affiliated Hospital
of USTC (approval no. 201907201445000472393). In total, 48 healthy
male Sprague-Dawley rats (age, 8 weeks; weight, 200–250 g) were
housed in under the following conditions: 22±1°C, 45–55% humidity
and 12 h light/12 h dark photoperiod and were given free access to
food and water. A week after adaptation, rats were randomly
assigned to four groups: Sham group, AF group, AF + lentivirus
negative control (AF + LV-NC) group and AF + LV-miR-101a-3p group
(n=12 each group).
293T cells were provided by Shanghai ZhongQiao
XinZhou Biotechnology Co., Ltd. Cells in the logarithmic growth
phase were co-transfected for 48 h with a second-generation
lentivirus vector (pSico; Addgene, Inc.; 5 µg) and pHelper vector
pMD2.G (Hunan Fenghui Biotechnology Co., Ltd.; 2 µg) and pHelper
vector psPAX2 (Hunan Fenghui Biotechnology Co., Ltd.; 3 µg) using
Lipofectamine 3000® reagent (cat. no. L3000015;
Invitrogen; Thermo Fisher Scientific, Inc.) in the accordance with
the manufacturer's protocol at 37°C in an incubator containing 5%
CO2. Next, green fluorescent protein expression used for
screening was observed under a fluorescence microscope at ×100
magnification. Next, the supernatants were harvested via
ultracentrifugation (72,000 × g) for 2 h at 4°C and then stored at
−80°C.
For infection of LV vectors, rats were anesthetized
with an intraperitoneal injection of pentobarbital sodium (50
mg/kg), immobilized, intubated and ventilated. After the breathing
was stable, thoracic surgery was performed between the third and
fourth intercostals space to expose the heart, and then 20 µl LV-NC
or LV-miR-101a-3p (108 TU), based on the following
sequences: miR-NC,
5′-TTCTCCGAACGTGTCACGTCGATTTCTCCGAACGTGTCACGTACCGGTTTCTCCGAACGTGTCACGTTCACTTCTCCGAACGTGTCACGTTTTTTT−3′;
and miR-101a-3p, 5′-UACAGUACUGUGAUAACUGAA−3′, was injected into the
right atrium of the rat, as described in previous studies (4,7). Sham
rats and AF rats were injected with equal volume of saline. After
48 h from LV injection, all rats were anesthetized. Then, 1 ml/kg
acetylcholine (Ach)-CaCl2 mixture (cat. no. A6625;
Sigma-Aldrich; Merck KGaA) containing 10 mg CaCl2 and 60
µg Ach in mixture per ml (CaCl2: Ach=1:0.006) was
injected into rats via the tail vein daily for 7 days.
Subsequently, the rats were euthanatized with an overdose (200
mg/kg) of pentobarbital sodium via intraperitoneal injection. The
experimental design was displayed in Fig. 1A.
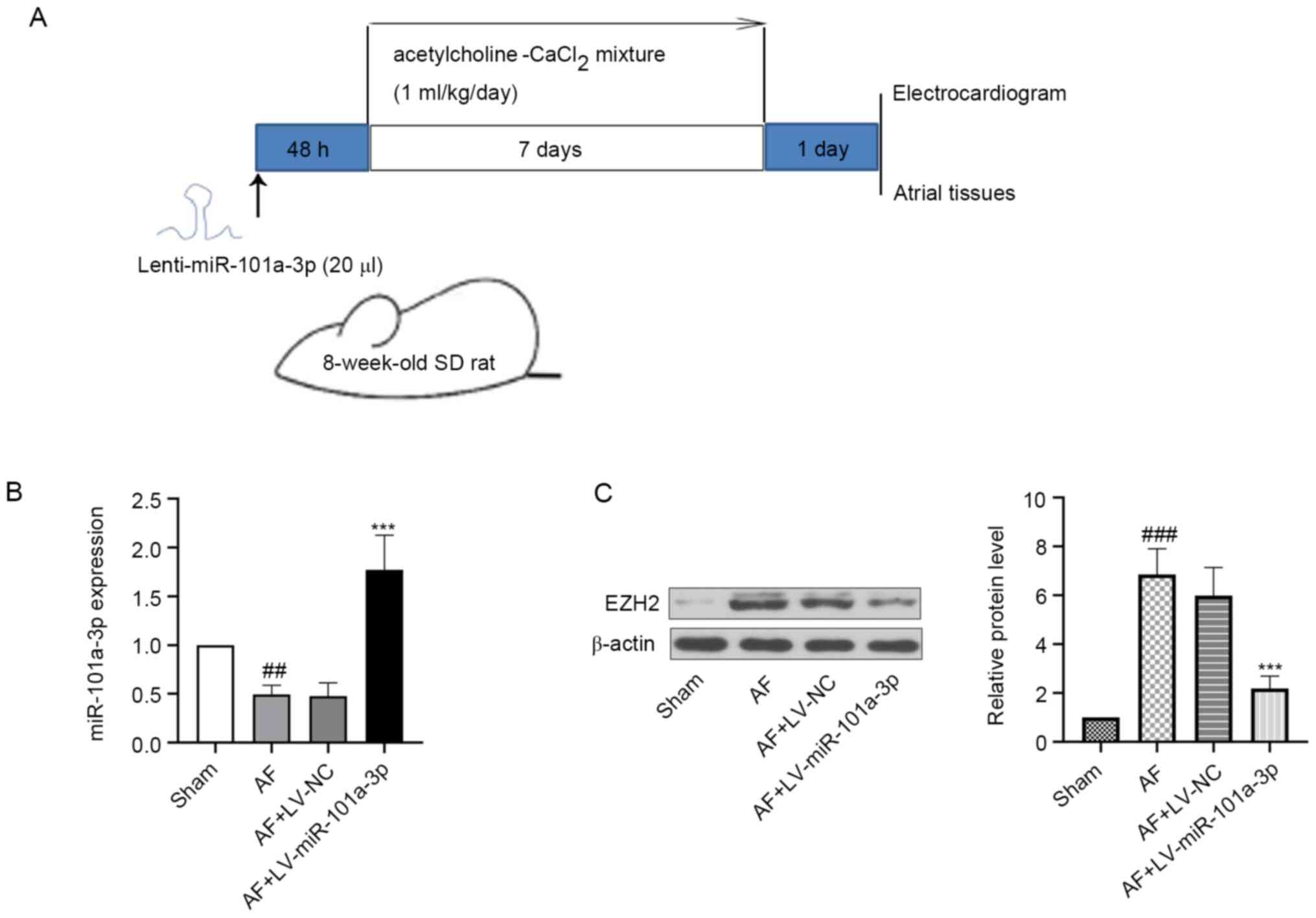 | Figure 1.Expression levels of miR-101a-3p and
EZH2 in atrial tissues of rats. The rats were anesthetized with an
intraperitoneal injection of pentobarbital sodium (50 mg/kg),
fixed, intubated and injected with LV-NC or LV-miR-101a-3p. After
injection for 48 h, all rats were anesthetized. Then, 1 ml/kg
acetylcholine-CaCl2 mixture via the tail vein was
injected into rats daily for 7 days. At day 8, rats were
anesthetized, injected with Ach-CaCl2 and sacrificed.
The atrial tissues of heart were collected for following
experiments. (A) Experimental protocol in rats. (B) miR-101a-3p
expression was detected via reverse transcription-quantitative PCR.
(C) Protein expression level of EZH2 was measured via western
blotting. β-actin was used as an internal reference. The results
are presented as the mean ± standard deviation (n=6).
##P<0.01 and ###P<0.001 vs. sham group;
***P<0.001 vs. AF + LV-NV group. LV-NC, lentivirus negative
control; AF, atrial fibrillation; miR, microRNA; EZH2, enhancer of
zeste 2 homolog 2; SD, Sprague-Dawley. |
Electrocardiogram (ECG) recording
At day 8, rats from each group were anesthetized
with pentobarbital sodium (50 mg/kg) via an intraperitoneal
injection. Saline was given to sham rats, while others were given a
mixture of Ach-CaCl2 via the tail vein. Then, standard
lead II ECG traces were recorded for evaluation of arrhythmias with
30-gauge subcutaneous needle electrodes (Grass Technologies)
utilizing the BL-420F bio-function experiments system (Chengdu
Techman Software Co., Ltd.; http://www.tme.com.cn/). With reference to a previous
article (20), ECG signals were
filtered (0.5–150 Hz) and amplified (20 mm/mV) with an FX8322
system (FUKUDA DENSHI). The sweep of ECG paper was performed at 50
mm/sec. Analog ECG recordings at baseline were made at 25–100
mm/sec paper speed and 1 mm/mV amplitude. Ach-CaCl2
induced the absence of P waves, irregular heartbeat and R-R
intervals from the ECG, which showed that the establishment of a
rat AF model was successful (21).
Electrophysiological study
After AF induction, the rats were anesthetized with
pentobarbital sodium, and the 2.0 F small animal
electrophysiological catheter (Nippoly; http://www.nippoly.com/) was inserted by the jugular
vein into the right atrial. The AERP was detected with S1-S2
programmed electrical stimulation (2X threshold at 2-msec duration)
as previously reported (4). The S2
extra-stimulus method using eight regularly paced beats was
delivered. AERP was measured at basic cycle lengths of 150 msec and
was defined as the longest S1-S2 coupling interval failing to
elicit an action potential. Moreover, AF duration was recorded, and
AF inducibility was calculated. Finally, rats were sacrificed, and
the atrial tissues were separated and stored at −70°C or fixed at
4°C in 4% paraformaldehyde until the following experiments.
H&E staining
Fixed atrial tissues of rats were dehydrated,
embedded in paraffin and cut into 5-µm slices. The sections were
deparaffinated and stained with hematoxylin (cat. no. H8070;
Beijing Solarbio Science & Technology Co., Ltd.) for 5 min at
room temperature. After being washed with distilled water, slices
were counterstained with eosin at room temperature for 3 min. The
pathological changes were visualized using a light microscope
(BX53; Olympus Corporation) at ×200 magnification.
Masson staining
A Masson staining kit (cat. no. DC0032; Beijing
Leagene Biotechnology; http://www.leagene.com/) was employed to assess the
degree of fibrosis in AF model rats according to the manufacturer's
instructions. In brief, dewaxed sections were stained with Reguad
hematoxylin dye at room temperature for 6 min. The slices were
immersed in 1% hydrochloric acid in ethanol for 3 sec and rinsed
with distilled water for 2 min. Ponceau-acid fuchsin was added to
each section and reacted for 1 min. After being washed with 0.2%
glacial acetic acid, 1% phosphomolybdic acid was added and slices
were counterstained with aniline blue for 5 min at room
temperature, following which they were imaged under a light
microscope. The size of fibrotic area was quantified as described
in previous study (22). Digital
images of five fields were randomly selected from each section and
the mean of the fibrosis area of atrial tissues was calculated
using Image-Pro Plus 6.0 analysis software (23,24).
Immunohistochemistry assay
Dewaxed slices were incubated with antigen retrieval
solution (mixing 9 ml citrate buffer solution, 41 ml sodium citrate
buffer solution and 450 ml distilled water; pH 6.0) for 10 min at
95°C in a water bath. After being washed with PBS three times, the
sections were incubated with 3% H2O2 (cat.
no. 10011218; Sinopharm Chemical Reagent Co., Ltd.) for 15 min at
room temperature and blocked with undiluted goat serum (cat. no.
SL038; Beijing Solarbio Science & Technology Co., Ltd.) for 15
min at room temperature. Next, slices were incubated with
antibodies against collagen I (cat. no. AF7001; 1:100; Affinity
Biosciences), collagen III (cat. no. AF0136; 1:100; Affinity
Biosciences) or EZH2 (cat. no. AF5150; 1:100; Affinity Biosciences)
at 4°C overnight. After being rinsed with PBS, sections were
cultured with HRP-labeled goat anti-rabbit IgG (cat. no. 31460;
1:500; Thermo Fisher Scientific, Inc.) at 37°C for 1 h.
Diaminobenzidine (cat. no. DA1010; Beijing Solarbio Science &
Technology Co., Ltd.) was added to each slice, and the sections
were counterstained with hematoxylin for 3 min at room temperature.
The staining results were observed with an Olympus light microscope
at ×400 magnification. Image-Pro Plus 6.0 software (23,24)
was utilized for quantitative analysis. In total, three
non-overlapping microscopic fields were selected and the number of
positive-stained cells was counted. Finally, the mean value was
calculated.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from atrial tissues of rats
using a commercial kit (cat. no. RP1201; BioTeke Corporation)
according to the manufacturer's instructions. After detection of
RNA purity and concentration, RNA was reverse transcribed into cDNA
using the RNA Reverse Transcription kit (Takara Bio, Inc.). The
reaction conditions were 37°C for 30 min, 42°C for 30 min and 70°C
for 10 min. For qPCR, cDNA templates, primers, SYBR GREEN (cat. no.
EP1602; BioTeke Corporation) and Taq HS Perfect mix (cat. no.
R300A; Takara Bio, Inc.) were mixed and incubated under standard
PCR conditions. The reactions were incubated at 94°C for 2 min,
94°C for 10 sec, 60°C for 15 sec and 72°C for 15 sec, followed by
40 cycles at 72°C for 5 min 30 sec, 40°C for 5 min 30 sec, and
melting from 60°C to 94°C, every 1°C/1 sec, and 25°C for 1 min. The
expression level of miR-101a-3p was assessed using the
2−ΔΔCq method (25). The
primers utilized were supplied by GenScript, and their sequences
were as follows: rno-miR-101a-3p forward,
5′-TACAGTACTGTGATAACTGAA−3′ and reverse: 5′-GCAGGGTCCGAGGTATTC−3′;
and 5S forward, 5′-GATCTCGGAAGCTAAGCAGG-3′ and reverse,
5′-TGGTGCAGGGTCCGAGGTAT-3′.
Western blotting
The atrial tissues of rats were lysed in RIPA buffer
(cat. no. P0013B; Beyotime Institute of Biotechnology) containing
1% PMSF (cat. no. ST506; Beyotime Institute of Biotechnology) on
ice for 5 min. After centrifugation (4°C, 10,000 × g, 3 min), the
supernatants were collected as total protein samples. The
concentration of protein was determined with a BCA kit (cat. no.
P0009; Beyotime Institute of Biotechnology). The samples (30 µg
protein) were separated via 10% SDS-PAGE at 80 V for 2.5 h and then
transferred onto PVDF membranes (cat. no. LC2005; Thermo Fisher
Scientific, Inc.). After reacting for 1.5 h at 80 V, the membranes
were blocked with 5% BSA (cat. no. BS043, Biosharp Life Sciences)
for 1 h at room temperature, and then incubated with primary
antibody at 4°C overnight. The membranes were cultured with
secondary antibody for 40 min at 37°C and washed with TBS-0.15%
Tween 20. The bands were observed using a gel imaging system
(WD-9413B, Beijing Liuyi Biotechnology Co., Ltd.) with ECL solution
(cat. no. E003; 7 Sea Biotech). The information for the antibodies
utilized in this study is as follows: EZH2 (85 kDa; cat. no.
AF7901; 1:1,000, Affinity Biosciences), TGF-β1 (12 kDa; cat. no.
BA0290; 1:1,000; Wuhan Boster Biological Technology, Ltd.), CTGF
(38 kDa; cat. no. DF7091; 1:500; Affinity Biosciences), fibronectin
(290 kDa; cat. no. A7488; 1:500; ABclonal Biotech Co., Ltd.), α-SMA
(42 kDa; cat. no. AF1032; 1:500; Affinity Biosciences), β-actin (42
kDa; cat. no. 60008-1-Ig; 1:2,000; ProteinTech Group, Inc.),
HRP-conjugated goat anti-rabbit IgG (cat. no. SA00001-2; 1:10,000;
ProteinTech Group, Inc.) and HRP-conjugated goat anti-mouse IgG
(cat. no. SA00001-1; 1:10,000; ProteinTech Group, Inc.).
Luciferase reporter gene assay
293T cells were cultured with DMEM (cat. no. D5648;
Sigma-Aldrich; Merck KGaA) containing 10% FBS (cat. no. F8067;
Sigma-Aldrich; Merck KGaA) in an incubator (37°C; 5%
CO2). The EZH2 wild-type (WT) sequences or mutant (MUT)
sequences in 3′UTR containing the miR-101a-3p binding site were
constructed and subcloned into the pmirGLO dual-luciferase miRNA
target expression plasmid (GenScript), herein referred as EZH2
3′UTR WT or EZH2 3′UTR MUT. When the density reached 70%, the cells
were seeded in a 12-well plate, and co-transfected with EZH2 3′UTR
WT or MUT EZH2 3′UTR and miR-101a-3p mimics or mimics NC (JTS
Scientific; http://www.jtsbio.com/) for 4 h in a
saturated and humidified 37°C incubator containing 5%
CO2. RNAs (75 pmol; 25 nmol/l) or plasmids (1.5 µg per
well; 0.5 µg/ml) were transfected into 293T cells using
Lipofectamine 2000® reagent (cat. no. 11668030;
Invitrogen; Thermo Fisher Scientific, Inc.), following the
manufacturer's instructions. Then, 48 h after transfection, cells
were lysed and luciferase activity was measured using a
Dual-Luciferase Reporter Assay system (cat. no. E1910; Promega
Corporation). Renilla luciferase activity was utilized to
normalize the reporter activity.
Statistical analysis
The data are presented as the mean ± standard
deviation and were analyzed using GraphPad Prism 8 software
(GraphPad Software, Inc.) with one-way ANOVA, followed by Tukey's
multiple comparisons test. All experiments were repeated
independently ≥3 times. P<0.05 was considered to indicate a
statistically significant difference.
Results
miR-101a-3p and EZH2 expression in
atrial tissues of rats
The expression levels of miR-101a-3p and EZH2 in
atrial tissues of AF model rats were evaluated using RT-qPCR and
western blotting. It was found that miR-101a-3p expression was
decreased in AF model rats when compared with the sham group. After
infection with LV-miR-101a-3p, the expression level of miR-101a-3p
was significantly increased (Fig.
1B). Moreover, it was suggested that the protein expression
level of EZH2 was higher in the AF model group compared with that
of sham rats (Fig. 1C). However,
miR-101a-3p overexpression reduced EZH2 expression.
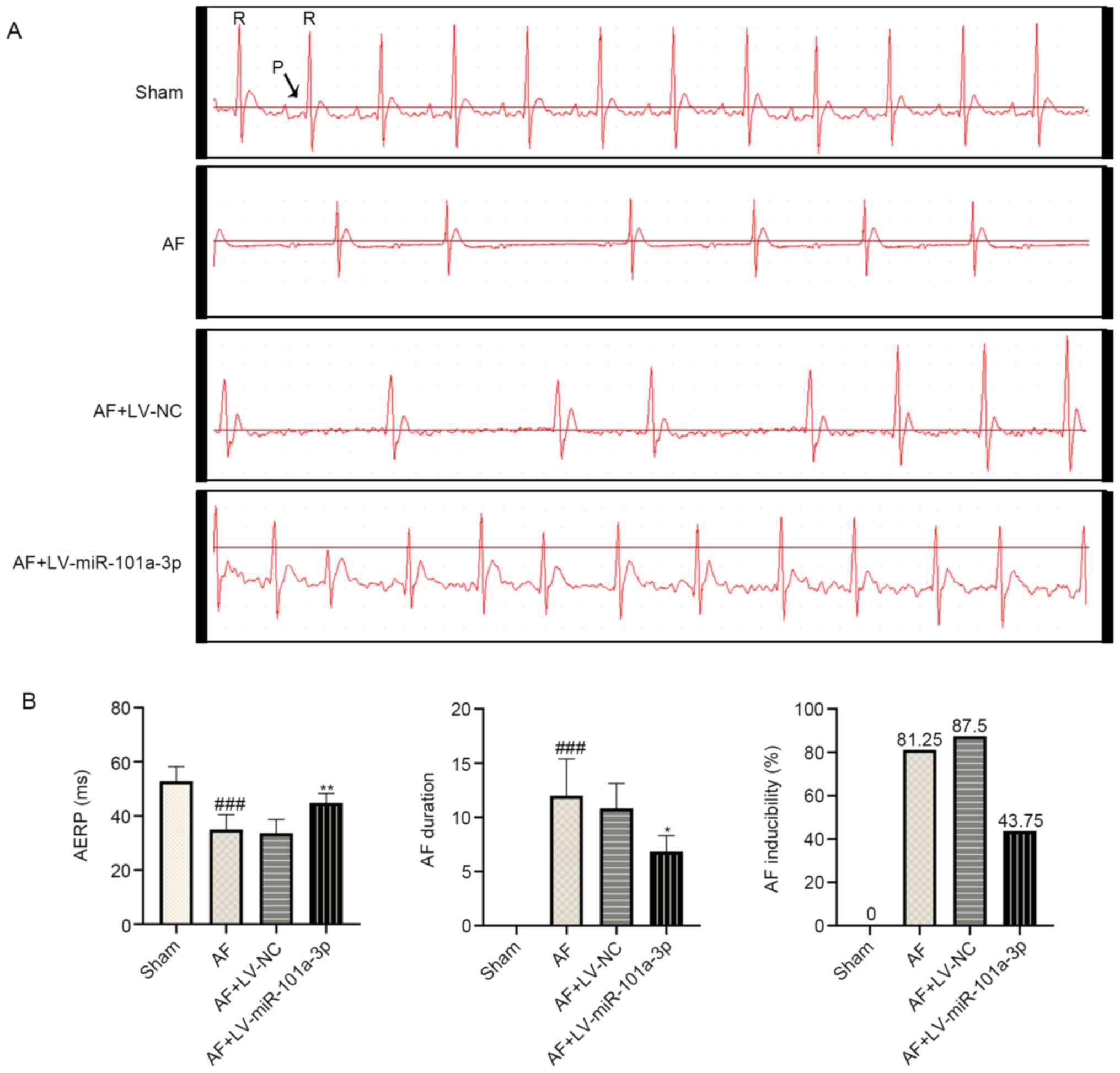
miR-101a-3p overexpression reduces the
incidence and duration of AF in rats
To evaluate whether the AF model in rats was
established successfully, electrophysiological changes were
monitored. As shown in Fig. 2A,
regular P wave was observed in the sham group, suggesting normal
sinus rhythm. However, disappeared P wave and irregular R-R
interphase changes were observed in AF model rats. In addition, a
decreased heartbeat was observed in AF model rats, most likely
owing to the continuous Ach-CaCl2 perfusion.
Interestingly, miR-101a-3p overexpression significantly inhibited
heart rhythm changes in rats with AF. As presented in Fig. 2B, the AERP was shorter in AF model
rats compared with that of sham rats. By contrast, miR-101a-3p
overexpression reversed this process. Additionally, a lowered
duration and incidence of AF was observed in AF + LV-miR-101a-3p
group compared with AF model rats infected with LV-NC. These
findings indicated the successful establishment of an AF rat
model.
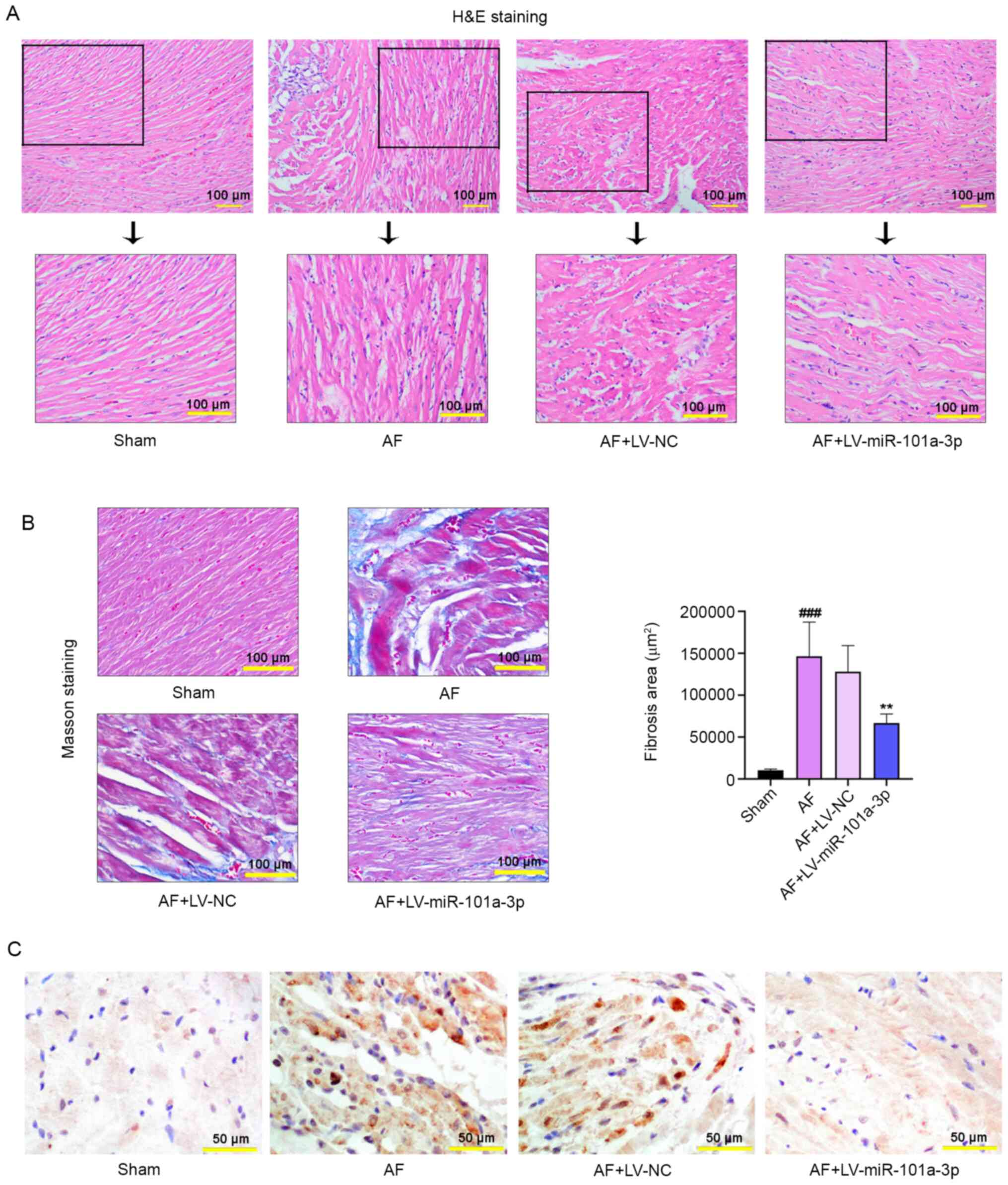
Pathological staining was performed to
evaluate the severity of AF in rats
H&E staining was conducted to evaluate the
pathological changes of atrial tissues in rats. As displayed in
Fig. 3A, miR-101a-3p overexpression
led to the reduction of Ach-CaCl2-triggered tissue
injury. Masson staining results also demonstrated that an increased
degree of atrial fibrosis was observed in AF model rats, whereas
miR-101a-3p overexpression abolished this process (Fig. 3B). The results of the quantitative
analysis of fibrosis were consistent with that of Masson staining.
Furthermore, immunohistochemistry analysis confirmed that
miR-101a-3p overexpression decreased the expression of EZH2 in
atrial tissues (Fig. 3C).
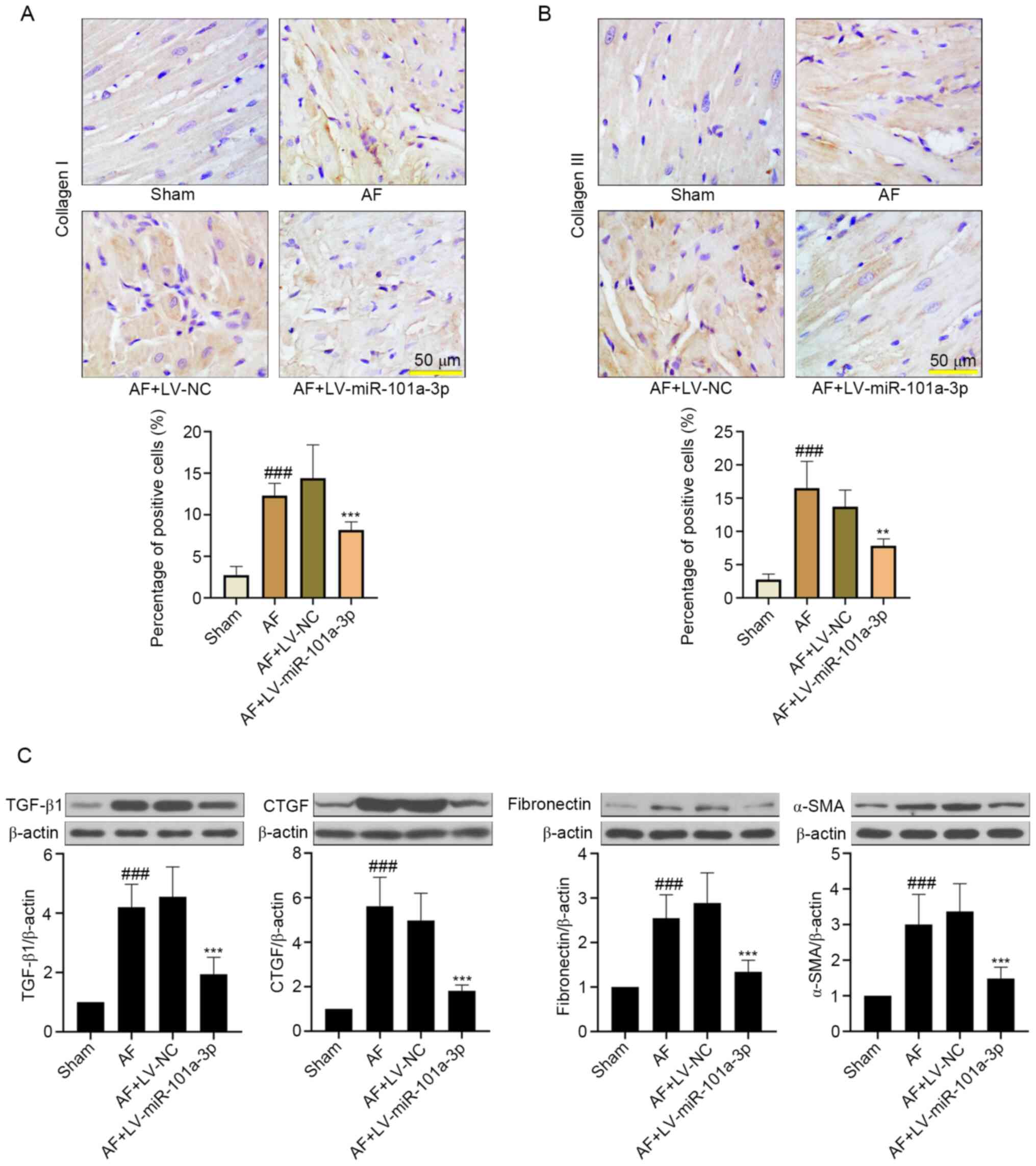
miR-101a-3p overexpression attenuates
atrial tissue fibrosis in rats with AF
To further examine the role of miR-101a-3p
overexpression in tissue fibrosis, the changes of collagen I and
collagen III were detected. As shown in Fig. 4A and B, Ach-CaCl2
significantly increased atrial fibrosis by upregulating collagen I
and collagen III expression. On the contrary, miR-101a-3p
overexpression decreased these protein expression levels. In
addition, the western blotting results indicated that
Ach-CaCl2-induced AF significantly increased the
expression levels of TGF-β1, CTGF, fibronectin and α-SMA, which
were downregulated by miR-101a-3p overexpression (Fig. 4C).
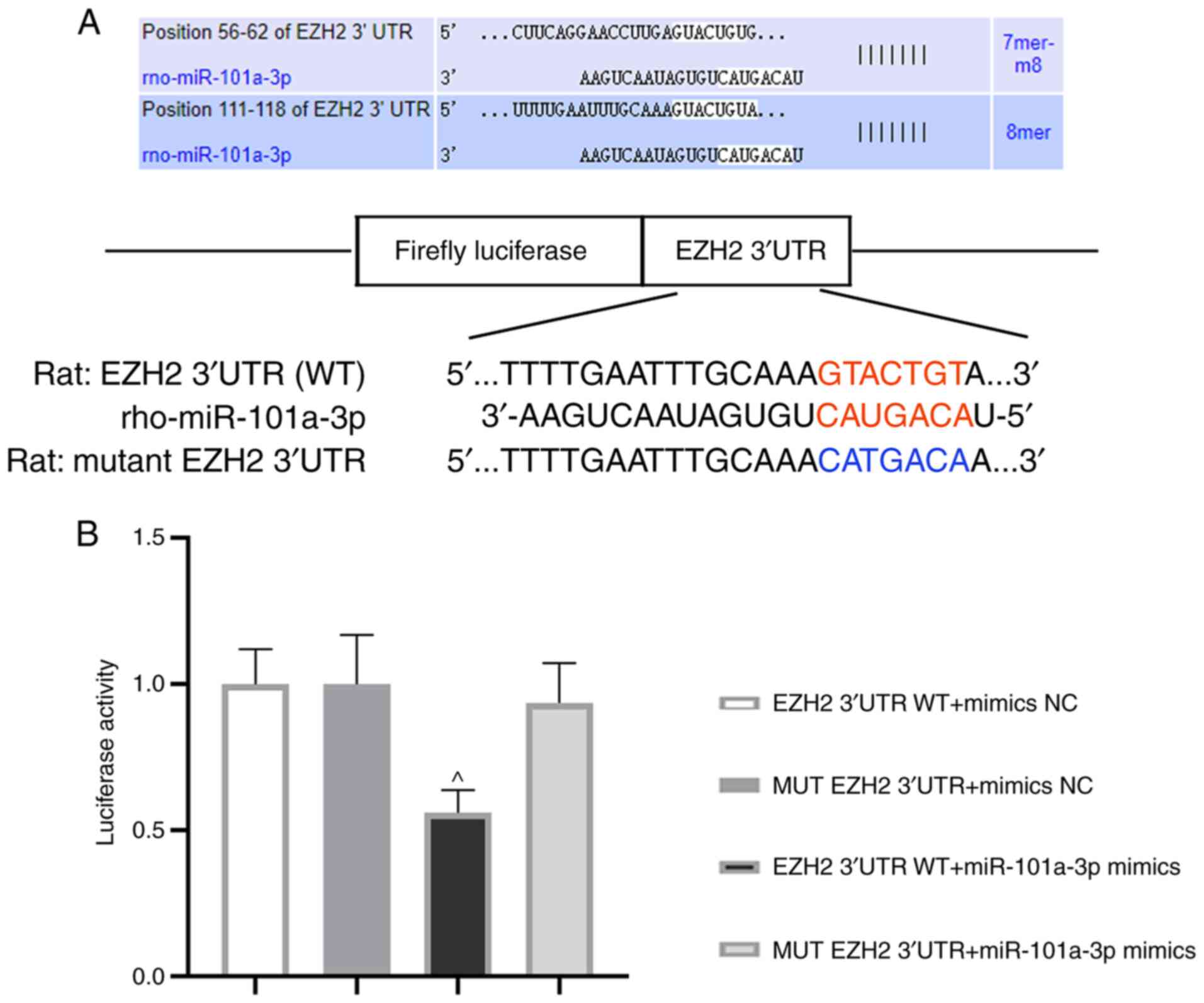
miR-101a-3p can bind to EZH2
3′UTR
Next, the relationship between miR-101a-3p and EZH2
was evaluated. The binding sequences of miR-101a-3p and EZH2 (WT or
MUT) are presented in Fig. 5A.
After co-transfection for 48 h, luciferase activity was assessed.
Of note, the luciferase activity in EZH2 3′UTR WT + miR-101a-3p
mimics group was decreased in comparison with MUT EZH2 3′UTR +
miR-101a-3p mimics group (Fig. 5B).
This result demonstrated that EZH2 was one of the target genes of
miR-101a-3p.
Discussion
The aim of the present study was to evaluate the
effect of miR-101a-3p overexpression on atrial fibrosis in
Ach-CaCl2-induced AF model rats. The results suggested
that miR-101a-3p expression was downregulated and the protein
expression level of EZH2 was upregulated in atrial tissues of AF
model rats. On the contrary, miR-101a-3p overexpression increased
the expression level of miR-101a-3p but decreased EZH2 expression.
Additionally, Ach-CaCl2-induced tissue injury was
significantly reduced by overexpression of miR-101a-3p.
At present, researchers have reported that AF can be
induced either pharmacologically or by vagal stimulation (26). For example, Zhang et al
(27) revealed that AF was induced
by infusion of angiotensin II (2,000 ng/kg per min) for 3 weeks in
male C57BL/6 mice. Moreover, Zou et al (18) reported that 1 ml/kg
Ach-CaCl2 exposure results in the absence of P waves,
irregular heartbeat and R-R intervals from the ECG, indicating the
successful establishment of the AF model. Thus, in our present
study, an AF model in rats was established by Ach-CaCl2
injection via the tail vein. Atrial fibrosis is one of the main
manifestations in the progression of AF. In the present study,
Masson staining was conducted to assess the degree of atrial tissue
fibrosis. It was found that AF-mediated tissue fibrosis in rats was
inhibited by miR-101a-3p overexpression, which was in agreement
with a previous study (10).
Moreover, the accumulation of extracellular matrix (ECM) proteins,
such as collagen I and collagen III, is able to enhance tissue
stiffness and cause cardiac diastolic dysfunction (28). As reported by Wang et al
(29), long non-coding RNA NRON,
functioned as a nuclear factor of activated T cells repressor,
improved atrial fibrosis via a reduction of collagen I and collagen
III expression. Consistent with the previous study, the current
experiments indicated that miR-101a-3p overexpression lowered these
protein expression levels, as determined by immunohistochemistry
staining. In order to further investigate the underlying mechanisms
of atrial fibrosis, the present study also detected the changes in
the protein expression levels of TGF-β1, CTGF, fibronectin and
α-SMA. Among them, TGF-β serves a crucial role in the development
of fibrosis, especially TGF-β1 existing in multi-tissues (15). CTGF, a downstream factor of TGF-β1,
has been considered a secreted protein and can facilitate the
synthesis of ECM (30). Similarly,
excessive fibronectin and α-SMA generation is a hallmark of atrial
fibrosis (31,32). In the present study, increased
TGF-β1, CTGF, fibronectin and α-SMA expression was observed in the
AF group. By contrast, the overexpression of miR-101a-3p reversed
these protein expression levels. To the best of our knowledge, this
finding that miR-101a-3p overexpression reduced atrial fibrosis in
rats administrated with Ach-CaCl2 via a decrease of
these biomarker protein expressions has not been reported
previously.
EZH2 is a histone-lysine N-methyltransferase enzyme,
participating in histone methylation. In recent years, it has been
reported that dysregulation of EZH2 is closely involved in the
pathogenesis and development of various cancer types, such as
hepatocellular carcinoma, head and neck cancer and triple-negative
breast cancer (33–35). It is well-known that EZH2 has a
vital role in modulation of cell differentiation (17). As evidenced by Xiao et al
(36), EZH2 promoted the
differentiation of pulmonary fibroblasts to myofibroblasts by
facilitating the nuclear translocation of Smad2/3. Moreover, EZH2
is responsible for regulating wound healing, fibrogenesis and
epithelial-mesenchymal transition (EMT) (37). A previous study indicated that
miR-214-3p suppressed fibrotic phenotype in cardiac myofibroblasts
by decreasing EZH2 expression (38). Another study reported that EZH2
triggered EMT in endometriosis (37). Additionally, it was shown that EZH2
was a target gene of miR-101 and the downregulation of miR-101
expression in cancer resulted in EZH2 overexpression, thereby
affecting cancer progression (36).
Similar to the previous study, the current luciferase assay results
demonstrated that EZH2 was one of the target genes of miR-101a-3p.
However, whether miR-101a-3p attenuated atrial fibrosis induced by
AF via regulation of EZH2 remains unknown and it is worthy of
research in the future.
Accumulating evidence has revealed the critical
roles of EZH2 in inducing tissue and organ fibrosis (39). Fibrosis aggravates the pathogenesis
of a variety of chronic disorders that affects the liver, renal,
lung and heart (40). Recently, the
role of EZH2 in renal fibrosis, liver fibrosis and peritoneal
fibrosis has been investigated (39–41).
However, its effect on atrial fibrosis in
Ach-CaCl2-administrated rats and the underlying
mechanisms are not fully understood. The present study found that
the protein expression level of EZH2 was increased, while
miR-101a-3p overexpression decreased EZH2 expression in AF model
rats, and miR-101a-3p overexpression attenuated AF-initiated atrial
fibrosis (Fig. S1). Although the
present study assessed the role of miR-101a-3p overexpression in
AF, the detailed molecular mechanisms are yet to be fully
elucidated. Therefore, targeting signaling pathways associated with
AF-induced atrial fibrosis is necessary. Emerging studies have
shown that the Wnt/β-catenin signaling pathway serves an important
part in regulation of EMT and has been considered as an effective
therapeutic strategy for fibrosis (39,42,43).
Inhibition of EZH2 mitigated angiotensin II-mediated fibroblast
activation via the TGF-β-Smad signaling pathway (17). Moreover, the NF-κB signaling pathway
is closely correlated with cardiac remodeling (17). As such, the aforementioned signaling
pathways deserve careful study in the future.
miRNAs can degrade or block the translation of the
target mRNA to suppress gene expression, while delivering these
RNAs to specific cells, which presents a significant challenge due
to widespread off-targeting (44,45).
In the current study, miR-101a-3p overexpression induced by a
lentivirus significantly upregulated miR-101a-3p expression and
downregulated that of EZH2 in atrial tissues of AF model rats. It
was identified that overexpression of miR-101a-3p effectively
reduced atrial fibrosis and mitigated AF. Moreover, EZH2 was a
target gene of miR-101a-3p. These data demonstrated that
lentivirus-mediated miR-101a-3p, delivered via intramyocardial
injection, affected EZH2 expression in atrial tissues, thereby
modulating the atrial fibrosis of rats. Although it is necessary to
further examine whether there is off-target blockage of EZH2 in
multiple bystander cells, the present study identified the effect
of miR-101a-3p/EZH2 axis on AF model rats. In addition, AF was
induced by injection of Ach-CaCl2, which was in
accordance with previous studies (18,26).
However, the majority of human AF may not be
Ach-CaCl2-induced AF. Therefore, the development a novel
animal model to better mimic human AF is necessary for further
research of the clinical implications.
In summary, the present results demonstrated that
miR-101a-3p expression was downregulated in AF model rats. In
addition, the in vivo investigations revealed that the
overexpression of miR-101a-3p mitigated AF by reducing atrial
fibrosis, suggesting that miR-101a-3p may be a potential target for
preventing and treating AF.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and JX conceptualized the study and designed the
experiments. JZ and NZ performed the experiments. JZ and NZ
analyzed the data and guaranteed the authenticity of the raw data.
JZ wrote the paper. JX revised the manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures were approved by the Ethics
Committee of The First Affiliated Hospital of USTC (Hefei, China;
approval no. 201907201445000472393).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yan Y, Shi R, Yu X, Sun C, Zang W and Tian
H: Identification of atrial fibrillation-associated microRNAs in
left and right atria of rheumatic mitral valve disease patients.
Genes Genet Syst. 94:23–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferrari R, Bertini M, Blomstrom-Lundqvist
C, Dobrev D, Kirchhof P, Pappone C, Ravens U, Tamargo J, Tavazzi L
and Vicedomini GG: An update on atrial fibrillation in 2014: From
pathophysiology to treatment. Int J Cardiol. 203:22–29. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carapetis JR, Steer AC, Mulholland EK and
Weber M: The global burden of group A streptococcal diseases.
Lancet Infect Dis. 5:685–694. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yao L, Zhou B, You L, Hu H and Xie R:
LncRNA MIAT/miR-133a-3p axis regulates atrial fibrillation and
atrial fibrillation-induced myocardial fibrosis. Mol Biol Rep.
47:2605–2617. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu Y, Zhang Y, Wang N, Pan Z, Gao X, Zhang
F, Zhang Y, Shan H, Luo X, Bai Y, et al: MicroRNA-328 contributes
to adverse electrical remodeling in atrial fibrillation.
Circulation. 122:2378–2387. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Santulli G, Iaccarino G, De Luca N,
Trimarco B and Condorelli G: Atrial fibrillation and microRNAs.
Front Physiol. 5:152014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li X, Wang B, Cui H, Du Y, Song Y, Yang L,
Zhang Q, Sun F, Luo D, Xu C, et al: Let-7e replacement yields
potent anti-arrhythmic efficacy via targeting beta 1-adrenergic
receptor in rat heart. J Cell Mol Med. 18:1334–1343. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Galenko O, Jacobs V, Knight S, Taylor M,
Cutler MJ, Muhlestein JB, Carlquist JL, Knowlton KU and Jared Bunch
T: The role of microRNAs in the development, regulation, and
treatment of atrial fibrillation. J Interv Card Electrophysiol.
55:297–305. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Briasoulis A, Sharma S, Telila T,
Mallikethi-Reddy S, Papageorgiou N, Oikonomou E and Tousoulis D:
MicroRNAs in atrial fibrillation. Curr Med Chem. 26:855–863. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lv X, Li J, Hu Y, Wang S, Yang C, Li C and
Zhong G: Overexpression of miR-27b-3p targeting Wnt3a regulates the
signaling pathway of Wnt/β-catenin and attenuates atrial fibrosis
in rats with atrial fibrillation. Oxid Med Cell Longev.
2019:57037642019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Q, Gao Y, Zhu J and Jia Q: MiR-101
attenuates myocardial infarction-induced injury by targeting DDIT4
to regulate autophagy. Curr Neurovasc Res. 17:123–130. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiao L, Gu Y, Sun Y, Chen J, Wang X, Zhang
Y, Gao L and Li L: The long noncoding RNA XIST regulates cardiac
hypertrophy by targeting miR-101. J Cell Physiol. 234:13680–13692.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dong H, Sun Y, Shan F, Sun Q and Yang B:
Down-regulation of miR-101 contributes to rheumatic heart disease
through up-regulating TLR2. Med Sci Monit. 21:1500–1506. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pan Z, Sun X, Shan H, Wang N, Wang J, Ren
J, Feng S, Xie L, Lu C, Yuan Y, et al: MicroRNA-101 inhibited
postinfarct cardiac fibrosis and improved left ventricular
compliance via the FBJ osteosarcoma oncogene/transforming growth
factor-β1 pathway. Circulation. 126:840–850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou Y, Shiok TC, Richards AM and Wang P:
MicroRNA-101a suppresses fibrotic programming in isolated cardiac
fibroblasts and in vivo fibrosis following trans-aortic
constriction. J Mol Cell Cardiol. 121:266–276. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ai S, Yu X, Li Y, Peng Y, Li C, Yue Y, Tao
G, Li C, Pu WT and He A: Divergent requirements for EZH1 in heart
development versus regeneration. Circ Res. 121:106–112. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Song S, Zhang R, Mo B, Chen L, Liu L, Yu
Y, Cao W, Fang G, Wan Y, Gu Y, et al: EZH2 as a novel therapeutic
target for atrial fibrosis and atrial fibrillation. J Mol Cell
Cardiol. 135:119–133. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zou D, Geng N, Chen Y, Ren L, Liu X, Wan
J, Guo S and Wang S: Ranolazine improves oxidative stress and
mitochondrial function in the atrium of
acetylcholine-CaCl2 induced atrial fibrillation rats.
Life Sci. 156:7–14. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Q, Lv Q, Feng M, Liu M, Feng Y, Lin
S, Yang J and Hu J: Taurine prevents the electrical remodeling in
Ach-CaCl(2) induced atrial fibrillation in rats. Adv Exp Med Biol.
975:821–830. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou Q, Chen B, Chen X, Wang Y, Ji J,
Kizaibek M, Wang X, Wu L, Hu Z, Gao X, et al: Arnebiae Radix
prevents atrial fibrillation in rats by ameliorating atrial
remodeling and cardiac function. J Ethnopharmacol. 248:1123172020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li Y, Song B and Xu C: Effects of Guanfu
total base on Bcl-2 and Bax expression and correlation with atrial
fibrillation. Hellenic J Cardiol. 59:274–278. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang Z, Ouyang Q, Huang Z, Lin L, Yu E and
Ferrari MW: Prenatal nicotine exposure induces gender-associated
left ventricular-arterial uncoupling in adult offspring. Mol Med
Rep. 12:410–418. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen B, Xu M and Li B: The clinical
experience for treating post-burn depigmentation with tiny
epidermal particles graft. Int Wound J. 14:165–171. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang
CF, Li Y and Zhang Z: Dexmedetomidine alleviated sepsis-induced
myocardial ferroptosis and septic heart injury. Mol Med Rep.
22:175–184. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gaspo R: The tachycardia-induced dog model
of atrial fibrillation. clinical relevance and comparison with
other models. J Pharmacol Toxicol Methods. 42:11–20. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang YL, Cao HJ, Han X, Teng F, Chen C,
Yang J, Yan X, Li PB, Liu Y, Xia YL, et al: Chemokine receptor
CXCR-2 initiates atrial fibrillation by triggering monocyte
mobilization in mice. Hypertension. 76:381–392. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dong Q, Li S, Wang W, Han L, Xia Z, Wu Y,
Tang Y, Li J and Cheng X: FGF23 regulates atrial fibrosis in atrial
fibrillation by mediating the STAT3 and SMAD3 pathways. J Cell
Physiol. 234:19502–19510. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y, Xu P, Zhang C, Feng J, Gong W, Ge
S and Guo Z: LncRNA NRON alleviates atrial fibrosis via promoting
NFATc3 phosphorylation. Mol Cell Biochem. 457:169–177. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qiao G, Xia D, Cheng Z and Zhang G:
miR-132 in atrial fibrillation directly targets connective tissue
growth factor. Mol Med Rep. 16:4143–4150. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen X, Zhang W, Wang Q, Du L, Yi Y, Liu
Y, Liu X and Duan S: Eplerenone inhibits atrial fibrosis in mutant
TGF-β1 transgenic mice. Sci China Life Sci. 59:1042–1047. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burstein B, Qi XY, Yeh YH, Calderone A and
Nattel S: Atrial cardiomyocyte tachycardia alters cardiac
fibroblast function: A novel consideration in atrial remodeling.
Cardiovasc Res. 76:442–452. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xiao G, Jin LL, Liu CQ, Wang YC, Meng YM,
Zhou ZG, Chen J, Yu XJ, Zhang YJ, Xu J and Zheng L: EZH2 negatively
regulates PD-L1 expression in hepatocellular carcinoma. J
Immunother Cancer. 7:3002019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou L, Mudianto T, Ma X, Riley R and
Uppaluri R: Targeting EZH2 enhances antigen presentation, antitumor
immunity, and circumvents Anti-PD-1 resistance in head and neck
cancer. Clin Cancer Res. 26:290–300. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yomtoubian S, Lee SB, Verma A, Izzo F,
Markowitz G, Choi H, Cerchietti L, Vahdat L, Brown KA, Andreopoulou
E, et al: Inhibition of EZH2 catalytic activity selectively targets
a metastatic subpopulation in triple-negative breast cancer. Cell
Rep. 30:755–770.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Xiao X, Senavirathna LK, Gou X, Huang C,
Liang Y and Liu L: EZH2 enhances the differentiation of fibroblasts
into myofibroblasts in idiopathic pulmonary fibrosis. Physiol Rep.
4:e129152016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Q, Dong P, Liu X, Sakuragi N and Guo
SW: Enhancer of Zeste homolog 2 (EZH2) induces
epithelial-mesenchymal transition in endometriosis. Sci Rep.
7:68042017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhu WS, Tang CM, Xiao Z, Zhu JN, Lin QX,
Fu YH, Hu ZQ, Zhang Z, Yang M, Zheng XL, et al: Targeting EZH1 and
EZH2 contributes to the suppression of fibrosis-associated genes by
miR-214-3p in cardiac myofibroblasts. Oncotarget. 7:78331–78342.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou X, Xiong C, Tolbert E, Zhao TC,
Bayliss G and Zhuang S: Targeting histone methyltransferase
enhancer of zeste homolog-2 inhibits renal epithelial-mesenchymal
transition and attenuates renal fibrosis. FASEB J.
32:fj201800237R2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cai X, Li Z, Zhang Q, Qu Y, Xu M, Wan X
and Lu L: CXCL6-EGFR-induced Kupffer cells secrete TGF-β1 promoting
hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2
pathway in liver fibrosis. J Cell Mol Med. 22:5050–5061. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shi Y, Tao M, Wang Y, Zang X, Ma X, Qiu A,
Zhuang S and Liu N: Genetic or pharmacologic blockade of enhancer
of zeste homolog 2 inhibits the progression of peritoneal fibrosis.
J Pathol. 250:79–94. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gonzalez DM and Medici D: Signaling
mechanisms of the epithelial-mesenchymal transition. Sci Signal.
7:re82014. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Duan J, Gherghe C, Liu D, Hamlett E,
Srikantha L, Rodgers L, Regan JN, Rojas M, Willis M, Leask A, et
al: Wnt1/β catenin injury response activates the epicardium and
cardiac fibroblasts to promote cardiac repair. EMBO J. 31:429–442.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Boudreau RL, Spengler RM and Davidson BL:
Rational design of therapeutic siRNAs: minimizing off-targeting
potential to improve the safety of RNAi therapy for Huntington's
disease. Mol Ther. 19:2169–2177. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Scaggiante B, Dapas B, Farra R, Grassi M,
Pozzato G, Giansante C, Fiotti N and Grassi G: Improving siRNA
bio-distribution and minimizing side effects. Curr Drug Metab.
12:11–23. 2011. View Article : Google Scholar : PubMed/NCBI
|