Introduction
Malignant melanoma (MM) is one of the tumors with
the highest mortality rate due to its strong invasiveness and drug
resistance. In the United States, the mortality of MM has decreased
upon use of new targeted therapies, but its incidence is rising
(1,2). Melanoma is considered as an
immunogenic tumor and its occurrence and development are affected
by the interaction of various immune cells in the tumor
microenvironment (TME) (3). There
are several immune cells in the TME, including CD8+ T
cells, NK cells and tumor-associated macrophages (TAMs), with TAMs
being the largest proportion (4).
Macrophages can be polarized to classically
activated macrophages (M1-type) or alternatively activated
macrophages (M2-type). M1-type macrophages have a strong antigen
presenting function that secretes a large amount of inflammatory
cytokines. Furthermore, they participate in the Th1-type immune
response, exert an anti-tumor immune function and inhibit tumor
development (5). TAMs are thought
to more closely resemble M2-type macrophages, which produce higher
levels of anti-inflammatory cytokines and angiogenic factors. These
factors inhibit the anti-tumor immune response and help tumor cells
escape from the immune system and reactivate epithelial-mesenchymal
transition (EMT) to promote tumor metastasis (6), resulting in the promotion of tumor
growth and metastasis. Therefore, altering the polarity of TAMs and
reprogramming them toward M1-type has become a potential target for
tumor immunotherapy.
Endostatin is one of the potential endogenous
angiogenesis inhibitors, which can effectively inhibit tumor
angiogenesis (7), but there are few
studies on its immunology. In China, the endostatin recombinant
protein, Endostar, has been listed as the preferred anti-tumor
angiogenesis-targeted drug for MM first-line treatment. Previously
we have found that an overexpression endostatin plasmid
(pEndostatin plasmid) induced M1-type polarization of mouse
RAW264.7 macrophages and inhibited the growth of breast cancer
(8). Therefore, it was hypothesized
that M1-type macrophages induced by pEndostatin plasmid would also
have an inhibitory effect on melanoma cells. To confirm this
hypothesis, normal RAW264.7 cells or M1-type macrophages induced by
pEndostatin plasmid were co-cultured with melanoma B16 cells and
the changes in the biological functions of the co-cultured B16
cells were observed.
Materials and methods
Cell lines and cell culture
The mouse melanoma cell line, B16 and the mouse
macrophage cell line, RAW264.7, were purchased from the Cell Bank
of the Chinese Academy of Sciences. B16 cells and RAW264.7 cells
were cultured in DMEM (HyClone; Cytiva) with 10% FBS (HyClone;
Cytiva) and penicillin-streptomycin (1,000 µg/ml). The two cell
lines were cultured at 37°C in a cell incubator with 5%
CO2.
Plasmid
To overexpress endostatin, the pcDNA3.1-ssEndostatin
(pEndostatin) plasmid was constructed by the Laboratory of
Pathophysiology, Basic Medical College of Jilin University, as
previously described (9). The
plasmid was transfected into RAW264.7 cells using
Lipofectamine® 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
RAW264.7 cells were collected 48 h after transfection and then
co-cultured with B16 cells.
Cell co-culture
B16 cells were inoculated into the lower chambers of
Transwell 6-well plates (Corning Inc.) at a density of
5×105 cells/well. RAW264.7 cells or M1-type macrophages
induced by pEndostatin plasmid were seeded in the upper chambers of
the Transwell plates at a density of 5×104 cells/well.
These cells were co-cultured for 48 h at 37°C in a 5%
CO2 cell culture incubator.
MTT assay
Co-cultured B16 cells were seeded in a 96-well plate
at a density of 1×104 cells/100 µl/well, with five replicate wells
in each group and continued to be cultured for 24, 48 and 72 h.
Then 20 µl MTT (5 mg/ml; Sangon Biotech Co., Ltd.) was added to
each well for 4 h in the incubator. The supernatant was removed and
150 µl/well of DMSO (Sangon Biotech Co., Ltd.) was added. The light
absorption of each well was measured at 490 nm by an ELISA reader
and the survival rate of B16 cells was calculated.
Colony-forming cell assay
Co-cultured B16 cells were seeded in a 6-well plate
at a density of 500 cells/well. After 7 days, the number of
colonies (containing ≥50 cells) in each well was counted by crystal
violet staining (Sangon Biotech Co., Ltd.). Colony-forming rate (%)
= (total number of colonies/total number of seeded cells) ×100.
Scratch wound assay
B16 cells co-cultured with different macrophages for
48 h were seeded in a 24-well plate and cultured overnight. When
the confluence of cells was 90–100%, a scratch was applied with a
200-µl pipette tip. The wells were then rinsed twice with PBS
(Hyclone, GE Healthcare Life Sciences) to wash away suspended cells
and debris and the remaining cells were further cultured in DMEM
with 2% FBS, which was used to maintain cell survival but prevent
cell proliferation. The change in scratch width at 0, 24 and 48 h
was monitored under a BX41-PHD-P11 light microscope (Olympus
Corporation). Data analyses were conducted using ImageJ (v1.8.0
National Institutes of Health). Transport rate (%) = (0 h scratch
width - scratch width after cultivation)/0 h scratch width
×100.
Transwell assay
Co-cultured B16 cells were inoculated
(1×103) with normal cell culture medium into the upper
chambers of Transwell plates (pore size, 8 µm), and Matrix gel
(Corning Inc.), which was used for precoating at 37°C for 2 h, and
normal cell culture medium was added to the lower chamber. After
culturing for 24 h at 37°C, the cells on the upper chamber membrane
were gently wiped with a cotton swab and then stained with crystal
violet at room temperature for 20 min. The number of cells passing
through the Transwell membrane was counted using a BX41-PHD-P11
light microscope (Olympus Corporation). The relative invasion rate
of B16 cells in the M1 group was calculated relative to the control
group. Relative invasion rate = Number of migrating cells in the M1
group/number of migrating cells in the control group.
Flow cytometry analysis
Co-cultured B16 cells were stained with an Annexin V
Apoptosis kit (Beckman Coulter, Inc.) according to the
manufacturer's protocol. The apoptotic ratio (late apoptosis) of
each group of cells was determined by an Epics-XL-MCL flow
cytometer (Beckman Coulter, Inc.). The cell cycle distribution of
the co-cultured B16 cells was determined by single staining with
100 µg/ml propidium iodide (Beckman Coulter, Inc.) at 37°C for 30
min. The fixative used for the cell cycle assay was 75% ice
ethanol, overnight at −20°C.
Western blot analysis
Co-cultured B16 cells were harvested and lysed with
RIPA lysis buffer (Takara Bio Inc.) and the supernatant was
collected. The protein concentration was measured by a Bio-Rad
protein concentration quantification method (Bio-Rad Laboratories
Inc.). The protein samples (30 µg) were electrophoresed on 12%
SDS-PAGE gels and transferred to PVDF membranes (Invitrogen; Thermo
Fisher Scientific, Inc.), blocked with 5% skimmed milk (EMD
Millipore) at room temperature for 1 h, incubated with primary
antibodies overnight at 4°C and then incubated with secondary
antibodies at room temperature for 1 h. Protein bands were
visualized with an ECL kit (GE Healthcare). The images were
captured using a Syngene Bio Imaging System (Synoptics). The
following primary antibodies were used: mouse monoclonal
anti-matrix metalloproteinase (MMP)-2 (1:200; cat. no. sc-13594),
mouse monoclonal anti-MMP-9 (1:200; cat. no. sc-21733; both Santa
Cruz Biotechnology, Inc.); mouse monoclonal anti-cleaved Caspase-8
(1:1,000; cat. no. 9429), rabbit polyclonal anti-cleaved Caspase-3
(1:1,000; cat. no. 9664), rabbit polyclonal anti-proliferating cell
nuclear antigen [(PCNA); 1:1,000; cat. no. 13110; all Cell
Signaling Technology, Inc.]; rabbit polyclonal anti-Bax (1:500;
cat. no. BS2538), rabbit polyclonal anti-Bcl-2 (1:500; cat. no.
BS1511), rabbit polyclonal anti-β-actin (1:5000; cat. no. AP0060)
and rabbit polyclonal anti-GAPDH (1:10,000; cat. no. AP0063; all
Bioworld Technology, Inc.). The secondary antibodies used were
anti-rabbit IgG (1:3,000; cat. no. sc-2357) and anti-mouse IgG
(1:2,000, sc-2005; both Santa Cruz Biotechnology, Inc.). Protein
expression was semi-quantified using ImageJ software (v1.8.0;
National Institutes of Health).
Statistical analysis
The results are expressed as mean ± standard
deviation Comparisons between two groups were analyzed using an
unpaired Student's t-test. Comparisons among more than two groups
were assessed using one-way ANOVA followed by Tukey's post hoc test
in pair-wise repetitive comparisons. Data analyses were conducted
using SPSS 11.0 (SPSS, Inc.). Graphs were prepared using GraphPad
Prism 8 (GraphPad Software, Inc.). Each experiment was repeated
three independent times. P<0.05 was considered to indicate a
statistically significant difference.
Results
The present study established a co-culture system
with Transwell chambers. B16 cells were co-cultured with normal
RAW264.7 cells (control group) or M1-type macrophages (M1 group)
induced by pEndostatin plasmid for 48 h and then collected to
examine their biological functions.
Endostatin-induced M1-type macrophages
inhibit the proliferation of B16 cells
B16 cells co-cultured with normal RAW264.7 cells or
M1-type macrophages for 48 h were collected and seeded in 96-well
plates. The proliferation of the co-cultured B16 cells was
determined by the MTT assay after 24, 48 and 72 h. The
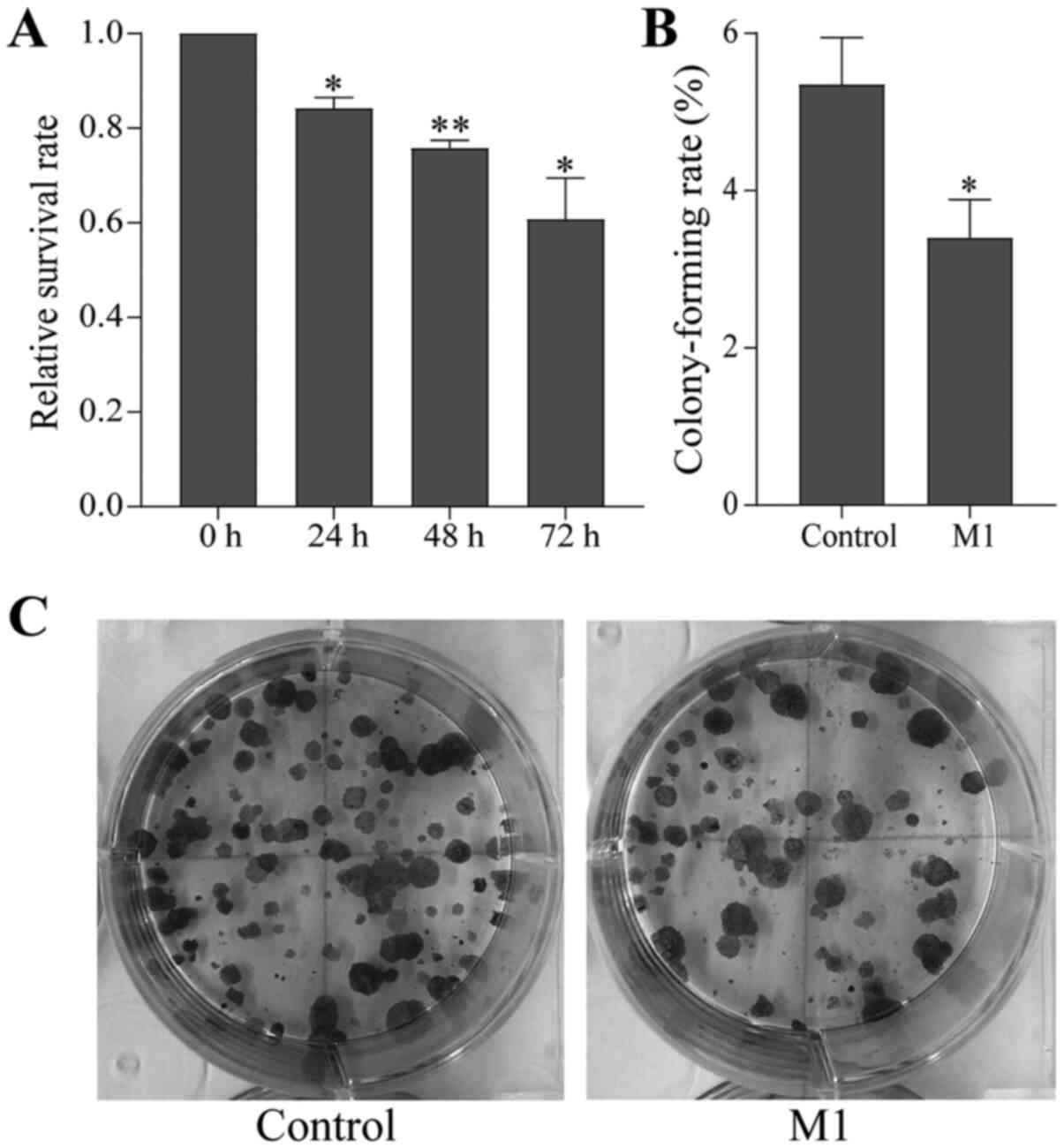
proliferative ability of B16 cells in the M1 group was
significantly weakened and the survival rate decreased in a
time-dependent manner compared with the control group (Fig. 1A).
Furthermore, the proliferation ability of individual
cells was tested by the colony-forming cell assay. It was found
that the colony formation ability of B16 cells in the M1 group was
also reduced compared with the control group (Fig. 1B and C). This result was consistent
with the MTT assay, indicating that M1-type macrophages induced by
the pEndostatin plasmid inhibited the proliferation of B16
cells.
Endostatin-induced M1-type macrophages
inhibit the migration and invasion of B16 cells
To examine the effect of M1-type macrophages induced
by the pEndostatin plasmid on the migration and invasion of B16
cells, scratch and Transwell assays were used. After 24 and 48 h of
culture, the number of B16 cells in the M1 group passing through
the Transwell membrane was significantly smaller than that in the
control group (Fig. 2A and B).
Additionally, the scratch width of the B16 cells in the M1 group
was significantly larger than that in the control group, i.e., the
migration distance was reduced (Fig. 2C
and D).
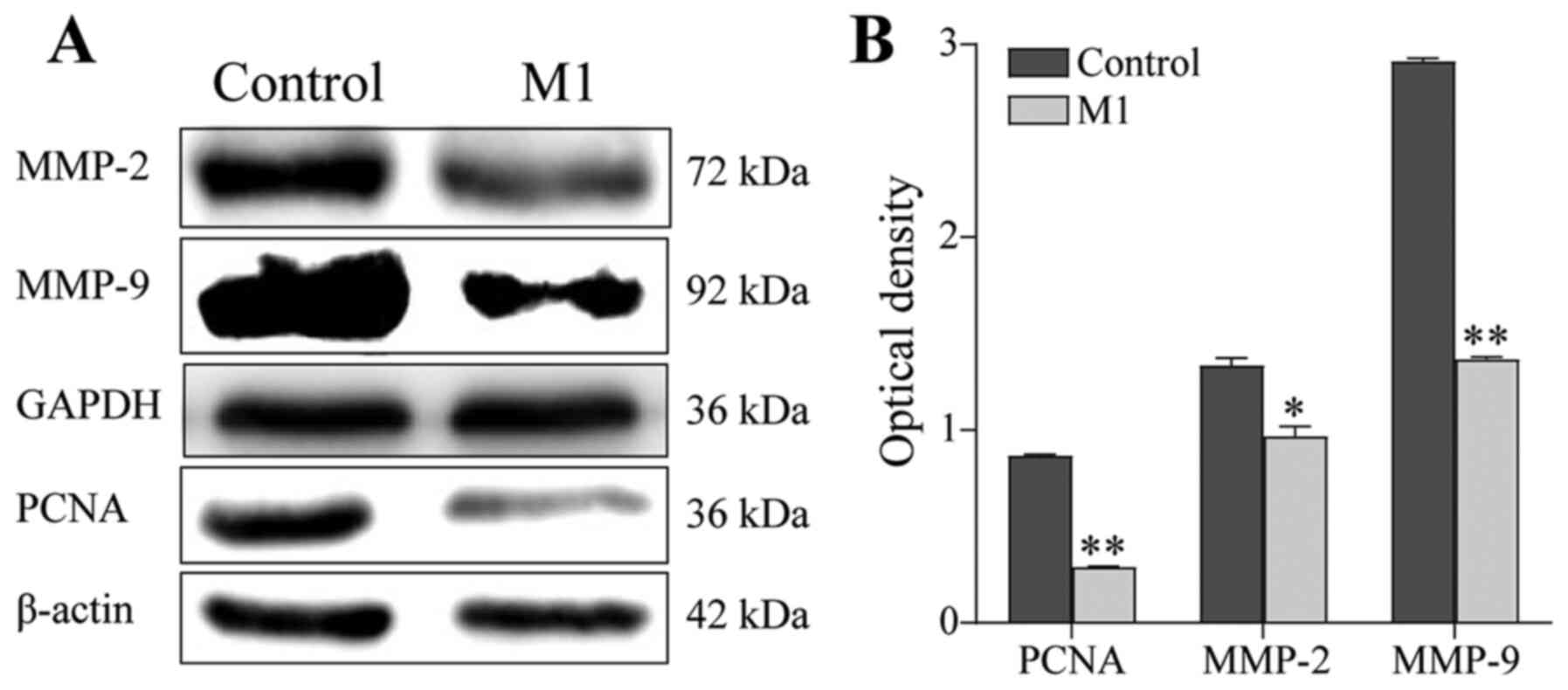
Next, the expression of MMPs was examined by western
blotting. Compared with the control group, the expression of PCNA,
MMP-2 and MMP-9 in B16 cells of the M1 group was downregulated
(P<0.05; Fig. 3). These results
suggested that M1-type macrophages induced by the pEndostatin
plasmid inhibited the migration and invasion of B16 cells by
downregulating the expression of PCNA, MMP-2 and MMP-9.
Endostatin-induced M1-type macrophages
promote apoptosis of B16 cells
To further elucidate the mechanism by which
endostatin-induced M1-type macrophages inhibit the proliferation of
B16 cells, the changes in the cell cycle distribution and apoptosis
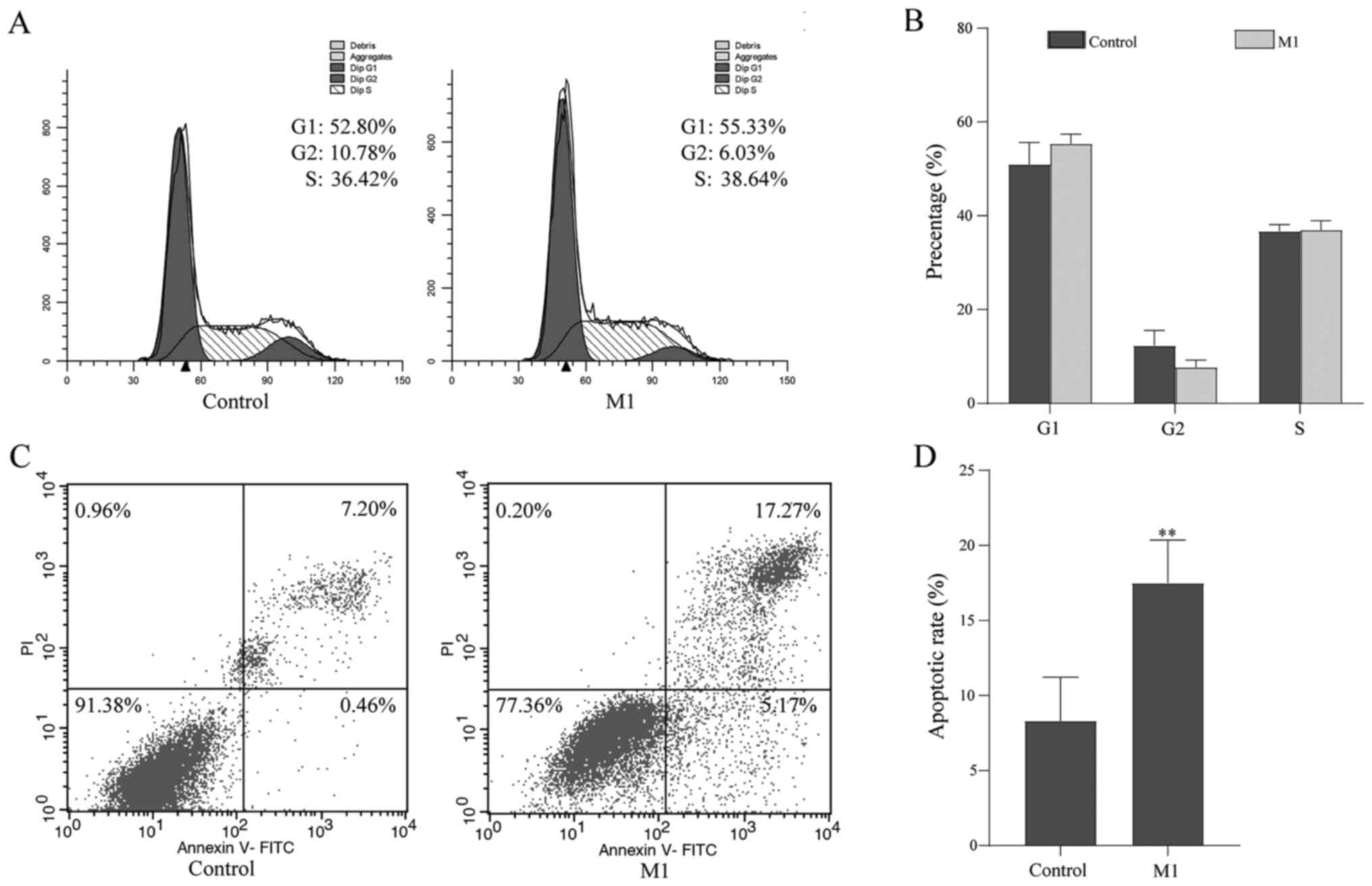
of B16 cells were examined by flow cytometry. The results showed
that the cell cycle distribution of B16 cells in the M1 group was
similar to that of the control group (Fig. 4A and B), but the proportion of
apoptotic cells was significantly increased (control group:
8.30±2.91% vs. M1 group: 17.48±2.89%; P<0.05; Fig. 4C and D).
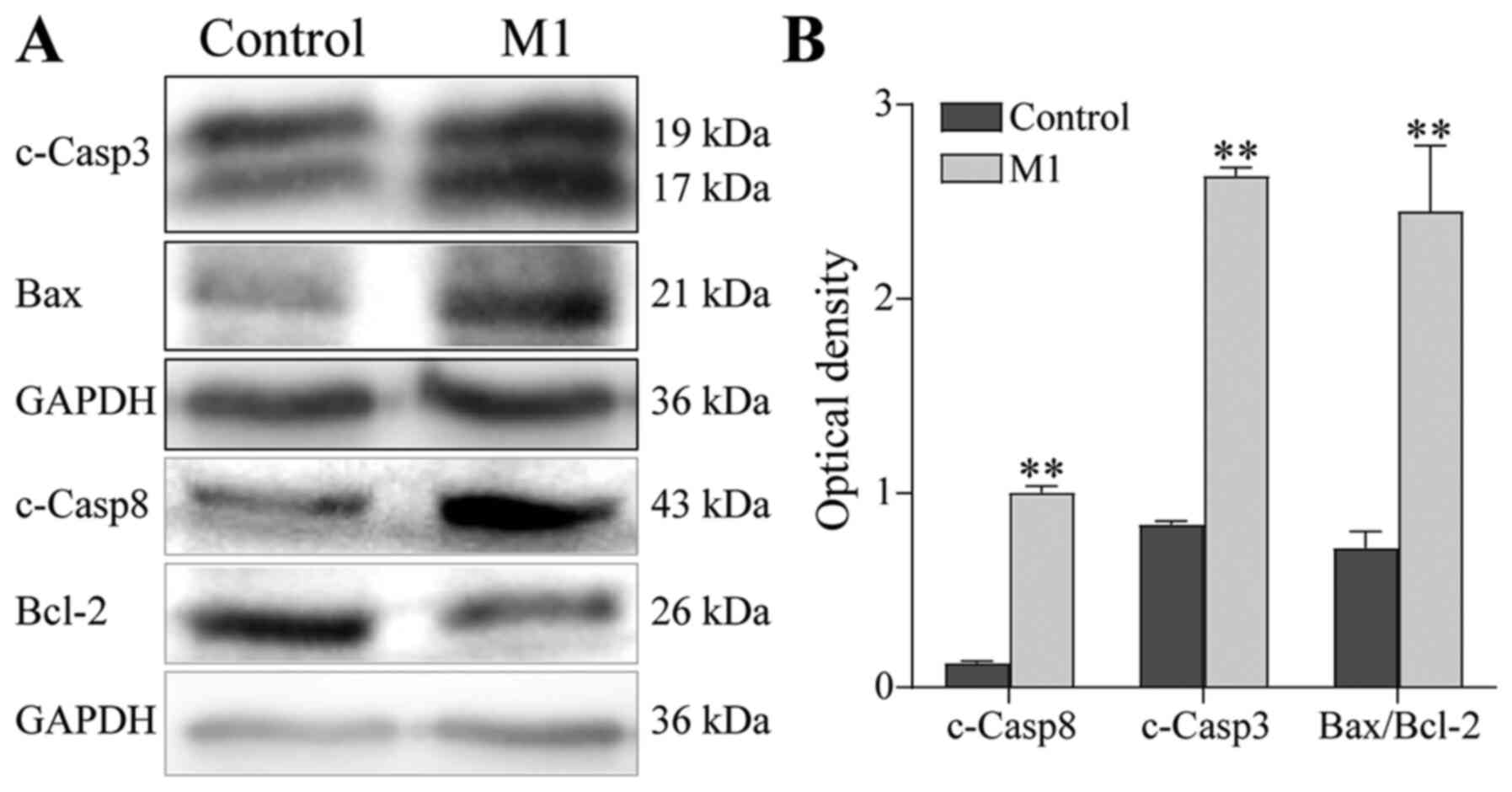
Using western blotting to determine the expression
of apoptosis-related proteins, it was found that the protein
expression of cleaved Caspase-3 and cleaved Caspase-8 was
significantly upregulated and the Bax/Bcl-2 ratio was significantly
increased in B16 cells of the M1 group, compared with the control
group (P<0.01; Fig. 5). These
results suggest that the M1-type macrophages induced by the
pEndostatin plasmid inhibited the proliferation of B16 cells by
promoting apoptosis, though they did not affect the cell cycle of
B16 cells.
Discussion
MM has become a major public health threat owing to
its high metastasis rate, drug resistance and poor prognosis
(10). Although much effort has
been put into the development of MM treatments in recent years,
effective treatment remains limited. MM is an immunogenic tumor and
its biological behavior is affected by host immune cells and
inflammatory cells in the TME (3).
The immunosuppressive TME composed of regulatory T cells, bone
marrow-derived suppressor cells and TAMs promote the immune escape
of melanoma (11,12), hence a number of researchers are
beginning to investigate to MM immunotherapy. As the main immune
cells in the TME of MM, TAMs play an important role in the
development and metastasis of MM (10). Therefore, reprogramming the polarity
of TAMs to inhibit the development of MM has become of great
interest to researchers. Melanoma cells release different
macrophage chemokines (including monocyte chemoattractant protein 1
and VEGF-C) to attract macrophages to the tumor site and induce
their activation into M2-type TAMs to exert pro-tumor effects
(10,13). In turn, M2-type TAMs secrete a
number of effector molecules (including IFN-γ, cyclooxygenase 2 and
IL-10) to promote the growth and metastasis of MM, thus forming a
vicious circle (10,14). Additionally, melanoma exosomes
directly induce macrophage polarization to an M1/M2 ‘mixed’
phenotype, which has multiple tumor-promoting functions (15). The immunosuppressive function of
TAMs derives from their kinase activity and secretion of
anti-inflammatory cytokines, including IL-10 and TGF-β, which have
an inhibitory effect on tumoricidal lymphocytes. TGF-β1 secreted by
M2-type TAMs inhibits the release of nitric oxide from M1
macrophages (16) and inhibits M1
polarization through matrix remodeling to increase the survival of
melanoma (17). Adrenomedullin
produced by TAMs induces nitric oxide production in endothelial
cells through a paracrine mechanism and polarizes macrophages
toward M2-type through an autocrine mechanism, thereby increasing
angiogenesis and tumor growth in melanoma (18). TAMs also directly activate the VEGF
pathway and IL-6 secreted by TAMs promotes the generation of tumor
microvessels by activating STAT3 to induce hypoxia-inducible factor
1-mediated VEGF-A transcription (19). Additionally, TAMs secrete MMPs
(MMP-2, MMP-9 and MMP-7) to regulate the decomposition and
reconstruction of the cell matrix, provide support for the
formation of blood vessels and promote the migration of endothelial
cells (20). Therefore, inducing
the polarity transformation of M2-type TAMs to M1-type with tumor
suppressor function in the TME can be used as one of the targets
for MM treatment. The present study found that M1 type macrophages
induced by the pEndostatin plasmid inhibited the proliferation and
migration of melanoma, indicating that it is feasible to treat
melanoma by changing the polarity of TAMs in the TME; however, the
specific mechanism requires further research.
Endostatin is an endogenous angiogenesis inhibitor
isolated from the conditioned medium of hemangioendothelioma cell
line (21). It suppresses tumor
development by inhibiting the proliferation, migration and invasion
of endothelial cells. In 2005, the State Food and Drug
Administration of China approved Endostar as an anti-vascular drug
for the clinical treatment of non-small cell lung cancer. As there
are almost no side effects and drug resistance rarely occurs,
Endostar is more suitable for patients with long-term antitumor
therapy and is more effective in suppressing tumor recurrence and
metastasis than other anti-vascular targeted therapies (22).
It has been demonstrated that Endostar inhibits the
growth of melanoma by inhibiting angiogenesis and the MAPK pathway
mediated by basic fibroblast growth factor (23). A combined treatment with Endostar
and chemotherapeutics significantly inhibits the proliferation of
melanoma cells and promotes their apoptosis and prolonged the
survival time of tumor-bearing mice (24). Clinical trials found that treatment
with Endostar + dacarbazine increased the one-year survival rate
(22.5 vs. 49.7%) and the two-year survival rate (14.3 vs. 22.2%) of
melanoma patients compared with the placebo + dacarbazine group and
reduced the risk of death in patients with mucosal melanoma (93%)
and acral melanoma (62%) (25,26).
Endostar combined with dacarbazine and cisplatin was effective for
MM without genetic mutations and the patient tolerated the
treatment well (27). Additionally,
endostatin may be used as a prognostic biomarker of immunotherapy
with ipilimumab for MM patients (28). These studies suggest that endostatin
can inhibit microvascular growth in melanoma and improve drug
resistance.
Although there are a number of studies about
endostatin inhibiting MM tumor microvessels, it remains unclear
whether it can inhibit MM growth by regulating the polarity of
macrophages. Some studies have found that endostatin enhanced the
anti-tumor immune response and increased the infiltration of
cytotoxic T lymphocytes to the TME and polarized the macrophages
that reached the tumor tissue to M1-type, resulting in an increased
proportion of M1-type TAMs and reduced proportion of M2-type TAMs,
thereby reversing the immunosuppressive environment in the TME
(8,29–31).
From the perspective of the change of macrophage polarity, the
present study briefly explored that the inhibition of endostatin on
B16 cells may be partly due to its immunological function, but the
specific mechanism requires further study. More in-depth studies on
its related pathways (including angiogenesis factor and oxidative
stress markers)are being conducted. The present study found that
M1-type macrophages induced by the pEndostatin plasmid inhibited
the proliferation of B16 cells by promoting apoptosis and inhibited
their migration and invasion ability by downregulating the
expression of MMP-2 and MMP-9. These results suggested that in
addition to directly inhibiting the formation of microvessels in
MM, the therapeutic effect of endostatin on MM was also related to
inducing a polarity change in TAMs.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural
Science Foundation of Zhejiang Province (grant nos. LQ20H160009 and
LY18H280004), the Natural Science Foundation of Ningbo (grant no.
2019A610318) and K.C. Wong Magna Fund in Ningbo University.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JGu participated in project design and technical
guidance. HG participated in project design and was a major
contributor to writing the manuscript and data analysis. LZ
performed the investigation of B16 cell proliferation, migration
and apoptosis. JGuo performed the western blotting. XH performed
the investigation of B16 cell invasion. XH and HG confirmed the
authenticity of all the raw data. All authors reviewed and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable
Patient consent for publication
Not applicable
Competing interests
The authors declare that they have no competing
interests
Glossary
Abbreviations
Abbreviations:
|
FBS
|
fetal bovine serum
|
|
M1
|
classical activation macrophage
|
|
M2
|
alternative activation macrophage
|
|
MM
|
malignant melanoma
|
|
MMP
|
Matrix metalloproteinase
|
|
MTT
|
3-(4, 5-dimethylthiazol-2-yl)-2,
5-dimethyltetrazolium bromide
|
|
PCNA
|
proliferating cell nuclear antigen
|
|
pEndostatin
|
the plasmid of over-express
endostatin
|
|
TME
|
tumor microenvironment
|
|
TAMs
|
tumor-associated macrophages
|
References
|
1
|
Henley SJ, Ward EM, Scott S, Ma J,
Anderson RN, Firth AU, Thomas CC, Islami F, Weir HK, Lewis DR, et
al: Annual report to the nation on the status of cancer, part I:
National cancer statistics. Cancer. 126:2225–2249. 2020. View Article : Google Scholar
|
|
2
|
Uprety D, Bista A, Chennamadhavuni A,
Niroula A, Jafri SIM, Smith A and Arjyal L: Survival trends among
patients with metastatic melanoma in the pretargeted and the
post-targeted era: A US population-based study. Melanoma Res.
28:56–60. 2018. View Article : Google Scholar
|
|
3
|
Ladányi A: Prognostic and predictive
significance of immune cells infiltrating cutaneous melanoma.
Pigment Cell Melanoma Res. 28:490–500. 2015. View Article : Google Scholar
|
|
4
|
Yang L and Zhang Y: Tumor-associated
macrophages: From basic research to clinical application. J Hematol
Oncol. 10:582017. View Article : Google Scholar
|
|
5
|
Mantovani A, Sica A, Sozzani S, Allavena
P, Vecchi A and Locati M: The chemokine system in diverse forms of
macrophage activation and polarization. Trends Immunol. 25:677–686.
2004. View Article : Google Scholar
|
|
6
|
Fuxe J and Karlsson MC: TGF-β-induced
epithelial-mesenchymal transition: A link between cancer and
inflammation. Semin Cancer Biol. 22:455–461. 2012. View Article : Google Scholar
|
|
7
|
Walia A, Yang JF, Huang YH, Rosenblatt MI,
Chang JH and Azar DT: Endostatin's emerging roles in angiogenesis,
lymphangiogenesis, disease, and clinical applications. Biochim
Biophys Acta. 1850:2422–2438. 2015. View Article : Google Scholar
|
|
8
|
Guo H, Liu Y, Gu J, Wang Y, Liu L, Zhang P
and Li Y: Endostatin inhibits the growth and migration of 4T1 mouse
breast cancer cells by skewing macrophage polarity toward the M1
phenotype. Cancer Immunol Immunother. 65:677–688. 2016. View Article : Google Scholar
|
|
9
|
Jia H, Li Y, Zhao T, Li X, Hu J, Yin D,
Guo B, Kopecko DJ, Zhao X, Zhang L, et al: Antitumor effects of
Stat3-siRNA and endostatin combined therapies, delivered by
attenuated Salmonella, on orthotopically implanted hepatocarcinoma.
Cancer Immunol Immunother. 61:1977–1987. 2012. View Article : Google Scholar
|
|
10
|
Wang H, Yang L, Wang D, Zhang Q and Zhang
L: Pro-tumor activities of macrophages in the progression of
melanoma. Hum Vaccin Immunother. 13:1556–1562. 2017. View Article : Google Scholar
|
|
11
|
Umansky V and Sevko A: Tumor
microenvironment and myeloid-derived suppressor cells. Cancer
Microenviron. 6:169–177. 2013. View Article : Google Scholar
|
|
12
|
Ruffell B and Coussens LM: Macrophages and
therapeutic resistance in cancer. Cancer Cell. 27:462–472. 2015.
View Article : Google Scholar
|
|
13
|
Skobe M, Hamberg LM, Hawighorst T,
Schirner M, Wolf GL, Alitalo K and Detmar M: Concurrent induction
of lymphangiogenesis, angiogenesis, and macrophage recruitment by
vascular endothelial growth factor-C in melanoma. Am J Pathol.
159:893–903. 2001. View Article : Google Scholar
|
|
14
|
Wang H and Zhang L, Yang L, Liu C, Zhang Q
and Zhang L: Targeting macrophage anti-tumor activity to suppress
melanoma progression. Oncotarget. 8:18486–18496. 2017. View Article : Google Scholar
|
|
15
|
Bardi GT, Smith MA and Hood JL: Melanoma
exosomes promote mixed M1 and M2 macrophage polarization. Cytokine.
105:63–72. 2018. View Article : Google Scholar
|
|
16
|
Vodovotz Y, Bogdan C, Paik J, Xie QW and
Nathan C: Mechanisms of suppression of macrophage nitric oxide
release by transforming growth factor beta. J Exp Med. 178:605–613.
1993. View Article : Google Scholar
|
|
17
|
Berking C, Takemoto R, Schaider H, Showe
L, Satyamoorthy K, Robbins P and Herlyn M: Transforming growth
factor-beta1 increases survival of human melanoma through stroma
remodeling. Cancer Res. 61:8306–8316. 2001.
|
|
18
|
Chen P, Huang Y, Bong R, Ding Y, Song N,
Wang X, Song X and Luo Y: Tumor-associated macrophages promote
angiogenesis and melanoma growth via adrenomedullin in a paracrine
and autocrine manner. Clin Cancer Res. 17:7230–7239. 2011.
View Article : Google Scholar
|
|
19
|
Xu Q, Briggs J, Park S, Niu G, Kortylewski
M, Zhang S, Gritsko T, Turkson J, Kay H, Semenza GL, et al:
Targeting Stat3 blocks both HIF-1 and VEGF expression induced by
multiple oncogenic growth signaling pathways. Oncogene.
24:5552–5560. 2005. View Article : Google Scholar
|
|
20
|
Murdoch C, Muthana M, Coffelt SB and Lewis
CE: The role of myeloid cells in the promotion of tumour
angiogenesis. Nat Rev Cancer. 8:618–631. 2008. View Article : Google Scholar
|
|
21
|
O'Reilly MS, Boehm T, Shing Y, Fukai N,
Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR and Folkman J:
Endostatin: An endogenous inhibitor of angiogenesis and tumor
growth. Cell. 88:277–285. 1997. View Article : Google Scholar
|
|
22
|
Li K, Shi M and Qin S: Current Status and
Study Progress of Recombinant Human Endostatin in Cancer Treatment.
Oncol Ther. 6:21–43. 2018. View Article : Google Scholar
|
|
23
|
Xiao L, Yang S, Hao J, Yuan X, Luo W,
Jiang L, Hu Y, Fu Z, Zhang Y and Zou C: Endostar attenuates
melanoma tumor growth via its interruption of b-FGF mediated
angiogenesis. Cancer Lett. 359:148–154. 2015. View Article : Google Scholar
|
|
24
|
Zheng AW, Jia DD, Xia LM, Jin G, Wu H and
Li T: Impact of carboplatin plus paclitaxel combined with endostar
against A375 melanoma cells: An in vitro and in vivo analysis.
Biomed Pharmacother. 83:1321–1326. 2016. View Article : Google Scholar
|
|
25
|
Cui C, Mao L, Chi Z, Si L, Sheng X, Kong
Y, Li S, Lian B, Gu K, Tao M, et al: A phase II, randomized,
double-blind, placebo-controlled multicenter trial of Endostar in
patients with metastatic melanoma. Mol Ther. 21:1456–1463. 2013.
View Article : Google Scholar
|
|
26
|
Cui C, Si L, Chi Z, Sheng X and Guo J:
Preliminary results of a phase II trial with continuous intravenous
infusion of rh-endostatin in combination with dacarbazine as the
first-line therapy for metastatic acral melanoma. Anticancer Res.
35:4350–4351. 2015.
|
|
27
|
Yang L, Xu Y, Luo P, Chen S, Zhu H and
Wang C: Baseline platelet counts and derived inflammatory
biomarkers: Prognostic relevance in metastatic melanoma patients
receiving Endostar plus dacarbazine and cisplatin. Cancer Manag
Res. 11:3681–3690. 2019. View Article : Google Scholar
|
|
28
|
Nyakas M, Aamdal E, Jacobsen KD, Guren TK,
Aamdal S, Hagene KT, Brunsvig P, Yndestad A, Halvorsen B, Tasken
KA, et al: Prognostic biomarkers for immunotherapy with ipilimumab
in metastatic melanoma. Clin Exp Immunol. 197:74–82. 2019.
View Article : Google Scholar
|
|
29
|
Liu X, Nie W, Xie Q, Chen G, Li X, Jia Y,
Yin B, Qu X, Li Y and Liang J: Endostatin reverses
immunosuppression of the tumor microenvironment in lung carcinoma.
Oncol Lett. 15:1874–1880. 2018.
|
|
30
|
Liang J, Liu X, Xie Q, Chen G, Li X, Jia
Y, Yin B, Qu X and Li Y: Endostatin enhances antitumor effect of
tumor antigen-pulsed dendritic cell therapy in mouse xenograft
model of lung carcinoma. Chin J Cancer Res. 28:452–460. 2016.
View Article : Google Scholar
|
|
31
|
Wang X, Zhan RY, Wang YY, Yan XI, Cao D,
Li Y, Wang YQ and Luo F: Endostatin improves cancer-associated
systemic syndrome in a lung cancer model. Oncol Lett. 9:2023–2030.
2015. View Article : Google Scholar
|