Introduction
In the past few years, cerebral vasospasm (CVS) has
been considered to be one of the complications leading to severe
neurological dysfunction after subarachnoid hemorrhage (SAH).
Numerous studies have explored the mechanisms of CVS following SAH
and have made several discoveries (1–4).
However, the pathophysiological mechanism remains elusive and no
effective treatment for CVS exists at present (5). Previous studies have revealed that
certain pathological processes, such as smooth muscle cell
contraction resulting from spasmogenic substances generated during
the lysis of the subarachnoid blood wall and endothelial damage,
are associated with the pathogenesis of CVS (6–8).
As has been previously reported, the phenotypic transformation of
vascular smooth muscle cells (VSMCs) occurs in the vessel wall of
CVS (9–11) and which is considered to
contribute to the CVS following SAH.
VSMCs retain extraordinary plasticity and possess
the quality of invertible phenotypic transition; they can convert
from the contractile phenotype to the synthetic phenotype in
response to various cellular stimulator substances (12,13). Synthetic phenotype VSMCs can
increase their rates of proliferation and oversynthesis and
excretion of extracellular matrix, which are the pivotal events of
vascular wall thickening and vascular lumen stenosis, eventually
leading to dysfunction of vascular autoregulation and CVS (14,15). CVS resulting from phenotypic
transformation of VSMCs impairs the cerebral blood flow
distribution (16) and leads to a
delayed ischemic neurological deficit. Notably, the phenotype of
VSMCs is distinguished mainly based on specific proteins; α-smooth
muscle actin (α-SMA) is chiefly expressed in the contractile
phenotype and embryonic smooth muscle myosin heavy chain (SMemb) is
an accurate marker of synthetic VSMCs (13,17).
Peroxisome proliferator-activated receptor β/δ
(PPARβ/δ) has been characterized as anti-inflammatory and
antiapoptotic and can modulate vascular cells proliferation
(18). In addition, it exerts
neuroprotective effects on acute and chronic injury of the central
nervous system (19,20). A previous study reported that
activation of PPARβ/δ can efficiently attenuate the inflammation of
VSMCs and decrease the release of inflammatory factors (21), which provides a potential line of
inquiry for vascular diseases of the central nervous system.
Phosphorylation of the ERK1/2 signaling pathway induces the
activation of intracellular signals via the phosphorylation of
regulatory targets, including extracellular proteins and
transcription factors (22,23). ETS domain-containing protein Elk-1
(Elk-1) and p90 ribosomal S6 kinase (p90RSK) are downstream targets
of ERK1/2, which are phosphorylated by ERK1/2 and associated with
gene transcription, protein synthesis and proliferation (24,25). There is compelling evidence that
subsequent activation of the ERK1/2 signaling pathway exerts an
efficiently effect in modulating VSMC proliferation and migration
(26). However, to the best of
the authors' knowledge, no studies have investigated the function
and possible mechanisms of PPARβ/δ in VSMCs phenotypic modulation,
which contributes to CVS following SAH.
Materials and methods
Animals and SAH model
establishment
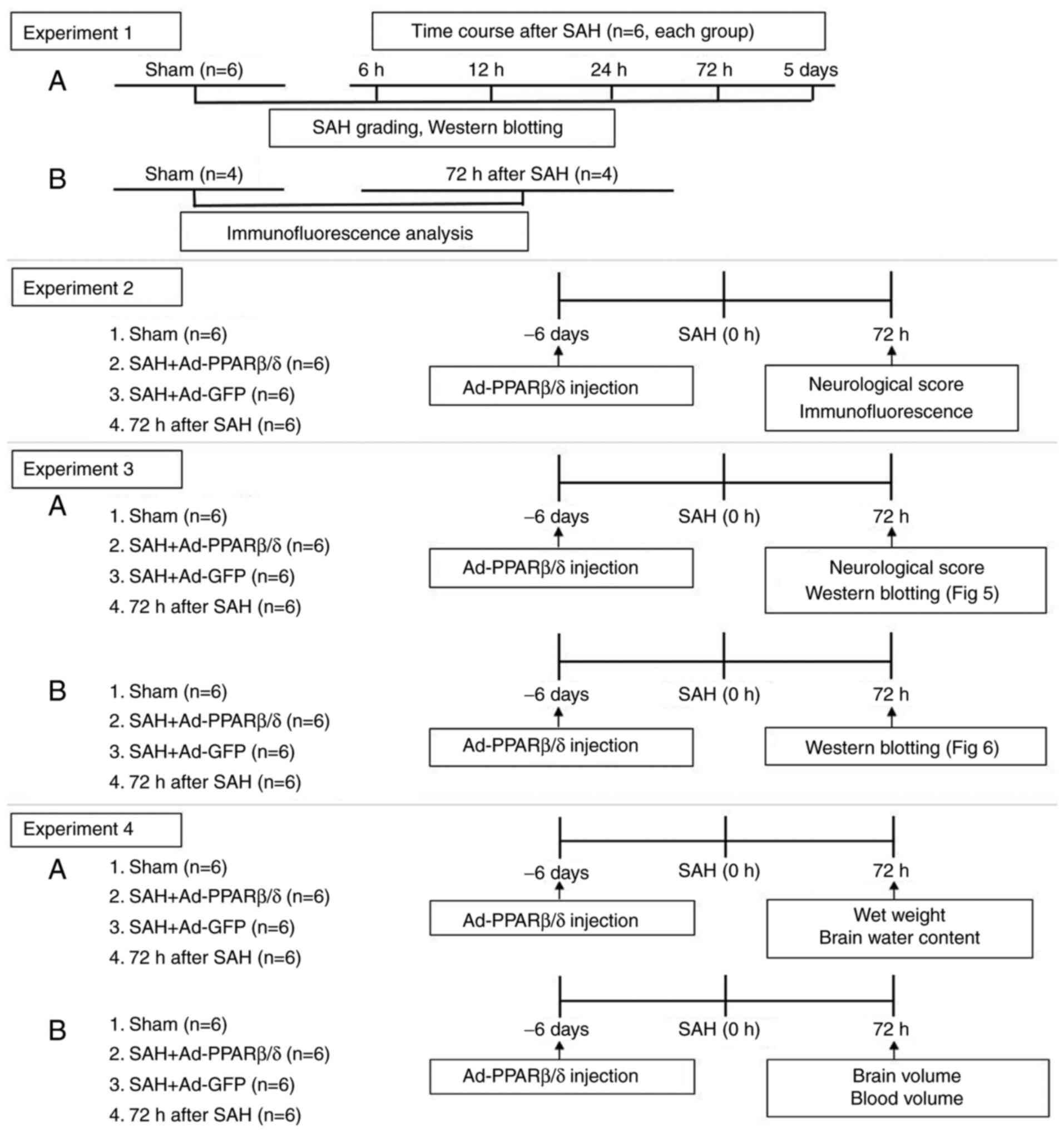
The experiments were performed as shown in the
schematic (Fig. 1). All
experimental procedures were approved by the Animal Experiments
Ethic Committee at The First Affiliated Hospital of Soochow
University (Suzhou, China; approval no. 2018-094) and were
performed in accordance with the Laboratory Animal Guidelines for
ethical review of animal welfare (standard no. GB/T 35892-2018)
(27). A total of 192 male
Sprague-Dawley (SD) rats purchased from the Animal Experiment
Center of Soochow University (age, 6–8 weeks; weight, 300–350 g)
were used in the present study and the endovascular perforation
model of SAH was performed as reported previously (28). Briefly, anesthesia was induced
with 5% isoflurane using a facemask and maintained with 1.5%
isoflurane. Subsequently, the rats were fixed in the supine
position. The internal carotid artery (ICA) and external carotid
artery (ECA) were carefully separated. Afterwards, a sharpened 4-0
monofilament nylon suture was inserted into the middle cerebral
artery through the ICA and gently pushed forward to pierce the
vessel. The thread was drawn out to restore blood supply. Sham
operated rats received the same procedure except for perforation.
Following surgery, the animals were kept in an incubator heated at
36°C for 1 h. The rats were housed under controlled environmental
conditions (12-h light/dark cycle; 23±1°C, 55±5% relative humidity)
with free access to water and food. Body weight and body
temperature was determined daily as a sensitive indicator for their
well-being. Likewise, neurological deficits were assessed on a
daily basis using the previously described modified Garcia scale
(29). In the present study, rats
clearly in pain, showing signs of severe and enduring distress, or
characterized as moribund were humanely sacrificed rather than
allowed to survive to the end of the scheduled study (at 72 h after
SAH). Animals were euthanized at 72 h after SAH when a loss of more
than 20% baseline body weight occurred or the following qualitative
humane endpoint criteria were observed during inspection: Paralysis
with absence of spontaneous movement, severe ataxia or loss of
postural reflexes, epileptic seizures, coma, reduction of general
health status with reduced grooming or refusal of food intake. Rats
were euthanized at 72 h after SAH by deep (5%) isoflurane followed
by cardiac perfusion with 30 ml phosphate-buffered saline,
decapitation was implemented under deep anesthesia after cardiac
perfusion and the brains were rapidly removed.
The basal cistern was divided into six segments and
scored from 0–3 points by a blinded observer according to the
amount of subarachnoid blood. Each segment was allotted a grade
between 0 and 3 depending on the amount of subarachnoid blood clot
in the segment as follows: i) Grade 0, no subarachnoid blood; ii)
1, minimal subarachnoid blood; iii) 2, moderate blood clot with
recognizable arteries; and iv) 3, blood clot obliterating all
arteries within the segment. The total score from all six segments
represented the severity of the SAH (29). Rats who received a score <7
were excluded from the present study. As in previous studies,
modified Garcia scale (score, 3–18; Table I) (29) and beam balance (score, 0–4)
(30) were used to assess
neurological impairment.
 | Table I.Neurological evaluation of animals at
72 h after SAH. |
Table I.
Neurological evaluation of animals at
72 h after SAH.
|
| Score |
|---|
|
|
|
|---|
| Test | 0 | 1 | 2 | 3 |
|---|
| Spontaneous
activity (in cage for 5 min) | No movement | Barely moves
position | Moves but does not
approach at least three sides of cage | Moves and
approaches at least three sides of cage |
| Spontaneous
movements of all limbs | No movement | Slight movement of
limbs | Moves all limbs but
slowly | Move all limbs same
as pre-SAH |
| Movements of
forelimbs (outstretching while held by tail) | No outreaching | Slight
outreaching | Outreach is limited
and less than pre-SAH | Outreach same as
pre-SAH |
| Climbing wall of
wire cage | N/A | Fails to climb | Climbs weakly | Normal
climbing |
| Reaction to touch
on both side of trunk | N/A | No response | Weak response | Normal
response |
| Response to
vibrissae touch | N/A | No response | Weak response | Normal
response |
Adenoviruses administration
A total of 36 rats were randomly divided into six
groups with six rats in each group as follows: i) Sham; ii) 6 h
after SAH; iii) 12 h after SAH; iv) 24 h after SAH; v) 72 h after
SAH; and vi) 5 days after SAH. Western blotting was used to detect
the expression of α-SMA and SMemb in the cerebral vessels of each
group at different time points.
Adenoviruses PPARβ/δ (Ad-PPARβ/δ) and adenoviruses
green fluorescent protein (Ad-GFP; as a control for Ad-PPARβ/δ)
(Shanghai GeneChem Co., Ltd.) were diluted to 1.3×1010
pfu/ml in enhanced transfection solution (Shanghai GeneChem Co.,
Ltd.) before use. According to a previous report (31), a small hole was drilled on the
skull according to the coordinates of 1.0 mm posterior of bregma
and 2.0 mm lateral of sagittal suture. A total of 10 µl diluted
Ad-PPARβ/δ or Ad-GFP were slowly injected into the lateral
ventricle on the 6th days before SAH (32) through the hole, at a depth of 4.0
mm. At 72 h after SAH, the rats were randomly divided into the
following four groups: i) Sham; ii) SAH + Ad-GFP; iii) SAH +
Ad-PPARβ/δ; and iv) SAH.
Brain water content
Rats were euthanized at 72 h after SAH by deep (5%)
isoflurane followed by cardiac perfusion with 30 ml
phosphate-buffered saline, decapitation was implemented under deep
anesthesia after cardiac perfusion and the brains were removed
rapidly. Brain water content was measured using the wet/dry method
(33). Briefly, brains were
removed from rats at 72 h after SAH and divided into four parts: i)
Right hemisphere; ii) left hemisphere; iii) brain stem; and iv)
cerebellum. Each part was weighed (wet weight) immediately on a
precise electronic scale and then placed into the oven for drying
for 48 h at 105°C (dry weight) and the percentage of brain water
content was calculated as follows: [(wet weight-dry weight)/wet
weight] ×100%.
Brain volume and cerebral blood
volume
Rats were euthanized at 72 h after SAH by deep (5%)
isoflurane followed by cardiac perfusion with 30 ml
phosphate-buffered saline, decapitation was implemented under deep
anesthesia after cardiac perfusion and the brains were removed
rapidly. Brain volume was measured as following previously
described (33). Briefly, plastic
10 ml vials containing 5.0 ml of saline were marked equally to the
level of liquid by using ultrathin marker. After meticulous clot
removal, the brain was put into the vial and the increased level of
liquid was marked and the brain volume was measured by titrated
saline infusion within marked levels. Vertical position of vials
upon measurements was maintained. Cerebral blood volume was
measured as previously described (33,34). The brain was then extracted and
phosphate-buffered saline (PBS) was added to it to reach a total
volume of 3 ml. This solution was homogenized for 30 sec. After
homogenization, ultrasound (20 KHz, ice bath) was applied for 1 min
to lyse erythrocytic membranes and then centrifuged for 30 min
(11,000 × g, 4°C). Several (minimum of three) samples of the 100 µl
supernatant were added to 400 µl Drabkins reagent (Sigma-Aldrich;
Merck KGaA) and allowed to stand for 15 min at room temperature.
Finally, optical density was measured and recorded at 540 nm with a
spectrophotometer. Then, additional hemispheric brain tissue was
obtained from normal rats subjected to complete cardiac perfusion.
Incremental volumes (0, 2, 4, 8, 16 and 32 µl) of homologous blood
which were obtained from a cardiac puncture after deep (5%)
isoflurane anesthesia were added to each hemispheric sample with
PBS to reach a total volume of 3 ml and then the above procedures
completed. These procedures yielded a linear relationship between
hemoglobin concentrations in brain tissue and blood volume. Using
the equation of the best-fitting linear regression line from these
data, the blood volume of SAH rats was determined based on
absorbance readings.
Immunofluorescence
Immunofluorescence staining was performed using
fixed frozen sections. At 72 h after SAH, deep anesthesia of rats
was induced and maintained with 5% isoflurane, followed by cardiac
perfusion with 30 ml phosphate-buffered saline. Decapitation was
implemented under deep anesthesia after cardiac perfusion and the
brains were rapidly removed. All fixed frozen sections were
incubated at 4°C overnight with primary antibodies: Mouse
anti-α-SMA (Abcam; 1:200; cat. no. ab7817) and rabbit anti-SMemb
(Abcam; 1:200; cat. no. ab230823) antibodies followed by incubation
with secondary antibodies (Abbkine Scientific Co., Ltd.; 1:200;
cat. nos. A22110 and A24421) for 2 h at room temperature.
Fluorescence images were effectively captured using a fluorescence
microscope (Leica Microsystems GmbH). As reported previously
(35,36), Image-Pro Plus v.6.0 software
(Media Cybernetics, Inc.) was used to measure the thickness of the
basilar artery and lumen diameter.
Western blotting
Rats were euthanized at 72 h after SAH by deep (5%)
isoflurane, decapitation was implemented under deep anesthesia and
the brains were rapidly removed. With the aid of a microscope, the
basilar arteries and circle of Willis arteries were separated out
of each brain, cleared of connective tissue and snap frozen in
liquid nitrogen until homogenization in RIPA buffer (Beyotime
Institute of Biotechnology) containing protease and phosphatase
inhibitors. The total protein concentration was measured using a
BCA Protein Assay Reagent (Beyotime Institute of Biotechnology).
The proteins (25 µg) were separated via 10% SDS-PAGE (Beyotime
Institute of Biotechnology), and then transferred onto a PVDF
membrane (Bio-Rad Laboratories, Inc.). The membranes were then
blocked with 5% skimmed milk in Tris-buffered saline containing
0.1% Tween-20 (Beijing Solarbio Science & Technology Co., Ltd.)
for 1 h at room temperature, and incubated overnight at 4°C with
primary antibodies against α-SMA (Abcam; 1:200; cat. no. ab7817),
SMemb (Abcam; 1:200; cat. no. ab230823), p-ERK (Cell Signaling
Technology, Inc.; 1:2,000; cat. no. 4370), ERK (Cell Signaling
Technology, Inc.; 1:1,000; cat. no. 9102), p-Elk-1 (Cell Signaling
Technology, Inc.; 1:1,000; cat. no. 9186), Elk-1 (Cell Signaling
Technology, Inc.; 1:1,000; cat. no. 9182), p-p90RSK (Cell Signaling
Technology, Inc.; 1:1,000; cat. no. 11989), p90RSK (Cell Signaling
Technology, Inc.; 1:1,000; cat. no. 9355) and GAPDH (ProteinTech
Group, Inc.; 1:2,000; cat. no. 60004-1-Ig). The membranes were
subsequently incubated with relative horseradish
peroxidase-conjugated IgG (1:1,000; Beyotime Institute of
Biotechnology; cat. no. A0216) for 1 h at 37°C and visualized by
using an ECL kit (Beyotime Institute of Biotechnology; cat. no.
P0018FS) through X-ray film. Optical densities of these bands were
quantified using Quantity One Software (v4.6.2; Bio-Rad
Laboratories, Inc.). GAPDH was used as an internal reference.
Statistical analysis
All experiments were repeated at least three times.
All data are presented as the mean ± standard deviation (SD) and
were analyzed using SPSS 19.0 software (IBM Corp.). Fisher's exact
test was used to compare the mortality rates among groups.
Significance of differences in neurologic scores was analyzed by
Kruskal-Wallis test followed by Dunn's post hoc test. Mean group
values were assessed using one-way analysis of variance followed by
Tukey's post hoc test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
Mortality and SAH severity
A total of 192 SD rats were used in this work and 28
animals succumbed due to severe SAH reactions within 24 h following
surgery; in the end, 164 rats were effectively applied to
subsequent experiments. The overall mortality of SAH in the present
study was 14.58% (28 out of 192 rats). A similar death rate is
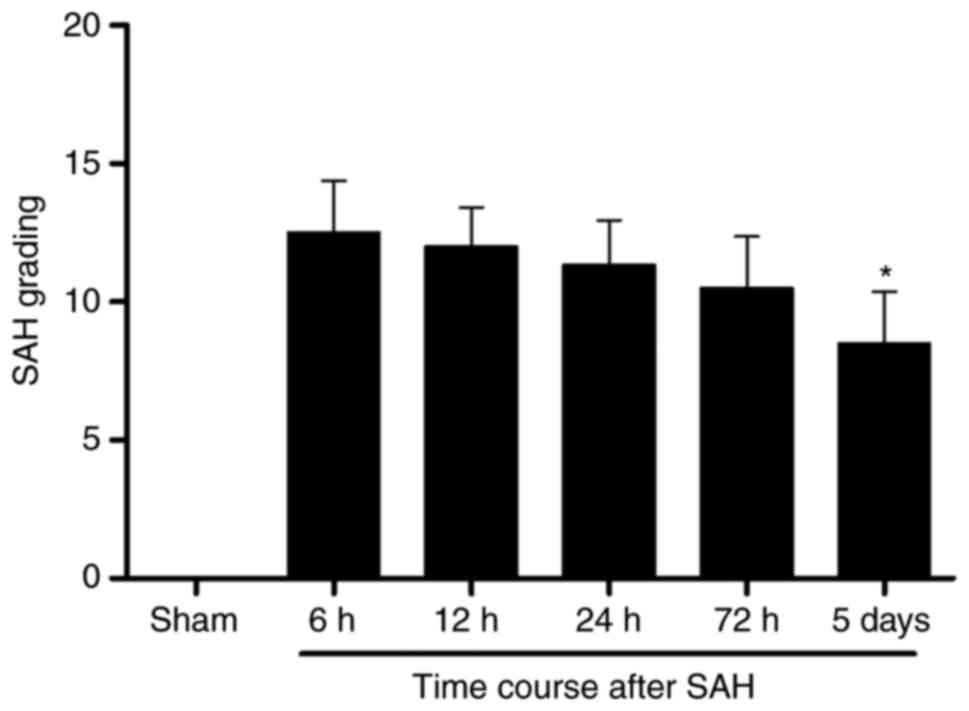
reported in a previous study (37). The average SAH grading score was
not significantly different among the 6, 12, 24 and 72 h groups
after SAH. The average SAH grading declined to some extent at 5
days following SAH compared with 6 h after SAH (Fig. 2).
Expression profile of marker proteins
of phenotypic change of VSMCs
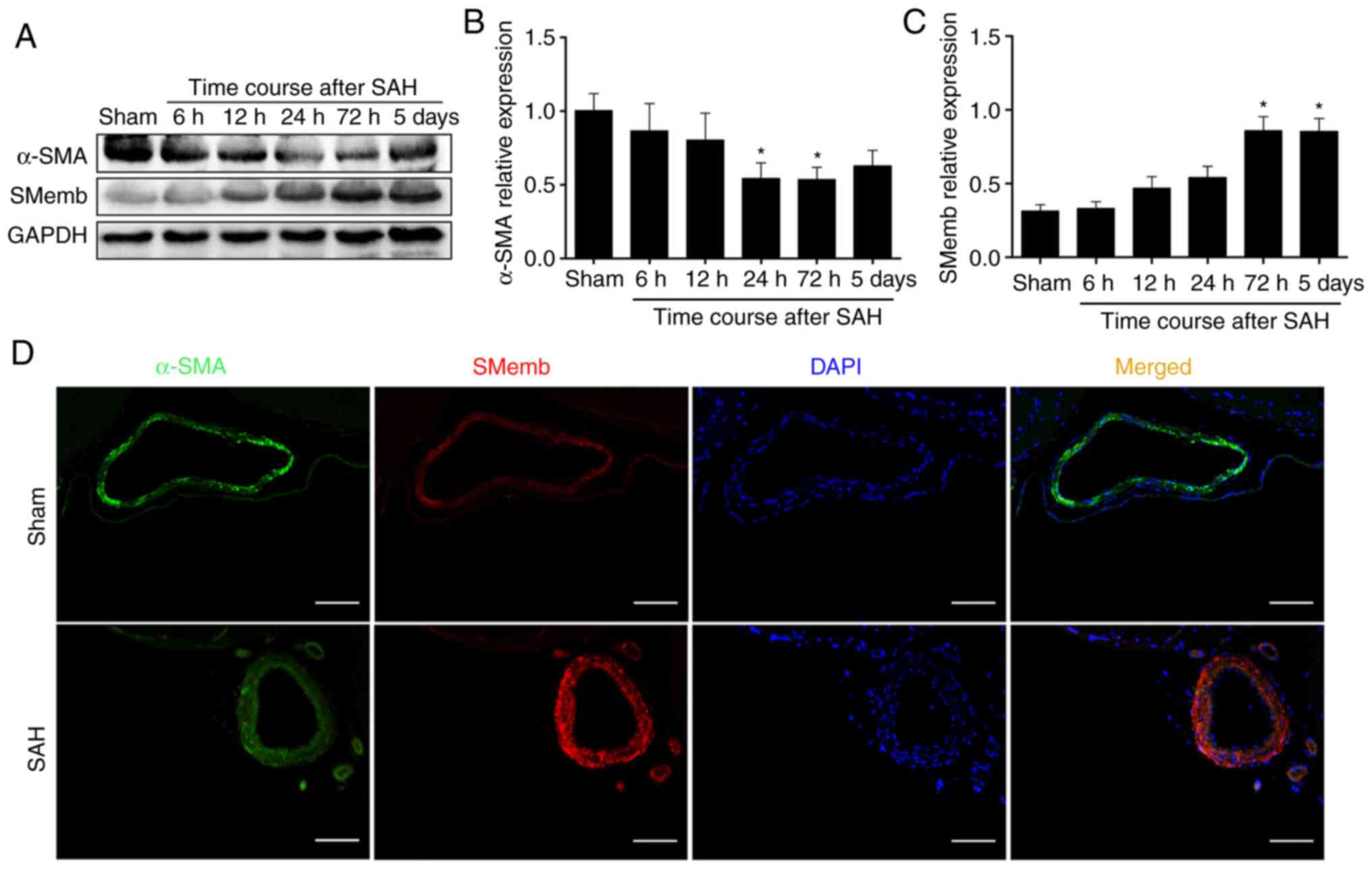
Our previous study revealed that the hemoglobin
could induce a cerebral VSMCs switch from the contractile to
synthetic phenotype in vitro (38). The present study first
investigated whether cerebral VSMCs would respond to SAH in
vivo. As expected, the western blotting results demonstrated
that the α-SMA expression decreased as early as at 6 h and was
significantly decreased at 72 h, whereas it exhibited no
significant difference at 5 days compared with Sham group (Fig. 3A and B). In addition, SMemb
expression variations occurred as early as at 6 h and it was
significantly increased at 72 h (Fig.
3C) compared with the expression levels in the Sham group. This
coincided with the time of CVS. According to this preliminary data,
the time point of 72 h (time of maximal SMemb expression) was
selected to perform the subsequent experiments.
To further confirm the changes of the expression
profile and morphology of cerebral vessels, double
immunofluorescence was performed to analyze α-SMA and SMemb
expression and localization in the basilar artery. As expected, the
results demonstrated that the SMemb fluorescence intensity was
increased on the wall of the basilar artery at 72 h after SAH,
whereas the α-SMA fluorescence intensity was decreased at 72 h
after SAH (Fig. 3D).
Overexpression of PPARβ/δ protein
induced by Ad-PPARβ/δ improves neurological deficits and attenuates
CVS at 72 h after SAH
To further ascertain whether PPARβ/δ could attenuate
the VSMCs phenotypic transformation following SAH in vivo,
additional experiments were conducted in which PPARβ/δ protein was
overexpressed using intracerebroventricular injection of Ad-PPARβ/δ
at day 6 before SAH.
Consistent with the results in Fig. 3D, immunofluorescence analysis
(Fig. 4A) of the basilar artery
revealed that α-SMA immunoreactivity decreased at 72 h and this was
accompanied by an increase in SMemb immunoreactivity. However, the
change in immunoreactivity could be reversed by Ad-PPARβ/δ. With
the use of Ad-PPARβ/δ, the immunoreactivity of α-SMA was notably
enhanced in the SAH + Ad-PPARβ/δ group. As shown in Fig. 4B, rats in the SAH and SAH + Ad-GFP
groups exhibited severe neurological impairments compared with rats
in the sham and SAH + Ad-PPARβ/δ groups. Furthermore, the
histopathological alterations, including the thickness of the
vascular wall and lumen stenosis in the basilar artery, were
significantly ameliorated by Ad-PPARβ/δ (Fig. 4C).
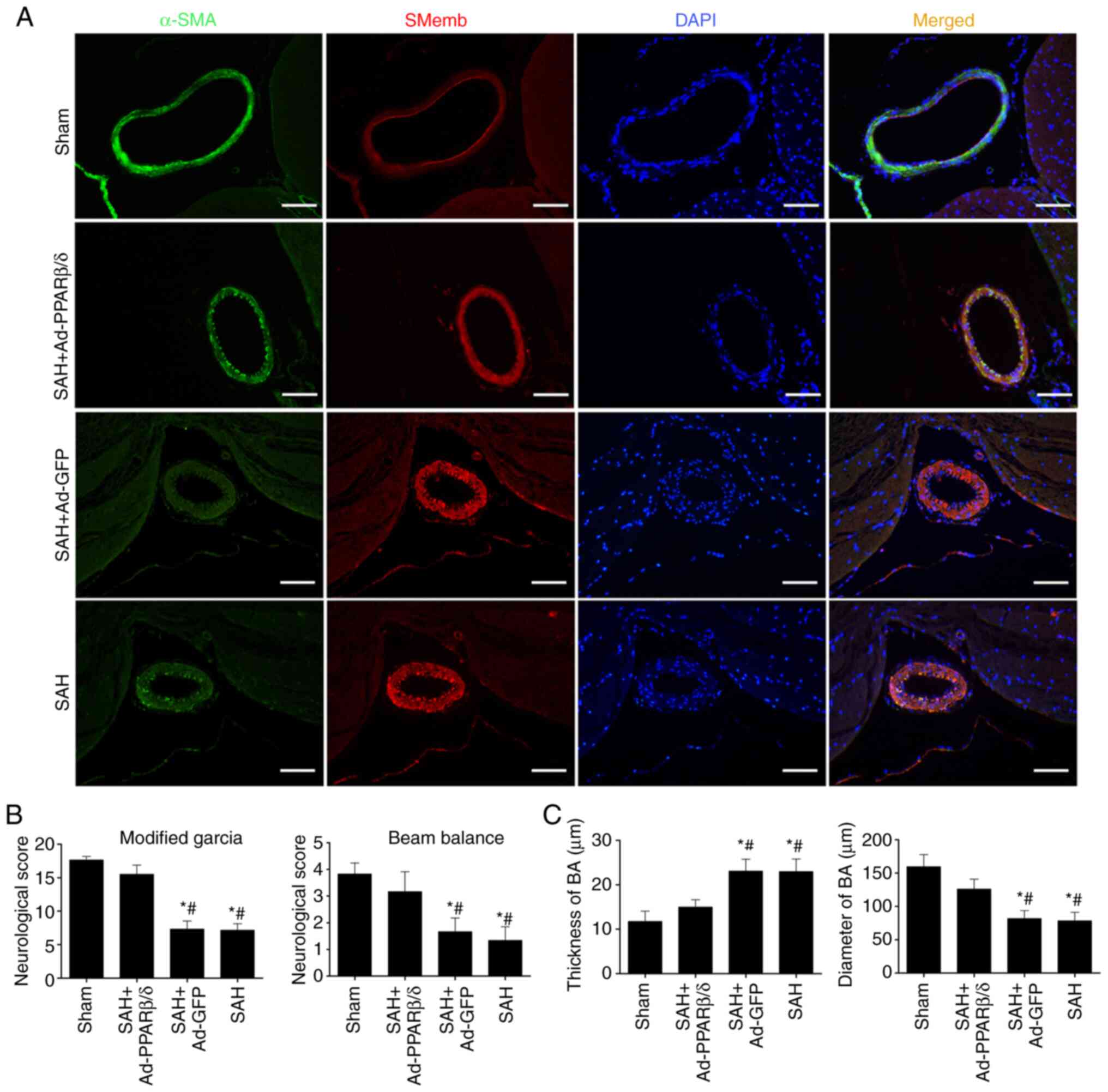 | Figure 4.Overexpression of PPARβ/δ protein
induced by Ad-PPARβ/δ improves the neurological deficits and
attenuates cerebral vasospasm. (A) Immunofluorescence analysis of
the BA with α-SMA (green), SMemb (red) and DAPI (blue) at 72 h
after SAH (n=6). Scale bar, 50 µm. (B) Rats in the SAH and SAH +
Ad-GFP groups exhibited severe neurological impairments, while
Ad-PPARβ/δ improved neurological impairments (n=6). (C)
Histopathological alterations, including the thickness of the
vascular wall and lumen stenosis in BA, were significantly
ameliorated by Ad-PPARβ/δ (n=6). *P<0.05 vs. Sham;
#P<0.05 vs. SAH + Ad-PPARβ/δ. PPARβ/δ, peroxisome
proliferator-activated receptor β/δ; Ad, adenovirus; α-SMA,
α-smooth muscle actin; SMemb, smooth muscle myosin heavy chain;
SAH, subarachnoid hemorrhage; GFP, green fluorescent protein; BA,
basilar artery. |
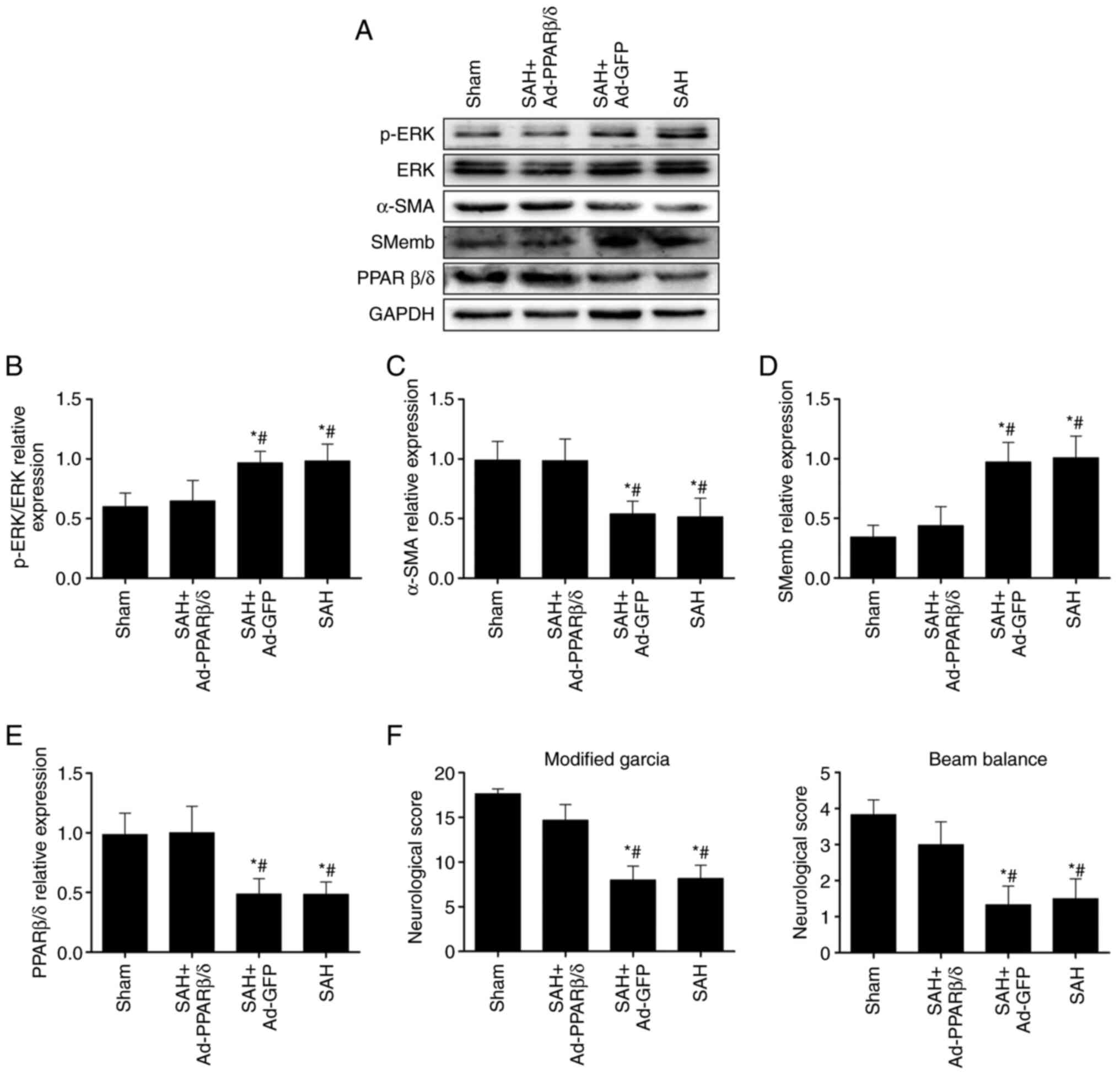
ERK signaling pathway is involved in
PPARβ/δ suppression of phenotypic modulation of VSMCs and CVS in
rats at 72 h after SAH
It was hypothesized that the effect of PPARβ/δ on
the phenotypic switch of VSMCs was regulated by the ERK signaling
pathway. Therefore, the levels of p-ERK was first detected using
western blotting at 72 h after SAH. As shown in Fig. 5A, activation of the ERK signaling
pathway was demonstrated by increased levels of p-ERK following
SAH. Furthermore, prior injection of Ad-PPARβ/δ similarly
administered into cerebral ventricles significantly increased
PPARβ/δ and α-SMA expression, while decreasing the levels of p-ERK
and SMemb following SAH (Fig.
5A-E) and this was accompanied by an improvement in
neurological functions (Fig. 5F).
Combined, these data revealed that the ERK signaling pathway served
a role in PPARβ/δ modulating phenotypic transformation of VSMCs and
attenuating CVS following SAH.
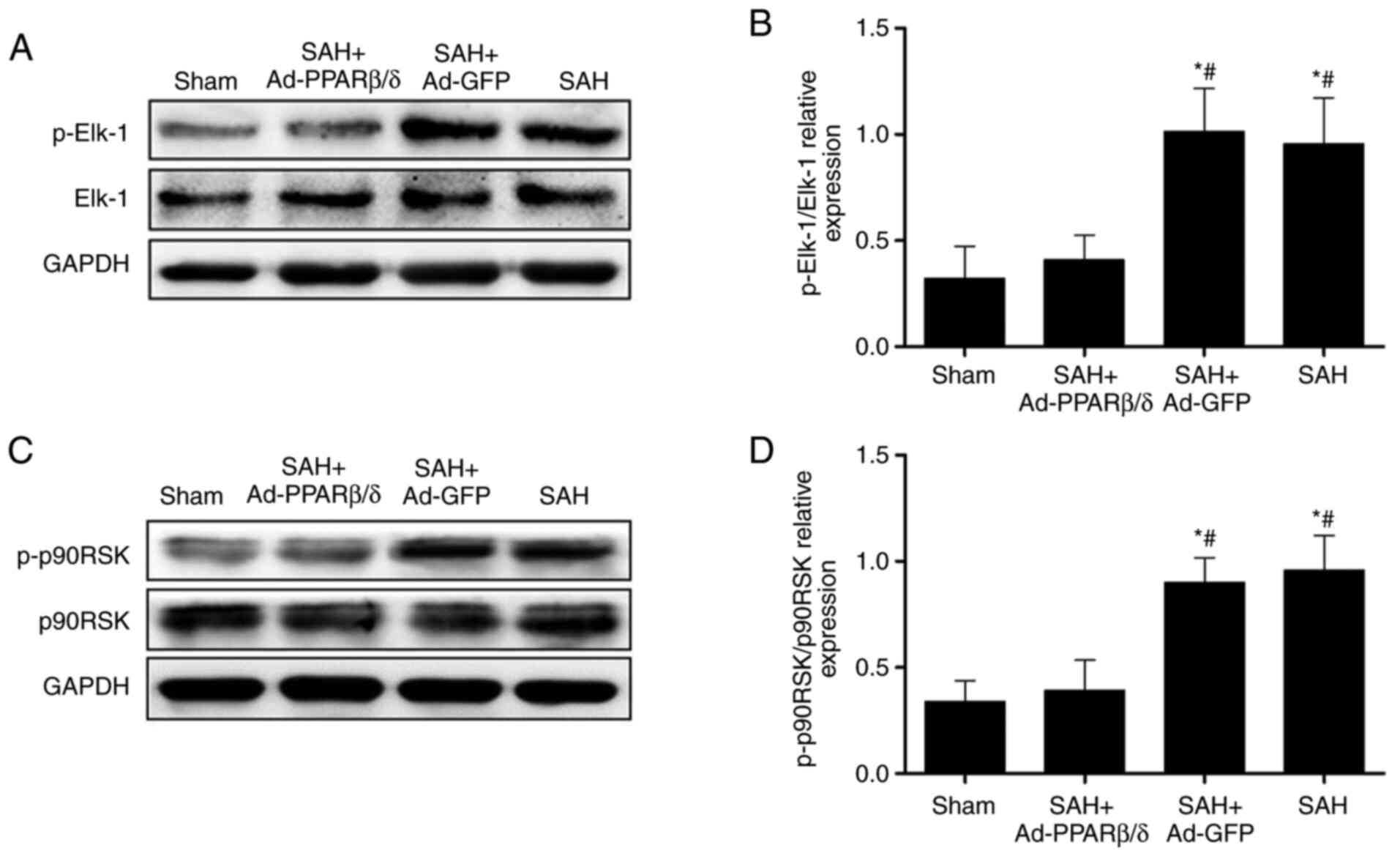
PPARβ/δ inhibits cerebral VSMCs
phenotypic switch and attenuates CVS via suppression of p-Elk-1 and
p-p90RSK expression
Elk-1 and p90RSK are downstream targets of ERK and
can be activated by phosphorylation of ERK1/2 and JNK MAPKs, which
are essential for gene regulation, cell migration and
proliferation, and protein synthesis (24–26).
In the present study, the expression of p-Elk-1 was
significantly increased at 72 h after SAH. However, the expression
p-Elk-1 was significantly downregulated by pretreatment of rats
with Ad-PPARβ/δ (Fig. 6A and B).
As with p-Elk-1, the expression of p-p90RSK was increased at 72 h
after SAH and these were markedly downregulated by addition of
Ad-PPARβ/δ (Fig. 6C and D). These
results indicated that PPARβ/δ maintained the contractile phenotype
of VSMCs and attenuated CVS by restraining the expression of
p-Elk-1 and p-p90RSK.
Ad-PPARβ/δ ameliorates brain swelling
and brain edema
Brain swelling was assessed based on the wet brain
weight, brain volume and cerebral blood volume. To ascertain the
effect of PPARβ/δ on brain swelling, Ad-PPARβ/δ was administrated
at 6 days before SAH. In the present study, brain wet weight and
brain volume were significantly increased at 72 h compared with
those of the Sham group (Fig. 7A and
B) and Ad-PPARβ/δ pretreatment significantly decreased the
brain wet weight and brain volume. Specifically, Ad-PPARβ/δ
pretreatment markedly decreased the water content in the bilateral
cerebral hemispheres and cerebellum at 72 h, whereas treatment with
Ad-GFP did not have the same effect (Fig. 7C). Additionally, the cerebral
blood volume in the Ad-PPARβ/δ pretreatment group at 72 h was
significantly lower than that of the SAH and SAH + Ad-GFP groups
(Fig. 7D).
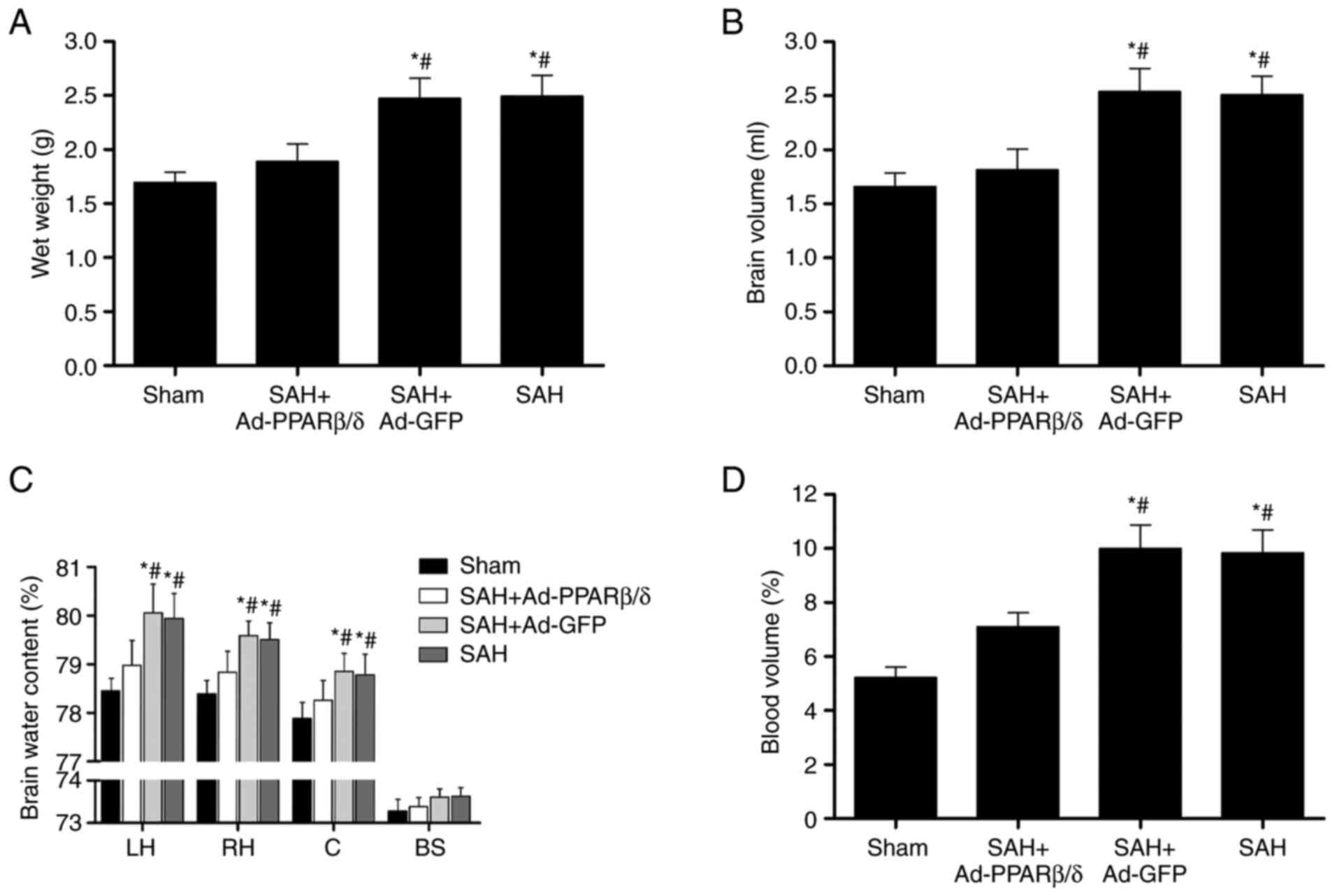 | Figure 7.Intracerebroventricular injection of
Ad-PPARβ/δ improves brain swelling and brain edema. Ad-PPARβ/δ
pre-treatment decreased the (A) brain wet weight, (B) brain volume,
(C) brain water content and (D) blood volume at 72 h after SAH
(n=6). *P<0.05 vs. Sham; #P<0.05 vs. SAH +
Ad-PPARβ/δ. Ad, adenovirus; PPARβ/δ, peroxisome
proliferator-activated receptor β/δ; SAH, subarachnoid hemorrhage;
LH, left hemisphere; RH, right hemisphere; C, cerebellum; BS, brain
stem; GFP, green fluorescent protein. |
Discussion
VSMCs are highly plastic and their phenotypic switch
is considered to be involved in the process of vessel remodeling
(11,39). The current study demonstrated that
the Ad-PPARβ/δ ameliorated CVS and impeded VSMCs phenotypic
transformation partially via suppression of ERK1/2 signaling
pathway and was associated with decreased levels of p-Elk-1 and
p-p90RSK. Furthermore, pre-administration of Ad-PPARβ/δ attenuated
the brain volume, brain water content and blood volume at 72 h
after SAH. Immunohistochemistry demonstrated that Ad-PPARβ/δ
markedly increased the lumen diameter in the basilar arteries at 72
h after SAH and clearly improved the neurologic deficit. Overall,
the findings suggested that the phenotypic conversion of VSMCs
participated in the process of CVS following SAH and Ad-PPARβ/δ
prevented VSMCs phenotypic transformation and attenuated CVS
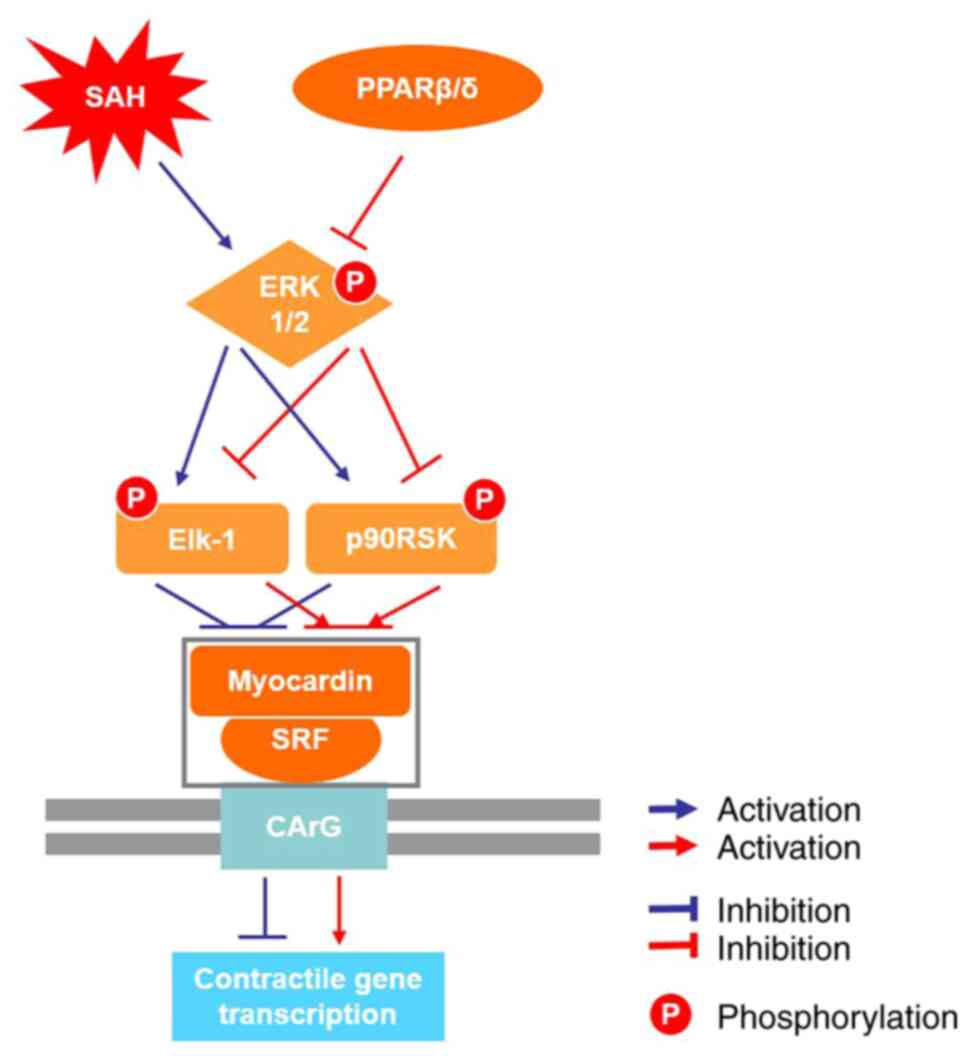
following SAH in rats via the ERK1/2 signaling pathway (Fig. 8).
A number of clinical studies have demonstrated that
angiographically detectable CVS is observed in patients with SAH
and it is hypothesized to be one of the most important
considerations that contribute to the neurological impairment and
lead to the increase of deformity and mortality following SAH
(40,41). Despite some achievements having
been made in CVS research, the precise underlying mechanism remains
to be elucidated. It has been reported that the phenotypic
transition of VSMCs is considered to be one of the pathogenesis of
CVS following SAH (42). VSMCs,
which are an essential component of the cerebral vascular neural
network, are involved in the pathophysiology of SAH, as previously
reported by Zhang et al (16) and subsequent studies have
demonstrated that VSMCs undergoes phenotypic transformation under
the conditions of local vascular inflammation or destruction and in
the presence of toxic metabolites in the blood following SAH
(15,43). After transforming into synthetic
cells, proliferation is abnormally increased and these cells
exhibit high extracellular matrix proteins synthesis efficiency,
ultimately leading to a decline in autoregulatory capacity
(15,44). The proliferative synthetic state
of VSMCs has been shown to exert a pivotal role in the impairment
of vascular regulation function and cerebral blood flow
distribution (15,44,45). The VSMC phenotypic switch leads to
the loss of arteriolar autoregulation and influences the integrity
of the blood-brain barrier (46).
Due to these characteristics, VSMCs might be an alternative therapy
target for CVS following SAH. In the present study, it was observed
that the existence of VSMC phenotypic transformation caused
stenosis of the basilar artery and increased the wall thickness in
the setting of SAH and this result was in line with previous
studies.
PPARβ/δ is a member of the PPARs nuclear hormone
receptor superfamily, which also includes PPARα, PPARβ/δ and PPARγ,
and is the most widely and highly expressed isotype in the central
nervous system compared with PPARα and PPARγ (19). PPARβ/δ is involved in regulating
the transcription of numerous target genes, modulating vascular
cell proliferation and the inflammatory process (47,48). However, the mechanisms of
PPARβ/δ-dependent gene regulation are complex. A large number of
inflammatory cytokines are released following SAH and may partially
contribute to the decrease of PPARβ/δ expression (49). Additionally, our previous study
demonstrated that PPARβ/δ expression decreases following SAH;
however, upregulated expression of PPARβ/δ markedly ameliorates the
early inflammation reaction (32). A previous study reported that the
PPARβ/δ exerts positive anti-inflammatory effects during the
attenuation of neointimal hyperplasia by inhibiting VSMC
proliferation and accelerating re-endothelization (50). These results indicate that the
PPARβ/δ has the capability to maintain the stability of vascular.
However, to the best of the authors' knowledge, there is no direct
evidence showing that PPARβ/δ has positive effects on modulating
the VSMC phenotypic switch in the central nervous system. In the
present study morphological examinations revealed that the
phenotypic switch occurred in the vascular wall following SAH.
Administration of Ad-PPARβ/δ could alleviate the neurological
deficit and impeded VSMC phenotypic transformation and CVS.
It has been documented that the ternary complex
formed by serum response factor (SRF) and its coactivator myocardin
binds with cis-acting elements CArG in the nucleus to regulate VSMC
differentiation and maintain the contractile phenotype (51,52). Takata et al (53) report that the PPARβ/δ exerts
anti-inflammatory effects in rat vascular tissues via suppression
of the ERK1/2 signaling pathway. In addition, PPARβ/δ prevents
endoplasmic reticulum stress and inflammation in skeletal muscle
cells by suppressing the ERK1/2 signaling pathway (54). These findings indicate that ERK1/2
signaling pathway may serve an important role in the regulation of
VSMC phenotype transformation. Elk-1 and p90RSK are important
targets of the MEK/ERK signaling pathway (24,55), p-Elk-1 competitively displaces
myocardin from SRF, leading to repression of VSMCs contractile
genes (56) and p90RSK takes
effect on phenotypic switch, cell differentiation by binding to
signal transduction molecules, gene transcription factors (24). In the present study, the
expression levels of p-ERK, p-Elk-1 and p-p90RSK were markedly
increased at 72 h after SAH. However, prior injection of Ad-PPARβ/δ
into cerebral ventricles significantly decreased the levels of
p-ERK, p-Elk-1 and p-p90RSK and improved the neurological functions
following SAH. These results revealed that PPARβ/δ modulates the
phenotypic transformation of VSMCs following SAH, at least partly,
via inhibition of the ERK1/2 signaling pathway and expression of
p-Elk-1 and p-p90RSK.
There are PPAR β/δ binding sites on the promoter of
NF-κB and activator protein 1 (AP1) and PPAR β/δ binding to these
promoters contribute to suppressing transcription of these target
genes (57). In addition, it is
reported that PPARβ/δ prevents TNF-a-induced NF-κB activation in
human HaCaT cells through AMP-activated protein kinase (AMPK) and
sirtuin1 (SIRT1) (58). In
addition, it is reported that inactivation of NF-κB or AP1 leads to
a decrease in p-ERK (59).
Therefore, it was hypothesized that the PPARβ/δ suppressed p-ERK
levels in a dual mode by directly binding to promoter of target
gene and by inhibiting other possible potential signal pathways.
However, the expression of NF-κB or AP1 were not detected, which is
a limitation of the present study. Additional experiments should be
conducted to investigate the potential mechanism of how PPARβ/δ
regulates the ERK1/2 signaling pathway.
The PPARγ agonists rosiglitazone or troglitazone
have been used to treat clinical diabetes mellitus and to study its
effects, mainly in ischemic stroke, for a number of years.
Activation of PPAR-γ in VSMCs by the antidiabetic agent
troglitazone (or rosiglitazone) inhibits VSMC proliferation and
migration (9,60). However, it is unclear whether
these agonists can also activate PPARβ/δ and inhibit VSMC
phenotypic transformation. Although the PPARβ/δ ligand L-165041
induces cell cycle arrest in VSMCs and inhibits their proliferation
and migration (18), PPARβ/δ
agonists are still in the preclinical research stage. Further
studies are required to account for the beneficial mechanisms and
to examine the effects of additional PPARβ/δ agonists. Nimodipine
is the standard and main treatment of patients with aneurysmal SAH
as it has been demonstrated to have a statistically significant
beneficial impact on clinical outcome (61). Nimodipine prevents increases in
intracellular calcium by blocking dihydropyridine sensitive
(L-type) calcium channels (62),
but its mechanism of effect in patients with SAH is unknown. It
does not appear to reduce angiographically detectable CVS. This
suggests that studies should focus more on inhibiting vascular
pathological changes following SAH.
An increasing number of studies have explored the
mechanisms of CVS following SAH, whereas the molecular mechanism
underlying it remains largely speculative. Both basic and clinical
researches focus on inflammation, disruption of blood-brain
barrier, neuronal cell death, apoptosis, endothelial injury during
in CVS course following SAH (1,2,63).
The cerebral vascular neural network, which includes VSMCs, is
involved in the pathophysiology of SAH (16). In addition, other studies
demonstrate that unbalanced contractile/synthetic VSMC phenotype
affects the size of the cerebral arteries and alters brain swelling
or cerebral edema (44,45,64). To date, however, few studies have
directly addressed the relationship between phenotype conversion of
VSMCs and CVS. The present study focused on the vascular
pathological changes induced by the phenotypic transformation of
VSMCs following SAH due to its association with CVS. There is more
widespread distribution and content of PPARβ/δ in the central
nervous system (19), however
little is known about the exact function of PPARβ/δ in central
nervous system disease. Similarly, no direct evidence indicates
that PPAR β/δ serves an active role in regulating the phenotypic
transformation of VSMCs following SAH. The present authors have
reported that PPARβ/δ inhibits hemoglobin-induced phenotypic
transformation of cultured VSMCs in vitro (38). The present study mainly explored
the mechanism of PPARβ/δ regulating VSMCs transformation and CVS
through in vivo experiments to provide ideas for future
clinical transformation research.
In conclusion, the present study demonstrated that
the phenotypic transformation of VSMC was involved in the
pathophysiology of CVS in experimental SAH, PPARβ/δ positively
regulated VSMC phenotypic modulation and CVS, partly via
suppression of ERK activation and downregulation of p-Elk-1 and
p-p90RSK expression levels. Further studies should be implemented
to evaluate whether PPARβ/δ would increase cerebral blood flow. The
results of the present study may provide new evidence on the
treatment of clinical vascular damage following SAH and clinical
translation.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation for Youth of China (grant no. 81801137),
the Jiangsu Provincial Medical Key Talent Grant (grant no.
ZDRCA2016040) and a grant (grant no. SYS2019045) from the Suzhou
Government.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HZ and CM designed the research protocols. LX and YL
performed the experiments. JW, LX and GC analyzed the study data.
LX and JW wrote the manuscript. HZ and CM confirm the authenticity
of all the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Animal Experiments Ethic Committee at The First Affiliated Hospital
of Soochow University (Suzhou, China; approval no. 2018-094).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pluta RM, Hansen-Schwartz J, Dreier J,
Vajkoczy P, Macdonald RL, Nishizawa S, Kasuya H, Wellman G, Keller
E, Zauner A, et al: Cerebral vasospasm following subarachnoid
hemorrhage: Time for a new world of thought. Neurol Res.
31:151–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen S, Feng H, Sherchan P, Klebe D, Zhao
G, Sun X, Zhang J, Tang J and Zhang JH: Controversies and evolving
new mechanisms in subarachnoid hemorrhage. Prog Neurobiol.
115:64–91. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rothoerl RD and Ringel F: Molecular
mechanisms of cerebral vasospasm following aneurysmal SAH. Neurol
Res. 29:636–642. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ciurea AV, Palade C, Voinescu D and Nica
DA: Subarachnoid hemorrhage and cerebral vasospasm-literature
review. J Med Life. 6:120–125. 2013.PubMed/NCBI
|
|
5
|
Crowley RW, Medel R, Kassell NF and Dumont
AS: New insights into the causes and therapy of cerebral vasospasm
following subarachnoid hemorrhage. Drug Discov Today. 13:254–260.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fassbender K, Hodapp B, Rossol S, Bertsch
T, Schmeck J, Schütt S, Fritzinger M, Horn P, Vajkoczy P, Kreisel
S, et al: Inflammatory cytokines in subarachnoid hemorrhage:
Association with abnormal blood flow velocities in basal cerebral
arteries. J Neurol Neurosurg Psychiatry. 70:534–537. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Osuka K, Watanabe Y, Yamauchi K, Nakazawa
A, Usuda N, Tokuda M and Yoshida J: Activation of the JAK-STAT
signaling pathway in the rat basilar artery after subarachnoid
hemorrhage. Brain Res. 1072:1–7. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dumont AS, Dumont RJ, Chow MM, Lin CL,
Calisaneller T, Ley KF, Kassell NF and Lee KS: Cerebral vasospasm
after subarachnoid hemorrhage: Putative role of inflammation.
Neurosurgery. 53:123–135. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cheng MF, Song JN, Li DD, Zhao YL, An JY,
Sun P and Luo XH: The role of rosiglitazone in the proliferation of
vascular smooth muscle cells after experimental subarachnoid
hemorrhage. Acta Neurochir (Wien). 156:2103–2109. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kolias AG, Sen J and Belli A: Pathogenesis
of cerebral vasospasm following aneurysmal subarachnoid hemorrhage:
Putative mechanisms and novel approaches. J Neurosci Res. 87:1–11.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohkuma H, Tsurutani H and Suzuki S:
Changes of beta-actin mRNA expression in canine vasospastic basilar
artery after experimental subarachnoid hemorrhage. Neurosci Lett.
311:9–12. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Davis-Dusenbery BN, Wu C and Hata A:
Micromanaging vascular smooth muscle cell differentiation and
phenotypic modulation. Arterioscler Thromb Vasc Biol. 31:2370–2377.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rensen SS, Doevendans PA and van Eys GJ:
Regulation and characteristics of vascular smooth muscle cell
phenotypic diversity. Neth Heart J. 15:100–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Beamish JA, He P, Kottke-Marchant K and
Marchant RE: Molecular regulation of contractile smooth muscle cell
phenotype: Implications for vascular tissue engineering. Tissue Eng
Part B Rev. 16:467–491. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shimamura N and Ohkuma H: Phenotypic
transformation of smooth muscle in vasospasm after aneurysmal
subarachnoid hemorrhage. Transl Stroke Res. 5:357–364. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang JH, Badaut J, Tang J, Obenaus A,
Hartman R and Pearce WJ: The vascular neural network-a new paradigm
in stroke pathophysiology. Nat Rev Neurol. 8:711–716. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang J, Wang L, Fu W, Wang C, Guo D,
Jiang J and Wang Y: Smooth muscle cell phenotypic diversity between
dissected and unaffected thoracic aortic media. J Cardiovasc Surg
(Torino). 54:511–521. 2013.PubMed/NCBI
|
|
18
|
Lim HJ, Lee S, Park JH, Lee KS, Choi HE,
Chung KS, Lee HH and Park HY: PPAR delta agonist L-165041 inhibits
rat vascular smooth muscle cell proliferation and migration via
inhibition of cell cycle. Atherosclerosis. 202:446–454. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Moreno S, Farioli-Vecchioli S and Cerù MP:
Immunolocalization of peroxisome proliferator-activated receptors
and retinoid X receptors in the adult rat CNS. Neuroscience.
123:131–145. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ehrenborg E and Krook A: Regulation of
skeletal muscle physiology and metabolism by peroxisome
proliferator-activated receptor delta. Pharmacol Rev. 61:373–393.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim HJ, Ham SA, Kim SU, Hwang JY, Kim JH,
Chang KC, Yabe-Nishimura C, Kim JH and Seo HG: Transforming growth
factor-beta1 is a molecular target for the peroxisome
proliferator-activated receptor delta. Circ Res. 102:193–200. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blenis J: Signal transduction via the MAP
kinases: Proceed at your own RSK. Proc Natl Acad Sci USA.
90:5889–5892. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Frödin M and Gammeltoft S: Role and
regulation of 90 kDa ribosomal S6 kinase (RSK) in signal
transduction. Mol Cell Endocrinol. 151:65–77. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kawai-Kowase K and Owens GK: Multiple
repressor pathways contribute to phenotypic switching of vascular
smooth muscle cells. Am J Physiol Cell Physiol. 292:C59–C69. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kang YH, Yang IJ and Shin HM: Herbal
formula HMC05 prevents human aortic smooth muscle cell migration
and proliferation by inhibiting the ERK1/2 MAPK signaling cascade.
J Nat Med. 66:177–184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
MacArthur Clark JA and Sun D: Guidelines
for the ethical review of laboratory animal welfare People's
Republic of China national standard GB/T 35892-2018 (issued 6
February 2018 effective from 1 September 2018). Animal Model Exp
Med. 3:103–113. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Muroi C, Fujioka M, Okuchi K, Fandino J,
Keller E, Sakamoto Y, Mishima K, Iwasaki K and Fujiwara M: Filament
perforation model for mouse subarachnoid hemorrhage:
Surgical-technical considerations. Br J Neurosurg. 28:722–732.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sugawara T, Ayer R, Jadhav V and Zhang JH:
A new grading system evaluating bleeding scale in filament
perforation subarachnoid hemorrhage rat model. J Neurosci Methods.
167:327–334. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Suzuki H, Hasegawa Y, Chen W, Kanamaru K
and Zhang JH: Recombinant osteopontin in cerebral vasospasm after
subarachnoid hemorrhage. Ann Neurol. 68:650–660. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen Y, Zhang Y, Tang J, Liu F, Hu Q, Luo
C, Tang J, Feng H and Zhang JH: Norrin protected blood-brain
barrier via frizzled-4/β-catenin pathway after subarachnoid
hemorrhage in rats. Stroke. 46:529–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Teng Z, Jiang L, Hu Q, He Y and Guo Z, Wu
Y, Huang Z, Cao F, Cheng C, Sun X and Guo Z: Peroxisome
proliferator-activated receptor β/δ alleviates early brain injury
after subarachnoid hemorrhage in rats. Stroke. 47:196–205. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ostrowski RP, Colohan AR and Zhang JH:
Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat
model of subarachnoid hemorrhage. J Cereb Blood Flow Metab.
25:554–571. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choudhri TF, Hoh BL, Solomon RA, Connolly
ES Jr and Pinsky DJ: Use of a spectrophotometric hemoglobin assay
to objectively quantify intracerebral hemorrhage in mice. Stroke.
28:2296–2302. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carpenter RC, Miao L, Miyagi Y, Bengten E
and Zhang JH: Altered expression of P(2) receptor mRNAs in the
basilar artery in a rat double hemorrhage model. Stroke.
32:516–522. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen D, Tang J, Khatibi NH, Zhu M, Li Y,
Wang C, Jiang R, Tu L and Wang S: Treatment with Z-ligustilide, a
component of Angelica sinensis, reduces brain injury after a
subarachnoid hemorrhage in rats. J Pharmacol Exp Ther. 337:663–672.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan R, Enkhjargal B, Camara R, Yan F, Gong
L, Shengtao Yao, Tang J, Chen Y and Zhang JH: Critical role of
EphA4 in early brain injury after subarachnoid hemorrhage in rat.
Exp Neurol. 296:41–48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang H, Jiang L, Guo Z, Zhong J, Wu J, He
J, Liu H, He Z, Wu H, Cheng C and Sun X: PPARβ/δ, a novel regulator
for vascular smooth muscle cells phenotypic modulation and vascular
remodeling after subarachnoid hemorrhage in rats. Sci Rep.
7:452342017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Maddahi A, Povlsen GK and Edvinsson L:
Regulation of enhanced cerebrovascular expression of
proinflammatory mediators in experimental subarachnoid hemorrhage
via the mitogen-activated protein kinase kinase/extracellular
signal-regulated kinase pathway. J Neuroinflammation. 9:2742012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Danura H, Schatlo B, Marbacher S, Kerkeni
H, Diepers M, Remonda L, Fathi AR and Fandino J: Acute angiographic
vasospasm and the incidence of delayed cerebral vasospasm:
Preliminary results. Acta Neurochir Suppl. 120:187–190.
2015.PubMed/NCBI
|
|
41
|
Shimamura N, Munakata A and Ohkuma H:
Current management of subarachnoid hemorrhage in advanced age. Acta
Neurochir Suppl. 110:151–155. 2011.PubMed/NCBI
|
|
42
|
Chou SH, Smith EE, Badjatia N, Nogueira
RG, Sims JR II, Ogilvy CS, Rordorf GA and Ayata C: A randomized,
double-blind, placebo-controlled pilot study of simvastatin in
aneurysmal subarachnoid hemorrhage. Stroke. 39:2891–2893. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Edvinsson LI and Povlsen GK: Vascular
plasticity in cerebrovascular disorders. J Cereb Blood Flow Metab.
31:1554–1571. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ohkuma H, Suzuki S and Ogane K: Phenotypic
modulation of smooth muscle cells and vascular remodeling in
intraparenchymal small cerebral arteries after canine experimental
subarachnoid hemorrhage. Neurosci Lett. 344:193–196. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wu J, Zhang Y, Yang P, Enkhjargal B,
Manaenko A, Tang J, Pearce WJ, Hartman R, Obenaus A, Chen G and
Zhang JH: Recombinant osteopontin stabilizes smooth muscle cell
phenotype via integrin receptor/integrin-linked kinase/rac-1
pathway after subarachnoid hemorrhage in rats. Stroke.
47:1319–1327. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Smyth LCD, Rustenhoven J, Scotter EL,
Schweder P, Faull RL, Park TI and Dragunow M: Markers for human
brain pericytes and smooth muscle cells. J Chem Neuroanat.
92:48–60. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Piqueras L, Reynolds AR, Hodivala-Dilke
KM, Alfranca A, Redondo JM, Hatae T, Tanabe T, Warner TD and
Bishop-Bailey D: Activation of PPARbeta/delta induces endothelial
cell proliferation and angiogenesis. Arterioscler Thromb Vasc Biol.
27:63–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee CH, Chawla A, Urbiztondo N, Liao D,
Boisvert WA, Evans RM and Curtiss LK: Transcriptional repression of
atherogenic inflammation: Modulation by PPARdelta. Science.
302:453–457. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chang CZ, Wu SC and Kwan AL: Glycyrrhizin
attenuates proinflammatory cytokines through a peroxisome
proliferator-activated receptor-γ-dependent mechanism and
experimental vasospasm in a rat model. J Vasc Res. 52:12–21. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hamaya R, Ogawa M, Suzuki J, Kobayashi N,
Hirata Y, Nagai R, Komuro I and Isobe M: A selective peroxisome
proliferator-activated receptor-β/δ agonist attenuates neointimal
hyperplasia after wire-mediated arterial injury. Expert Opin.
Investig. Drugs. 22:1095–1106. 2013.PubMed/NCBI
|
|
51
|
Pipes GC, Creemers EE and Olson EN: The
myocardin family of transcriptional coactivators: Versatile
regulators of cell growth, migration, and myogenesis. Genes Dev.
20:1545–1556. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Parmacek MS: Myocardin-related
transcription factors: Critical coactivators regulating
cardiovascular development and adaptation. Circ Res. 100:633–644.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Takata Y, Liu J, Yin F, Collins AR, Lyon
CJ, Lee CH, Atkins AR, Downes M, Barish GD, Evans RM, et al:
PPARdelta-mediated antiinflammatory mechanisms inhibit angiotensin
II-accelerated atherosclerosis. Proc Natl Acad Sci USA.
105:4277–4282. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Salvadó L, Barroso E, Gómez-Foix AM,
Palomer X, Michalik L, Wahli W and Vázquez-Carrera M: PPARβ/δ
prevents endoplasmic reticulum stress-associated inflammation and
insulin resistance in skeletal muscle cells through an
AMPK-dependent mechanism. Diabetologia. 57:2126–2135. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hsieh HL, Wu CY and Yang CM: Bradykinin
induces matrix metalloproteinase-9 expression and cell migration
through a PKC-delta-dependent ERK/Elk-1 pathway in astrocytes.
Glia. 56:619–632. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang Z, Wang DZ, Hockemeyer D, McAnally J,
Nordheim A and Olson EN: Myocardin and ternary complex factors
compete for SRF to control smooth muscle gene expression. Nature.
428:185–189. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Toral M, Romero M, Pérez-Vizcaíno F,
Duarte J and Jiménez R: Antihypertensive effects of peroxisome
proliferator-activated receptor-β/δ activation. Am J Physiol Heart
Circ Physiol. 312:H189–H200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Barroso E, Eyre E, Palomer X and
Vázquez-Carrera M: The peroxisome proliferator-activated receptor
β/δ (PPARβ/δ) agonist GW501516 prevents TNF-α-induced NF-κB
activation in human HaCaT cells by reducing p65 acetylation through
AMPK and SIRT1. Biochem Pharmacol. 81:534–543. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zarzuelo MJ, Jiménez R, Galindo P, Sánchez
M, Nieto A, Romero M, Quintela AM, López-Sepúlveda R, Gómez-Guzmán
M, Bailón E, et al: Antihypertensive effects of peroxisome
proliferator-activated receptor-β activation in spontaneously
hypertensive rats. Hypertension. 58:733–743. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Law RE, Goetze S, Xi XP, Jackson S, Kawano
Y, Demer L, Fishbein MC, Meehan WP and Hsueh WA: Expression and
function of PPARgamma in rat and human vascular smooth muscle
cells. Circulation. 101:1311–1318. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dorhout Mees SM, Rinkel GJE, Feigin VL,
Algra A, van den Bergh WM, Vermeulen M and van Gijn J: Calcium
antagonists for aneurysmal subarachnoid hemorrhage. Stroke.
39:514–515. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Pandey AS, Elias AE, Chaudhary N, Thompson
BG and Gemmete JJ: Endovascular treatment of cerebral vasospasm:
Vasodilators and angioplasty. Neuroimaging Clin N Am. 23:593–604.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Hansen-Schwartz J: Cerebral vasospasm: A
consideration of the various cellular mechanisms involved in the
pathophysiology. Neurocrit Care. 1:235–246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Edvinsson L, Larsen SS, Maddahi A and
Nielsen J: Plasticity of cerebrovascular smooth muscle cells after
subarachnoid hemorrhage. Transl Stroke Res. 5:365–376. 2014.
View Article : Google Scholar : PubMed/NCBI
|