Introduction
Obstructive sleep apnea (OSA) is a common disorder
characterized by episodic arterial oxygen desaturation due to
recurrent closure of the upper airway during sleep, namely
intermittent hypoxia (IH), which can trigger excessive oxidative
stress, systemic inflammation, sympathetic activation and increased
renin generation, leading to abnormal vascular reactivity (1,2).
IH-related vascular dysfunction involves impaired acetylcholine
(ACh)-induced endothelium-dependent relaxation (3,4) and
increased contractile responses to vasoconstrictors, such as
angiotensin II (Ang II) (5,6) and endothelin-1 (ET-1) (7–9), which
underlie the initiation and progression of hypertension. In our
previous study, rats exposed to chronic IH (CIH) were found to
exhibit enhanced Ang II-induced vasoconstriction, which was
possibly attributed to the overactivation of ERK1/2 signaling
through the upregulation of myosin phosphatase targeting subunit
(MYPT1) and myosin light chain (MLC) phosphorylation (5). Other studies have shown that IH
increases ET-1-mediated constriction by increasing Rho-kinase
activation (7,8) or through a protein kinase C
(PKC)-mitochondrial oxidative pathway (9), leading to the increase in
Ca2+ sensitization and sensitivity of vascular smooth
contractile machinery.
O-linked-β-N-acetylglucosamine (O-GlcNAc)
modifications on serine/threonine (Ser/Thr) residues of proteins,
known as O-GlcNAcylation, are a critical regulatory
post-translational modification (PTM), analogous to
phosphorylation, that can induce functional changes in numerous
target proteins, including protein kinases, phosphatases, cellular
signaling proteins and transcriptional factors (10,11).
O-GlcNAcylation is controlled by two conserved enzymes, namely
O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). OGT catalyzes the
addition of O-GlcNAc to the Ser/Thr residues of target proteins,
whereas OGA is responsible for the hydrolytic cleavage of O-GlcNAc
from post-translationally modified proteins (10). O-GlcNAc modifications actively
participate in the regulation of cardiovascular pathological
processes, such as hypertension, cardiac hypertrophy,
ischemia-reperfusion (I/R) injury and vascular inflammatory injury
(10,12,13).
In our previous study, it was found that the O-GlcNAc protein and
cellular signaling involving ERK1/2 and p38 MAPK may act as
potential regulators in IH-related cardiac remodeling and left
ventricular function (14). Lima
et al (15) reported that
global O-GlcNAc proteins were elevated in the arteries of
deoxycorticosterone acetate and salt (DOCA-salt)-induced
hypertensive rats. Increasing O-GlcNAc levels resulted in impaired
endothelium-dependent relaxation and increased reactivity to
constrictor stimuli, which are hallmarks of hypertension. A
reduction in phosphorylated Akt and endothelial NO synthase (eNOS),
possibly due to direct competition with O-GlcNAc, decreased NO
production and therefore contributed to vascular dysfunction
(15–17). These findings support a contributing
role of O-GlcNAcylation in regulating vascular reactivity, which
underlies the pathogenesis of hypertension.
Since both O-GlcNAcylation and phosphorylation
modify the same or neighboring Ser/Thr residues of substrate
proteins, an extensive interplay, termed PTM crosstalk, exists
between them, including competitive site occupancy, alternative
occupancy at adjacent sites and simultaneous occupancy at the same
protein (18). OGA and OGT often
occur in the same protein complexes as kinases and phosphatases,
suggesting that a protein can quickly cycle between being
phosphorylated and O-GlcNAcylated (10,18).
An increasing number of kinases have been found to be
O-GlcNAcylated, resulting in conformational changes that promote
downstream functional effects (11). The important regulatory role of
phosphorylation in a subset of proteins associated with
transcriptional factor activation, signaling pathways and
neurotransmitter synthesis has been identified in OSA-related
autonomic abnormalities (5,8,19). It
could be suggested that intracellular protein O-GlcNAcylation may
also play a regulatory role in the OSA-related pathophysiological
processes.
Considering the role of O-GlcNAcylation in
modulating vascular reactivity and the pathophysiological
association between IH and vascular function, the aim of the
present study was to investigate whether O-GlcNAcylation exerted
regulatory effects on IH exposure-related vascular dysfunction. In
addition, previous evidence has shown that the acute activation of
O-GlcNAcylation protects against cellular injury, and that
sustained increases in O-GlcNAc proteins may confer deleterious
effects on cardiovascular function (10,12,20).
Acute IH (AIH), with a hypoxia duration of a few hours, resembles
acute I/R injury (1), and the
transient elevation of O-GlcNAcylation protects cells against I/R
injury (13). On the other hand,
chronically-increased levels of O-GlcNAc protein acts as a general
mechanism underlying the negative effects of hypertension on
vascular function (15). Therefore,
the vascular effects of O-GlcNAc modifications on aortic and
mesenteric arteries (MAs) subjected to both AIH and CIH were
investigated. The potential regulatory role of O-GlcNAcylation in
p38 MAPK, ERK1/2 and Ca2+/calmodulin-dependent kinase II
(CaMKII) signaling associated with vascular contractile responses
during IH exposure were further examined, since these pathways have
been found to be targets of O-GlcNAcylation and have been
implicated in arterial hypertension-related vascular dysfunction
(5,19,21–28).
The aim of the present study was to determine whether
O-GlcNAcylation contributed to IH-related vascular dysfunction
through the modulation of the MAPK and CaMKII signaling pathways.
It is hoped that this study could provide novel insights into the
molecular mechanisms underlying CIH-associated vascular
dysfunction.
Materials and methods
Animals
All animal experiments were conducted on a total of
90 male Wistar rats provided by the Experimental Animal Center of
Tongji Hospital, Huazhong University of Science and Technology
(Wuhan, China). Rats were housed with a 12-h light/dark cycle at
22±1°C and 55±10% humidity and fed a standard chow diet with ad
libitum access to water. All procedures were performed in
accordance with the Guide for the Care and Use of Laboratory
Animals (29) and approved by the
Institutional Animal Care and Use Committee of Huazhong University
of Science and Technology. Animal experiments were conducted in the
Experimental Animal Center of Tongji Hospital or Tongji Medical
College, Huazhong University of Science and Technology.
Reagents and antibodies
Ang II (cat. no. ALX-151-039-M025) and KN-93 (CaMKII
inhibitor; cat. no. BML-EI268) were obtained from Enzo Life
Sciences, Inc. ST045849 (OGT inhibitor; cat. no. MFCD03308174) was
purchased from TimTec, Inc. PD98059 (MEK inhibitor; cat. no. 1213)
was from Tocris Bioscience and SB203580 (p38 MAPK inhibitor; cat.
no. 13067) from Cayman Chemical Company. Ach (cat. no. A2661),
phenylephrine (PE; cat. no. P6126), the NOS blocker
Nω-nitro-L-arginine methyl ester (L-NAME; cat. no. N5751), sodium
nitroprusside (SNP; cat. no. S0501) and other high-quality reagents
were obtained from Sigma-Aldrich (Merck KGaA). PugNAc (OGA
inhibitor; cat. no. sc-204415), primary antibodies for O-GlcNAc
(mouse monoclonal; cat. no. sc-59623; 1:800), CaMKII (rabbit
polyclonal; cat. no. sc-9035; 1:800) and phosphorylated (p)-CaMKII
(mouse monoclonal; cat. no. sc-32289; 1:800), as well as secondary
anti-mouse (cat. no. sc-358914; 1:4,000) and anti-rabbit IgG
antibodies (cat. no. sc-2004; 1:4,000) were purchased from Santa
Cruz Biotechnology, Inc. Antibodies against OGT (rabbit polyclonal;
cat. no. 11576-2-AP; 1:1,000) and OGA (rabbit polyclonal; cat. no.
14711-1-AP; 1:400) were obtained from ProteinTech Group, Inc.
Antibodies against p38 MAPK (rabbit polyclonal; cat. no. 9212;
1:1,000), p-p38 MAPK (rabbit monoclonal; cat. no. 4511; 1:1,000),
ERK1/2 (rabbit monoclonal; cat. no. 4695; 1:1,000), p-ERK1/2
(rabbit monoclonal; cat no. 4376; 1:1,000) and GAPDH (rabbit
monoclonal; cat. no. 5174; 1:3,000) were purchased from Cell
Signaling Technology, Inc. SNP solution was freshly prepared. Stock
solutions were prepared either in distilled water (L-NAME, ACh, Ang
II and PE) or DMSO (PugNAc, ST045849, SB203580, PD98059 and KN-93),
and then frozen in small aliquots.
AIH exposure
Briefly, a total of 40 12-week-old Wistar rats
(weight, 330–370 g) were anesthetized with urethane (1.2 g/kg;
i.p.) and euthanized by exsanguination by extracting ~5 ml blood
samples with a small needle and syringe following an inferior vena
cava puncture. Blood was allowed to flow from the puncture points
until the rats lost spontaneous breath and an abdominal aorta pulse
(within 3 min). The hearts were harvested and immediately frozen in
liquid nitrogen. Thoracic aortas and primary branches of the
superior MA (15) were isolated and
placed in ice-cold Krebs-Ringer buffer, and connective tissue was
removed (5). Next, 4-mm arterial
segments were incubated in DMEM (Sigma-Aldrich; Merck KGaA)
containing 1% fetal calf serum (Sigma-Aldrich; Merck KGaA), 120
U/ml penicillin and 120 µg/ml streptomycin, as previously described
(20). To determine the potential
regulatory role of O-GlcNAcylation on vascular function, arterial
rings were treated with DMSO (vehicle), PugNAc (100 µM) or ST045849
(50 µM) and exposed to either 3-h normoxic exposure (AON) or 3-h
AIH conditions. The incubation time and concentration of ST045849
were determined from pilot studies. The normoxic rings were
maintained at atmospheric oxygen concentrations (21% O2,
5% CO2, 37°C). AIH rings were subjected to 15
alternating cycles of 7 min hypoxia (5% O2, 5%
CO2, 37°C) and 5-min reoxygenation in the chambers
attached to a computerized controller (OxyCycler C42; BioSpherix,
Ltd.). Thereafter, vessels were either submitted to isometric
tension recordings or immediately frozen in liquid nitrogen and
stored at −80°C for further protein assays.
CIH procedures
In another set of experiments, a total of 50
8-week-old Wistar rats (weight, 200–250 g) were subjected to either
normoxic [21% O2; control (CON)] or CIH conditions
between 8:00 a.m. and 4:00 p.m. for 4 weeks, as previously
described (5). During each IH
cycle, the oxygen concentration was briefly decreased from 21 to
6–8% over 40 sec and maintained at 6–8% for 2 min, which was then
increased to 21% over 20 sec and maintained under normoxia for
another 2 min. Normoxia was achieved by compressed air distribution
at the same flow rate. After measuring blood pressure, rats were
anesthetized with urethane (1.2 g/kg, i.p.) and euthanized by
exsanguination using inferior vena cava puncture, as described
above. Next, rat thoracic aortas and primary MAs were excised, cut
into 4-mm segments and subjected to 3-h normoxic incubation with
vehicle or antagonists (PugNAc or ST045849).
Vascular reactivity studies
Vascular reactivity was assessed in vessels from the
following groups: i) AOD, arterial rings treated with DMSO
(vehicle) and 3-h normoxia exposure; ii) AOP, arterial rings
treated with PugNAc and 3-h normoxia exposure; iii) AOS, arterial
rings treated with ST045849 and 3-h normoxia exposure; iv) AID,
arterial rings treated with DMSO and 3-h IH exposure; v) AIP,
arterial rings treated with PugNAc and 3-h IH exposure; vi) AIS,
arterial rings treated with ST045849 and 3-h IH exposure; vii) COD,
arterial rings from control rats treated with DMSO; viii) COP,
arterial rings from control rats treated with PugNAc; ix) COS,
arterial rings from control rats treated with ST045849; x) CID,
arterial rings from CIH rats treated with DMSO; xi) CIP, arterial
rings from CIH rats treated with PugNAc; and xii) CIS, arterial
rings from CIH rats treated with ST045849. The vascular isometric
force was recorded by a CO pod and PowerLab/4SP data acquisition
system (AD Instruments), as previously described (3–5,30). In
brief, rings were steadily stretched to optimal passive force
(aorta 2 g, MA 1 g) established from force-active tension curves to
PE (1 µM), equilibrated for 45 min and depolarized with
high-K+ Krebs-Ringer buffer (aorta, 120 mM KCl; MA, 80
mM KCl). The optimal depolarizing concentration of KCl was
determined using a concentration-response curve (CRC) to KCl
(aorta, 20–140 mM; MA, 20–100 mM) and the contraction generated was
regarded as 100%. After a 45-min washout period, rings were
stimulated with PE (1 µM) followed by ACh (10 µM) to assess the
integrity of the endothelium. Next, following a 45-min
stabilization period, ACh (0.001–10 µM) was added cumulatively to
rings with a steady-state maximal contraction induced by PE (1 µM).
Meanwhile, endothelium-independent relaxation was tested using SNP
(0.001–3 µM) in rings pre-incubated with L-NAME (100 µM, 30
min).
In another subset of segments, CRCs to PE (0.001–10
µM) were generated in the presence of DMSO (vehicle) or inhibitors
(SB049859, PD98059 or KN-93; 10 µM) added to the Krebs-Ringer
buffer for 30 min before PE stimulation. Each ring was used to
generate only one CRC of PE. The endothelium-denuded rings were
pretreated with DMSO or the aforementioned inhibitors for 30 min,
followed by stimulation with Ang II (0.1 µM). Since Ang II might
cause tachyphylaxis, a single dose of Ang II was added to each ring
(31).
Arterial morphometry and
immunohistochemistry (IHC)
The tissues were fixed in 4% formalin for 24 h at
room temperature, processed and embedded in paraffin.
Paraffin-embedded sections (5 µm) were stained with hematoxylin and
eosin (hematoxylin for ~5 min at room temperature; eosin for ~2 min
at room temperature) and elastin Van Gieson (EVG; ~5 min at room
temperature) to assess arterial wall structure. The vessel luminal
diameter (LD), intima-to-media thickness (IMT) and IMT to LD ratio
(IMT/LD) were calculated using Image-Pro Plus 7.0 software (IPP;
version 7.0; Media Cybernetics, Inc.) (5). EVG stain was used in order to identify
elastic fibers by staining them black. The continuity of elastic
fibers was observed with a light microscope (magnification, ×200).
Then, elastic fiber content was determined by calculating the
percentage of the positive area to the total cross-sectional vessel
wall area using IPP software. An IHC assay was performed using a
3,3′diaminobenzidine (DAB) kit (Agilent Technologies, Inc.),
according to the manufacturer's instructions. Briefly, slices were
deparaffinized, hydrated, treated with 3% hydrogen peroxide,
blocked in 3% BSA (Sigma-Aldrich; Merck KGaA) in phosphate-buffered
saline (pH 7.4) for 30 min at room temperature, and then incubated
with primary antibodies against OGA (1:100) or OGT (1:100)
overnight at 4°C. Following washing, the sections were incubated
with HRP-conjugated secondary antibodies (1:200) for 50 min at room
temperature. Slides were washed, developed with DAB, counterstained
with hematoxylin for 2 min at room temperature and mounted with
crystal mount. Stained sections were visualized by light microscopy
(magnification, ×200) and all the images were analyzed using IPP
software according to previous study (32). The measurement parameters used to
assess the immunostaining quantification were density mean, area
sum, and integrated optical density (IOD). The area of interest
(AOI) was set for the parameter measurement. The IOD and area sum
of AOI was calibrated. Density mean was calculated as the
percentage of IOD to area sum and compared between groups.
Western blot analysis
Vascular tissues were homogenized with RIPA buffer
containing PMSF (1 mM), protease inhibitor cocktail and phosphatase
inhibitor cocktail. Protein concentration was determined using the
Enhanced BCA Protein Assay kit (Beyotime Institute of
Biotechnology). Protein extracts (60 µg) were separated on 10%
sodium dodecyl sulphate-polyacrylamide gel electrophoresis and
transferred onto polyvinylidene fluoride membranes (0.45 µm; EMD
Millipore) using a Bio-Rad Scientific Instrument (Bio-Rad
Laboratories, Inc.). Membranes were blocked with 5% non-fat milk or
3% BSA in Tris-buffered saline containing 0.05% Tween-20 (pH 7.6)
for 1 h at room temperature, followed by incubation with primary
antibodies specific to O-GlcNAc (1:800), OGT (1:1,000), OGA
(1:400), CaMKII (1:800), p-CaMKII (1:800), ERK1/2 (1:1,000),
p-ERK1/2 (1:1,000), p38 MAPK (1:1,000), p-p38 MAPK (1:1,000) and
GAPDH (1:3,000) at 4°C overnight with gentle shaking. Membranes
were incubated with HRP-conjugated secondary antibodies (1:4,000)
for 1 h at room temperature. The bands were detected using an
enhanced chemiluminescence detection kit (Advansta, Inc.). The
protein level of GAPDH served as the internal reference. In order
to re-use the membrane, stripping buffer (Beyotime Institute of
Biotechnology) was used to remove antibodies from the blotted
membranes and the membranes were re-probed for other target
proteins with similar molecular weight as previously described
(33,34). The intensity of bands was
semi-quantified using AlphaEaseFC software (version 6.0;
ProteinSimple).
Statistical analysis
CRCs to agonists were fitted by non-linear
regression using SigmaPlot software (version 11.0; SPSS, Inc.). The
maximal responses (Emax) and the negative logarithm of
the concentration at which agonists produce 50% of the maximum
response were calculated. Statistical analysis was performed using
PASW version 18.0 software (SPSS, Inc.). Data are expressed as the
mean ± SD of ≥3 independent repeats. Differences within groups were
compared using an unpaired Student's t-test or one-way analysis of
variance (ANOVA) followed by LSD post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
CIH causes elevations in mean arterial
blood pressure (MABP) and vascular morphological changes
Rats exposed to CIH exhibited a higher MABP compared
with CON rats (Fig. 1B). The IMT
and IMT/LD of MAs were greater in the CIH group compared with the
CON group. No differences in the IMT and IMT/LD of aortas was
observed between the CON and CIH groups (Fig. 1D and E). The elastic lamella of MAs
was disrupted by CIH exposure (Fig.
1A). No significant differences in elastic fiber content were
observed between the two groups (Fig.
1C). CIH treatment increased the expression of OGT protein but
decreased that of OGA in both aortic and mesenteric tissue
(Fig. 1F and G).
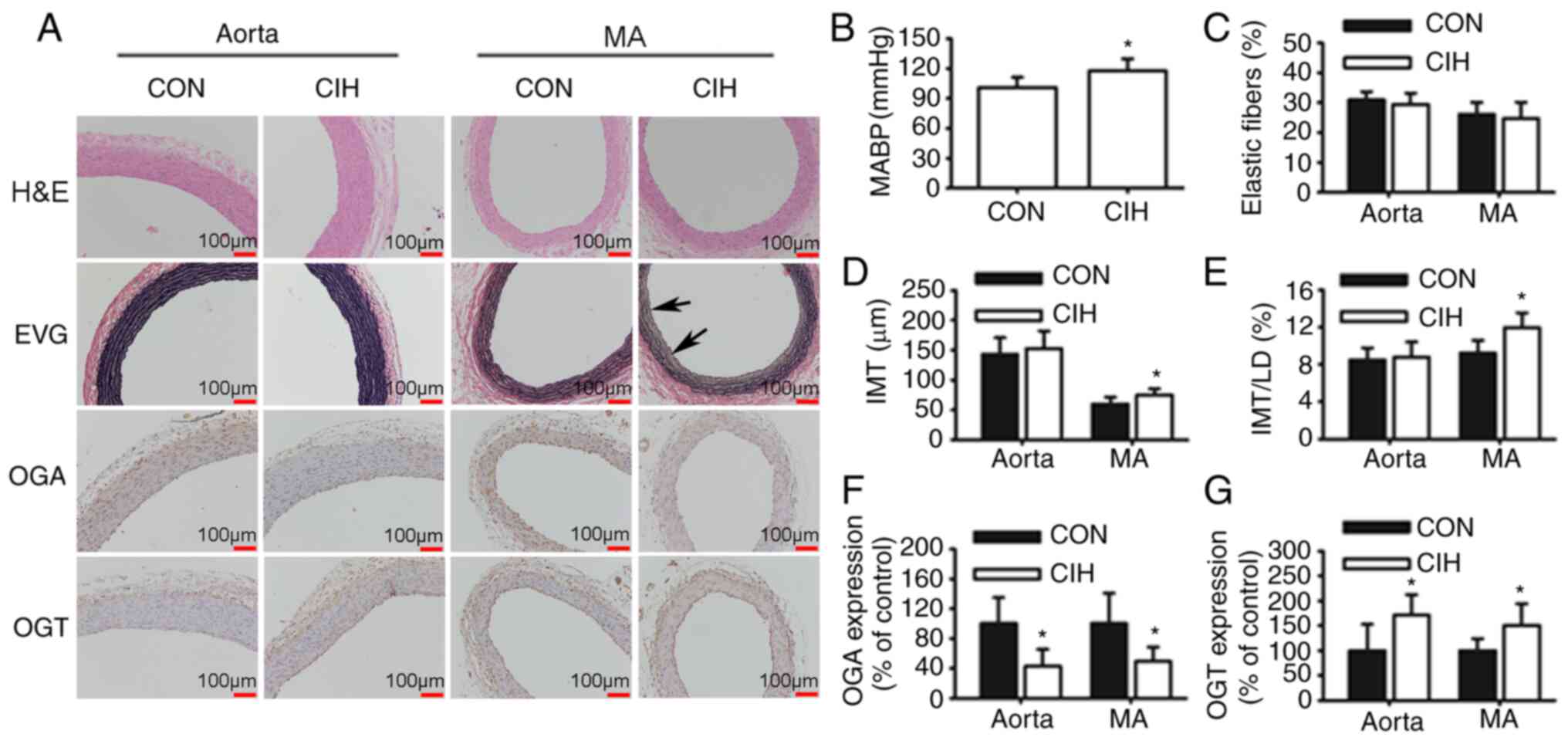 | Figure 1.CIH elevates MABP and induces changes
in vascular morphology and protein expression. (A) Representative
H&E and EVG staining of vessel walls. Immunohistochemical
staining for OGA and OGT. Arrowheads indicate disrupted elastic
lamella. (B) MABP was increased by CIH. (C) No differences in the
content of elastic fibers were observed. (D and E) Changes in
vascular wall thickness. Changes in the protein expression levels
of (F) OGA and (G) OGT. Scale bar, 100 µm. The groups were as
follows: i) CON, normoxia (21% O2); and ii) CIH,
intermittent hypoxia cycles (6–8% O2 for 2 min and 21%
O2 for another 2 min). Data are presented as the mean ±
SD (n=5-20). *P<0.05 between groups. CIH, chronic intermittent
hypoxia; MABP, mean arterial blood pressure; H&E, hematoxylin
and eosin; EVG, elastin Van Gieson; OGA, O-GlcNAcase; OGT, O-GlcNAc
transferase; CON, control; MA, mesenteric artery; LD, luminal
diameter; IMT, intima-to-media thickness. |
Protein O-GlcNAc levels influence
endothelium-dependent vasodilation during IH exposure
The endothelium-dependent vasodilatory responses of
both aortas (Fig. 2A and B) and MAs
(Fig. 2I and J) to ACh were
significantly inhibited by AIH exposure, which could be prevented
by OGA inhibition with PugNAc. CIH impaired the ACh-induced
vasodilation of mesenteric rings, which could be partly improved by
OGT inhibition with ST045849 (Fig. 2K
and L), but did not have a noticeable effect on the aortas
(Fig. 2C and D). ST045849 was found
to attenuate the ACh-induced relaxation in aortas and MAs that had
undergone either normoxia or AIH treatments, except those from the
CIH group. SNP-induced endothelium-independent relaxation of aortas
(Fig. 2E-H) and MAs (Fig. 2M-P) was similar among all
groups.
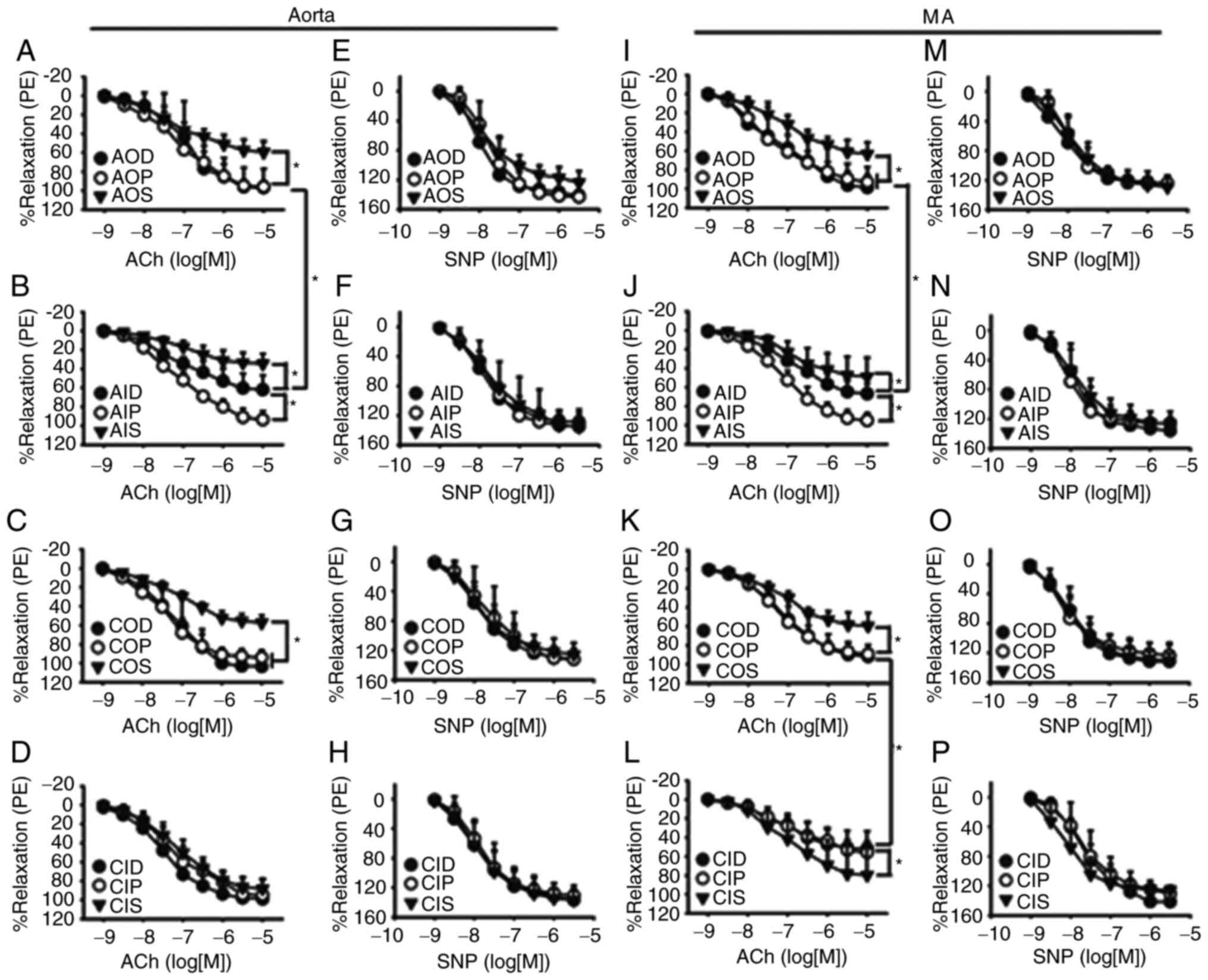 | Figure 2.Vasodilatory responses to ACh and SNP
of aortas and MAs. O-GlcNAc levels influenced ACh-induced relaxant
responses during IH exposure in the (A-D) aorta and (I-L) MA. The
vasodilatory responses of both (A and B) aortas and (I and J) MAs
to ACh were significantly inhibited by AIH exposure, which was
prevented by PugNAc. CIH impaired ACh-induced vasodilation of
mesenteric rings, which was partly improved by (K and L) ST045849
but did not have a noticeable effect on the (C and D) aortas.
SNP-induced relaxation was similar among all groups in the (E-H)
aorta and (M-P) MA. Neither AIH nor CIH treatment altered
SNP-induced relaxation of (F and H) aortas and (N and P) MAs. The
groups were as follows: i) AOD, arterial rings treated with DMSO
(vehicle) and 3-h normoxia exposure; ii) AOP, arterial rings
treated with PugNAc and 3-h normoxia exposure; iii) AOS, arterial
rings treated with ST045849 and 3-h normoxia exposure; iv) AID,
arterial rings treated with DMSO and 3-h IH exposure; v) AIP,
arterial rings treated with PugNAc and 3-h IH exposure; vi) AIS,
arterial rings treated with ST045849 and 3-h IH exposure; vii) COD,
arterial rings from control rats treated with DMSO; viii) COP,
arterial rings from control rats treated with PugNAc; ix) COS,
arterial rings from control rats treated with ST045849; x) CID,
arterial rings from CIH rats treated with DMSO; xi) CIP, arterial
rings from CIH rats treated with PugNAc; and xii) CIS, arterial
rings from CIH rats treated with ST045849. Data are presented as
the mean ± SD (n=5). *P<0.05. ACh, acetylcholine; SNP, sodium
nitroprusside; O-GlcNAc, O-linked-β-N-acetylglucosamine; IH,
intermittent hypoxia; CIH, chronic intermittent hypoxia; PE,
phenylephrine; MA, mesenteric artery. |
O-GlcNAcylation modulates contractile
responses to Ang II during IH exposure
Neither AIH nor CIH treatment altered vascular
reactivity to PE in aortas and MAs. Incubation with PugNAc or
ST045849 did not affect the PE-induced contraction of arteries from
any of the studied groups. Of note, SB203580 and PD98059 blocked
contractile responses to PE in PugNAc-incubated arteries from the
AON, AIH and CON groups. No inhibitory effects of KN-93 on vascular
reactivity to PE were observed (Fig.
3).
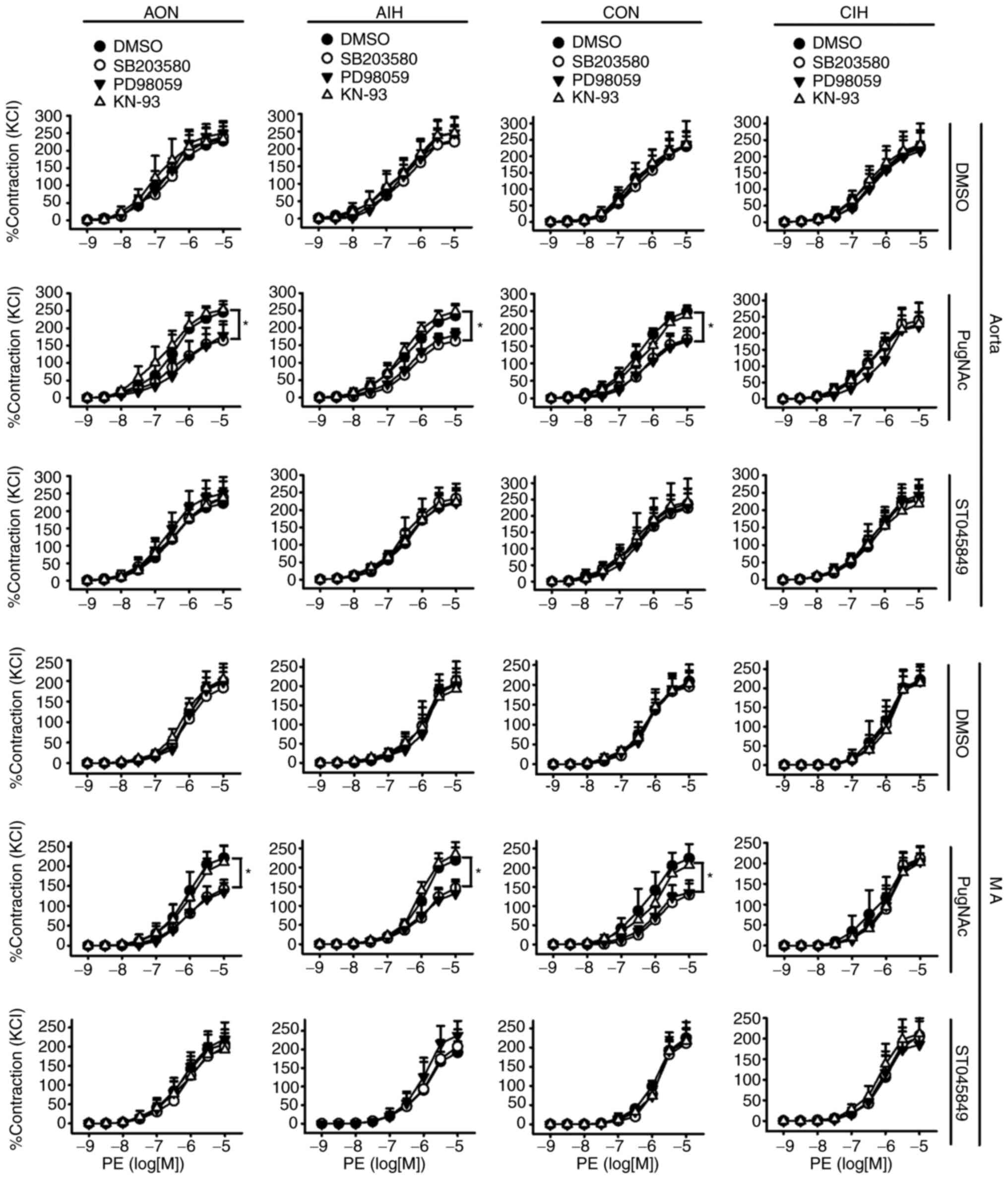 | Figure 3.Effects of SB203580, PD98059 and
KN-93 on contractile responses to PE in aortas and MAs treated with
DMSO, PugNAc or ST045849. Vessels were treated under AON and AIH
conditions or isolated from CON and CIH group rats. The groups were
as follows: i) AON, 3-h normoxia treatment; ii) AIH, 3-h
intermittent hypxia treatment; iii) CON, normoxic (21%
O2) condition; and iv) CIH, intermittent hypoxia cycles
(6–8% O2 for 2 min and 21% O2 for another 2
min). Data are presented as the mean ± SD (n=5). *P<0.05. PE,
phenylephrine; AIH, acute intermittent hypoxic; CON, control; CIH,
chronic intermittent hypoxia; MA, mesenteric artery. |
AIH treatment did not alter contractile responses to
Ang II in aortas and MAs, whereas CIH arteries displayed a
significantly increased contraction in response to Ang II. The
magnitude of Ang II-induced vasoconstriction was not affected by
PugNAc intervention. In CIH arteries that had received vehicle or
PugNAc treatment, Ang II-induced contraction was markedly blocked
by SB203580 and PD98059, but not by KN-93. A smaller decrease was
observed following treatment with PugNAc. Of note, Ang II-induced
contraction of mesenteric rings from all the non-ST045849-treated
groups was inhibited by SB203580 and PD98059, but not by KN-93,
with higher inhibitory effects observed in CIH arteries. ST045849
markedly suppressed the contractile responses to Ang II in arteries
from all groups, with greater decreases observed in the CIH rings.
The inhibitory effects of SB203580 and PD98059 on vascular
contraction to Ang II were abolished by ST045849 (Fig. 4).
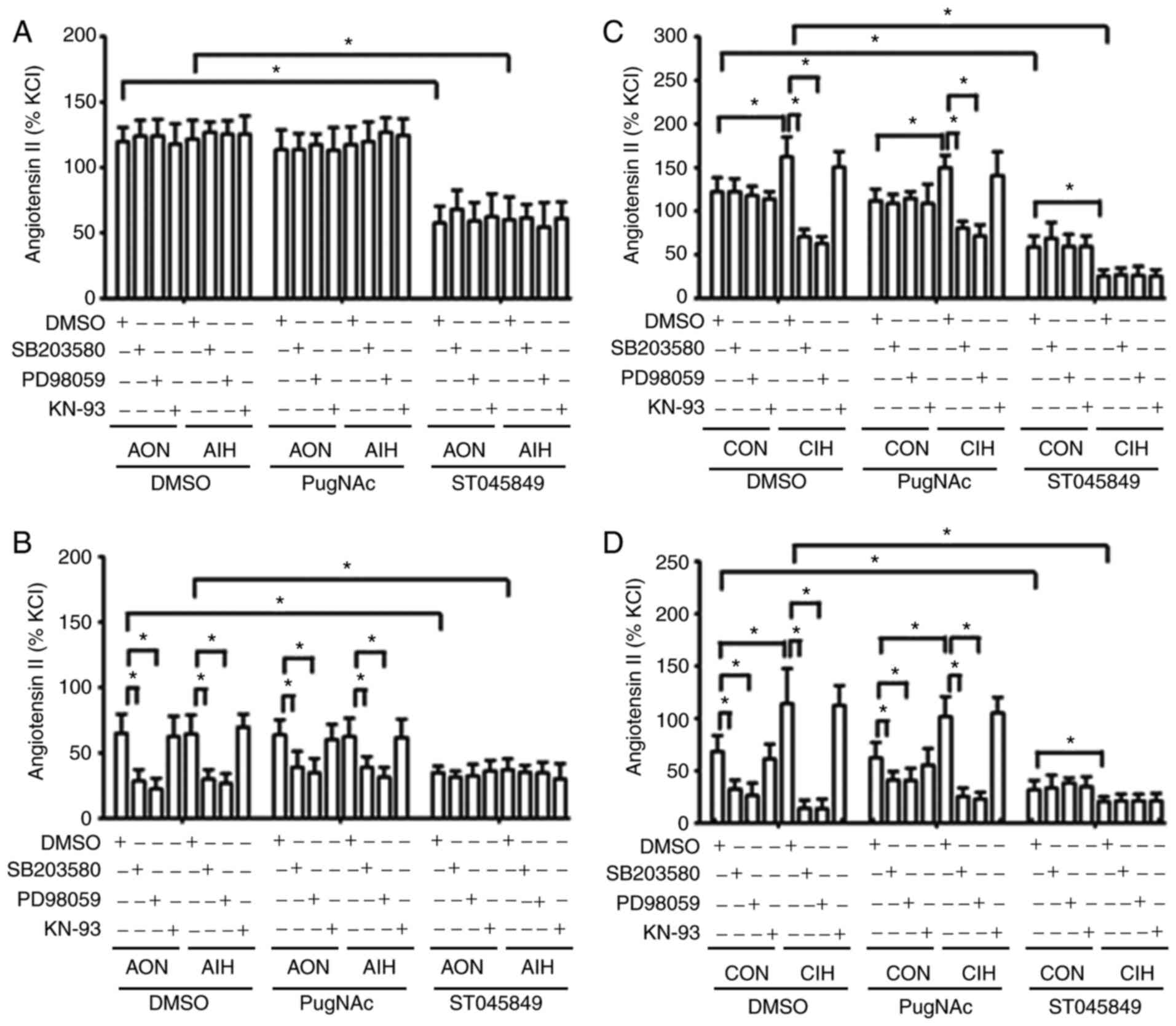 | Figure 4.Effects of SB203580, PD98059 and
KN-93 on contractile responses to Ang II. Contractile responses to
Ang II during AON and AIH exposure in (A) aortic rings and (B) MAs
treated with DMSO, PugNAc or ST045849. Ang II-induced contraction
of vessels from CON and CIH rats in (C) aortic rings and (D) MAs
treated with DMSO, PugNAc or ST045849. The groups were as follows:
i) AON, 3-h normoxia treatment; ii) AIH, 3-h intermittent hypxia
treatment; iii) CON, normoxic (21% O2) condition; and
iv) CIH, intermittent hypoxia cycles (6–8% O2 for 2 min
and 21% O2 for another 2 min). Data are presented as the
mean ± SD (n=5). *P<0.05. Ang II, angiotensin II; AIH, acute
intermittent hypoxia; CON, control; CIH, chronic intermittent
hypoxia; MA, mesenteric artery. |
O-GlcNAcylation interferes with the
phosphorylation of p38 MAPK, ERK1/2 and CaMKII
CIH significantly increased the expression of
O-GlcNAc and OGT proteins, but reduced the expression of OGA in
both aortic and mesenteric tissues (Figs. 1F and G, 5, 6B,
S1 and S2B). No statistically significant
differences in protein O-GlcNAc levels of cultured aortic rings
exposed to AIH were observed (Fig.
6A and S2A). CIH significantly
increased the phosphorylation of p38 MAPK and ERK1/2 but did not
exert significant effects on CaMKII phosphorylation levels
(Figs. 5, 6B, S1 and
S2B). Incubation with PugNAc
significantly increased vascular global O-GlcNAcylation, which was
accompanied by an increase in the phosphorylation of p38 MAPK and
ERK1/2, as well as a slight non-significant elevation in p-CaMKII
levels in all groups. On the other hand, ST045849 treatment
markedly downregulated O-GlcNAcylation levels, which resulted in a
decrease in p38 MAPK and ERK1/2 phosphorylation and no changes in
CaMKII phosphorylation (Figs. 6 and
S2). No differences in the total
expression of p38 MAPK, ERK1/2 and CaMKII were observed among the
groups (Figs. 5, 6, S1 and
S2).
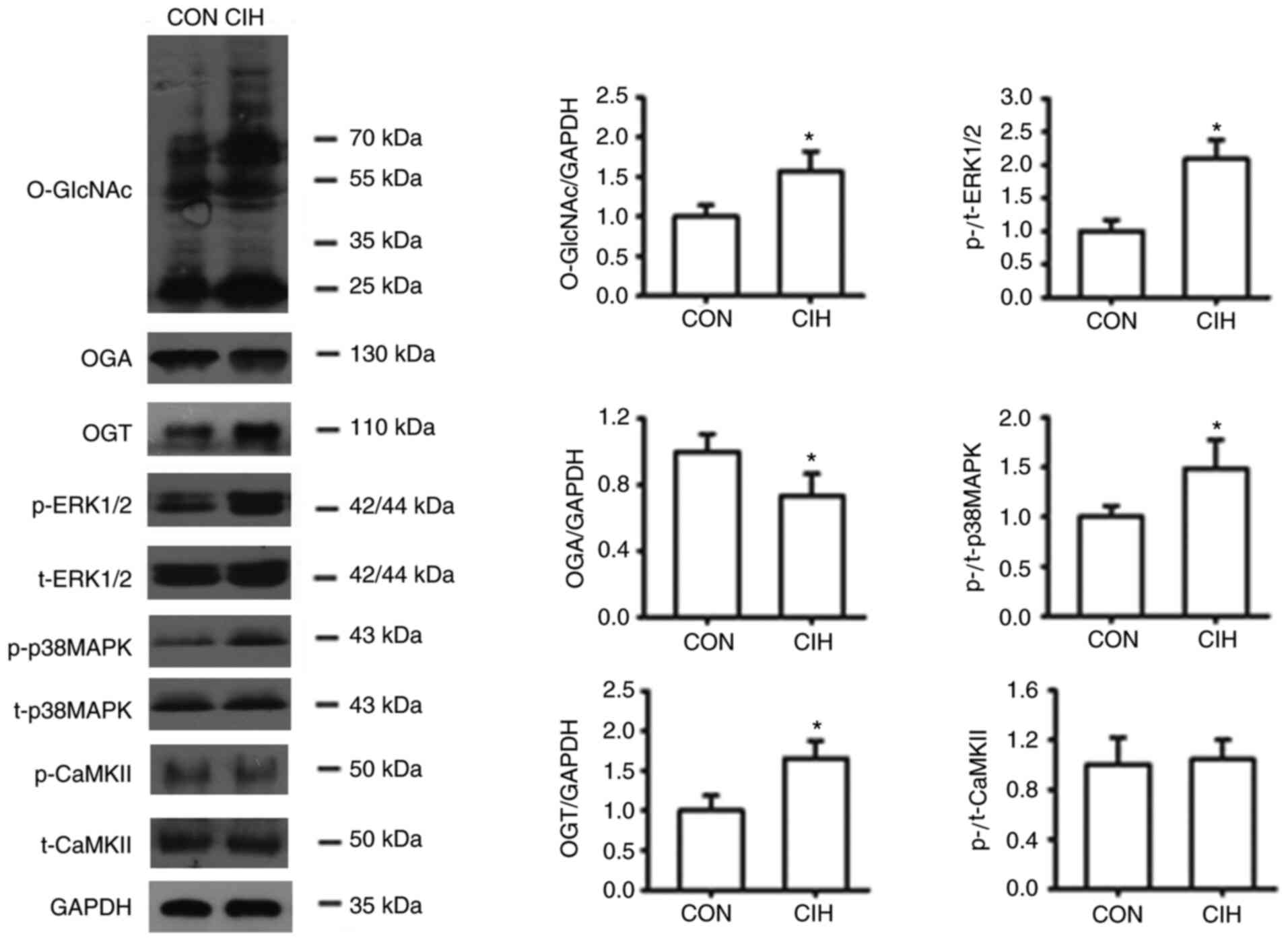 | Figure 5.CIH enhances O-GlcNAc levels, and the
expression levels of OGT, p-p38 MAPK and p-ERK1/2 levels, but
decreases OGA expression in mesenteric arteries. The groups were as
follows: i) CON, normoxic (21% O2) condition; and ii)
CIH, intermittent hypoxia cycles (6–8% O2 for 2 min and
21% O2 for 2 min). Data are presented as the mean ± SD
(n=3-5). *P<0.05 vs. CON group. CIH, chronic intermittent
hypoxia; O-GlcNAc, O-linked-β-N-acetylglucosamine; OGT, O-GlcNAc
transferase; OGA, O-GlcNAcase; p-, phosphorylated; CON, control;
CaMKII, Ca2+/calmodulin-dependent kinase II; t-, total
protein. |
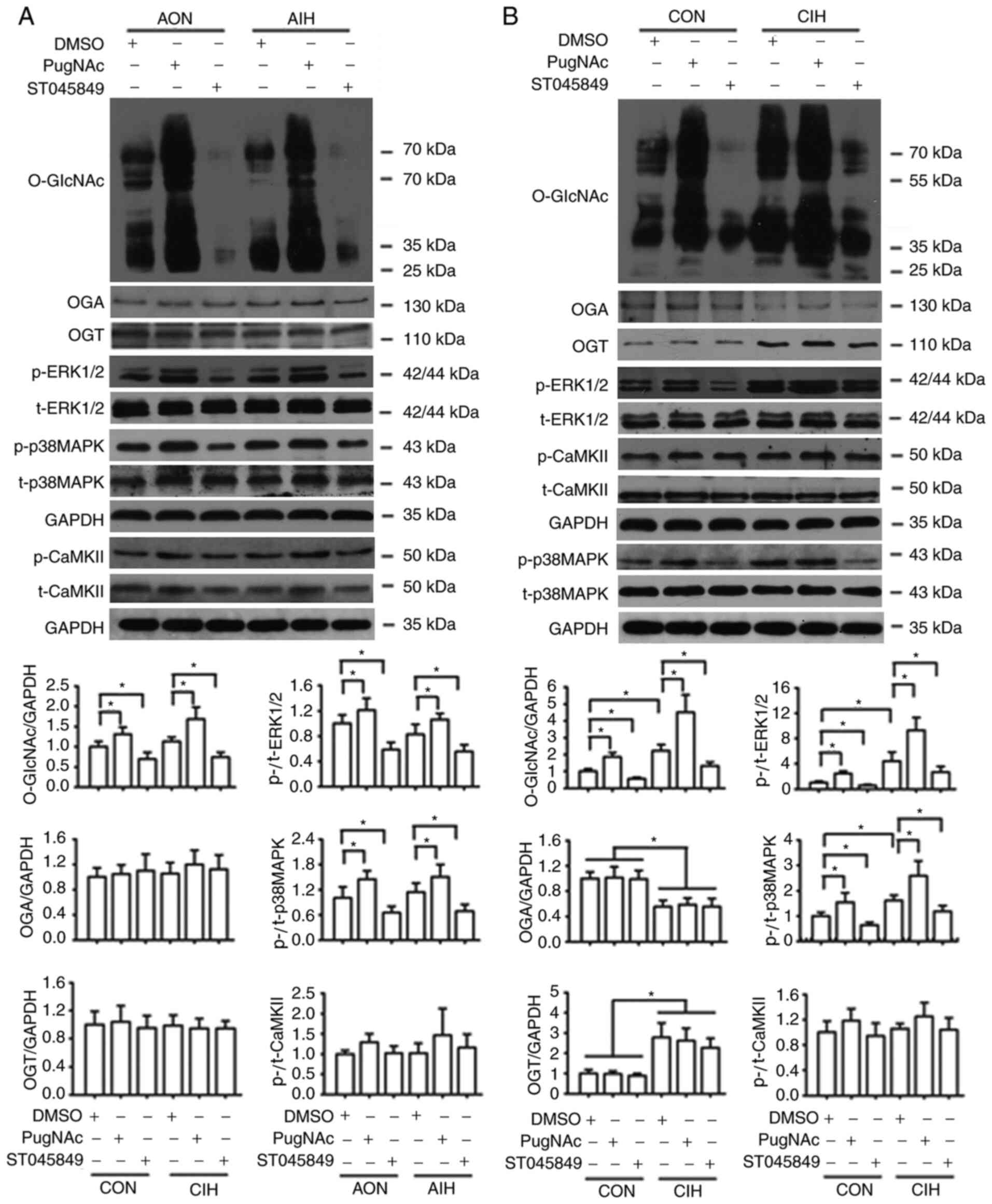 | Figure 6.Protein O-GlcNAc levels interfere
with the phosphorylation of p38 MAPK, ERK1/2 and CaMKII. (A)
Protein levels of O-GlcNAc, OGA, OGT, p-p38 MAPK, p-ERK1/2 and
p-CaMKII in cultured aortas that received either AON or AIH
treatment in the presence of DMSO, PugNAc or ST045849. (B) Protein
levels of O-GlcNAc, OGA, OGT, p-p38 MAPK, p-ERK1/2 and p-CaMKII in
cultured aortas from the CON or CIH rats in the presence of DMSO,
PugNAc or ST045849. Experimental groups were as follows: i) AON,
3-h normoxia treatment; ii) AIH, 3-h intermittent hypxia treatment;
iii) CON, normoxic (21% O2) condition; and iv) CIH,
intermittent hypoxia cycles (6–8% O2 for 2 min and 21%
O2 for 2 min). Data are presented as the mean ± SD
(n=3). *P<0.05. O-GlcNAc, O-linked-β-N-acetylglucosamine;
CaMKII, Ca2+/calmodulin-dependent kinase II; OGA,
O-GlcNAcase; OGT, O-GlcNAc transferase; AIH, acute intermittent
hypoxia; CON, control; CIH, chronic intermittent hypoxia; p-,
phosphorylated; t-, total protein. |
Discussion
The overall objective of the present study was to
identify the regulatory role of O-GlcNAcylation in modulating
signaling pathways involved in vascular function during IH
exposure. Herein, it was mainly found that protein O-GlcNAc and OGT
levels, and the phosphorylation of p38 MAPK and ERK1/2, were
increased in aortas and MAs from rats with CIH-induced
hypertension. In addition, the OGA levels were decreased, but no
changes were observed in p-CaMKII levels. IH exposure could impair
endothelial function and vasoconstrictor reactivity. The modulation
of global O-GlcNAcylation by pharmacological approaches could
interfere with the endothelium-dependent relaxation and vascular
contractility, at least partly through MAPK-mediated, but not
CaMKII-mediated, signaling pathways.
To the best of our knowledge, the present study was
the first to reveal that CIH exposure significantly increased the
content of O-GlcNAc proteins in the vasculature, which was
accompanied by upregulated protein levels of OGT and reduced levels
of OGA. In addition, elevations in the protein expression levels of
O-GlcNAc by CIH coincided with a decreased endothelium-dependent
relaxation in MAs and increased responsiveness to Ang II in aortas
and MAs. Similarly, Lima et al (15) reported that O-GlcNAcylation was
increased in DOCA-salt hypertensive rats and proposed that
sustained increases in O-GlcNAcylation comprise a fundamental
mechanism underlying the negative effects of hypertension on
vascular function, thereby contributing to hypertension-related
vascular dysfunction. In addition, it was discovered that OGA
inhibition with the specific inhibitor PugNAc enhanced global
O-GlcNAc levels in cultured aortas from all experimental groups,
whereas the blockade of OGT with ST045849 had the opposite effects.
In addition, PugNAc treatment could protect endothelial cells from
AIH-related relaxation injury, but the acute reduction in
O-GlcNAcylation by ST045849 tended to restore the relaxant response
to ACh in CIH MAs and caused a greater reduction in Ang II-induced
contractile responses in CIH vessels. Collectively, these findings
indicated that IH-related vascular dysfunction was, to a certain
extent, regulated by O-GlcNAcylation modifications.
In the present study, it was found that CIH markedly
attenuated ACh-induced vasodilation, which was accompanied by
increases in the wall thickness of MAs, but not in aortas, which
might indicate that the endothelial cells of resistance arteries in
the animal models were more susceptible to CIH insults. CIH-induced
endothelial dysfunction most likely results from the decreased
generation and bioavailability of NO caused by the unbalanced
endothelial expression of eNOS and arginase-1, oxidative stress
injury, inflammatory injury, endothelial cell apoptosis and
impaired repair process (3,4,30,35).
Consistent with these findings, our previous studies revealed that
IH exposure could induce endothelial cell apoptosis due to a
disturbance in the Ca2+ homeostasis and endoplasmic
reticulum function through the misregulation of the phospholipase C
pathway (36,37). It was also demonstrated in the
present study that AIH could impair ACh-induced relaxant responses
in both aortic and mesenteric rings, which was prevented by PugNAc
incubation. The differences in the functional effects on the
endothelium between AIH and CIH could be attributed to the
relatively weaker anti-oxidant defense and anti-inflammatory
capacity of AIH vessels cultured in vitro. In addition,
previous data have revealed that acute rises in protein O-GlcNAc
levels could improve cellular tolerance to stresses, such as I/R
injury, inflammation and oxidative stress (10,12,20).
Similarly, as shown in the present study, the increase in protein
O-GlcNAc levels could protect human corneal endothelial cells from
oxidative stress by attenuating the loss of mitochondrial membrane
potential, reducing mitochondrial Ca2+ overload and
intracellular reactive oxygen species formation through the Akt
signaling pathway (38). Based on
these findings, it was hypothesized that the protective effect of
PugNAc on the endothelium against AIH injury might be associated
with its ability to increase O-GlcNAcylated proteins. Of note,
reducing global O-GlcNAcylation markedly weakened ACh-induced
relaxant responses in vessels under conditions of normoxia and AIH,
but tended to improve the relaxation of MAs from CIH rats, in which
vascular O-GlcNAc proteins were chronically increased. Likewise, it
has also been revealed that chronically increased O-GlcNAc
modification could impair endothelial function by inhibiting eNOS
activity and the subsequent release of NO, due to the impaired
activation of the PI3K/Akt pathway (25). This, in turn, has been shown to lead
to endothelial dysfunction (10,15,25).
Therefore, these findings further supported the notion that
O-GlcNAc formation exerts both protective and adverse effects on
vascular function, depending on whether it occurs under acute or
chronic stress conditions, respectively, which is most likely
through the modulation of different downstream signaling pathways
(13,20). Possible regulatory mechanisms
involved in the regulation of vascular effects of O-GlcNAcylation
during IH exposure require further study.
Regarding the functional consequences of IH on
vasoconstriction and signaling mechanisms, it was discovered that
CIH exposure elevated contractile responses to Ang II in both
aortas and MAs, but without affecting PE-induced contraction.
Similarly, impaired vascular reactivity to different constrictor
stimuli, such as Ang II and ET-1, has been reported in the aorta,
as well as small mesenteric, cerebral and pulmonary arteries of CIH
animal models (3,5,7,9,39).
In individuals with OSA, arterial stiffness, as evaluated by the
vasoconstrictor effects of intravenous Ang II challenge, was
increased, which could be corrected by continuous positive airway
pressure treatment (6). It was also
found that the phosphorylation of p38 MAPK and ERK1/2 was increased
by CIH, and the blockade of these kinases reduced Ang II-mediated
contractions in both CIH aortic and mesenteric rings. This could be
evidenced by previous studies suggesting that MAPKs act as
important regulators of vascular reactivity in animal models of
hypertension. The activation of ERK1/2 contributes to elevations in
blood pressure, which are most likely associated with the ability
of ERK1/2 to modulate vascular oxidative stress, and regulate
Ca2+ sensitivity and the downstream contractile protein
(5,23,40,41).
Furthermore, MAPK inhibition has been reported to block Ang
II-induced contraction in MAs from non-ST045849-treated groups
(42,43), with higher inhibitory effects
observed in CIH vessels; this might indicate a greater contribution
of MAPKs to Ang II-mediated vasoconstriction in small resistance
arteries from CIH rats. These findings supported the present study
hypothesis that MAPK pathway activation contributed to CIH-related
vascular dysfunction. However, despite evidence that IH could
affect CaMKII activity (44,45)
and a similar finding showing an increased phosphorylated
modification of CaMKII by increased O-GlcNAc levels (11,22),
CaMKII blockade did not alter vascular contractile responses to PE
or Ang II in any of the groups studied, indicating the lack of
CaMKII involvement in the regulation of IH-related
vasoconstriction.
A number of kinases or phosphatases can be modified
by O-GlcNAcylation, including the CaMK, CK1, tyrosine kinases (TK)
and Sterile (STE) kinase families (11). It was further observed in the
present study that p38 MAPK and ERK1/2 were phosphorylated in
response to enhanced O-GlcNAc levels, whereas reducing
O-GlcNAcylation tended to decrease the phosphorylation of these
kinases, which was consistent with earlier research showing that
O-GlcNAc levels could affect MAPK phosphorylation dynamics. As
previously reported, increases in the phosphorylation level of
ERK1/2 and p38 MAPK are positively correlated with the increase in
O-GlcNAcylation (26,27). Of note, a positive correlation has
also been found between activated MAPK/ERK signaling and
hyper-O-GlcNAcylation in various cancer types (28). Unlike changes in phosphorylated
MAPKs induced by O-GlyNAcylation alterations, the phosphorylation
of CaMKII was only slightly enhanced by PugNAc, but this result was
not significant, and was not affected by ST045849 in the present
study. These findings were consistent with the results of vascular
contractile responses. The reason for these findings may have been
the differences between the target protein substrates of OGA and
those of OGT (10,11,18).
However, no studies have directly addressed how
O-GlcNAcylation affects contractile responses in IH-treated
vessels. In the current study, it was discovered that Ang
II-induced contractions of CIH arteries were attenuated by SB203580
and PD98059, with a lower magnitude of reduction by these
inhibitors in vessels treated with PugNAc, possibly due to the
relatively higher activity of the kinases following PugNAc
incubation. Conversely, ST045849 abolished the inhibitory effects
of SB203580 and PD98059 on vasoconstriction to Ang II, suggesting
the involvement of other signaling proteins in the regulation of
vasoconstrictor sensitivity (46).
Based on these observations, it could be concluded that alterations
in global O-GlcNAc trigger functional changes in the vasculature
during IH exposure, at least partly by modulating MAPK-mediated
signaling pathways. Of note, although PugNAc did not alter
PE-induced contraction, MAPK blockade attenuated the contractile
response, suggesting the contribution of MAPKs to PE-stimulated
vasoconstriction in PugNAc-treated arteries. The alterations in OGA
activity may interfere with the contractile apparatus required for
PE-induced contraction via MAPK signaling, since MAPK blockade had
no effects on vasoconstrictor sensitivity to PE in the vessels that
had not received PugNAc treatment. This could be proven by
preliminary data showing that PugNAc increases contractions to PE
through the activation of the Ras homolog family member A
(RhoA)/Rho-kinase pathway (47). In
addition, an interaction between ERK1/2 and Rho-kinase has been
reported, where ERK1/2 activation depends on Rho-kinase signaling
(48). However, further research is
required to confirm this.
The current understanding of the unique molecular
O-GlcNAcylation signaling in regulating vascular reactivity is
incomplete. With respect to arterial hypertension and the
associated vascular abnormality, several specific pathways, such as
PKC, MAPK, PI3K/Akt and RhoA/Rho-kinase, are targets for
O-GlcNAcylation (16,17,21).
It has been proposed that O-GlcNAc modification of contractile
regulators downstream of those specific cascades, such as MLC
kinase, MYPT1 and MLC, can interfere with their phosphorylated
activities, thus directly influencing vascular smooth muscle
function (16,17,47,49).
The present study was not without its limitations.
First, changes in the protein levels of OGA, OGT and the
phosphorylation of p38 MAPK, ERK1/2 and CaMKII were not
investigated in MAs following stimulation with PugNAc or ST045849,
but only in aortic tissues, due to the insufficient amount of
protein that could be extracted from the MAs. Secondly, the
different regulatory role of O-GlcNAcylation in PE-induced and Ang
II-induced vasoconstriction could not be determined, possibly due
to the agonist-specific signaling transduction cascades (7,46).
Thirdly, given the unavailability of specific antibodies for
O-GlcNAcylated proteins, which identify the O-GlcNAc-modified
sites, the exact O-GlcNAcylation levels of MAPKs and CaMKII were
not evaluated. Finally, although the in vitro findings
suggested pathophysiological links between increased
O-GlcNAcylation and CIH-induced vascular dysfunction, future
studies using non-toxic pharmacological agents for in vivo
experiments, and even transgenic animal models, are required to
clarify the exact mechanisms of the regulation of IH-related
vascular abnormalities by O-GlcNAcylation.
In conclusion, the present findings demonstrated
that CIH increased global O-GlcNAc proteins in the vasculature and
O-GlcNAcylation participated in the regulation of misregulated
vascular reactivity during IH exposure, at least partly through
MAPK, but not CaMKII signaling pathways. Acute increases in protein
O-GlcNAc levels exerted vaso-protective effects against AIH
insults, whereas sustained elevations in O-GlcNAc proteins promoted
the detrimental effects of CIH on the vasculature. The modulation
of protein O-GlcNAcylation may serve as a therapeutic target for
the treatment of impaired vascular function and the associated
cardiovascular disorders in patients with OSA.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Qinghua Hu
from the Department of Pathophysiology, Huazhong University of
Science and Technology (Wuhan, China) for the use of the CO pod and
PowerLab data acquisition systems.
Funding
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81570080 and
81770088) and the Medical and Health Guidance Project of Xiamen
(grant no. 3502Z20199018).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HL, XG and YD contributed to the research design and
drafting of the manuscript. XG, YD, LZ and JS performed the
experiments. XG, YD and LZ conducted the analysis and
interpretation of the data. HL, XG and LZ confirmed the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All animal procedures were performed in accordance
with the Guiding Principles in the Care and Use of Animals and
approved by the Institutional Animal Care and Use Committee of
Huazhong University of Science and Technology (Wuhan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Foster GE, Poulin MJ and Hanly PJ:
Intermittent hypoxia and vascular function: Implications for
obstructive sleep apnoea. Exp Physiol. 92:51–65. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cai AP, Wang L and Zhou YL: Hypertension
and obstructive sleep apnea. Hypertens Res. 39:391–395. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Krause BJ, Casanello P, Dias AC, Arias P,
Velarde V, Arenas GA, Preite MD and Iturriaga R: Chronic
intermittent hypoxia-induced vascular dysfunction in rats is
reverted by-acetylcysteine supplementation and arginase inhibition.
Front Physiol. 9:901–912. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang YN, Zhang CL, Li HO and Hou JD:
Down-regulation of vascular PPAR-γ contributes to endothelial
dysfunction in high-fat diet-induced obese mice exposed to chronic
intermittent hypoxia. Biochem Biophys Res Commun. 492:243–248.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo XL, Deng Y, Shang J, Liu K, Xu YJ and
Liu HG: ERK signaling mediates enhanced angiotensin II-induced rat
aortic constriction following chronic intermittent hypoxia. Chin
Med J (Engl). 126:3251–3258. 2013.PubMed/NCBI
|
|
6
|
Nicholl DDM, Hanly PJ, Zalucky AA, Mann
MC, MacRae JM, Poulin MJ, Handley GB, Sola DY and Ahmed SB: CPAP
therapy delays cardiovagal reactivation and decreases arterial
renin-angiotensin system activity in humans with obstructive sleep
apnea. J Clin Sleep Med. 14:1509–1520. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Allahdadi KJ, Walker BR and Kanagy NL: ROK
contribution to endothelin-mediated contraction in aorta and
mesenteric arteries following intermittent hypoxia/hypercapnia in
rats. Am J Physiol Heart Circ Physiol. 293:H2911–H2918. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Durgan DJ, Crossland RF, Lloyd EE,
Phillips SC and Bryan RM: Increased cerebrovascular sensitivity to
endothelin-1 in a rat model of obstructive sleep apnea: A role for
endothelin receptor B. J Cereb Blood Flow Metab. 35:402–411. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Snow JB, Norton CE, Sands MA, Weise-Cross
L, Yan S, Herbert LM, Sheak JR, Gonzalez Bosc LV, Walker BR, Kanagy
NL, et al: Intermittent hypoxia augments pulmonary vasoconstrictor
reactivity through PKCβ/mitochondrial oxidant signaling. Am J
Respir Cell Mol Biol. 62:732–746. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chatham JC, Zhang JH and Wende AR: Role of
O-linked N-acetylglucosamine (O-GlcNAc) protein modification in
cellular (patho)physiology. Physiol Rev. 101:427–493. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schwein PA and Woo CM: The O-GlcNAc
modification on kinases. ACS Chem Biol. 15:602–617. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wright JN, Collins HE, Wende AR and
Chatham JC: O-GlcNAcylation and cardiovascular disease. Biochem Soc
Trans. 45:545–553. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jensen RV, Andreadou I, Hausenloy DJ and
Bøtker HE: The role of O-GlcNAcylation for protection against
ischemia-reperfusion injury. Int J Mol Sci. 20:404–424. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo XL, Shang J, Deng Y, Yuan X, Zhu D and
Liu HG: Alterations in left ventricular function during
intermittent hypoxia: Possible involvement of O-GlcNAc protein and
MAPK signaling. Int J Mol Med. 36:150–158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lima VV, Giachini FR, Choi H, Carneiro FS,
Carneiro ZN, Fortes ZB, Carvalho MH, Webb RC and Tostes RC:
Impaired vasodilator activity in deoxycorticosterone acetate-salt
hypertension is associated with increased protein O-GlcNAcylation.
Hypertension. 53:166–174. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lima VV, Giachini FR, Hardy DM, Webb RC
and Tostes RC: O-GlcNAcylation: A novel pathway contributing to the
effects of endothelin in the vasculature. Am J Physiol Regul Integr
Comp Physiol. 300:R236–R250. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lima VV, Giachini FR, Carneiro FS,
Carvalho MH, Fortes ZB, Webb RC and Tostes RC: O-GlcNAcylation
contributes to the vascular effects of ET-1 via activation of the
RhoA/Rho-kinase pathway. Cardiovasc Res. 89:614–622. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
van der Laarse SAM, Leney AC and Heck AJR:
Crosstalk between phosphorylation and O-GlcNAcylation: friend or
foe. FEBS J. 285:3152–3167. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kumar GK and Prabhakar NR:
Post-translational modification of proteins during intermittent
hypoxia. Respir Physiol Neurobiol. 164:272–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hilgers RHP, Xing DQ, Gong KZ, Chen YF,
Chatham JC and Oparil S: Acute O-GlcNAcylation prevents
inflammation-induced vascular dysfunction. Am J Physiol Heart Circ
Physiol. 303:H513–H522. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lima VV, Rigsby CS, Hardy DM, Webb RC and
Tostes RC: O-GlcNAcylation: A novel post-translational mechanism to
alter vascular cellular signaling in health and disease: Focus on
hypertension. J Am Soc Hypertens. 3:374–387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Erickson JR, Pereira L, Wang L, Han G,
Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM and
Bers DM: Diabetic hyperglycaemia activates CaMKII and arrhythmias
by O-linked glycosylation. Nature. 502:372–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Giachini FR, Sullivan JC, Lima VV,
Carneiro FS, Fortes ZB, Pollock DM, Carvalho MHC, Webb RC and
Tostes RC: Extracellular signal-regulated kinase 1/2 activation,
via downregulation of mitogen-activated protein kinase phosphatase
1, mediates sex differences in desoxycorticosterone acetate-salt
hypertension vascular reactivity. Hypertension. 55:172–179. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yousif MHM, Akhtar S, Walther T and Benter
IF: Role of Ca2+/calmodulin-dependent protein kinase II
in development of vascular dysfunction in diabetic rats with
hypertension. Cell Biochem Funct. 26:256–263. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Federici M, Menghini R, Mauriello A,
Hribal ML, Ferrelli F, Lauro D, Sbraccia P, Spagnoli LG, Sesti G
and Lauro R: Insulin-dependent activation of endothelial nitric
oxide synthase is impaired by O-linked glycosylation modification
of signaling proteins in human coronary endothelial cells.
Circulation. 106:466–472. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Goldberg H, Whiteside C and Fantus IG:
O-linked β-N-acetylglucosamine supports p38 MAPK activation by high
glucose in glomerular mesangial cells. Am J Physiol Endocrinol
Metab. 301:E713–E726. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang MZ, Qiu ZY, Zhang S, Fan X, Cai XQ,
Xu B, Li XW, Zhou JF, Zhang XY, Chu Y, et al: Elevated
O-GlcNAcylation promotes gastric cancer cells proliferation by
modulating cell cycle related proteins and ERK 1/2 signaling.
Oncotarget. 7:61390–61402. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang XL, Ma LN, Qi JQ, Shan H, Yu WG and
Gu YC: MAPK/ERK signaling pathway-induced hyper-O-GlcNAcylation
enhances cancer malignancy. Mol Cell Biochem. 410:101–110. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Institute for Laboratory Animal and
Research, . Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press; Washington, DC: 2011
|
|
30
|
Li JR, Zhao YS, Chang Y, Yang SC, Guo YJ
and Ji ES: Fasudil improves endothelial dysfunction in rats exposed
to chronic intermittent hypoxia through RhoA/ROCK/NFATc3 pathway.
PLoS One. 13:e01956042018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ishihata A, Tasaki K and Katano Y:
Involvement of p44/42 mitogen-activated protein kinases in
regulating angiotensin II- and endothelin-1-induced contraction of
rat thoracic aorta. Eur J Pharmacol. 445:247–256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang CJ, Zhou ZG, Holmqvist A, Zhang H, Li
Y, Adell G and Sun XF: Survivin expression quantified by Image
Pro-Plus compared with visual assessment. Appl Immunohistochem Mol
Morphol. 17:530–535. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu W, Mao Z, Zhu C, Li M, Cao C, Guan Y,
Yuan J, Xie G and Guan X: Adolescent exposure to cocaine increases
anxiety-like behavior and induces morphologic and neurochemical
changes in the hippocampus of adult rats. Neuroscience.
313:174–183. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Xu Y, Zhu Q, Zhao F, Luo J, Zhang
X and Wang X: Upregulation of dysbindin in temporal lobe epileptic
foci of human and experimental animals. Synapse. 66:622–629. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng J, Zhang D and Chen BY: Endothelial
mechanisms of endothelial dysfunction in patients with obstructive
sleep apnea. Sleep Breath. 16:283–294. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang YY, Shang J and Liu HG: Role of
endoplasmic reticular stress in aortic endothelial apoptosis
induced by intermittent/persistent hypoxia. Chin Med J (Engl).
126:4517–4523. 2013.PubMed/NCBI
|
|
37
|
Ren J, Liu W, Deng Y, Li GC, Pan YY, Xie
S, Jin M and Liu HG: Losartan attenuates aortic endothelial
apoptosis induced by chronic intermittent hypoxia partly via the
phospholipase C pathway. Sleep Breath. 21:679–689. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yoon CK, Yoon SY, Hwang JS and Shin YJ:
O-GlcNAc signaling augmentation protects human corneal endothelial
cells from oxidative stress via AKT pathway activation. Curr Eye
Res. 45:556–562. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Friedman JK, Nitta CH, Henderson KM,
Codianni SJ, Sanchez L, Ramiro-Diaz JM, Howard TA, Giermakowska W,
Kanagy NL and Gonzalez Bosc LV: Intermittent hypoxia-induced
increases in reactive oxygen species activate NFATc3 increasing
endothelin-1 vasoconstrictor reactivity. Vascul Pharmacol.
60:17–24. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kim B, Kim J, Bae YM, Cho SI, Kwon SC,
Jung JY, Park JC and Ahn HY: p38 mitogen-activated protein kinase
contributes to the diminished aortic contraction by endothelin-1 in
DOCA-salt hypertensive rats. Hypertension. 43:1086–1091. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ding LL, Chapman A, Boyd R and Wang HD:
ERK activation contributes to regulation of spontaneous contractile
tone via superoxide anion in isolated rat aorta of angiotensin
II-induced hypertension. Am J Physiol Heart Circ Physiol.
292:H2997–H3005. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Escano CS, Keever LB, Gutweiler AA and
Andresen BT: Angiotensin II activates extracellular
signal-regulated kinase independently of receptor tyrosine kinases
in renal smooth muscle cells: Implications for blood pressure
regulation. J Pharmacol Exp Ther. 324:34–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Matrougui K, Eskildsen-Helmond YE,
Fiebeler A, Henrion D, Levy BI, Tedgui A and Mulvany MJ:
Angiotensin II stimulates extracellular signal-regulated kinase
activity in intact pressurized rat mesenteric resistance arteries.
Hypertension. 36:617–621. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang K, Ma ZW, Song C, Duan XR, Yang Y
and Li GP: Role of ion channels in chronic intermittent
hypoxia-induced atrial remodeling in rats. Life Sci.
254:1177972020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yuan GX, Nanduri J, Bhasker CR, Semenza GL
and Prabhakar NR: Ca2+/calmodulin kinase-dependent
activation of hypoxia inducible factor 1 transcriptional activity
in cells subjected to intermittent hypoxia. J Biol Chem.
280:4321–4328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Do KH, Kim MS, Kim JH, Rhim BY, Lee WS,
Kim CD and Bae SS: Angiotensin II-induced aortic ring constriction
is mediated by phosphatidylinositol 3-kinase/L-type calcium channel
signaling pathway. Exp Mol Med. 41:569–576. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lima VV, Lobato NS, Filgueira FP, Webb RC,
Tostes RC and Giachini FR: Vascular O-GlcNAcylation augments
reactivity to constrictor stimuli by prolonging phosphorylated
levels of the myosin light chain. Braz J Med Biol Res. 47:826–833.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Matrougui K, Tankó LB, Loufrani L, Gorny
D, Levy BI, Tedgui A and Henrion D: Involvement of Rho-kinase and
the actin filament network in angiotensin II-induced contraction
and extracellular signal-regulated kinase activity in intact rat
mesenteric resistance arteries. Arterioscler Thromb Vasc Biol.
21:1288–1293. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pedowitz NJ, Batt AR, Darabedian N and
Pratt MR: MYPT1 O-GlcNAc modification regulates
sphingosine-1-phosphate mediated contraction. Nat Chem Biol.
17:169–177. 2021. View Article : Google Scholar : PubMed/NCBI
|














