Introduction
Retinal pigment epithelial (RPE) cells form a
monolayer between the retina and choriocapillaris. In response to
changes in the microenvironment, RPE cells may be activated to
produce autocrine/paracrine growth factors, which can affect not
only their microenvironment but also adjacent compartments, such as
the choroid plexus and retinal neurons (1–3).
Neovascularization in diabetic retinopathy and other retinal
vasculopathies has long been known to be associated with retinal
capillary nonperfusion and retinal hypoxia (4,5).
Notably, retinal hypoxia has been experimentally evidenced in pig
and monkey retinas following laser vein occlusion (6). A previous study indicated that
angiogenic factors, including vascular endothelial cell growth
factor (VEGF), are detectable in vitreous fluid and epiretinal
membrane samples derived from patients with proliferative diabetic
retinopathy (PDR) and proliferative vitreoretinopathy (PVR)
(7). Therefore, these factors may
be involved in the pathogenesis of proliferative membranes;
however, their pathogenic mechanisms have yet to be fully
understood.
Transforming growth factor-β (TGF-β) is a
multifunctional cytokine that regulates RPE cell proliferation
(8,9), differentiation (10), migration (11,12)
and extracellular matrix (ECM) production (12,13).
Vitreous TGF-β2 levels have been reported to be increased in
patients with PDR and other vitreoretinal disorders, and this
increase has been shown to be correlated with the severity of
fibrous proliferation (14).
Oxidative stress can induce the synthesis of TGF-β2 and increase
ECM gene expression in cultured RPE cells; this cellular response
may contribute to the deposition of ECM in the aging macula
(15). There is growing evidence
that RPE cells are able to synthesize TGF-β2 (16,17)
and the release of TGF-β2 may regulate VEGF production in human RPE
cells (18–20). Moreover, TGF-β2 has been reported to
activate RPE cell transformation by upregulating the expression of
α-smooth muscle actin (α-SMA) (21). The main signaling response induced
by TGF-β is mediated via Smad proteins; however, evidence has
suggested that TGF-β is capable of signaling independently of
Smads. While TGF-β has been found to activate non-Smad signaling
pathways, including p38 mitogen-activated protein kinase (MAPK),
c-Jun N-terminal kinase and extracellular-regulated kinase (ERK),
the specific pathway involved appears to vary depending on the cell
type and tissue function (22,23).
Since the breakdown of the blood-retinal barrier
(BRB) is symptomatic of several sight-threatening diseases
(24), intravitreally injected
triamcinolone acetonide (TA), a corticosteroid agent, has long been
proposed for the stabilization of the inner BRB and to prevent
macular edema in retinopathies (25–27).
Thus, it is considered a promising treatment modality for a variety
of proliferative, edematous, neovascular and inflammatory ocular
disorders (28). Since the
upregulation of VEGF is known to be associated with the presence of
diabetic macular edema and PDR (29), the pharmacological mechanisms of TA
treatment include modulation of intercellular adhesion molecule-1
expression and cellular permeability in endothelial cells (25), reduction of VEGF production in RPE
cells under oxidative stress (30),
and VEGF receptor-associated signaling (31). These findings suggest that TA may
exhibit an anti-angiogenic effect against choroidal
neovascularization. In addition, TA has been reported to abrogate
the regulatory loop between VEGF and matrix metalloproteinase
(MMP)-9 in RPE cells (32), and its
intravitreal injection was shown to improve wound healing in mice
with laser retinal photocoagulation (33). However, the pharmacological action
of TA on TGF-β2 signaling in RPE cells and the corresponding
cellular behaviors have yet to be fully defined. Therefore, the
present study aimed to investigate whether TA may modulate
TGF-β2-mediated angiogenic and tissue-remodeling effects in
cultured human RPE cells.
Materials and methods
Chemicals and reagents
TA (cat. no. T6501) was purchased from
MilliporeSigma and maintained in methanol (MeOH) at 10 mM.
Recombinant human TGF-β2 (cat. no. 302-B2) was purchased from
R&D Systems, Inc. Oligonucleotide primers for PCR analyses were
synthesized by Genomics BioSci & Tech Co., Ltd. Selective
kinase inhibitors of p38 MAPK (SB203580; cat. no. S8307), MEK1/MEK2
(PD98059; cat. no. P215), the selective inhibitor for TGF-β type I
receptor (SB431542; cat. no. S4317) and the mouse monoclonal
antibody against β-actin (cat. no. MAB1501) were purchased from
MilliporeSigma. Rabbit monoclonal antibodies against total Smad2
(cat. no. 5339s), phosphorylated (p)-Smad2 (Ser465/467; cat. no.
3108s), total ERK1/2 (cat. no. 4695s), p-ERK1/2 (Thr202/185 and
Tyr204/187; cat. no. 9101s), total p38 MAPK (cat. no. 8690s) and
p-p38 MAPK (Thr180/Tyr182; cat. no. 4631) were purchased from Cell
Signaling Technology, Inc.
Cell culture and drug treatment
A spontaneously immortalized human RPE R-50 cell
line established from a donor eye with informed consent was
provided by Professor D.N. Hu (New York Eye and Ear Infirmary of
Mount Sinai, New York, USA), according to previously described
methods (34,35). Briefly, R-50 cells were grown in
Ham's F-12 nutrient medium supplemented with 10% FBS (both HyClone;
Cytiva), 4 mM glutamine, 100 IU/ml penicillin, 100 µg/ml
streptomycin and 250 ng/ml amphotericin (MP Biomedicals, Inc.) in a
humidified atmosphere containing 5% CO2 at 37°C.
R-50 cells were dissociated by incubating with 0.25%
trypsin at 37°C for 2 min and seeded on either dishes or 96-well
plates at an appropriate cell density of 2×104
cells/cm2 growth area, and were incubated overnight
until they attached. The cells were treated with TA (0.1–100 µM),
TGF-β2 (10 ng/ml) or with the different kinase inhibitors (10 µM)
at 37°C. The specimens collected after a 6-h incubation were used
for gene expression study, the cells collected after a 24-h
incubation were used for the cell viability assay and protein
expression analyses, and the supernatants collected (obtained by
centrifugation at 1,1000 × g; 5 min; 4°C) after 48 h exposure were
used for ELISAs. Cells cultured in medium without supplementation
were used as negative controls, and those incubated with 0.1% MeOH
or 0.1% DMSO were used as solvent controls for TA or kinase
inhibition experiments. The morphology of the treated cells was
examined using a phase-contrast light microscope (Axio Vert.A1;
Carl Zeiss AG) and documented using a digital imaging system
(AxioVision; v4.8.2.0; Carl Zeiss AG).
Cell viability assay
A commercial cell viability assay kit (CellTiter™
Aqueous One Solution; cat. no. G3582; Promega Corporation) using
MTS reagent as the chromogenic substrate was used to determine the
effect of TA on RPE cell viability in the presence or absence of
TGF-β2. The group treated with 0.1% MeOH was used as a solvent
control equivalent to TA treatment at 10 µM. The assay was
performed according to the manufacturer's protocol, as described
previously (36). The end-point
optical density was measured using a microplate reader (Sunrise™;
Tecan Group, Ltd.). The relative cell viability was normalized to
that of the negative control (without drug treatment).
Rat tail tendon collagen preparation
and 3-dimensional (3D) collagen gel contraction assay
Rat tails were collected from adult male
Sprague-Dawley rats under the approval and supervision of the
Institutional Animal Care and Use Committee at E-Da Hospital
(approval no. IACUC-102016; Kaohsiung, Taiwan). All procedures were
performed in accordance with the Guide for the Care and Use of
Laboratory Animals (NIH publication no. 85–23, revised 1996)
(37). The rat tails (n=5) were
excised from the sacrificed animals of a previous study (38), in which deep anesthesia was induced
and maintained with 2.5 and 2% isoflurane, respectively, and
supplemental oxygen was administered using a calibrated vaporizer.
Animal death was caused by collagenase solution perfusion and
subsequent removal of the liver for hepatocyte isolation. The
extraction of collagen from rat tail tendons and 3D collagen gel
contraction assays were performed as previously described (39). Briefly, 1.5×105 R-50
cells were repopulated in a 3D lattice collagen gel by rapidly
mixing 1 ml culture medium containing 20 mM HEPES, 0.5 ml cell
suspension containing freshly trypsinized RPE cells, and 2.5 mg rat
tail collagen solution in a 35-mm dish. After complete gelling by
incubating for 5 min in a CO2 incubator at 37°C, the
gels were detached and allowed to float in dishes. The gel area was
directly measured at 24-h intervals. Data are expressed as the
percentage of the area compared to the original size of the gel
measured at 0 h.
Western blotting
Total cellular protein extracts from RPE cells were
obtained by lysing the cells in ice-cold RIPA buffer (150 mM NaCl,
1% Nonidet P-40, 1 mM EDTA, 0.25% sodium deoxycholate and 0.1% SDS
in 50 mM Tris pH 7.4) in the presence of a protease inhibitor
cocktail (Roche Applied Science) and phosphatase inhibitors (1 mM
sodium fluoride and 1 mM sodium orthovanadate). Total protein was
quantified using a bicinchoninic acid protein assay kit (Pierce;
Thermo Fisher Scientific, Inc.) and 40 µg total protein per lane
was separated by SDS-PAGE, using 8% acrylamide gels under reducing
conditions. After electrophoresis and electrotransfer onto a PVDF
membrane, the blots were blocked in 5% skim milk in PBS-0.1%
Tween-20 (PBST) at room temperature for 1 h, followed by incubation
with one of the following antibodies at 4°C overnight: Anti-β-actin
(1:10,000), anti-Smad2 (1:1,000), anti-p-Smad2 (1:1,000),
anti-ERK1/2 (1:1,000), anti-p-ERK1/2 (1:1,000), anti-p38 MAPK
(1:1,000), anti-p-p38 MAPK (1:1,000), anti-COL1A1 (1:1,000),
anti-α-SMA (1:1,000), anti-MMP-2 (1:1,000) or anti-MMP-9 (1:1,000).
After washing with PBST, the membranes were incubated with
HRP-conjugated goat anti-rabbit IgG (1:10,000; cat. no. 7074; Cell
Signaling Technology, Inc) or goat anti-mouse IgG (1:10,000; cat.
no. 115-035-003; Jackson ImmunoResearch Laboratories, Inc.)
secondary antibody at room temperature for 1 h. The immunoreactive
signals were visualized using an enhanced chemiluminescence
detection system (MilliporeSigma), documented on a digital imaging
system and densitometrically analyzed using ImageJ software (1.48v;
National Institutes of Health). Relative protein expression levels
were presented as ratios of the densities between target proteins
and actin or compared to their total protein levels, and further
normalized to that of the untreated negative control. The
normalized density data were expressed as the mean ± standard
deviation (SD) from three independent experiments.
Total RNA extraction,
semi-quantitative reverse transcription (RT)-PCR and
RT-quantitative PCR (RT-qPCR)
RNA extraction, RT and semi-quantitative multiplex
PCR detection of VEGF isoform gene transcripts were performed as
previously described (36).
Briefly, total RNA was extracted from 1×106 R50 cells
using TRIzol® (Invitrogen; Thermo Fisher Scientific,
Inc.) and cDNA was generated using avian myeloblastosis virus
reverse transcriptase (cat. no. M5108; Promega Corporation) and
oligo-dT primers (cat. no. C1101; Promega Corporation) in the
presence of a ribonuclease inhibitor (RNasin; cat. no. N2111;
Promega Corporation). For the multiplex amplification of VEGF gene
transcripts, the following primers were synthesized: VEGF sense,
5′-CGAAGTGGTGAAGTTCATGGATG-3′ and anti-sense,
5′-ATATCCATCACACTGGCGGCCGC-3′; and β-actin sense,
5′-GCCAAGGTCATCCATGACAAC-3′ and anti-sense,
5′-GTCCACCACCCTGTTGCTGTA-3′. PCR was performed according to a
previously described protocol (36). The PCR products were titrated to
establish standard curves for documenting linearity and permitting
semi-quantitative analysis using a densitometrical analysis system
(UVITEC Imaging system; UVITEC). The relative gene expression
levels were expressed as the ratio of densities between the PCR
products and β-actin. For RT-qPCR detection of the
VEGF165 transcript levels, the RNA extraction and RT
reaction were performed as described previously for
semi-quantitative PCR. The cDNA was used for qPCR analysis on a
real-time PCR machine (Eco Real-Time PCR system; Illumina, Inc.)
according to a previously described protocol (40). Briefly, VEGF165 mRNA was
amplified using a SYBR Green PCR Master Mix kit (Thermo Fisher
Scientific, Inc.) and specific primers for detecting
VEGF165 (forward, 5′-TTGCCTTGCTGCTCTACCTC-3′ and
reverse, 5′-AAATGCTTTCTCCGCTCTGA-3′) and the housekeeping β-actin
gene (forward, 5′-TCCTGTGGCATCCACGAAACT-3′ and reverse,
5′-GAAGCATTTGCGGTGGACGAT-3′). The following thermocycling
conditions were used for the qPCR reaction: Initial denaturation at
95°C for 10 min; followed by 40 cycles of 95°C for 15 sec, 60°C for
60 sec and 72°C for 30 sec. After amplification, a final melting
curve protocol was used to confirm the specificity of PCR products.
Data were analyzed using the 2−∆∆Cq method (41) and normalized to β-actin expression
levels.
ELISA detection of VEGF
Cultured R-50 cells were treated with 10 ng/ml
TGF-β2 at 37°C in the presence of either TA (1 or 10 µM), kinase
inhibitors (10 µM) or equivalent solvent control (0.1% MeOH for TA
or 0.1% DMSO for kinase inhibitor treatments). The supernatants
were collected after 48 h of incubation by centrifugation at 1,000
× g for 5 min at 4°C. Soluble VEGF release from RPE cells was
assessed using a commercially available ELISA kit (cat. no. DVE00;
R&D Systems, Inc.) according to the manufacturer's
instructions.
Gelatin zymography
To determine the level of gelatinolytic activities
of MMP-2 and MMP-9 in the conditioned media from the drug-treated
cells, non-reducing SDS-PAGE and gelatin zymography were performed
as previously described (42).
Briefly, 10 µl conditioned medium was mixed with SDS-PAGE sample
buffer without reducing agent and boiling. After electrophoresis
via an 8% polyacrylamide gel containing 2 mg/ml gelatin
(Sigma-Aldrich; Merck KGaA), the gel was soaked in 50 mM Tris-HCl
buffer (pH 7.4) containing 0.25% Triton X-100 for 30 min at room
temperature, followed by an incubation with a digestion buffer (50
mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 mM CaCl2, 2 µM
ZnSO4, 0.01% Brij-35 and 5 mM phenylmethylsulfonyl
fluoride) at 37°C overnight. After washing with distilled water,
the gelatin-digested gels were stained with 0.25% Coomassie
brilliant blue R-250 in 50% methanol and 10% acetic acid at room
temperature for 2 h, and destained with 10% methanol and 10% acetic
acid at room temperature for 20 min. The gelatinolytic activities
shown as clear bands in the gels were determined by analyzing the
band densities using ImageJ software.
Statistical analysis
All data are expressed as the mean ± SD from three
independent experiments. Statistical analysis was performed using
JMP Version 5.1.2 statistical software (SAS Institute, Inc.).
Unpaired Student's t-test or one-way ANOVA and Bonferroni post hoc
test were used for statistical analysis. P<0.05 was considered
to indicate a statistically significant difference among the groups
tested.
Results
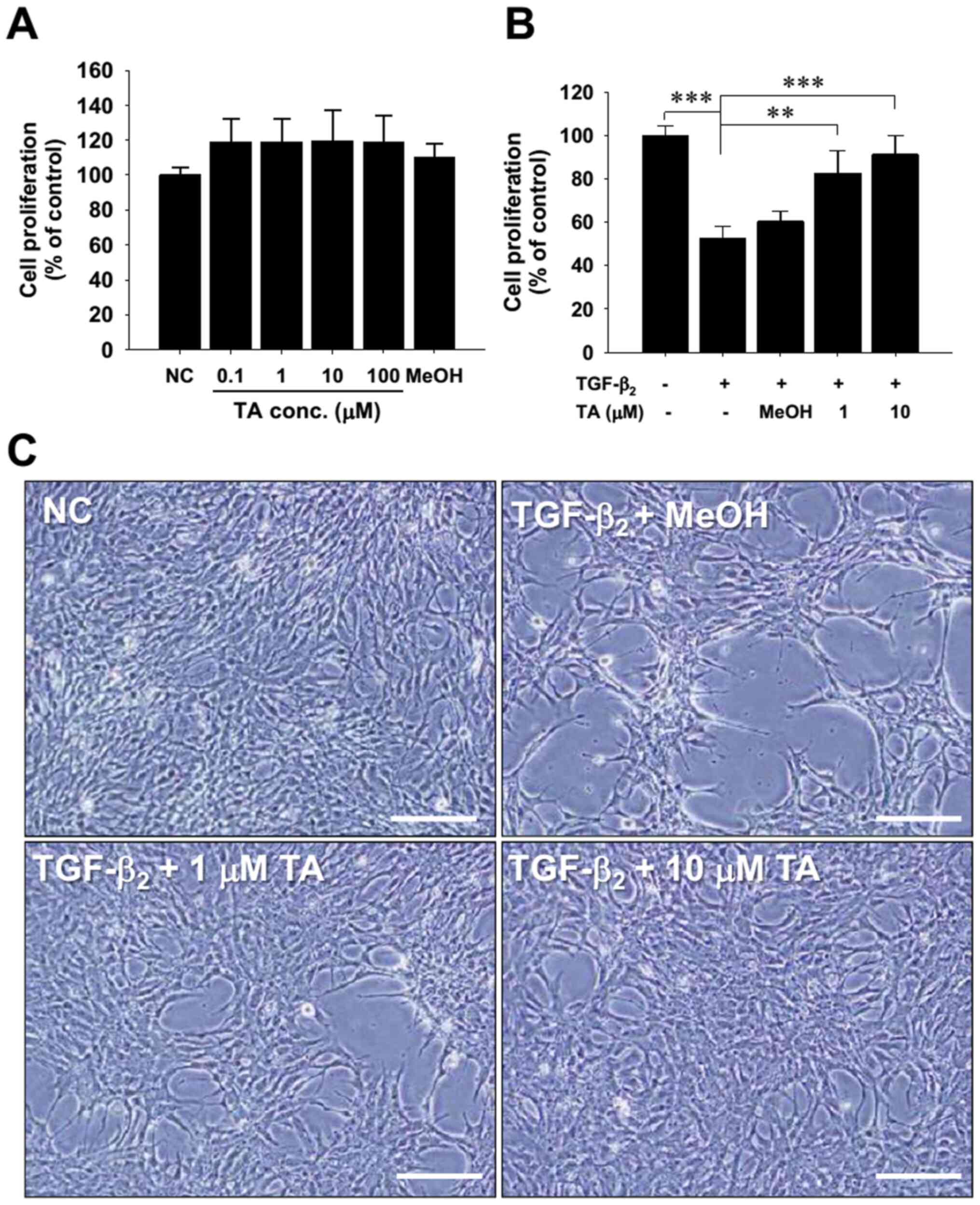
TA antagonizes the cytostatic effect
of TGF-β2 on RPE cells
Preservatives (such as MeOH) in commercial TA
solution, but not TA itself, were previously revealed to be
cytotoxic to RPE cells (43). To
confirm that TA did not have a cytotoxic effect on cultured human
RPE cells, various concentrations of TA (0–100 µM) were used to
evaluate cell viability using the MTS cell viability assay. After
24 h of treatment, no alteration in cell morphology was observed
(data not shown) and cell growth was not affected by treatment with
TA or MeOH, a solvent control (Fig.
1A). These results demonstrated that TA treatment exhibited no
cytotoxicity on cultured human RPE cells in vitro.
TGF-β has long been known to induce arrest of RPE
cell proliferation (44). Thus, the
present study further examined whether TA at doses not affecting
RPE cell proliferation had the ability to modulate TGF-β2-induced
growth arrest. TA was added to cultured cells in the presence of 10
ng/ml TGF-β2 and MTS viability assay was performed to evaluate the
viability of RPE cells. The results indicated that TA significantly
attenuated TGF-β2-induced RPE cell growth arrest (Fig. 1B). Microscopic morphological
observations supported the conclusion that the lower confluence of
TGF-β2-treated RPE cells was reversed by TA treatment (Fig. 1C). These findings strongly suggested
that TA may pharmacologically antagonize the biological effects of
TGF-β2.
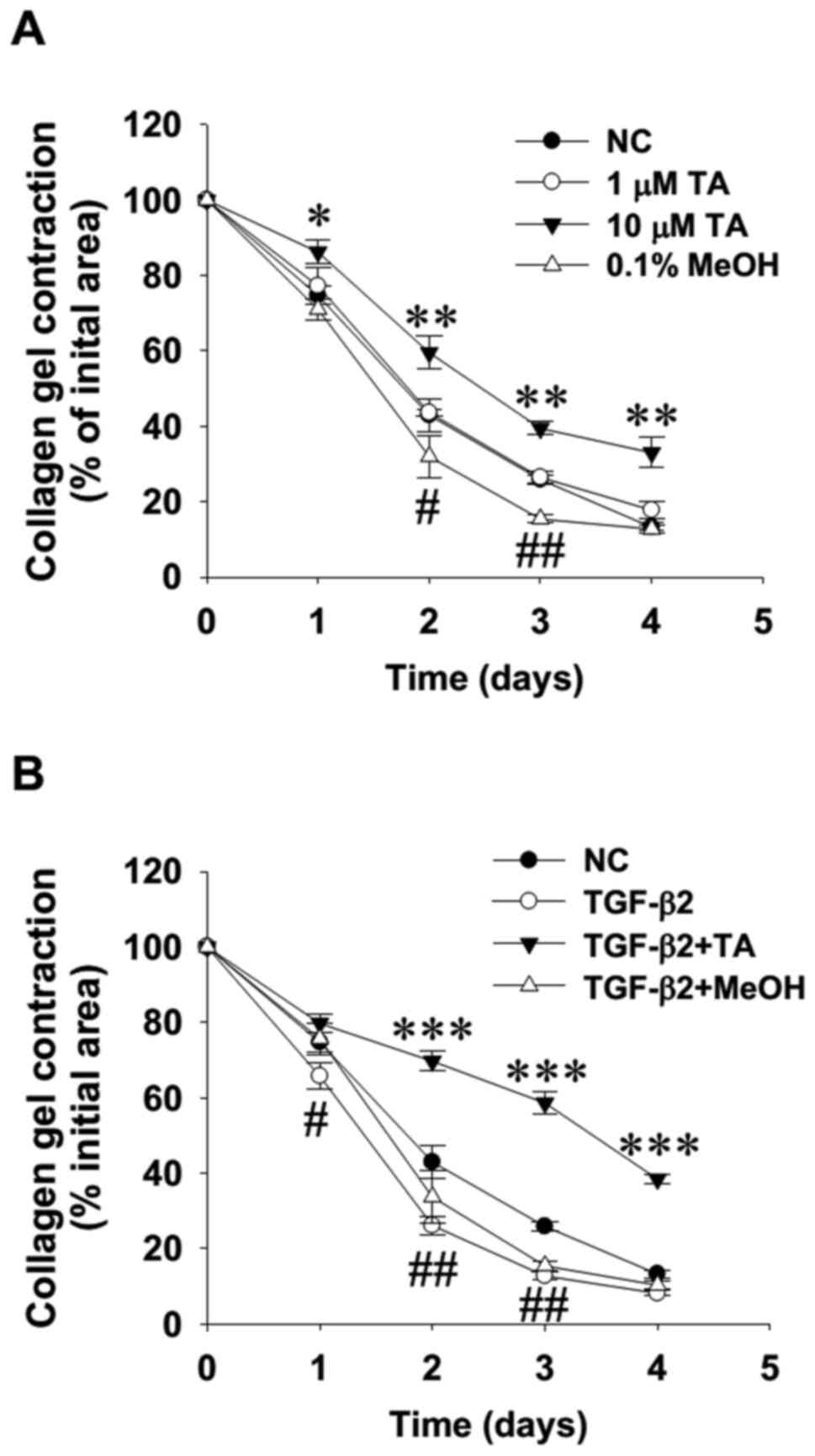
TA suppresses TGF-β2-enhanced RPE cell
contractility
TGF-β2 is known to upregulate α-SMA expression in
RPE cells (21), and has also been
shown to stimulate RPE cell-mediated collagen gel contraction
(45). Therefore, the present study
examined the direct effect and antagonism of TA on RPE cell
contractility using a 3D collagen gel contraction assay. The
results of direct gel-size measurement at 24-h intervals indicated
that the addition of TA at 10 µM in RPE cell-populated gels
significantly delayed gel contraction, in clear contrast to the
enhanced contractility in solvent control cells treated with MeOH
(Fig. 2A; representative images not
shown due to a lack of documentation). Moreover, the addition of TA
at 10 µM markedly suppressed the TGF-β2-induced increase in RPE
cell contractility (Fig. 2B),
suggesting that TA is able to suppress RPE cell contraction, most
likely by interfering with TGF-β2-driven signaling.
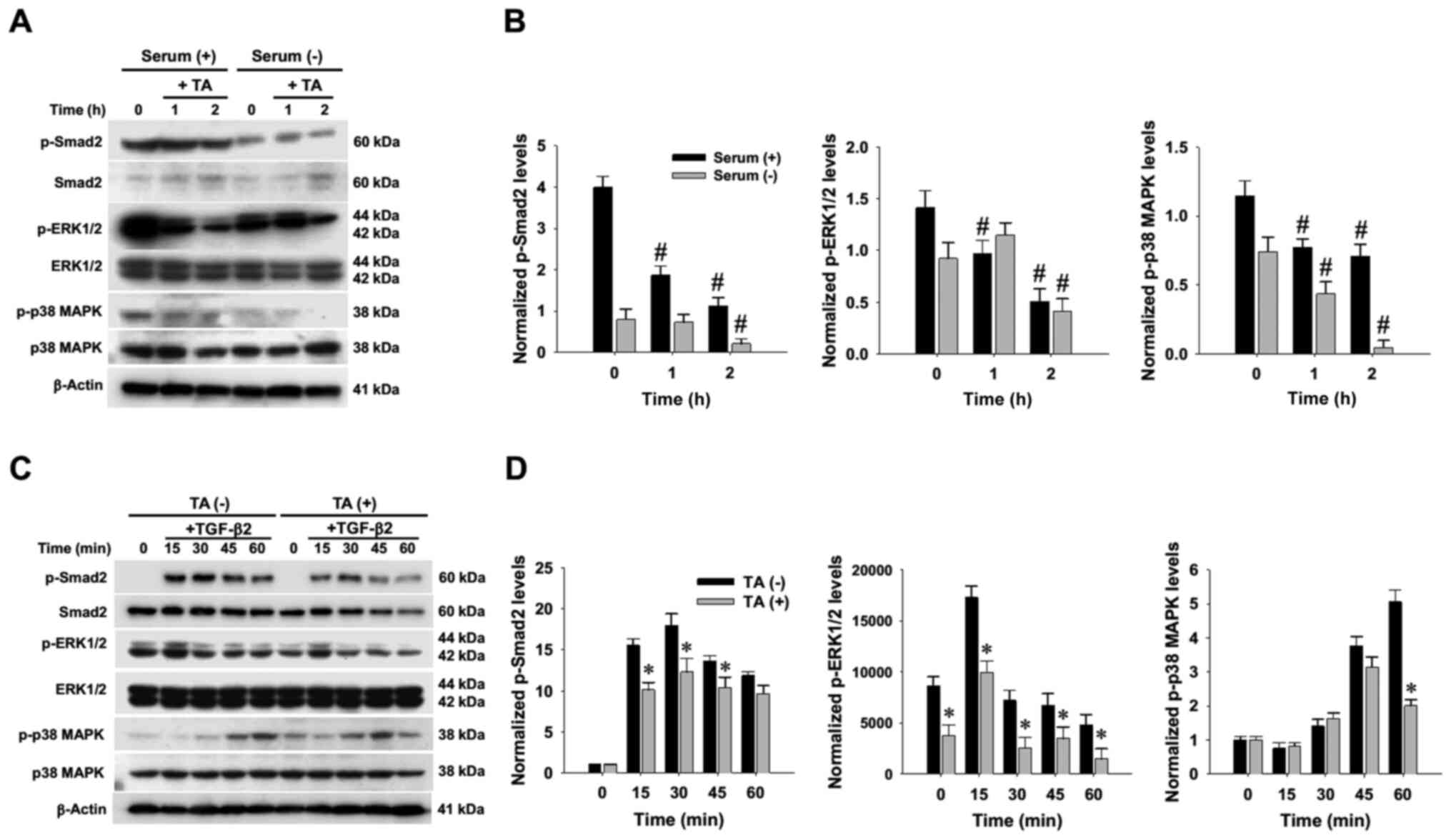
TA mitigates TGF-β2-induced Smad2,
ERK1/2 and p38 MAPK hyperphosphorylation
To characterize the pharmacological effect of TA on
the signaling machinery in RPE cells under serum-supplemented and
-deprived conditions, R-50 cells were grown in media with or
without serum supplementation, and then further treated with TA at
10 µM for different durations up to 2 h. Western blotting and
densitometry indicated that, under serum-deprived conditions, TA
exposure markedly reduced not only phosphorylation of Smad2, but
also that of non-Smad signaling mediators, including ERK1/2 and p38
MAPK (Fig. 3A and B). To determine
whether TA attenuated the signaling response of RPE cells to TGF-β2
stimulation, serum-starved R-50 cells were stimulated with TGF-β2
in the presence or absence of TA. The results clearly confirmed
that TA mitigated the TGF-β2-induced hyperphosphorylation of Smad2,
ERK1/2 and p38 MAPK proteins (Fig. 3C
and D).
TA suppresses TGF-β2-induced VEGF
expression in RPE cells
Since TGF-β signaling is of critical importance for
normal vascular development and physiology, and since aberrant
TGF-β signaling forms the basis for several disorders due to
neovascularization (46), the
present study aimed to determine whether TGF-β2 upregulated VEGF
production in cultured RPE cells and whether TA modulated the
TGF-β2-induced upregulation of VEGF. The measurement of VEGF
isoform transcripts by multiplex PCR indicated that TGF-β2 at 10
ng/ml significantly increased the expression levels of both
VEGF121 and VEGF165 transcripts in R50 cells.
By contrast, this upregulation was significantly reduced by TA
treatment in a dose-dependent manner (Fig. 4A and B). In parallel, RT-qPCR
analysis demonstrated that TA exerted a dose-dependent suppression
on the mRNA expression levels of VEGF165 in
TGF-β2-stimulated cells, and that, compared with the TA treatment
group, a less obvious suppression of VEGF165 was
observed in the cells pretreated with SB431542, an inhibitor of
TGF-β2 receptor kinase that abolishes Smad2 phosphorylation
(Fig. 4C). Consistent with the
changes detected in mRNA expression levels, TGF-β2 markedly
increased the release of soluble VEGF165 peptides from
R-50 cells after 48-h treatment, and the increase in VEGF peptide
release was significantly alleviated by TA treatment (Fig. 4D). As TGF-β2-induced p38 MAPK
signaling activation has been demonstrated to regulate VEGF
expression in RPE cells and critically contributes to its
pro-angiogenic effect (19), we did
not monitor the phosphorylation status of signaling mediators due
to kinetic fluctuations after long-term exposure. Consistent with
the previous observation, treatment of R-50 cells with kinase
inhibitors demonstrated that blockade of p38 MAPK activity by
SB203580 not only significantly suppressed TGF-β2-induced VEGF
expression, but also reduced constitutive VEGF expression.
Moreover, abolishment of Smad2 phosphorylation by SB431542 markedly
attenuated TGF-β2-induced VEGF overproduction (Fig. 4E). These findings suggested that
both TGF-β2 canonical and non-canonical signaling pathways may be
essential for VEGF expression in RPE cells.
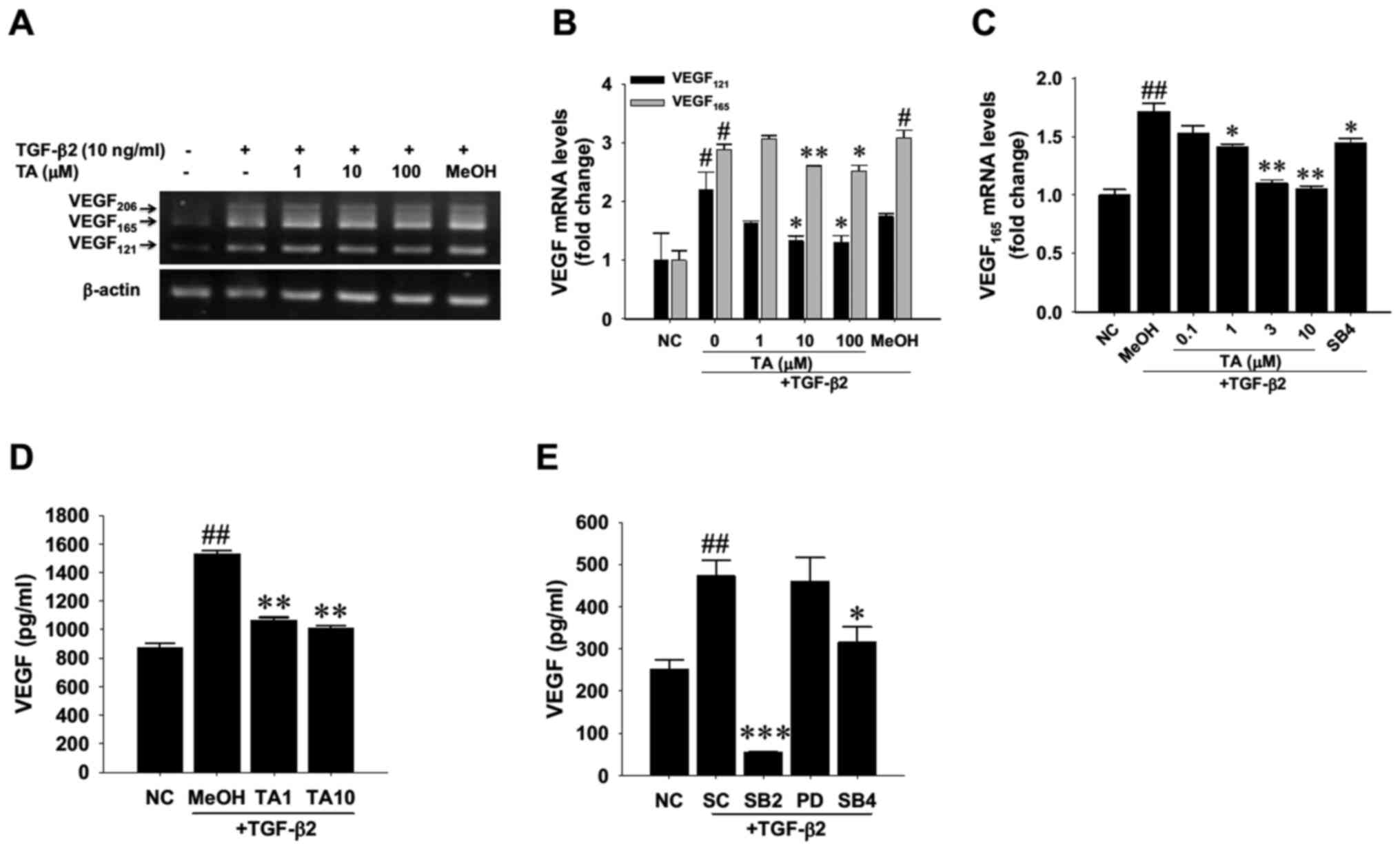 | Figure 4.Suppressive effect of TA on
TGF-β2-induced de novo synthesis of VEGF in cultured human retinal
pigment epithelial cells. (A) Human R-50 cells were treated with
TGF-β2 for 6 h in the presence of TA at the indicated doses or 0.1%
MeOH as a SC. Total RNA was extracted and a multiplex reverse
transcription-PCR was performed to simultaneously detect three
isoforms of VEGF transcripts. (A) Representative gel showing VEGF
isoform PCR products. (B) Densitometric analysis of the two major
VEGF121 and VEGF165 transcripts. The density
values of genes were normalized to those of the respective internal
control, β-actin. Data are presented as the mean ± SD. (C) Reverse
transcription-quantitative PCR analysis of the mRNA expression
levels of VEGF165. TGF-β receptor blocker SB4 at 10 µM
was used to block receptor kinase activity. (D) ELISA of R-50 cells
receiving 10 ng/ml TGF-β2 for 48 h in the presence of 1 µM (TA1) or
10 µM (TA10) TA. (E) ELISA of R-50 cells receiving 10 ng/ml TGF-β2
for 24 h in the presence of p38 mitogen-activated protein kinase
inhibitor SB2, MEK inhibitor PD, TGF-β receptor blocker SB4 at 10
µM or equivalent DMSO as a SC. Data are presented as the mean ± SD
from three independent experiments. #P<0.05,
##P<0.01 vs. NC; *P<0.05, **P<0.01,
***P<0.001 vs. MeOH or vs. DMSO SC. TA, triamcinolone acetonide;
TGF-β2, transforming growth factor-β2; VEGF, vascular endothelial
growth factor; NC, negative control; SC, solvent control; SB4,
SB431542; SB2, SB203580; PD, PD98059. |
TA modulates TGF-β2-induced
tissue-remodeling effects on RPE cells
Given that TGF-β2 induced a tissue-remodeling effect
on RPE cells (2), the present study
explored which signaling pathway participated in this remodeling
effect. After R-50 RPE cells were treated with 10 ng/ml TGF-β2 for
48 h with or without the addition of selective kinase inhibitors,
the protein lysates were subjected to western blotting (Fig. 5A). The results consistently revealed
that TGF-β2 treatment significantly upregulated the protein
expression levels of COL1A1, α-SMA and MMP-2 in the RPE cells.
Treatment with kinase inhibitors demonstrated that the activities
of both the MEK/ERK and p38 MAPK pathways were involved in
TGF-β2-induced upregulation of COL1A1 (Fig. 5B), whereas only the MEK/ERK pathway
appeared to be essential for TGF-β2-induced α-SMA upregulation
(Fig. 5C). Notably, neither pathway
was involved in MMP-9 and MMP-2 protein expression in cultured RPE
cells (Fig. 5D and E). Western
blotting and densitometric analysis were also used to determine
whether TA interfered with this remodeling effect; the results
revealed that TA exposure significantly abrogated the
TGF-β2-induced upregulation of COL1A1 (Fig. 5F and G), α-SMA (Fig. 5H), MMP-9 (Fig. 5I) and MMP-2 (Fig. 5J) in R-50 cells. A discrepancy in
the TGF-β2-modulated cellular MMP-9 abundance is likely due to the
fact that the solvent DMSO used for kinase inhibitor treatment
might exhibit an unexpected blunting effect (Fig. 5D). Notably, TGF-β2 receptor kinase
blockade significantly suppressed the protein expression levels of
these remodeling mediators.
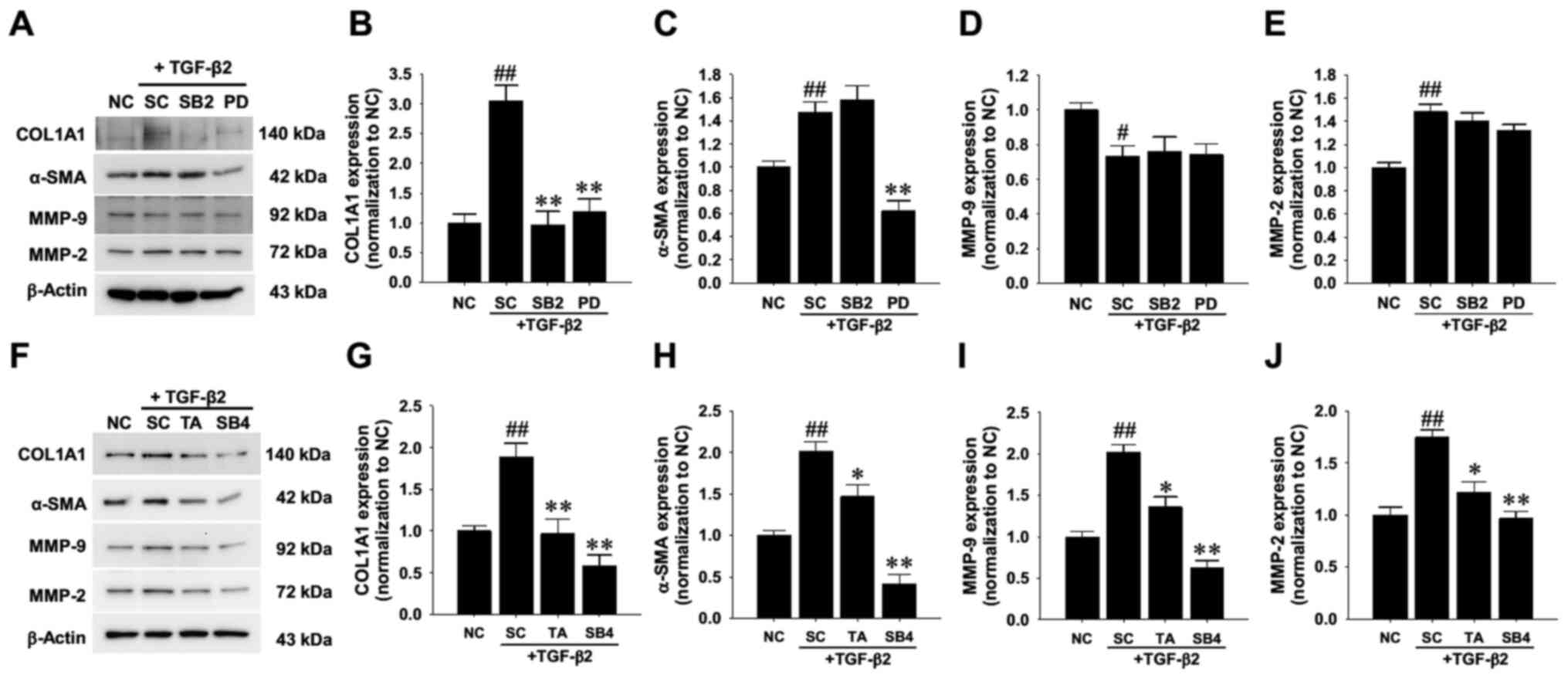 | Figure 5.Effect of TA treatment on
TGF-β2-driven ECM remodeling in cultured human retinal pigment
epithelial cells. (A) Western blot analysis of signaling pathway
proteins involved in TGF-β2-driven ECM remodeling. Human R-50 cells
were treated with 10 ng/ml TGF-β2 for 24 h in the presence of
either 10 µM p38 mitogen-activated protein kinase inhibitor SB2,
MEK inhibitor PD or equivalent DMSO as a SC. Bands were
densitometrically semi-quantified and the relative expression
levels of (B) COL1A1, (C) α-SMA, (D) MMP-9 and (E) MMP-2 were
normalized to internal actin expression levels. (F) R-50 cells were
treated with 10 ng/ml TGF-β2 for 48 h in the presence of 10 µM TA,
TGF-β receptor blocker SB4 or 0.1% methanol as a SC. The relative
expression levels of (G) COL1A1, (H) α-SMA, (I) MMP-9 and (J) MMP-2
were densitometrically semi-quantified and normalized to β-actin.
Fold changes are expressed as the mean ± SD from three independent
experiments. #P<0.05, ##P<0.01 vs. NC;
*P<0.05, **P<0.01 vs. SC. TA, triamcinolone acetonide;
TGF-β2, transforming growth factor-β2; α-SMA, α-smooth muscle
actin; MMP, matrix metalloproteinase; NC, negative control; SC,
solvent control; SB4, SB431542; SB2, SB203580; PD, PD98059; COL1A1,
collagen type I α1 chain. |
To confirm the modulatory effect of TA, gelatin
zymography was used to detect the gelatinolytic activities of both
MMPs. The results revealed that, despite no detectable MMP-2
gelatinolytic activity in the conditioned media (data not shown),
TA at a higher concentration (such as 10 µM) significantly
attenuated the TGF-β2-induced increase in MMP-9 gelatinolytic
activity (Fig. 6A). Furthermore,
pretreatment with PD98059 markedly reduced the TGF-β2-induced MMP-9
activation (Fig. 6B), suggesting
the involvement of the MEK1/MEK2 pathway in TGF-β2-induced MMP-9
upregulation.
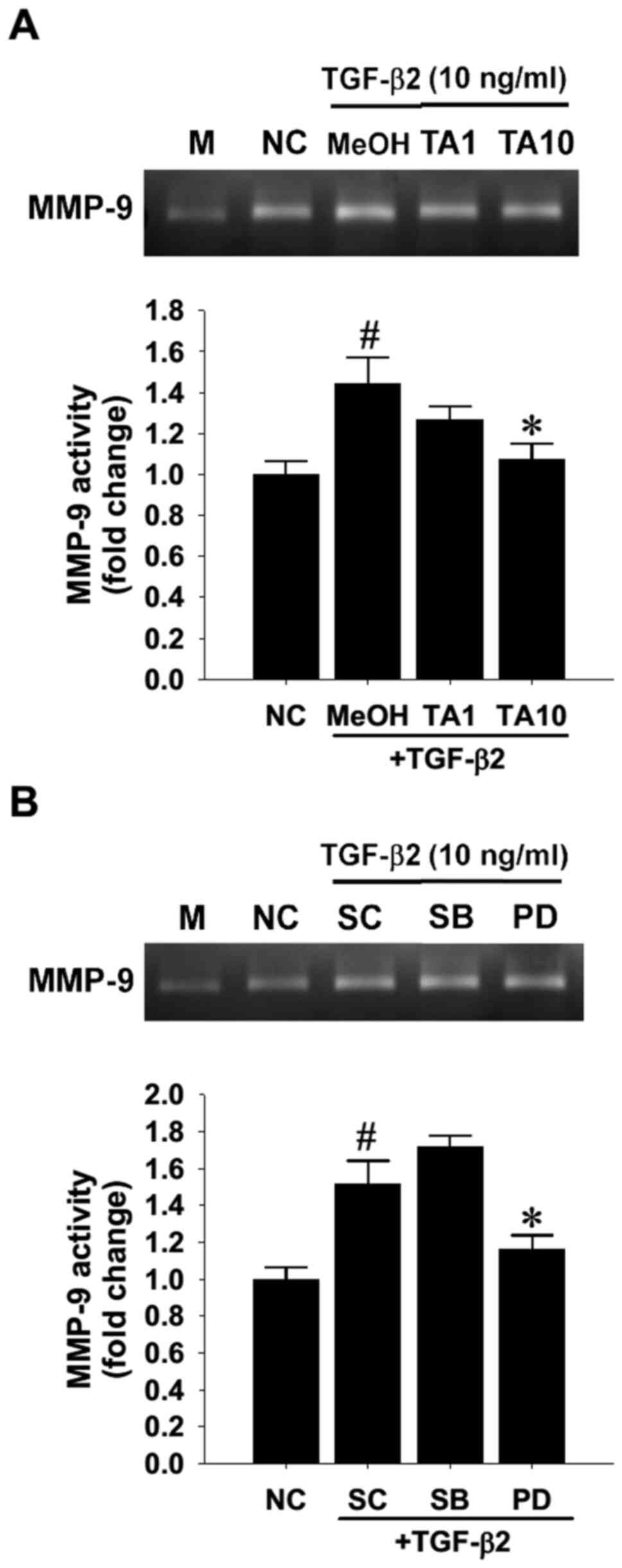 | Figure 6.Effect of TA treatment on
TGF-β2-driven MMP-9 gelatinolytic activity in cultured human
retinal pigment epithelial cells. (A) Human R-50 cells were treated
with TGF-β2 for 48 h in the presence of 1 µM (TA1) or 10 µM (TA10)
TA or 0.1% MeOH as a SC. The supernatants were collected and
subjected to gelatin zymography detection of MMP-9 activity. Bands
were densitometrically semi-quantified and the scanned densities
were subtracted from the background levels in the M group. (B) R50
cells were treated with TGF-β2 for 24 h in the presence of either
10 µM p38 mitogen-activated protein kinase inhibitor SB, MEK
inhibitor PD or equivalent DMSO as a SC. Fold changes presented as
the mean ± SD were obtained from three independent experiments.
#P<0.05 vs. NC; *P<0.05 vs. MeOH or SC. TA,
triamcinolone acetonide; TGF-β2, transforming growth factor-β2;
MMP, matrix metalloproteinase; NC, negative control; SC, solvent
control; M, medium; MeOH, methanol; SB, SB203580; PD, PD98059. |
Discussion
The present study demonstrated that the ERK and p38
MAPK signaling pathways may have a crucial role in the therapeutic
mechanism of TA for the treatment of TGF-β2-associated intraocular
angiogenesis and tissue remodeling, which are known to be
associated with retinal disorders, such as PVR and PDR. TA was
shown to markedly antagonize TGF-β2-induced RPE cell growth arrest
and contractility, without exhibiting prominent cytotoxicity. In
the context of TGF-β2-induced activation, TA significantly
mitigated TGF-β2-elicited Smad2, ERK1/2, and p38 MAPK
hyperphosphorylation, whereas inhibition of TGF-β receptor- and p38
MAPK-mediated signaling activities attenuated TGF-β2-induced
upregulation of VEGF mRNA and peptide release. Furthermore,
TGF-β2-induced ERK1/2 and p38 MAPK activation was involved in COLA1
overproduction, while only the ERK1/2 signaling pathway was
observed to participate in the α-SMA upregulation induced by
TGF-β2. Similar to the blunting effects of Smad2 inhibition, TA
effectively mitigated TGF-β2-induced upregulation of COLA1, α-SMA,
MMP-2 and MMP-9 expression and that of MMP-9 gelatinolytic activity
in R-50 RPE cells. Taken together, TA treatment may prevent
pathogenic signaling activation that contributes to angiogenesis
and tissue remodeling.
It is well known that the sequential process of
fibroblast-like transformation and differentiation of RPE cells is
a pathological feature in the genesis of PVR and associated
vitreoretinal diseases (47). The
contraction of fibrotic and scarring membranes may lead to retinal
detachment, increasing the risk of vision deterioration or
blindness (48). TGF-β2 is
considered a key component in the development of abnormal fibrosis
in the pathological environment. Various studies have confirmed
TGF-β2 as a dominant factor in the development of age-related
macular degeneration and macular fibrosis or atrophy (49,50).
The present results demonstrated that TA not only enhanced cell
viability in TGF-β2-treated RPE cells, but also suppressed
contractility in collagen gels. Furthermore, TA altered the
TGF-β2-driven ECM remodeling process in cultured RPE cells, as the
protein expression levels of COL1A1, α-SMA, MMP-2 and MMP-9 were
revealed to be partially attenuated. Clinically, these results
indicated that TA may cause substantial changes in the composition,
structure and mechanics of fibrotic vitreoretinal conditions, such
as PVR and PDR.
Although TGF-β2 is widely considered a major
constituent of the fibrotic transformation process (50), to the best of our knowledge, the
specificity of the effects of TA on TGF-β2-driven molecular
behaviors remain elusive. Interleukin-2 interacts with TGF-β2,
which may lead to the excessive expression of ECM proteins, further
aggravating the fibrosis process (49). In the present study, TA was revealed
to significantly attenuate TGF-β2-elicited Smad2, ERK1/2 and p38
MAPK phosphorylation, effectively blocking the signaling cascade.
According to the present results, PD98059 may prevent
TGF-β2-induced COL1A1 and α-SMA upregulation, while SB203580 also
inhibit the increase in COL1A1 expression, suggesting the
involvement of both the ERK and p38 MAPK signaling pathways.
VEGF is known to be a potent vasopermeability
factor, which is involved in the development of diabetic macular
edema and PDR (51). The
development of diabetic macular edema is multifactorial, and
intravitreal injections of VEGF inhibitors and steroids may help to
reduce central retinal thickness and therefore improve visual
acuity (52). Treatment with VEGF
inhibitors requires multiple repeated injections, especially in the
first and second years of therapy (53). Compared with the currently available
anti-VEGF agents, steroids are advantageous because of their high
potency and selectivity, along with a lower release rate, providing
a high enough intraocular concentration to achieve therapeutic
efficacy in the macular region (54). In a study of pseudophakic eyes at
baseline, visual acuity improvement was observed in the combined TA
and prompt or deferred laser photocoagulation group compared with
in the VEGF-antagonizing ranibizumab group (55,56).
At present, TA is used for the treatment of various neovascular and
inflammatory ocular disorders with the risk of intraocular pressure
elevation, cataract formation and infection (52). The present study confirmed that TA
blocked the increase in VEGF transcription caused by TGF-β2 and
that TA attenuated the TGF-β2-stimulated p38 activation. These
results indicated that the attenuated effect was associated with
the ablation of p38 signaling activity.
In conclusion, the findings of the present study may
have important and novel clinical implications. The results
revealed that the pharmacological inhibition via the use of TA may
have a role in the inhibition of RPE cell transformation and
differentiation through contractility and ECM regulation,
suppressing the associated fibrosis in vitreoretinal diseases. In
addition, it was hypothesized that both the ERK and p38 MAPK
pathways may be applied as strategic targets in utilizing TA to
prevent retinal diseases associated with angiogenesis and tissue
remodeling.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the
National Science Council, the Executive Yuan, Taiwan (grant no.
NSC95-2314-B-037-042), Kaohsiung Medical University Hospital (grant
no. KMUH96-6R11) and E-Da Hospital (grant no. EDAHT105045).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
WCW and YHK obtained the funds, conceived the study
and supervised all experiments. CCH, YCC and YHK confirm the
authenticity of all the raw data. CCH and YCC interpreted the data
and drafted the manuscript. YTH, PHC and MCH performed the
experiments. YTH and PHC performed all statistical analyses. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Rat tails were collected from adult male
Sprague-Dawley rats under the approval and supervision of the
Institutional Animal Care and Use Committee at E-Da Hospital
(approval no. IACUC-102016; Kaohsiung, Taiwan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nomura M, Yamagishi S, Harada S, Hayashi
Y, Yamashima T, Yamashita J and Yamamoto H: Possible participation
of autocrine and paracrine vascular endothelial growth factors in
hypoxia-induced proliferation of endothelial cells and pericytes. J
Biol Chem. 270:28316–28324. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schlingemann RO: Role of growth factors
and the wound healing response in age-related macular degeneration.
Graefes Arch Clin Exp Ophthalmol. 242:91–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kijlstra A, La Heij E and Hendrikse F:
Immunological factors in the pathogenesis and treatment of
age-related macular degeneration. Ocul Immunol Inflamm. 13:3–11.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Henkind P: Ocular neovascularization. The
Krill memorial lecture. Am J Ophthalmol. 85:287–301. 1978.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Campochiaro PA: Retinal and choroidal
neovascularization. J Cell Physiol. 184:301–310. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pournaras CJ, Miller JW, Gragoudas ES,
Husain D, Munoz JL, Tolentino MJ, Kuroki M and Adamis AP: Systemic
hyperoxia decreases vascular endothelial growth factor gene
expression in ischemic primate retina. Arch Ophthalmol.
115:1553–1558. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aiello LP, Avery RL, Arrigg PG, Keyt BA,
Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, et
al: Vascular endothelial growth factor in ocular fluid of patients
with diabetic retinopathy and other retinal disorders. N Engl J
Med. 331:1480–1487. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Enzmann V, Hollborn M, Wiedemann P and
Kohen L: Minor influence of the immunosuppressive cytokines IL-10
and TGF-beta on the proliferation and apoptosis of human retinal
pigment epithelial (RPE) cells in vitro. Ocul Immunol Inflamm.
9:259–266. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kishi H, Kuroda E, Mishima HK and
Yamashita U: Role of TGF-beta in the retinoic acid-induced
inhibition of proliferation and melanin synthesis in chick retinal
pigment epithelial cells in vitro. Cell Biol Int. 25:1125–1129.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Esser P, Heimann K, Bartz-schmidt KU,
Fontana A, Schraermeyer U, Thumann G and Weller M: Apoptosis in
proliferative vitreoretinal disorders: Possible involvement of
TGF-beta-induced RPE cell apoptosis. Exp Eye Res. 65:365–378. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mitsuhiro MR, Eguchi S and Yamashita H:
Regulation mechanisms of retinal pigment epithelial cell migration
by the TGF-beta superfamily. Acta Ophthalmol Scand. 81:630–638.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eichler W, Friedrichs U, Thies A, Tratz C
and Wiedemann P: Modulation of matrix metalloproteinase and TIMP-1
expression by cytokines in human RPE cells. Invest Ophthalmol Vis
Sci. 43:2767–2773. 2002.PubMed/NCBI
|
|
13
|
Itoh Y, Kimoto K, Imaizumi M and Nakatsuka
K: Inhibition of RhoA/Rho-kinase pathway suppresses the expression
of type I collagen induced by TGF-beta2 in human retinal pigment
epithelial cells. Exp Eye Res. 84:464–472. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirase K, Ikeda T, Sotozono C, Nishida K,
Sawa H and Kinoshita S: Transforming growth factor beta2 in the
vitreous in proliferative diabetic retinopathy. Arch Ophthalmol.
116:738–741. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu AL, Fuchshofer R, Kook D, Kampik A,
Bloemendal H and Welge-Lüssen U: Subtoxic oxidative stress induces
senescence in retinal pigment epithelial cells via TGF-beta
release. Invest Ophthalmol Vis Sci. 50:926–935. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SC, Seong GJ, Kim SH and Kwon OW:
Synthesized TGF-beta s in RPE regulates cellular proliferation.
Korean J Ophthalmol. 13:16–24. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matsumoto M, Yoshimura N and Honda Y:
Increased production of transforming growth factor-beta 2 from
cultured human retinal pigment epithelial cells by
photocoagulation. Invest Ophthalmol Vis Sci. 35:4245–4252.
1994.PubMed/NCBI
|
|
18
|
Nagineni CN, Samuel W, Nagineni S,
Pardhasaradhi K, Wiggert B, Detrick B and Hooks JJ: Transforming
growth factor-beta induces expression of vascular endothelial
growth factor in human retinal pigment epithelial cells:
Involvement of mitogen-activated protein kinases. J Cell Physiol.
197:453–462. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bian ZM, Elner SG and Elner VM: Regulation
of VEGF mRNA expression and protein secretion by TGF-beta2 in human
retinal pigment epithelial cells. Exp Eye Res. 84:812–822. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bian ZM, Elner SG and Elner VM:
Thrombin-induced VEGF expression in human retinal pigment
epithelial cells. Invest Ophthalmol Vis Sci. 48:2738–2746. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kurosaka D, Muraki Y, Inoue M and Katsura
H: TGF-beta 2 increases alpha-smooth muscle actin expression in
bovine retinal pigment epithelial cells. Curr Eye Res.
15:1144–1147. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang YE: Non-Smad pathways in TGF-beta
signaling. Cell Res. 19:128–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mu Y, Gudey SK and Landström M: Non-Smad
signaling pathways. Cell Tissue Res. 347:11–20. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ando N, Sen HA, Berkowitz BA, Wilson CA
and de Juan E Jr: Localization and quantitation of blood-retinal
barrier breakdown in experimental proliferative vitreoretinopathy.
Arch Ophthalmol. 112:117–122. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Penfold PL, Wen L, Madigan MC, Gillies MC,
King NJ and Provis JM: Triamcinolone acetonide modulates
permeability and intercellular adhesion molecule-1 (ICAM-1)
expression of the ECV304 cell line: Implications for macular
degeneration. Clin Exp Immunol. 121:458–465. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Penfold PL, Wen L, Madigan MC, King NJ and
Provis JM: Modulation of permeability and adhesion molecule
expression by human choroidal endothelial cells. Invest Ophthalmol
Vis Sci. 43:3125–3130. 2002.PubMed/NCBI
|
|
27
|
Khairallah M, Zeghidi H, Ladjimi A, Yahia
SB, Attia S, Zaouali S and Messaoud R: Primary intravitreal
triamcinolone acetonide for diabetic massive macular hard exudates.
Retina. 25:835–839. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Jonas JB, Kreissig I and Degenring R:
Intravitreal triamcinolone acetonide for treatment of intraocular
proliferative, exudative, and neovascular diseases. Prog Retin Eye
Res. 24:587–611. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Witmer AN, Vrensen GF, Van Noorden CJ and
Schlingemann RO: Vascular endothelial growth factors and
angiogenesis in eye disease. Prog Retin Eye Res. 22:1–29. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matsuda S, Gomi F, Oshima Y, Tohyama M and
Tano Y: Vascular endothelial growth factor reduced and connective
tissue growth factor induced by triamcinolone in ARPE19 cells under
oxidative stress. Invest Ophthalmol Vis Sci. 46:1062–1068. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Edelman JL, Lutz D and Castro MR:
Corticosteroids inhibit VEGF-induced vascular leakage in a rabbit
model of blood-retinal and blood-aqueous barrier breakdown. Exp Eye
Res. 80:249–258. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hollborn M, Stathopoulos C, Steffen A,
Wiedemann P, Kohen L and Bringmann A: Positive feedback regulation
between MMP-9 and VEGF in human RPE cells. Invest Ophthalmol Vis
Sci. 48:4360–4367. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dot C, Behar-Cohen F, BenEzra D, Doat M,
Jonet L, May F and Jeanny JC: Influence of triamcinolone
intravitreal injection on retinochoroidal healing processes. Exp
Eye Res. 84:1081–1089. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wu WC, Kao YH and Hu DN: Relationship
between outcome of proliferative vitreoretinopathy and results of
tissue culture of excised preretinal membranes. Kaohsiung J Med
Sci. 16:614–619. 2000.PubMed/NCBI
|
|
35
|
Gentile RC, Hu DN and McCormick SA: Cell
culture of retinal membranes from proliferative vitreoretinopathy
and age-related macular degeneration. Invest Ophthalmol Vis Sci.
37:S3871996.
|
|
36
|
Wu WC, Kao YH, Hu PS and Chen JH:
Geldanamycin, a HSP90 inhibitor, attenuates the hypoxia-induced
vascular endothelial growth factor expression in retinal pigment
epithelium cells in vitro. Exp Eye Res. 85:721–731. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Committee for the Update of the Guide for
the Care and Use of Laboratory Animals, . Guide for the Care and
Use of Laboratory Animals. The National Academies Press (US);
Washington, DC: 1996
|
|
38
|
Tsai MS, Lee PH, Sun CK, Chiu TC, Lin YC,
Chang IW, Chen PH and Kao YH: Nerve growth factor upregulates
sirtuin 1 expression in cholestasis: A potential therapeutic
target. Exp Mol Med. 50:e4262018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chang YC, Kao YH, Hu DN, Tsai LY and Wu
WC: All-trans retinoic acid remodels extracellular matrix and
suppresses laminin-enhanced contractility of cultured human retinal
pigment epithelial cells. Exp Eye Res. 88:900–909. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chang YC, Lin CW, Chang YS, Chen PH, Li
CY, Wu WC and Kao YH: Monounsaturated oleic acid modulates
autophagy flux and upregulates angiogenic factor production in
human retinal pigment epithelial ARPE-19 cells. Life Sci.
259:1183912020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kao YH, Jawan B, Goto S, Pan MC, Lin YC,
Sun CK, Hsu LW, Tai MH, Cheng YF, Nakano T, et al: Serum factors
potentiate hypoxia-induced hepatotoxicity in vitro through
increasing transforming growth factor-beta1 activation and release.
Cytokine. 47:11–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chang YS, Wu CL, Tseng SH, Kuo PY and
Tseng SY: Cytotoxicity of triamcinolone acetonide on human retinal
pigment epithelial cells. Invest Ophthalmol Vis Sci. 48:2792–2798.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee J, Choi JH and Joo CK: TGF-β1
regulates cell fate during epithelial-mesenchymal transition by
upregulating survivin. Cell Death Dis. 4:e7142013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Carrington L, McLeod D and Boulton M:
IL-10 and antibodies to TGF-beta2 and PDGF inhibit RPE-mediated
retinal contraction. Invest Ophthalmol Vis Sci. 41:1210–1216.
2000.PubMed/NCBI
|
|
46
|
Wang X, Ma W, Han S, Meng Z, Zhao L, Yin
Y, Wang Y and Li J: TGF-β participates choroid neovascularization
through Smad2/3-VEGF/TNF-α signaling in mice with Laser-induced wet
age-related macular degeneration. Sci Rep. 7:96722017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang IH, Lee JJ, Wu PC, Kuo HK, Kuo YH and
Huang HM: Oxidative stress enhanced the transforming growth
factor-β2-induced epithelial-mesenchymal transition through
chemokine ligand 1 on ARPE-19 cell. Sci Rep. 10:40002020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim SJ, Kim YS, Kim JH, Jang HY, Ly DD,
Das R and Park KS: Activation of ERK1/2-mTORC1-NOX4 mediates
TGF-β1-induced epithelial-mesenchymal transition and fibrosis in
retinal pigment epithelial cells. Biochem Biophys Res Commun.
529:747–752. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jing R, Qi T, Wen C, Yue J, Wang G, Pei C
and Ma B: Interleukin-2 induces extracellular matrix synthesis and
TGF-β2 expression in retinal pigment epithelial cells. Dev Growth
Differ. 61:410–418. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang K, Li H, Sun R, Liu C, Luo Y, Fu S
and Ying Y: Emerging roles of transforming growth factor β
signaling in wet age-related macular degeneration. Acta Biochim
Biophys Sin (Shanghai). 51:1–8. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Simó R, Carrasco E, García-Ramírez M and
Hernández C: Angiogenic and antiangiogenic factors in proliferative
diabetic retinopathy. Curr Diabetes Rev. 2:71–98. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Haritoglou C, Maier M, Neubauer AS and
Augustin AJ: Current concepts of pharmacotherapy of diabetic
macular edema. Expert Opin Pharmacother. 21:467–475. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wells JA, Glassman AR, Ayala AR, Jampol
LM, Aiello LP, Antoszyk AN, Arnold-Bush B, Baker CW, Bressler NM,
Browning DJ, et al Diabetic Retinopathy Clinical Research Network,
: Aflibercept, bevacizumab, or ranibizumab for diabetic macular
edema. N Engl J Med. 372:1193–1203. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Edelman JL: Differentiating intraocular
glucocorticoids. Ophthalmologica. 224 (Suppl 1):25–30. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Elman MJ, Aiello LP, Beck RW, Bressler NM,
Bressler SB, Edwards AR, Ferris FL III, Friedman SM, Glassman AR,
Miller KM, et al Diabetic Retinopathy Clinical Research Network, :
Randomized trial evaluating ranibizumab plus prompt or deferred
laser or triamcinolone plus prompt laser for diabetic macular
edema. Ophthalmology. 117:1064–1077.e35. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Elman MJ, Ayala A, Bressler NM, Browning
D, Flaxel CJ, Glassman AR, Jampol LM and Stone TW; Diabetic
Retinopathy Clinical Research Network, : Intravitreal Ranibizumab
for diabetic macular edema with prompt versus deferred laser
treatment: 5-year randomized trial results. Ophthalmology.
122:375–381. 2015. View Article : Google Scholar : PubMed/NCBI
|