Tumor initiation and development depend on
epithelial tissue and are closely associated with the surrounding
tumor microenvironment (TME). Although no consensus has been
reached concerning the definition of the TME, which is mainly
composed of tumor cells, peripheral immune and inflammatory cells,
tumor-related fibroblasts, peripheral interstitial tissue,
microvessels and various cytokines and chemokines, it has become an
important research focus to understand tumor development and to
identify target for cancer treatment (1). Metabolic transformation is not only
a sign of cancer, but also a key goal of cancer treatment. Due to
the influence of cancer cells, the physiological characteristics of
the TME (such as metabolism, secretion and immunity, amongst
others) are no longer regulated by the body, which has a
far-reaching impact on the progress of cancer. Remodeling of the
TME has therefore become a cancer treatment strategy. However, how
changes in the TME metabolic levels affect cancer metabolism and
behavior remains unclear (2,3).
Moreover, whether adipocytes (cells abundant in the TME) play a key
role in the malignant progression of tumors remains to be
determined. Considering the relationship between tumor progression
and obesity, adipocytes are considered to be essential components
of the TME (4). Currently, from
the available in vitro, in vivo and clinical data, it can be
demonstrated that the characteristics of adipocyte-derived factors
change during tumor progression and tumor cells can also
significantly affect the surrounding adipocytes. When peritumoral
adipocytes display an altered phenotype and specific biological
features (such as the decrease in differentiation markers of mature
adipocytes, production of a large number of fat-derived factors and
the promotion of the metabolic reprogramming of cancer cells), they
are termed cancer-associated adipocytes (CAAs) (5). CAAs are considered to serve an
important role in the TME. Previous studies reported that
adipocytes co-cultivated with cancer cells exhibited considerable
morphological and functional alterations, including reduced lipid
content and decreased adipocyte markers, such as adiponectin,
leptin and fatty acid-binding protein (FABP)2 (6–8).
Overexpression of interleukin (IL)-6, IL-1 and matrix
metalloproteinase (MMP)-11 is also detected in activated
adipocytes, which are also referred to as CAAs. Notably, in a tumor
subset, growth and metastasis predominantly occur adjacent to
adipocytes, for example, in breast cancer (BC), or at anatomical
sites where cancer cells are close to the adipose tissue (AT),
including in gastric, colorectal and ovarian cancer (9). Adipocytes participate in a highly
complex inflammatory cycle that is regulated by tumor cells to
promote tumor development. Moreover, the malignant functions of
CAAs may amplify this cycle and therefore CAAs may serve as an
obstacle to tumor treatment. A previous study has demonstrated that
tumor drug resistance is an important driver of disease progression
and could be a potential target for new therapies. The role of
adipocytes in tumor drug resistance has been overlooked. CAAs cause
drug resistance in oncological treatments, including chemotherapy,
radiotherapy, hormone therapy and immunotherapy, which may lead to
the persistence of tumor remnants and increase the risk of tumor
recurrence (8). However, to the
best of our knowledge the role of CAAs and how they evolve from
adipocytes during tumor progression has previously not been
described in detail. The present review has focused on the
adipocyte-cancer cell circle in terms of metabolism, adipokines and
drug resistance. The potential mechanisms underlying the dynamic
communication between CAAs and numerous types of cancer, including
BC, ovarian cancer and colorectal cancer (CRC), especially in the
co-occurrence of obesity, which may result in neoteric therapy,
have also been investigated.
AT comprises a highly complex and heterogeneous
group of cellular components, mostly consisting of adipocytes.
There are also numerous other types of stromal cells in AT,
including endothelial cells, pericytes, macrophages and adipocyte
progenitor cells. Both adipocytes and stromal vascular cells in AT
contribute with several other factors towards tumor growth and
maintenance (10–12). AT is an important and complex
tissue that regulates energy balance and is distributed in almost
all compartments of the human body. Histologically, there are three
main types of AT: i) White AT (WAT), which accounts for >95% of
the fat mass; ii) brown AT, which accounts for 1–2% of the fat
mass; and iii) beige AT, which is difficult to quantify as it is
scattered under the skin near the spine and clavicle in adults and
cannot be taken out as a whole. WAT is the most dynamic AT in the
human body, accounting for 2–70% of the body weight (13). Mature adipocytes are the main cell
type in WAT, containing a small number of mitochondria (14). Mature adipocytes function by
secreting various adipocytokines, and are characterized by a round
shape with only one large lipid droplet (15). Compared with mature adipocytes,
CAAs possess an irregular morphology, with small lipid droplets and
a small volume. Furthermore, tumor-associated adipocytes usually
have some changes (such as differentiation markers), as later
described. Following lipolysis, differentiation markers of mature
adipocytes decrease, such as adipocyte p2 and FABP4 (16,17). In the presence of cancer cells,
especially at the tumor invasive front, CAAs generate
fibroblast-like cells named adipocyte-derived fibroblasts after
undergoing a process via which adipocytes decompose their lipids
into glycerol and free fatty acids, and adopting a phenotypic
change. These adipocyte-derived fibroblasts, as part of the
cancer-associated fibroblast (CAF) population, are also involved in
the malignant progression of tumors. It has also been indicated
that CAAs may represent an intermediate form through which CAFs can
arise (18). Moreover, CAAs have
also been found to produce a number of adipose-derived factors via
endocrine and paracrine signaling pathways, including chemokine
(C-C motif) ligand (CCL)2, CCL5, IL-6, tumor necrosis factor α
(TNFα), vascular endothelial growth factor (VEGF) and leptin;
however, lower levels of adiponectin are produced (5,6).
In terms of metabolic changes, CAAs promote numerous catabolic
processes releasing high-energy metabolites, including lactic acid,
pyruvic acid, free fatty acids and ketone bodies (5). Furthermore, CAAs provide sufficient
energy for cancer cells by secreting exogenous fatty acids, leading
to the metabolic reprogramming of cancer cells (19). Tumor cell-derived signaling
molecules induce the lipolysis of adipocytes and promote the
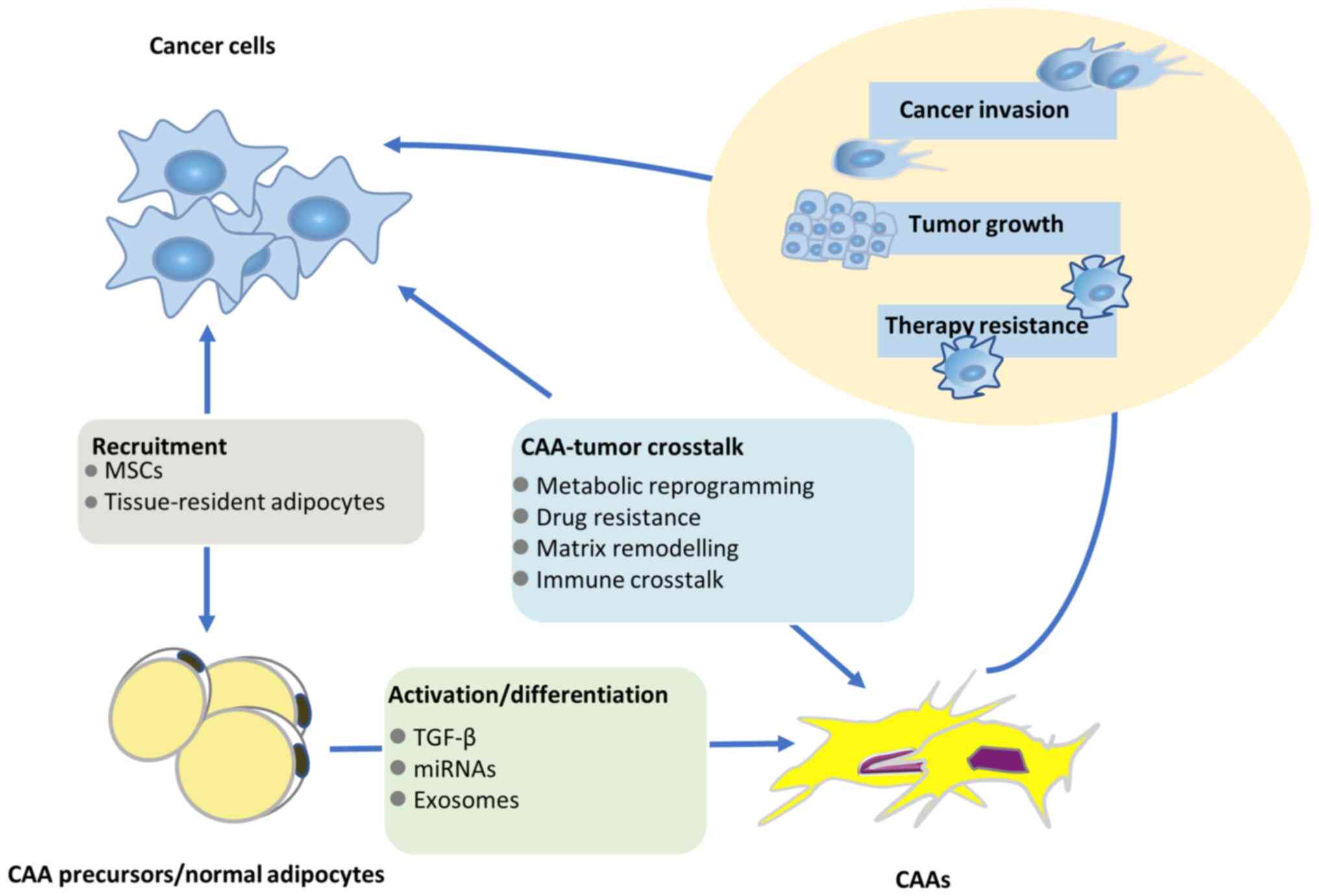
evolution of adipocytes to CAAs (20). Fig.
1 displays the complex relationship among tumors, adipocytes
and CAAs. Therefore, CAAs are thought to serve a paramount role in
tumor invasion and progression via the endocrine and paracrine
signaling pathways.
Obesity and CRC are both global health issues and
epidemiological data has demonstrated that obesity is positively
correlated with CRC (21).
Obesity serves a direct and independent role in the development of
CRC. Previous studies have reported that cancer cells can increase
carnitine palmitoyltransferase 1A (CPT1A) expression by interacting
with CAAs, which in turn upregulates mitochondrial fatty acid
oxidation and enables cancer cells to tolerate a hypoxic
environment (21,22). Furthermore, crosstalk between
tumor resident adipocytes and CRC cells is considered to be a
contributing factor in promoting cancer progression (23). Adipocyte-derived medium can
directly promote CRC progression, which also further suggests that
CAAs are a positive factor in carcinogenesis (24). CAAs serve a vital role in
promoting inflammation and angiogenesis. By comparing inflammation
and angiogenesis in lean and obese patients with or without CRC, it
was determined that the plasma levels of proinflammatory and
angiogenic factors were at least partially elevated in obese
patients with CRC. This study therefore revealed that peritumoral
visceral AT is crucial for the development of CRC (25). Furthermore, to avoid the influence
of individual differences on the experiment, in vitro
experiments were conducted to compare the inflammatory reactions
adjacent to and distant from the tumor site in the same patient. It
was confirmed that the difference was due to the special influence
of the tumor environment rather than due to the differences between
individuals. However, this experimental result has not been
confirmed in vivo (26).
Another study demonstrated that the average levels of
proinflammatory cytokines IL-6, IL-4 and granulocyte-macrophage
colony stimulating factor in patients with obesity and CRC, were
significantly higher than those in lean patients with CRC and
patients with obesity only. The levels of other pro-inflammatory
markers in the plasma displayed different trends; for example, IL-8
was only upregulated in patients with CRC and not obesity, whereas
interferon γ and TNFα were upregulated in patients with obesity and
not CRC. Plasma levels of numerous proinflammatory cytokines,
including IL-1α, IL-6, TNFα, CCL-2 and plasminogen activator
inhibitor-1 were increased in both patients with obesity and
patients with CRC; however, there was no significant difference
between obese and lean patients. Plasma VEGF levels were also
upregulated in response to both CRC and obesity, which is
consistent with results reported in a previous study (27). It has also been reported that the
expression of inducible nitric oxide synthase (iNOS) increases
under inflammatory conditions, such as pancreatitis or obesity
(28). Under inflammatory
conditions, nitric oxide (NO) is produced by iNOS, an enzyme
primarily expressed by macrophages and to a lesser extent by
adipocytes. In previous studies, the concentrations of nitrite and
nitrate in medium were used as indicators to evaluate the release
of NO under inflammatory conditions (27,28). Consequently, the inflammatory
characteristics of adipocytes away from and around the tumor site
in both lean patients and patients with obesity and CRC were
compared. The release of nitrite and nitrate in adipocytes around
the tumor site was increased in lean patients with CRC compared
with that in patients with obesity. However, adipocytes far from
the tumor site did not show increased nitrite and nitrate
secretions (29).
Previous studies have demonstrated that the TME has
pro-inflammatory properties, which may contribute to the occurrence
and progression of tumors (30).
It can therefore be hypothesized that these pro-inflammatory
factors in the TME may not only be due to increased macrophage
infiltration as previously reported (31), but may also be derived from
tumor-associated adipocytes. It has also been reported that the
expression levels of cyclooxygenase-2 (COX-2) and peroxisome
proliferator-activated receptor (PPAR)-γ are increased in the
cancerous tissues of patients with CRC. Similarly, the gene
expression of COX-2 and PPAR-γ is increased in tumor-associated
adipocytes. A previous study has reported that the expression
levels of COX-2 and PPAR-γ increase in the peritumoral tissues of
patients with CRC. The gene expression levels of COX-2 and PPAR-γ
are also increased in tumor-associated adipocytes (32).
Overall, the aforementioned studies indicate that
the plasma levels of different pro-inflammatory and angiogenic
factors in patients with CRC are increased, some of which may come
from tumor-associated adipocytes. Tumor-associated adipocytes are
considered a vital source of pro-inflammatory and angiogenic
factors, which may affect the progress of tumor biology and the
clinical prognosis of the patients. A recent study has reported
that an imbalance of intestinal microbiota caused by obesity
increases harmful microbiota and metabolites and decreases
beneficial microbes (including Akkermansia muciniphila) and
metabolites (short-chain fatty acids) (33). In bile acid metabolism, bile acids
promote the progression of CRC, especially in obese patients. Bile
acid-dependent inhibition of the farnesoid X receptor (FXR) can
promote the occurrence of CRC, which provides a theoretical basis
for FXR to become an antitumor target in CRC in the future
(33). The potential mechanism of
obesity-promoting CRC therefore needs to be explored further.
BC is one of the most common cancer types worldwide
and is the second highest cause of cancer-related mortality in
women (34). The TME is a
heterogeneous ecosystem composed of infiltrating immune cells,
mesenchymal support cells and a matrix that promotes tumor
progression. Adipocytes are the main cellular component of the BC
microenvironment. Recent evidence demonstrates that adipocytes
promote tumor progression via the interaction and dynamic
communication between tumor cells and adipocytes (35). AT serves a key role as an energy
reservoir, in which endocrine cells can produce various bioactive
substances (14). A recent study
has determined that the characteristics of adipocyte-derived
cytokines change during tumor progression, which has been confirmed
in human BC samples (36).
Furthermore, an increasing number of studies have confirmed that
adipocytes adjacent to invasive cancer cells, namely CAAs,
participate in BC progression (17,36). Moreover, inflammatory factors
secreted by CAAs can influence the behavior of BC cells, changing
the characteristics and phenotypes of BC cells to increase
invasiveness (36). Therefore, it
can be hypothesized that abnormal AT, especially that adjacent to
BC, is a highly complex participant in the interaction between the
tumor and microenvironment and is regulated by BC cells. This
interaction may be amplified as a result of the malignant function
of CAAs (16).
In the microenvironment of BC, the expression and
secretion profiles of CAA-mediated inflammatory factors are
altered. The secretion of CCL2, CCL5 (36), IL-1β, IL-6 (35), TNFα, VEGF and leptin (37) is increased in the BC
microenvironment, further promoting the proliferation, invasion and
angiogenesis of tumor cells. Therefore, CAAs promote the
tumorigenesis, invasion and metastasis of BC. Furthermore, CAAs in
the treatment of BC such as chemotherapy, radiotherapy, hormone
therapy and immunotherapy can result in tumor resistance, leading
to the persistence of residual tumors and an increased risk of
tumor recurrence (16). Moreover,
when mature adipocytes transform into CAAs, tumor cells become
metabolic parasites by ingesting metabolites such as ketones,
pyruvate, fatty acids and lactic acid from CAAs. Consequently,
tumor cells transition from relying on anaerobic glycolysis to
preferentially using fatty acids for fatty acid oxidation to
provide energy for themselves (38–40). The regulatory mechanisms of CAAs
in BC are complex, including adipokine secretion, metabolic
reprogramming and extracellular matrix (ECM) remodeling. Several
hypotheses have been proposed stating that obesity promotes the
development and progression of BC (39,40). However, the underlying mechanisms
remain unclear due to a lack of sufficient models to study the
relationship between obesity and BC.
The biological characteristics of ovarian cancer are
different from those of other types of the cancer, as ovarian
cancer can exhibit hematogenous metastasis, which is rare and often
limited to the AT-rich omentum (41). Although experiments have confirmed
that the adipocytes in the omentum are the key to inducing ovarian
cancer cell omental metastasis, the specific mechanism remains
unclear. According to recent studies, secretory acidic cysteine
rich protein (SPARC) may serve a role in this process (42,43). SPARC is an ECM protein, which
serves an important role in maintaining the homeostasis of the
surrounding tissue environment. SPARC can serve a role in tissue
remodeling, including in the regulation of adipocyte
differentiation. SPARC can also inhibit adipocyte production,
reduce the production of CAAs, interfere with the interaction
between cancer cells and mesothelial cells, and normalize the TME
(42). Previous studies have
demonstrated that the protein and lipid metabolomics of ovarian
cancer cells change significantly following coculture with
adipocytes. Among them, fat chaperone protein FABP4 was induced in
tumor cells by adipocytes, and makes the cancer cells more invasive
and metastatic, which serves a key regulatory role in changes to
lipid metabolism in cancer cells. Furthermore, targeting FABP4 can
reduce the metastasis of cancer cells to the greater omentum and
improves cell sensitivity to carboplatin (42–44). Another study demonstrated that the
selective metastasis of ovarian cancer to the greater omentum is
caused by the CCL2 receptor, C-C chemokine receptor type (CCR)2, on
ovarian cancer cells (44). CCL-2
is produced by the binding of omental adipocytes to homologous
receptor CCR2 on ovarian cancer cells, which promotes cancer cell
migration and omental metastasis by activating related signaling
pathways such as the phosphatidylinositol 3-kinase (PI3K)/protein
kinase B (AKT)/mammalian target of rapamycin (mTOR) signaling
pathway. Metformin can specifically inhibit the secretion of CCL-2
and therefore serves a role in the treatment of ovarian cancer
(44). To help understand the
cause of omental metastasis in ovarian cancer, a previous study has
verified that primary omental adipocytes can induce the
proliferation and invasion of ovarian cancer cells both in
vivo and in vitro (45). CD36 is a transmembrane
glycoprotein belonging to the class B scavenger receptor family.
CD36 promotes the uptake of fatty acids and cholesterol and
transmits intracellular signals to regulate fatty acid metabolism
in tumor cells. Furthermore, CD36 serves a crucial role in the
bioenergy adaptation of OVCA cells in an adipocyte-rich
microenvironment. Omental adipocytes can reprogram tumor metabolism
by upregulating oocyte CD36 expression (46). Functional studies have
demonstrated that miR-21 combined with apoptotic protease
activating factor-1, a new direct therapeutic target, could inhibit
the apoptosis of ovarian cancer cells and induce chemotherapy
resistance (47). Furthermore,
evidence suggests that the tumor-promoting effect of CAAs results
from both systemic and local effects of hormones involved in lipid
hemostasis mediated by direct contact, paracrine factors or both
(47). In the microenvironment,
CAA will affect the transformation of the metabolic phenotype of
ovarian cancer, which plays an important role in the occurrence and
development of ovarian cancer (48).
Although there is increasing evidence to suggest
that CAAs are positively correlated with certain types of cancer,
the underlying molecular mechanisms remain unclear. CAA induces
abnormal metabolic reprogramming (among which lipids serve an
unusual role in the process of tumor progression, as well as being
a metabolic substrate), chemoresistance and the secretion of
various adipokines, which may serve important roles in the complex
process of carcinogenesis.
Metabolic reprogramming to meet the bioenergetic and
biosynthetic demands, and maintain redox balance and cell
proliferation, is a hallmark of cancer (49). From benign growth to malignant and
invasive lesions, the changes of cell metabolic requirements are
complex and dynamic. With uncontrolled proliferation, cancer cells
need increasing amounts of biomolecules to produce new sister
cells. The metabolic substrates of these cancer cells change when
they invade the surrounding stromal tissue and interact with new
cell types. Furthermore, when tumors metastasize, cancer cells face
a series of new metabolic challenges in different environments.
Therefore, cancer cells need to adjust their metabolic programs to
adapt to a constantly changing situation.
Adipocytes adapt to their own metabolic processes
through dynamic interactions with tumors, which further supports
the proliferation of tumor cells (50). Furthermore, to meet the extreme
energy requirements of cancer cells, CAA metabolic reprogramming
that occurs involves alterations in the metabolism of
macronutrients, including carbohydrates, lipids and amino acids
(51). In terms of glycolysis,
the tricarboxylic acid cycle, amino acids, nucleotides and lipid
metabolism, the metabolic processes in cancerous and healthy
tissues are substantially different (39). As described by the ‘Warburg
effect’, cancer cells prefer to produce adenosine triphosphate by
glycolysis despite the presence of oxygen (52,53). However, in order to adapt to the
environmental changes of hypoxia and acidosis, the metabolic
pattern of cancer cells changes to rely on lipid metabolism,
especially fatty acid oxidation (54). Furthermore, cancer exhibits
heterogeneity in genetic and microenvironmental parameters that
influence cell metabolism. For instance, tumor metabolism
influences, and is influenced by, the metabolite composition of the
TME, while the interorgan and intratumor microenvironments define
the metabolic properties of tumor cells. The TME serves an
irreplaceable role in tumor metabolism. The ‘reverse Warburg
effect’ describes how glycolytic metabolism in the cancer-related
matrix supports adjacent cancer cells. The catabolic products
include lactate monocarboxylate, pyruvate and ketone body. The
transfer of these catabolic products can induce metabolic coupling
of interstitial carcinoma, inducing cancer cells to produce ATP,
increase their proliferation and reduce cell death. The ‘reverse
Warburg effect’ is also important when cancer cells utilize the
energy generated from stromal cells in the TME (2). As one of the main components of the
TME, tumor cells inevitably use free fatty acids and glycerol
produced by fat cell catabolism as energy sources (55,56). Tumor lipid metabolism is regulated
by genetic and epigenetic changes in the tumor cells, which affects
the process of receiving lipid from CAAs as a metabolic substrate.
Fatty acids can be released from lipid droplets via the adipose
triglyceride lipase (ATGL)-dependent lipolysis pathway. ATGL
expression in cancer cells is upregulated by the interaction
between adipocytes and cancer cells (38). Through the co-culture of
adipocytes and cancer cells, it has been determined that free fatty
acids in adipocytes can be transferred to cancer cells. This
process can upregulate CPT1A and other various protease expression
levels acting on the electron transfer chain, which promotes fatty
acid metabolism in cancer cells (57). Tumor cells can not only increase
the expression of ATGL to accelerate the utilization of
adipocyte-derived lipids, but also enhance the intracellular
transport of fatty acids by increasing the expression of FABP5
(58). CD36 and fatty acid
transporter-1 (FATP1) are mainly expressed in cancer cells adjacent
to AT (5). Zaoui et al
(59) reported that cancer cells
could regulate their metabolic reprogramming via exogenous fatty
acid uptake mediated by CD36 on the cell surface. Moreover,
adipocyte-induced expression of CD36 and FATP1 promotes tumor
progression by facilitating signal transduction in tumor cells
(60–62). Wang et al (63) reported the role of Janus kinase
(JAK) and signal transducer and activator of transcription 3
(STAT3) signaling pathways in regulating lipid metabolism.
Inhibition of the JAK/STAT3 signaling pathway inhibits the
expression of numerous lipid metabolism genes, including CPT1B,
which encodes a key enzyme of fatty acid oxidation (FAO).
Adipocytes also affect immune cell metabolism. Adipocyte-derived
leptin can change the metabolic pattern of CD8+ T cells
via activation of STAT3-FAO and inhibiting glycolysis, leading to
the downregulation of the effector function of CD8+ T
cells (64).
Although previous studies have shown that lipids are
mainly involved in the metabolic dynamics of the interaction
between tumor cells and stromal cells, lipids can also play an
additional role in the TME as a material source of cell membrane
synthesis and an energy source of cancer cells (45,65,66). Certain tumor-associated stromal
cells have been demonstrated to secrete autotoxin (ATX) (67). ATX hydrolyzes
lysophosphatidylcholine to lysophosphatidic acid (LPA). LPA can be
used as a signaling molecule in cancer cell mitosis and migration
(68). Furthermore,
adipose-derived stem cells and adipocytes in the TME can also
produce ATX. Once cancer cells begin to invade surrounding tissues,
inhibition of ATX has no significant effect on the growth of the
primary tumor. However, disruption of the LPA/ATX axis helps to
reduce cancer cell metastasis (69–71). Moreover, studies on metabolic
determinants supporting the metastatic potential of cancer cells
have determined that lipid metabolism is a major promoter (72). It is now widely recognized that
cancer cells with high metastatic potential often express CD36 and
that mice fed with a high-fat diet exhibit an increase in the size
and quantity of tumor lymph node metastases in a CD36-dependent
manner (73). Moreover, numerous
types of cancer, including melanoma, BC and prostate cancer, first
pass through lymphatic vessels to the proximal lymph nodes, where
lipid metabolism can be used as a driver for lymphangiogenesis
(72,73). Furthermore, a previous study on
the role of neutrophils in supporting lung colonization by
metastatic BC cells, revealed that the destruction of leukotriene
producing enzyme arachidonic acid 5-lipoxygenase, could
significantly inhibit the pre-metastasis activity of neutrophils.
These results suggested that lipid metabolism is not only involved
in the initiation of cancer cell metastasis, but also in the
colonization of peripheral metastasis sites (74).
In the TME, the ECM is composed of non-cellular
polymer proteins and helper molecules. The ECM is an important
component in the TME that determines cancer progression (75). Metastasis is the main cause of
cancer-related mortality, but the metastasis of cancer cells into a
new environment requires a cellular adaptation process. Cancer
cells can achieve their own TME homeostasis via an interaction with
the ECM. This functional adaptability needed in order to survive in
a changing environment is called cancer cell phenotypic plasticity
(76). ECM remodeling consists of
two stages (the transformation from epithelial cells to mesenchymal
cells, and the transformation from mesenchymal cells to epithelial
cells) among which the transformation from epithelial to
mesenchymal cells is established; however, there are few studies on
this transformation process (77). The reason cancer cells can
multiply indefinitely without being regulated by the body is that
they can dedifferentiate and acquire stem cell-like
characteristics. When the surrounding environment is relatively
stable, cancer cells are also in a dormant state. However, in the
case of external abnormalities, cancer cells can initiate TME
remodeling and epithelial-mesenchymal transition (EMT), obtaining a
stronger motor ability (77,78). The erosion and metastasis of
malignant tumor is a dynamic and continuous process. Tumor cells
first migrate from the primary site, invade the ECM, adhere to some
molecules in the basement membrane and intercellular matrix,
activate cells to synthesize and secrete various degrading enzymes,
as well as assist tumor cells to enter blood vessels via the ECM.
Then, tumor cells operate under the action of some factors,
penetrate the blood vessel wall to the secondary site where they
continue to proliferate and undergo metastasis. In short,
abscission, adhesion, degradation, movement and proliferation occur
through the whole process of malignant tumor erosion and
metastasis. Therefore, it is not difficult to observed that the
tumor will obtain a stronger metastatic ability when the tumor
cells undergo the EMT process (77,78). However, a previous study has
demonstrated that EMT is related to epithelial cell integration at
the distal metastatic site of cancer cells (78). The reason why ECM has such a great
impact on the TME remodeling of cancer cells remains to be fully
elucidated.
Previous studies have, however, demonstrated that
CAAs affect tumor ECM remodeling (75,79). CAA-induced TGF-β expression can
inhibit AT angiogenesis. Inhibition of angiogenesis can lead to
hypoxia and fibrosis in AT, thereby mediating the occurrence of the
EMT in BC cells (79). Adipocytes
extracted from AT around prostate tumors were found to highly
express proteins that regulate ECM structure, including TNFα,
osteopontin and MMP9 (80). CAAs
can also promote the invasiveness of renal cell carcinoma cells via
leptin (81). Furthermore,
adipocytes secrete and process type VI collagen, which provides
survival promoting signals in the early stages of breast tumor
growth. The type VI collagen cleavage product, endothelin, also
serves an important role in the subsequent occurrence and
development of BC (82). Overall,
these observations suggest that CAAs may act as proto-cancerous
stromal cells to remodel the ECM and create a tumor tolerant
environment.
CAAs in the TME serve a dynamic and complex role in
the drug response and promotion of tumor growth (83). Both adipocyte and tumor lipid
metabolism can affect the tumor drug response. CAAs provide
metabolic substrates, growth factors and cytokines to tumor cells.
CAAs can transdifferentiate into other stromal cells to change the
growth environment and adjust the drug response of the tumor. In a
variety of solid and non-solid types of cancer, adipocyte and lipid
metabolism-mediated chemotherapy resistance involves a host of
complex mechanisms (83,84). To date, there are two types of
drugs used for targeted cancer therapy: i) Drugs targeting cancer
cells; and ii) drugs targeting cellular and molecular components in
the TME. Among the drugs targeting the cellular and molecular
components of the TME, targeting tumor vessels with anti-angiogenic
drugs (AADs) has become key to the treatment of numerous types of
cancer (84). These drugs are
usually combined with conventional chemotherapy drugs (85). In the present review, mainly the
role of CAAs and lipid metabolism in anti-vascular drug resistance
is discussed. Epidemiological evidence suggests that patients with
obesity have poorer clinical outcomes than non-obese patients who
receive the same treatment (86,87). However, in general, obese patients
with cancer are in the advanced stages at the point of diagnosis
(85–87). The drug resistance properties of
advanced cancer are therefore the main reasons why it is difficult
to treat.
Previous studies have demonstrated that BC, ovarian
cancer and prostate cancer rich in AT grow faster and more
aggressively than those with less AT (88,89). Research into the reasons for this
phenomenon has revealed a number of results. Adipocyte volume is
increased in patients with obesity, resulting in an increased
distance between capillaries. Owing to the relatively low density
of microvessels the blood perfusion in the AT of patients with
obesity is insufficient (90).
Therefore, in patients with obesity, AT undergoes mild hypoxia,
which affects the cellular and molecular composition of the adipose
microenvironment. These changes inevitably affect the drug response
of tumors growing in the vicinity of obese AT and may limit the
distribution of anticancer drugs. Although tumor tissue is rich in
blood vessels, the growth of tumors in anoxic obese AT may be due
to decreased blood vessels and poor perfusion of disordered,
tortuous and leaky vessels in the tumor itself, which results in
restricted drug perfusion (91).
A previous study has determined that tumors implanted with the same
gene at different sites exhibit different responses to AAD
treatment, which provides evidence that the tissue environment
outside the tumor serves an important role in AAD resistance
(92). Implantation of pancreatic
ductal adenocarcinoma and CRC tumors in AT results in intrinsic AAD
resistance, whereas the same tumors growing in non-AT are sensitive
to the same AAD treatment (92).
Similarly, AAD-sensitive hepatocellular carcinoma (HCC) growing in
steatotic livers is resistant to anti-angiogenic therapy, whereas
HCC in non-steatotic livers remains sensitive. These preclinical
findings are thought to be associated with AAD resistance and the
AT environment (84,92). Unexpectedly, for the stromal
component of tumors, the microvessels of almost fat-free tumors and
fat-rich tumors are equally sensitive to AAD treatment (92). Tumor growth depends on active
angiogenesis, and the inhibition of angiogenesis weakens tumor
growth. However, tumors in the adipose environment can continue to
grow into large masses, even with a small amount of intratumoral
microvascular tissue. Furthermore, angiogenesis has been shown to
inhibit tumor growth in non-ATs (93). Thus, the unexpected discovery of
tumors growing in AT distinguishes anti-angiogenic responses from
tumor growth. Adipose vessels seem to be dependent on VEGFs. For
anti-VEGF targeted treatment, inhibiting VEGF leads to significant
vascular degeneration (94,95). Therefore, tumors growing in AT
experience more severe hypoxia than those growing in non-AT.
AAD-induced vascular reduction and hypoxia can limit the supply of
circulating glucose, resulting in an impaired glucose-dependent
metabolism (96). To survive and
proliferate, cancer cells must use alternative energy mechanisms to
generate energy. AAD-triggered hypoxia results in the following
three lipid metabolic processes: i) The release of free fatty acids
and glycerol as metabolites from adipocytes; ii) cancer cells
increase free fatty acid uptake by upregulating CD36; and iii)
cancer cells undergo metabolic pathway reprogramming, which
activates the β-oxidation pathway resulting in free fatty acid
metabolism-dependent energy production, supporting tumor growth and
metastasis (17,97). These processes are illustrated in
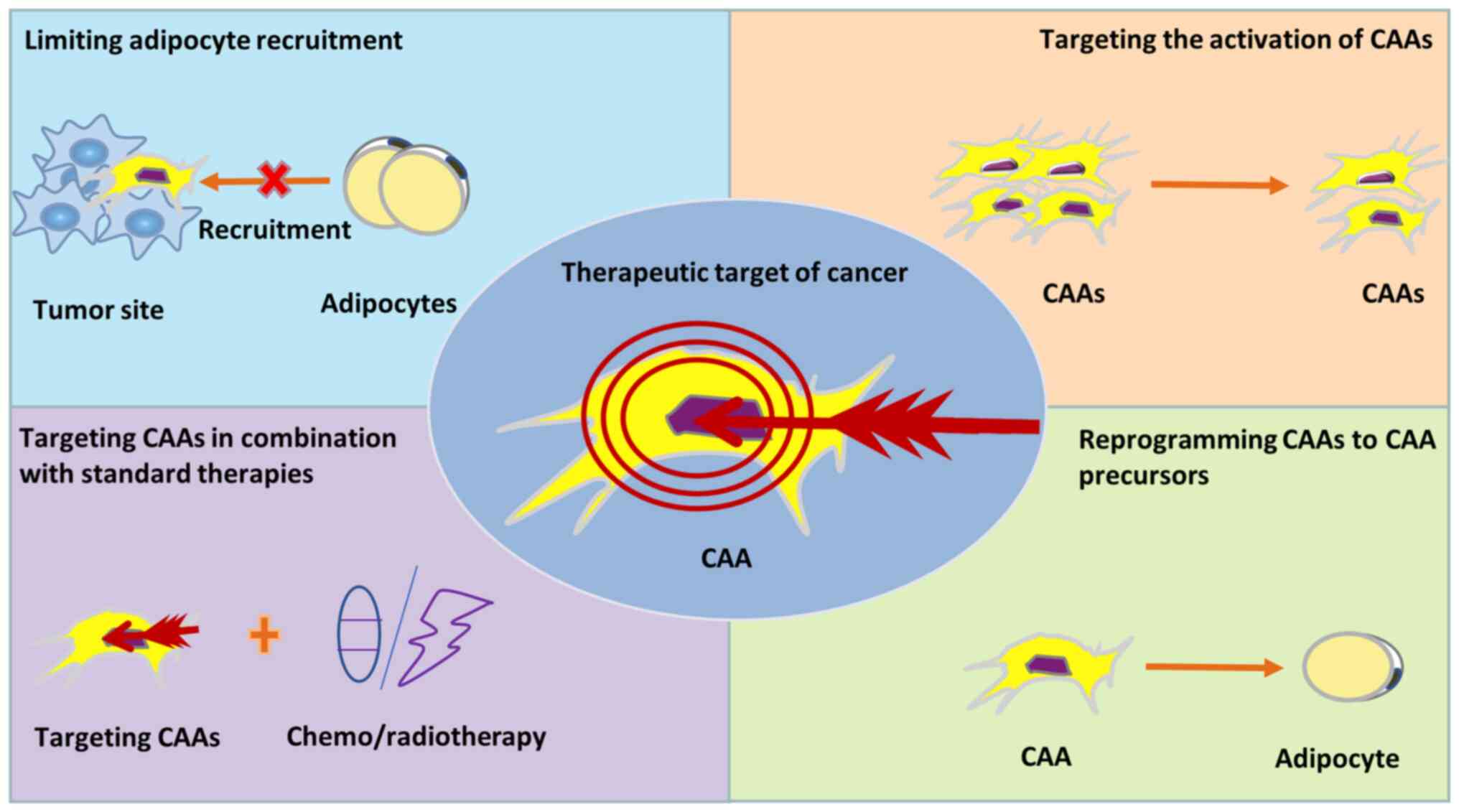
Fig. 2. Based on these findings,
targeting CAAs and lipid metabolism will provide an attractive
approach for cancer therapy. Compared with the use of a single
drug, the combination of AAD and β-oxidation inhibitors (for
example, etomoxir, a CPT1 inhibitor) has greater anticancer effects
in animal models of HCC grown in steatotic livers (92). Furthermore, clinical studies have
demonstrated that obesity is negatively associated with clinical
benefits, such as decreased sensitivity of chemotherapeutic drugs
and poor prognosis, in patients treated with anti-angiogenic
therapies (98). Hypoxia induced
by anti-VEGF treatment results in the high expression of IL-6 and
fibroblast growth factor 2 (FGF2) (99). Similarly, anti-VEGF treatment can
induce high expression levels of IL-6 and FGF2 in patients with BC,
which contribute to AAD resistance (98,99). Furthermore, the number of
adipocytes in tumors was directly and positively correlated with
adverse AAD reactions (100).
Adipocytes can secrete >600 metabolites, hormones
and cytokines, collectively known as adipokines (5). Although large-scale Mendelian
randomization analyses have been performed to assess the possible
causal relationship between adipokine concentrations and the risk
of obesity-related cancer, including CRC and ovarian cancer, the
results have demonstrated that there is no causal relationship
(101). Adipokines serve an
active role in regulating a variety of biological mechanisms,
including insulin secretion, fat distribution, inflammatory
response and energy consumption (5). The crosstalk between adipocytes and
BC cells favors tumor proliferation, survival and metastasis
(102). In terms of adipokines,
CAA secretes more chemokines, such as CCL2, CCL5, IL-1β, IL-6,
TNFα, VEGF and leptin, compared with relatively normal adipocytes
(37), which can promote tumor
invasion and metastasis (40,103). CAAs can also promote tumor
growth and metastasis, compared with distant mature adipocytes,
through fewer physical barriers (such as thickness and distance)
and more active adipokine secretions (5). Furthermore, in addition to the
aforementioned adipokines, the interaction between CAAs and cancer
cells involves a variety of specialized adipokines, including
resistin, insulin-like growth factor (IGF)-1, hepatocyte growth
factor, platelet-derived growth factor BB, IL-1, IGF binding
protein-2 and granulocyte colony stimulating factor (104–106). In the subsequent sections of the
present review, focus is given to the emerging roles of CAA-derived
leptin, adiponectin, IL-6, CCL2 and CCL5 adipokines in cancer.
Understanding the mechanisms of CAA-derived adipokines is important
for understanding the behavior of tumor cells and formulating new
therapeutics (Table I).
Leptin is a hormone with a molecular weight of 16
kDa, encoded by the LEP gene on human chromosome 7, and is mainly
synthesized and secreted by adipocytes (107). Leptin is also produced by cancer
cells (108). Leptin mediates
multiple biological functions, including proliferation,
differentiation, inflammation and nutrient absorption via the
leptin receptor (OBR) (107),
serving a role in the development and proliferation of normal and
malignant tissues. It has been reported that increased leptin serum
levels are positively correlated with cancer risk and with
increased plasma leptin levels in patients with cancer. High leptin
levels are also correlated with a high cancer grade, advanced tumor
stage and invasive cancer subtypes, such as in BC and CRC (107). Compared with distant tumor
sites, the expression of leptin in AT at the infiltration front is
higher (109). Furthermore, the
production and secretion of leptin is higher in CAAs than that in
mature adipocytes (107).
Therefore, leptin is involved in the interaction between adipocytes
and cancer cells (16). Leptin
may regulate numerous aspects of the occurrence, development and
metastasis of BC via autocrine, endocrine and paracrine signaling
pathways (5,16). Leptin can activate the estrogen
receptor, JAK/STAT3 and PI3K/AKT signaling pathways to promote the
proliferation of BC cells (91).
Leptin accelerates the cell cycle of BC cells by increasing the
expression of cyclinD1 and cyclin-dependent kinase 2 (110). Moreover, leptin can upregulate
the mRNA and protein expression levels of IL-1/IL-1 receptor
(IL-1R), further promoting the expression of VEGF and VEGFR, which
promote angiogenesis (111). The
role of leptin in BC invasion and metastasis has been thoroughly
researched. Wei et al (112) reported that leptin promoted the
EMT of BC cells via two main mechanisms: i) By upregulating the
expression of pyruvate kinase M2; and ii) by activating the
PI3K/AKT signaling pathway (112). Leptin also activates focal
adhesion kinase to enhance the secretion of ECM remodelers via the
steroid receptor coactivator-1/STAT3 signaling pathway, which
suggests that leptin is associated with the establishment of a more
invasive phenotype in BC cells (113). Previous studies have
demonstrated that tumor cell proliferation and tumor growth are
significantly reduced in leptin-deficient tumors; however,
leptin-deficient and leptin receptor-deficient mice are severely
obese. Moreover, OBR expression levels are significantly increased
in colon tumors compared with those in the normal epithelium.
Colonic leptin signaling-mediated CRC growth mainly occurs via the
OBR/STAT3 signaling pathway (114). A previous study has demonstrated
that adipocyte-derived leptin and IL-6 promote the local invasion
and metastasis of tumor cells, which occurs by activating
procollagen-lysine and 2-oxoglutarate 5-dioxygenase (lysine
hydroxylase) 2 when tumor cells are co-cultured with mature
adipocytes. Furthermore, leptin may promote BC invasion and
metastasis via the PI3K/AKT/activating transcription factor-2
signaling pathway in tumor cells (115,116). Leptin also regulates the
function of immune cells. Leptin can promote the progression of BC
by activating STAT3-FAO, inhibiting glycolysis and downregulating
the effector function of CD8+ T cells (64).
Adiponectin, a hormone with a molecular weight of
~30 kDa, is encoded by the ADIPOQ gene (117,118). Adiponectin serves a protective
role in tumor progression by binding to adiponectin receptor 1 and
adiponectin receptor 2. The decrease in adiponectin secretion in
CAAs (108) suggests a
relationship between adiponectin and its related signaling proteins
in antitumor activity. Adiponectin negatively regulates the cancer
cell proliferation by regulating inflammatory signaling molecules,
such as extracellular signal-regulated kinase (ERK)1/2, AKT, TNFα,
IL-1β, nuclear factor (NF)-κB, IL-6, IL-8 and CCL2 (118). Autophagy is a fundamental
vacuolar lysosomal degradation process known to help prevent the
accumulation of damaged proteins and organelles, recycle
cytoplasmic components and maintain intracellular homeostasis
(119). Adiponectin secreted by
adipocytes is an effective inducer of cytotoxic autophagy. CAAs are
functionally diverse and can induce adipocyte senescence via the
adiponectin-dependent autophagy signaling pathway, thereby
enhancing malignant behavior (120). Adenosine 5′-monophosphate
(AMP)-activated protein kinase (AMPK) is an important energy sensor
that can be regulated by cytokines, such as leptin and adiponectin,
promoting autophagy, regulating cellular metabolism and maintaining
energy balance (121). Chung
et al (119) proposed
that adiponectin can induce cancer autophagy and is involved in the
regulation of the serine/threonine kinase 11 and AMPK/Unc-51 like
autophagy activating kinase (ULK1) axes. ULK1 is an
autophagy-initiating kinase. Under nutrient sufficient conditions,
mTOR disrupts the interaction between ULK1 and AMPK by inhibiting
the activation of ULK1 (122).
Moreover, adiponectin can inhibit the growth and invasion of BC
cells and induce apoptosis by triggering the AMPK signaling pathway
and inhibiting the PI3K/AKT signaling pathway (107). In patients with obesity, this
relationship uses the adiponectin to leptin ratio, compared with
patients who are not obese, and poses a significant problem.
Theriau et al (123)
revealed that a low ratio of adiponectin to leptin in patients with
obesity can rapidly induce MCF-7 BC cells to enter the cell cycle
(123). Therefore, this increase
in adiponectin levels and the decrease in the leptin-to-adiponectin
ratio are related to a decrease in BC growth. In CRC, AT in the TME
leads to systemic low-grade and subclinical inflammation, which is
conducive to the carcinogenesis of various tissues in the body.
However, AT with endocrine activity influences the risk of systemic
carcinogenesis directly related to inflammation and by
significantly reducing the expression of adipokines, especially
adiponectin (124). Adiponectin
and its receptors, can induce the activation of various mechanisms,
thus exerting a cancer-preventive effect. Adiponectin participates
in the AMPK signaling pathway to regulate the cell cycle by
regulating the activity of various transcription factors, including
p53, and has been reported to inhibit signaling pathways involved
in proliferation, such as the Mtor (125), Wnt/GSK3β (126) and JAK/STAT signaling pathways
(127). Furthermore, a previous
study reported that adiponectin can inhibit cell proliferation
(128). The low mRNA expression
levels of COX-2 and high expression levels of T-cadherin in CRC
HCT116 cells suggest that adiponectin may directly inhibit tumor
growth via COX-2 (129).
The chemokine CCL2 is encoded by the CCL2 gene and
is also referred to as MCP-1 (130). In the TME, different cells,
including cancer cells, endothelial cells and fibroblasts, can
secrete CCL2 into the extracellular environment. CCL2 binds to G
protein-coupled receptors CCR2 and CCR4, and acts as a
chemoattractant to recruit CCR2-expressing immune cells to
inflammatory regions (130). A
previous study found that CCL2 expression levels increased in BC
e0771 cells, which recruited more adipocytes and
monocytes/macrophages (39). High
expression levels of CCL2 are associated with decreased survival in
patients with BC (131). Tsuyada
et al (132) reported
that BC cells secrete cytokines by activating the STAT3 promoter
and consequently the STAT3 signaling pathway in fibroblasts,
resulting in increased CCL2 expression and secretion. CCL2 can also
induce Notch1 expression and downstream signaling pathways in BC
cells, thereby inducing cancer stem cell (CSC) activity.
Furthermore, CCL2 expression is significantly correlated with
angiogenesis (133). CCL2 may
also serve an important role in the crosstalk between adipocytes
and macrophages. Studies have found that increased expression
levels of CCL2 and IL-1β in AT induce macrophages to secrete
chemokine (C-x-C motif) ligand 12 (CXCL12), which is associated
with obesity (134,135). It has also been demonstrated
that mammary epithelial cells surrounding AT recruit macrophages
and form a crown-like structure by secreting CCL2, which is related
to the malignant progression of BC. CCL2 has been proposed as a
therapeutic target for metastatic BC (134–136). In the presence of BC cells,
adipocytes can revert to an immature proliferation phenotype,
increasing the production of adipocyte-derived CCL2 and promoting
cell migration through adipokines such as IL-6 and CCL2 (15,136). Furthermore, neutralization
experiments using anti-IL-6 or CCL2 antibodies have demonstrated
that the migration enhancing effect of CAA-conditioned media can be
abolished (15). A recent study
has determined that chronic inflammation induced by CCL2 can
significantly promote tumor growth and connective tissue matrix
formation via the early entry of macrophages and fibroblasts into
the TME (137).
Chemokine CCL5 has a molecular weight of 8 kDa and
is located on chromosome 17q12; it is an effective chemokine that
attracts leukocytes and is a multifunctional inflammatory mediator
that can be expressed by BC cells (138). CCL5 is highly expressed in BC
and can also be produced by a variety of cells, including
mesenchymal stem cells (139).
CCL5 overexpression is associated with ERK phosphorylation in tumor
cells and with low 5-year disease-free survival and overall cancer
survival in patients with early human epidermal growth factor
receptor-2 (HER2)-positive BC (140). The abundance of CCL5 in the
peritumoral AT of patients with triple negative BC (TNBC) is also
associated with parallel low tumor metastasis and overall survival.
Song et al (141)
demonstrated that when adipocytes were cocultured with a TNBC cell
line, adipocytes could enhance the EMT effect by secreting more
CCL5. Karnoub et al (103) reported that BC cells stimulate
the secretion of CCL5 and that paracrine CCL5 reversibly binds to
CCR5 on the membrane surface of human BC MDA-MB-231 cells to
enhance migration, invasion and metastasis (103). Another study demonstrated that
when MDA-MB-231 TNBC cells were co-cultured with human adipocytes,
CCL5 levels were increased in the surrounding tissues, resulting in
the enhancement of MDA-MB-231 cell invasion and metastasis ability
(36). Therefore, antagonizing
CCL5 using specific CCL5 small molecule inhibitors can reduce the
invasion of BC cells (36).
Numerous studies have demonstrated that the CCL5/CCR5 axis is
highly activated in TNBC and HER2-positive BC. The invasive ability
of CCR5+ BC cells that responded to CCL5 was 40× higher
compared with that of CCR5− cells that did not respond
to CCR5 (142–144). In the mTOR-dependent mechanism,
MCF-7 cells overexpressing CCR5 have a stronger proliferation
ability (145). Moreover, it has
been shown that inhibition of CCL5/CCR5 signaling in endothelial
cells leads to the defective activation of the AKT/mTOR signaling
pathway and abnormal vascular and tumor growth in vitro and
in vivo (36). Therefore,
stromal cells may secrete CCL5 into the TME, where CCL5 may
activate the AKT/mTOR signaling pathway to promote tumor metastasis
by binding to CCR5. Moreover, CCL5 partially impairs triglyceride
synthesis in adipocytes via its cognate receptors by downregulating
the production of lipase and sterol regulatory element binding
protein-1 (146). CCL5 also
recruits macrophages (147) and
T helper 1 and T helper 17 cells (148) in the TME, which can induce and
maintain the striated structure of the matrix in the inflammatory
microenvironment. This structure allows carcinogenic cell behavior
and is destroyed by blocking CCL5 in vitro during matrix
deposition (149). Therefore,
CCL5 could be a potential target for BC precision therapy, but its
specific mechanism requires further investigation.
The level of IL-6 secreted by adipocytes is
significantly increased under the pathological conditions of
obesity and cancer. IL-6 is a cytokine involved in multiple
biological activities, including hematopoiesis, immune regulation
and tumorigenesis (150).
Following coculture with BC cells, the expression and secretion of
IL-6 in adipocytes increases (151). Among the proinflammatory
cytokines involved in obesity-related inflammation, IL-6 has been
determined to be the most important in CRC pathogenesis (15). IL-6 serves an important role in
CRC, which may be related to the expression of the IL-1β/IL-6
network in the TME. The significance of this finding stems from the
regulation of IL-6 synthesis by IL-1β and its synergistic
pro-inflammatory effects. Lee et al (152) demonstrated that mouse 3T3-L1
adipocytes indirectly cocultured with BC cells upregulated the
expression of IL-6 and pentraxin 3, which is consistent with the
overexpression of IL-6 observed in CAAs of human BC tissue
(152). Furthermore, IL-6 acts
as an independent poor prognostic factor for overall survival and
is associated with poor 5-year disease-free survival in patients
with steroid-refractory metastatic BC (153). IL-6 promotes tumor cell
proliferation, survival and angiogenesis by regulating the
JAK/STAT3 signaling pathway (154). In HER2-positive BC, IL-6 can
also induce the generation and maintenance of BC CSCs via the NF-κB
and STAT3 signaling pathways to promote tumor progression (155). IL-6 also regulates the
self-renewal of BC CSCs and promotes the survival and proliferation
of stem cells when the Notch, Wnt and TGF-β signaling pathways are
activated (156). Nickel et
al (157) demonstrated that
the migration ability of TNBC cells was significantly increased and
the secretion of IL-6 was increased following coculture with
adipocytes (157). Moreover,
adipocyte-derived IL-6 can also enhance the invasive behavior of BC
cells and induce the EMT phenotype (158). IL-6-induced BC metastasis is
reversed when the IL-6 receptor (IL-6R) is blocked by an anti-IL-6R
antibody (159,160). Blocking IL-6 signaling in BC
cells alters the expression of EMT regulatory genes, disrupts the
stability of focal adhesions and decreases the mobility of cells
(161).
CAAs significantly affect tumor growth, metastasis
and drug sensitivity via multiple mechanisms, including paracrine,
juxtacrine and endocrine signaling, metabolites and metabolic
reprogramming. This effect is greatest in tumors that are closely
related to adipocytes, such as BC, CRC and ovarian cancer.
Therefore, targeting CAAs and lipid metabolism would be a potential
approach for cancer treatment. Furthermore, adipocytes are
excellent candidates for altering tumor behavior via heterotypic
signal transduction processes that secrete adipokines, such as
hormones, growth factors, cytokines and other molecules. In the
TME, the expression and secretion profiles of inflammatory
mediators in adipocytes are altered. The secretion of chemokines,
such as CCL5, CCL2, IL-6 and leptin, further promotes the
proliferation, invasion and angiogenesis of tumor cells.
In summary, the present review mainly focused on
the role of CAAs in cancer. The link between CAAs and cancer is
important, and great progress has been made to elucidate the
underlying biological mechanisms of CAA and the pathogenesis of
cancer. CAAs induce leptin, IL-6, TNFα, CCL2 and CCL5 expression
and disturb gut microbiota and bile acid homeostasis. These changes
promote carcinogenesis, mediated by downstream signaling pathways.
Based on the current understanding of this mechanism, several
promising methods can be proposed for CAA in the treatment of
tumors. However, several challenges remain for CAA identification.
Certain CAA-related genes and protein markers have been identified,
but there are no uniform criteria for the authentication of CAAs.
An increased understanding of the link between CAA risk factors and
carcinogenic processes will help develop more promising therapeutic
targets and approaches for CAA-related cancer treatment in the
future.
Not applicable.
The present review was supported by the National
Science Foundation (grant no. BK20191172), the Project of Gusu
Medical Key Talent (grant no. GSWS2020005) and the Project of New
Pharmaceutics and Medical Apparatuses (grant no. SLJ2021007).
Not applicable.
The concept, design and drafting of the review were
performed by HY and SH. Data collection, involving searching,
analyzing and summarizing the literature, was carried out by HY and
manuscript revision was performed by SH. All authors read and
approved the final manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Laplane L, Duluc D, Larmonier N, Pradeu T
and Bikfalvi A: The multiple layers of the tumor environment.
Trends Cancer. 4:802–809. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Elia I and Haigis MC: Metabolites and the
tumour microenvironment: From cellular mechanisms to systemic
metabolism. Nat Metab. 3:21–32. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chen Q, Liu G, Liu S, Su H, Wang Y, Li J
and Luo C: Remodeling the tumor microenvironment with emerging
nanotherapeutics. Trends Pharmacol Sci. 39:59–74. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lazar I, Clement E, Attane C, Muller C and
Nieto L: A new role for extracellular vesicles: How small vesicles
can feed tumors' big appetite. J Lipid Res. 59:1793–1804. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu Q, Li B, Li Z, Li J and Sun S and Sun
S: Cancer-associated adipocytes: Key players in breast cancer
progression. J Hematol Oncol. 12:952019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dirat B, Bochet L, Dabek M, Daviaud D,
Dauvillier S, Majed B, Wang YY, Meulle A, Salles B, Le Gonidec S,
et al: Cancer-associated adipocytes exhibit an activated phenotype
and contribute to breast cancer invasion. Cancer Res. 71:2455–2465.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vaupel H, Schmidberger A and Mayer A: The
Warburg effect: Essential part of metabolic reprogramming and
central contributor to cancer progression. Int J Radiat Biol.
95:912–919. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hoxhaj G and Manning BD: The PI3K-AKT
network at the interface of oncogenic signalling and cancer
metabolism. Nat Rev Cancer. 20:74–88. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nieman KM, Romero IL, Van Houten B and
Lengyel E: Adipose tissue and adipocytes support tumorigenesis and
metastasis. Biochim Biophys Acta. 1831:1533–1541. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iyengar P, Combs TP, Shah SJ, Gouon-Evans
V, Pollard JW, Albanese C, Flanagan L, Tenniswood MP, Guha C,
Lisanti MP, et al: Adipocyte-secreted factors synergistically
promote mammary tumorigenesis through induction of anti-apoptotic
transcriptional programs and proto-oncogene stabilization.
Oncogene. 22:6408–6423. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Park J, Morley TS, Kim M, Clegg DJ and
Scherer PE: Obesity and cancer-mechanisms underlying tumour
progression and recurrence. Nat Rev Endocrinol. 10:455–465. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Donohoe CL, Lysaght J, O'sullivan J and
Reynolds JV: Emerging concepts linking obesity with the hallmarks
of cancer. Trends Endocrinol Metab. 28:46–62. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kahn CR, Wang G and Lee KY: Altered
adipose tissue and adipocyte function in the pathogenesis of
metabolic syndrome. J Clin Invest. 129:3990–4000. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scheja L and Heeren J: The endocrine
function of adipose tissues in health and cardiometabolic disease.
Nat Rev Endocrinol. 15:507–524. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fujisaki K, Fujimoto H, Sangai T,
Nagashima T, Sakakibara M, Shiina N, Kuroda M, Aoyagi Y and
Miyazaki M: Cancer-mediated adipose reversion promotes cancer cell
migration via IL-6 and MCP-1. Breast Cancer Res Treat. 150:255–263.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao C, Wu M, Zeng N, Xiong M, Hu W, Lv W,
Yi Y, Zhang Q and Wu Y: Cancer-associated adipocytes: Emerging
supporters in breast cancer. J Exp Clin Cancer Res. 39:1562020.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Choi J, Cha YJ and Koo JS: Adipocyte
biology in breast cancer: From silent bystander to active
facilitator. Prog Lipid Res. 69:11–20. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rybinska I, Agresti R, Trapani A,
Tagliabue E and Triulzi T: Adipocytes in breast cancer, the thick
and the thin. Cells. 9:5602020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pérez-Escuredo J, Van Hée VF, Sboarina M,
Falces J, Payen VL, Pellerin L and Sonveaux P: Monocarboxylate
transporters in the brain and in cancer. Biochim Biophys Acta.
1863:2481–2497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang E, Wang X, Gong Z, Yu M, Wu H and
Zhang D: Exosome-mediated metabolic reprogramming: The emerging
role in tumor microenvironment remodeling and its influence on
cancer progression. Signal Transduct Target Ther. 5:2422020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiong X, Wen YA, Fairchild R, Zaytseva YY,
Weiss HL, Evers BM and Gao T: Upregulation of CPT1A is essential
for the tumor-promoting effect of adipocytes in colon cancer. Cell
Death Dis. 11:7362020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schlaepfer IR, Rider L, Rodrigues LU,
Gijón MA, Pac CT, Romero L, Cimic A, Sirintrapun SJ, Glodé LM,
Eckel RH and Cramer SD: Lipid catabolism via CPT1 as a therapeutic
target for prostate cancer. Mol Cancer Ther. 13:2361–2371. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tabuso M, Homer-Vanniasinkam S, Adya R and
Arasaradnam RP: Role of tissue microenvironment resident adipocytes
in colon cancer. World J Gastroenterol. 23:5829–5835. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ko JH, Um JY, Lee SG, Yang WM, Sethi G and
Ahn KS: Conditioned media from adipocytes promote proliferation,
migration, and invasion in melanoma and colorectal cancer cells. J
Cell Physiol. 234:18249–18261. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fontana L, Eagon JC, Trujillo ME, Scherer
PE and Klein S: Visceral fat adipokine secretion is associated with
systemic inflammation in obese humans. Diabetes. 56:1010–1013.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Catalán V, Gómez-Ambrosi J, Rodríguez A,
Ramírez B, Silva C, Rotellar F, Hernández-Lizoain JL, Baixauli J,
Valentí V, Pardo F, et al: Up-regulation of the novel
proinflammatory adipokines lipocalin-2, chitinase-3 like-1 and
osteopontin as well as angiogenic-related factors in visceral
adipose tissue of patients with colon cancer. J Nutr Biochem.
22:634–641. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Peterson JE, Zurakowski D, Italiano JE Jr,
Michel LV, Connors S, Oenick M, D'Amato RJ, Klement GL and Folkman
J: VEGF, PF4 and PDGF are elevated in platelets of colorectal
cancer patients. Angiogenesis. 15:265–273. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lumeng CN, Deyoung SM, Bodzin JL and
Saltiel AR: Increased inflammatory properties of adipose tissue
macrophages recruited during diet-induced obesity. Diabetes.
56:16–23. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu H, Barnes GT, Yang Q, Tan G, Yang D,
Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA and Chen H:
Chronic inflammation in fat plays a crucial role in the development
of obesity-related insulin resistance. J Clin Invest.
112:1821–1830. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Harvey AE, Lashinger LM and Hursting SD:
The growing challenge of obesity and cancer: An inflammatory issue.
Ann N Y Acad Sci. 1229:45–52. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wagner M, Bjerkvig R, Wiig H,
Melero-Martin JM, Lin RZ, Klagsbrun M and Dudley AC: Inflamed
tumor-associated adipose tissue is a depot for macrophages that
stimulate tumor growth and angiogenesis. Angiogenesis. 15:481–495.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Amor S, Iglesias-de la Cruz MC, Ferrero E,
García-Villar O, Barrios V, Fernandez N, Monge L, García-Villalón
AL and Granado M: Peritumoral adipose tissue as a source of
inflammatory and angiogenic factors in colorectal cancer. Int J
Colorectal Dis. 31:365–375. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ye P, Xi Y, Huang Z and Xu P: Linking
obesity with colorectal cancer: Epidemiology and mechanistic
insights. Cancers (Basel). 12:14082020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statistics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lapeire L, Hendrix A, Lambein K, Van
Bockstal M, Braems G, Van Den Broecke R, Limame R, Mestdagh P,
Vandesompele J, Vanhove C, et al: Cancer-associated adipose tissue
promotes breast cancer progression by paracrine oncostatin M and
Jak/STAT3 signaling. Cancer Res. 74:6806–6819. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
D'Esposito V, Liguoro D, Ambrosio MR,
Collina F, Cantile M, Spinelli R, Raciti GA, Miele C, Valentino R,
Campiglia P, et al: Adipose microenvironment promotes triple
negative breast cancer cell invasiveness and dissemination by
producing CCL5. Oncotarget. 7:24495–24509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Palma M, Biziato D and Petrova TV:
Microenvironmental regulation of tumour angiogenesis. Nat Rev
Cancer. 17:457–474. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wang YY, Attané C, Milhas D, Dirat B,
Dauvillier S, Guerard A, Gilhodes J, Lazar I, Alet N, Laurent V, et
al: Mammary adipocytes stimulate breast cancer invasion through
metabolic remodeling of tumor cells. JCI Insight. 2:e874892017.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Attane C, Milhas D, Hoy AJ and Muller C:
Metabolic remodeling induced by adipocytes: A new Achille heels in
invasive breast cancer? Curr Med Chem. 27:3984–4001. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bussard KM, Mutkus L, Stumpf K,
Gomez-Manzano C and Marini FC: Tumor associated stromal cells as
key contributors to the tumor microenvironment. Breast Cancer Res.
18:842016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lengyel E: Ovarian cancer development and
metastasis. Am J Pathol. 177:1053–1064. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
John B, Naczki C, Patel C, Ghoneum A,
Qasem S, Salih Z and Said N: Regulation of the bi-directional
cross-talk between ovarian cancer cells and adipocytes by SPARC.
Oncogene. 38:4366–4383. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mukherjee A, Chiang CY, Daifotis HA,
Nieman KM, Fahrmann JF, Lastra RR, Romero IL, Fiehn O and Lengyel
E: Adipocyte-induced FABP4 expression in ovarian cancer cells
promotes metastasis and mediates carboplatin resistance. Cancer
Res. 80:1748–1761. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun C, Li X, Guo E, Li N, Zhou B, Lu H,
Huang J, Xia M, Shan W, Wang B, et al: MCP-1/CCR-2 axis in
adipocytes and cancer cell respectively facilitates ovarian cancer
peritoneal metastasis. Oncogene. 39:1681–1695. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nieman KM, Kenny HA, Penicka CV, Ladanyi
A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB,
Hotamisligil GS, et al: Adipocytes promote ovarian cancer
metastasis and provide energy for rapid tumor growth. Nat Med.
17:1498–1503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Coburn CT, Knapp FF Jr, Febbraio M, Beets
AL, Silverstein RL and Abumrad NA: Defective uptake and utilization
of long chain fatty acids in muscle and adipose tissues of CD36
knockout mice. J Biol Chem. 275:32523–32529. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Au Yeung CL, Co NN, Tsuruga T, Yeung TL,
Kwan SY, Leung CS, Li Y, Lu ES, Kwan K, Wong KK, et al: Exosomal
transfer of stroma-derived miR21 confers paclitaxel resistance in
ovarian cancer cells through targeting APAF1. Nat Commun.
7:111502016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Renehan AG, Tyson M, Egger M, Heller RF
and Zwahlen M: Body-mass index and incidence of cancer: A
systematic review and meta-analysis of prospective observational
studies. Lancet. 371:569–578. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sun X, Wang M, Wang M, Yu X, Guo J, Sun T,
Li X, Yao L, Dong H and Xu Y: Metabolic reprogramming in
triple-negative breast cancer. Front Oncol. 10:4282020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Santander AM, Lopez-Ocejo O, Casas O,
Agostini T, Sanchez L, Lamas-Basulto E, Carrio R, Cleary MP,
Gonzalez-Perez RR and Torroella-Kouri M: Paracrine interactions
between adipocytes and tumor cells recruit and modify macrophages
to the mammary tumor microenvironment: The role of obesity and
inflammation in breast adipose tissue. Cancers (Basel). 7:143–178.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cairns RA, Harris IS and Mak TW:
Regulation of cancer cell metabolism. Nat Rev Cancer. 11:85–95.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vander Heiden MG, Cantley LC and Thompson
CB: Understanding the Warburg effect: The metabolic requirements of
cell proliferation. Science. 324:1029–1033. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Corn KC, Windham MA and Rafat M: Lipids in
the tumor microenvironment: From cancer progression to treatment.
Prog Lipid Res. 80:1010552020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pallegar NK and Christian SL: Adipocytes
in the tumour microenvironment. Adv Exp Med Biol. 1234:1–13. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dias AS, Almeida CR, Helguero LA and
Duarte IF: Metabolic crosstalk in the breast cancer
microenvironment. Eur J Cancer. 121:154–171. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Balaban S, Shearer RF, Lee LS, van
Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC,
Fazakerley DJ, Grewal T, et al: Adipocyte lipolysis links obesity
to breast cancer growth: Adipocyte-derived fatty acids drive breast
cancer cell proliferation and migration. Cancer Metab. 5:12017.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang D, Li Y, Xing L, Tan Y, Sun J, Zeng
B, Xiang T, Tan J, Ren G and Wang Y: Utilization of
adipocyte-derived lipids and enhanced intracellular trafficking of
fatty acids contribute to breast cancer progression. Cell Commun
Signal. 16:322018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zaoui M, Morel M, Ferrand N, Fellahi S,
Bastard JP, Lamazière A, Larsen AK, Béréziat V, Atlan M and Sabbah
M: Breast-associated adipocytes secretome induce fatty acid uptake
and invasiveness in breast cancer cells via CD36 independently of
body mass index, menopausal status and mammary density. Cancers
(Basel). 11:20122019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ladanyi A, Mukherjee A, Kenny HA, Johnson
A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag
A, et al: Adipocyte-induced CD36 expression drives ovarian cancer
progression and metastasis. Oncogene. 37:2285–2301. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zhang M, Di Martino JS, Bowman RL,
Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal
C, Yong-Gonzales V, et al: Adipocyte-derived lipids mediate
melanoma progression via FATP proteins. Cancer Discov. 8:1006–1025.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lopes-Coelho F, Andre S, Felix A and Serpa
J: Breast cancer metabolic cross-talk: Fibroblasts are hubs and
breast cancer cells are gatherers of lipids. Mol Cell Endocrinol.
462:93–106. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wang T, Fahrmann JF, Lee H, Li YJ,
Tripathi SC, Yue C, Zhang C, Lifshitz V, Song J, Yuan Y, et al:
JAK/STAT3-regulated fatty acid β-oxidation is critical for breast
cancer stem cell self-renewal and chemoresistance. Cell Metab.
27:136–150.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhang C, Yue C, Herrmann A, Song J,
Egelston C, Wang T, Zhang Z, Li W, Lee H, Aftabizadeh M, et al:
STAT3 activation-induced fatty acid oxidation in CD8+ T
effector cells is critical for obesity-promoted breast tumor
growth. Cell Metab. 31:148–161.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cawthorn WP, Scheller EL, Learman BS,
Parlee SD, Simon BR, Mori H, Ning X, Bree AJ, Schell B, Broome DT,
et al: Bone marrow adipose tissue is an endocrine organ that
contributes to increased circulating adiponectin during caloric
restriction. Cell Metab. 20:368–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ye H, Adane B, Khan N, Sullivan T,
Minhajuddin M, Gasparetto M, Stevens B, Pei S, Balys M, Ashton JM,
et al: Leukemic stem cells evade chemotherapy by metabolic
adaptation to an adipose tissue niche. Cell Stem Cell. 19:23–37.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Auciello FR, Bulusu V, Oon C, Tait-Mulder
J, Berry M, Bhattacharyya S, Tumanov S, Allen-Petersen BL, Link J,
Kendsersky ND, et al: A stromal lysolipid-autotaxin signaling axis
promotes pancreatic tumor progression. Cancer Discov. 9:617–627.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Benesch MG, Tang X, Maeda T, Ohhata A,
Zhao YY, Kok BP, Dewald J, Hitt M, Curtis JM, McMullen TP and
Brindley DN: Inhibition of autotaxin delays breast tumor growth and
lung metastasis in mice. FASEB J. 28:2655–2666. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Yang L, Yu X and Yang Y: Autotaxin
upregulated by STAT3 activation contributes to invasion in
pancreatic neuroendocrine neoplasms. Endocr Connect. 7:1299–1307.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Azare J, Doane A, Leslie K, Chang Q,
Berishaj M, Nnoli J, Mark K, Al-Ahmadie H, Gerald W, Hassimi M, et
al: Stat3 mediates expression of autotaxin in breast cancer. PLoS
One. 6:e278512011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Schmid R, Wolf K, Robering JW, Strauß S,
Strissel PL, Strick R, Rübner M, Fasching PA, Horch RE, Kremer AE,
et al: ADSCs and adipocytes are the main producers in the
autotaxin-lysophosphatidic acid axis of breast cancer and healthy
mammary tissue in vitro. BMC Cancer. 18:12732018. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Corbet C and Feron O: Emerging roles of
lipid metabolism in cancer progression. Curr Opin Clin Nutr Metab
Care. 20:254–260. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pascual G, Avgustinova A, Mejetta S,
Martín M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A,
Hueto JA, et al: Targeting metastasis-initiating cells through the
fatty acid receptor CD36. Nature. 541:41–45. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wculek SK and Malanchi I: Neutrophils
support lung colonization of metastasis-initiating breast cancer
cells. Nature. 528:413–417. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Poltavets V, Kochetkova M, Pitson SM and
Samuel MS: The role of the extracellular matrix and its molecular
and cellular regulators in cancer cell plasticity. Front Oncol.
8:4312018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bacac M and Stamenkovic I: Metastatic
cancer cell. Annu Rev Pathol. 3:221–247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Quail DF and Joyce JA: Microenvironmental
regulation of tumor progression and metastasis. Nat Med.
19:1423–1437. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ribeiro RJ, Monteiro CP, Cunha VF, Azevedo
AS, Oliveira MJ, Monteiro R, Fraga AM, Príncipe P, Lobato C, Lobo
F, et al: Tumor cell-educated periprostatic adipose tissue acquires
an aggressive cancer-promoting secretory profile. Cell Physiol
Biochem. 29:233–240. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lee SG, Kim JS, Kim HJ, Schlaepfer DD, Kim
IS and Nam JO: Endothelial angiogenic activity and adipose
angiogenesis is controlled by extracellular matrix protein TGFBI.
Sci Rep. 11:96442021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Campo-Verde-Arbocco F, López-Laur JD,
Romeo LR, Giorlando N, Bruna FA, Contador DE, López-Fontana G,
Santiano FE, Sasso CV, Zyla LE, et al: Human renal adipose tissue
induces the invasion and progression of renal cell carcinoma.
Oncotarget. 8:94223–94234. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Iyengar P, Espina V, Williams TW, Lin Y,
Berry D, Jelicks LA, Lee H, Temple K, Graves R, Pollard J, et al:
Adipocyte-derived collagen VI affects early mammary tumor
progression in vivo, demonstrating a critical interaction in the
tumor/stroma microenvironment. J Clin Invest. 115:1163–1176. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Bochet L, Meulle A, Imbert S, Salles B,
Valet P and Muller C: Cancer-associated adipocytes promotes breast
tumor radioresistance. Biochem Biophys Res Commun. 411:102–106.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tennant DA, Durán RV and Gottlieb E:
Targeting metabolic transformation for cancer therapy. Nat Rev
Cancer. 10:267–277. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Ueda S, Saeki T, Osaki A, Yamane T and
Kuji I: Bevacizumab induces acute hypoxia and cancer progression in
patients with refractory breast cancer: Multimodal functional
imaging and multiplex cytokine analysis. Clin Cancer Res.
23:5769–5778. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Levitsky A, Brismar K, Hafström I,
Hambardzumyan K, Lourdudoss C, van Vollenhoven RF and Saevarsdottir
S: Obesity is a strong predictor of worse clinical outcomes and
treatment responses in early rheumatoid arthritis: Results from the
SWEFOT trial. RMD Open. 3:e0004582017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Castillo JJ, Mulkey F, Geyer S, Kolitz JE,
Blum W, Powell BL, George SL, Larson RA and Stone RM: Relationship
between obesity and clinical outcome in adults with acute myeloid
leukemia: A pooled analysis from four CALGB (alliance) clinical
trials. Am J Hematol. 91:199–204. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Laurent V, Guérard A, Mazerolles C, Le
Gonidec S, Toulet A, Nieto L, Zaidi F, Majed B, Garandeau D,
Socrier Y, et al: Periprostatic adipocytes act as a driving force
for prostate cancer progression in obesity. Nat Commun.
7:102302016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Osman MA and Hennessy BT: Obesity
correlation with metastases development and response to first-line
metastatic chemotherapy in breast cancer. Clin Med Insights Oncol.
9:105–112. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Engin A: Obesity-associated breast cancer:
Analysis of risk factors. Adv Exp Med Biol. 960:571–606. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Cao Y: Tumor angiogenesis and molecular
targets for therapy. Front Biosci (Landmark Ed). 14:3962–3973.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Iwamoto H, Abe M, Yang Y, Cui D, Seki T,
Nakamura M, Hosaka K, Lim S, Wu J, He X, et al: Cancer lipid
metabolism confers antiangiogenic drug resistance. Cell Metab.
28:104–117.e5. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hedlund EM, Yang X, Zhang Y, Yang Y,
Shibuya M, Zhong W, Sun B, Liu Y, Hosaka K and Cao Y: Tumor
cell-derived placental growth factor sensitizes antiangiogenic and
antitumor effects of anti-VEGF drugs. Proc Natl Acad Sci USA.
110:654–659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Honek J, Seki T, Iwamoto H, Fischer C, Li
J, Lim S, Samani NJ, Zang J and Cao Y: Modulation of age-related
insulin sensitivity by VEGF-dependent vascular plasticity in
adipose tissues. Proc Natl Acad Sci USA. 111:14906–14911. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yang Y, Zhang Y, Iwamoto H, Hosaka K, Seki
T, Andersson P, Lim S, Fischer C, Nakamura M, Abe M, et al:
Discontinuation of anti-VEGF cancer therapy promotes metastasis
through a liver revascularization mechanism. Nat Commun.
7:126802016. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Levine AJ and Puzio-Kuter AM: The control
of the metabolic switch in cancers by oncogenes and tumor
suppressor genes. Science. 330:1340–1344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Park JH, Vithayathil S, Kumar S, Sung PL,
Dobrolecki LE, Putluri V, Bhat VB, Bhowmik SK, Gupta V, Arora K, et
al: Fatty acid oxidation-driven src links mitochondrial energy
reprogramming and oncogenic properties in triple-negative breast
cancer. Cell Rep. 14:2154–2165. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Trédan O, Lacroix-Triki M, Guiu S,
Mouret-Reynier MA, Barrière J, Bidard FC, Braccini AL, Mir O,
Villanueva C and Barthélémy P: Angiogenesis and tumor
microenvironment: Bevacizumab in the breast cancer model. Target
Oncol. 10:189–198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Incio J, Ligibel JA, McManus DT, Suboj P,
Jung K, Kawaguchi K, Pinter M, Babykutty S, Chin SM, Vardam TD, et
al: Obesity promotes resistance to anti-VEGF therapy in breast
cancer by up-regulating IL-6 and potentially FGF-2. Sci Transl Med.
10:eaag09452018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Casanovas O, Hicklin DJ, Bergers G and
Hanahan D: Drug resistance by evasion of antiangiogenic targeting
of VEGF signaling in late-stage pancreatic islet tumors. Cancer
Cell. 8:299–309. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Dimou NL, Papadimitriou N, Mariosa D,
Johansson M, Brennan P, Peters U, Chanock SJ, Purdue M, Bishop DT,
Gago-Dominquez M, et al: Circulating adipokine concentrations and
risk of five obesity-related cancers: A mendelian randomization
study. Int J Cancer. 148:1625–1636. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Cha YJ, Kim ES and Koo JS:
Tumor-associated macrophages and crown-like structures in adipose
tissue in breast cancer. Breast Cancer Res Treat. 170:15–25. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Karnoub AE, Dash AB, Vo AP, Sullivan A,
Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R and Weinberg
RA: Mesenchymal stem cells within tumour stroma promote breast
cancer metastasis. Nature. 449:557–563. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Lee JO, Kim N, Lee HJ, Lee YW, Kim SJ,
Park SH and Kim HS: Resistin, a fat-derived secretory factor,
promotes metastasis of MDA-MB-231 human breast cancer cells through
ERM activation. Sci Rep. 6:189232016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Xiong Y, Russell DL, McDonald LT, Cowart
LA and LaRue AC: Hematopoietic stem cell-derived adipocytes promote
tumor growth and cancer cell migration. Int J Cancer Res Mol Mech.
3:102017.PubMed/NCBI
|
|
106
|
Wang C, Gao C, Meng K, Qiao H and Wang Y:
Human adipocytes stimulate invasion of breast cancer MCF-7 cells by
secreting IGFBP-2. PLoS One. 10:e01193482015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Sánchez-Jiménez F, Pérez-Pérez A, de la
Cruz-Merino L and Sánchez-Margalet V: Obesity and breast cancer:
Role of leptin. Front Oncol. 9:5962019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Ando S, Barone I, Giordano C, Bonofiglio D
and Catalano S: The multifaceted mechanism of leptin signaling
within tumor microenvironment in driving breast cancer growth and
progression. Front Oncol. 4:3402014.PubMed/NCBI
|
|
109
|
Gnerlich JL, Yao KA, Fitchev PS,
Goldschmidt RA, Bond MC, Cornwell M and Crawford SE: Peritumoral
expression of adipokines and fatty acids in breast cancer. Ann Surg
Oncol. 20 (Suppl 3):S731–S738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Okumura M, Yamamoto M, Sakuma H, Kojima T,
Maruyama T, Jamali M, Cooper DR and Yasuda K: Leptin and high
glucose stimulate cell proliferation in MCF-7 human breast cancer
cells: Reciprocal involvement of PKC-alpha and PPAR expression.
Biochim Biophys Acta. 1592:107–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zhou W, Guo S and Gonzalez-Perez RR:
Leptin pro-angiogenic signature in breast cancer is linked to IL-1
signalling. Br J Cancer. 104:128–137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Wei L, Li K, Pang X, Guo B, Su M, Huang Y,
Wang N, Ji F, Zhong C, Yang J, et al: Leptin promotes
epithelial-mesenchymal transition of breast cancer via the
upregulation of pyruvate kinase M2. J Exp Clin Cancer Res.
35:1662016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Juárez-Cruz JC, Zuñiga-Eulogio MD,
Olea-Flores M, Castañeda-Saucedo E, Mendoza-Catalán MÁ,
Ortuño-Pineda C, Moreno-Godínez ME, Villegas-Comonfort S,
Padilla-Benavides T and Navarro-Tito N: Leptin induces cell
migration and invasion in a FAK-Src-dependent manner in breast
cancer cells. Endocr Connect. 8:1539–1552. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Endo H, Hosono K, Uchiyama T, Sakai E,
Sugiyama M, Takahashi H, Nakajima N, Wada K, Takeda K, Nakagama H
and Nakajima A: Leptin acts as a growth factor for colorectal
tumours at stages subsequent to tumour initiation in murine colon
carcinogenesis. Gut. 60:1363–1371. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
He JY, Wei XH, Li SJ, Liu Y, Hu HL, Li ZZ,
Kuang XH, Wang L, Shi X, Yuan ST and Sun L: Adipocyte-derived IL-6
and leptin promote breast cancer metastasis via upregulation of
Lysyl Hydroxylase-2 expression. Cell Commun Signal. 16:1002018.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Li K, Wei L, Huang Y, Wu Y, Su M, Pang X,
Wang N, Ji F, Zhong C and Chen T: Leptin promotes breast cancer
cell migration and invasion via IL-18 expression and secretion. Int
J Oncol. 48:2479–2487. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Maroni P: Leptin, adiponectin, and Sam68
in bone metastasis from breast cancer. Int J Mol Sci. 21:10512020.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Sultana R, Kataki AC, Borthakur BB,
Basumatary TK and Bose S: Imbalance in leptin-adiponectin levels
and leptin receptor expression as chief contributors to triple
negative breast cancer progression in Northeast India. Gene.
621:51–58. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Chung SJ, Nagaraju GP, Nagalingam A,
Muniraj N, Kuppusamy P, Walker A, Woo J, Győrffy B, Gabrielson E,
Saxena NK and Sharma D: ADIPOQ/adiponectin induces cytotoxic
autophagy in breast cancer cells through STK11/LKB1-mediated
activation of the AMPK-ULK1 axis. Autophagy. 13:1386–1403. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Wu Q, Li B and Sun S and Sun S: Unraveling
adipocytes and cancer links: Is there a role for senescence? Front
Cell Dev Biol. 8:2822020. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Hardie DG: AMP-activated/SNF1 protein
kinases: Conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Theriau CF, Sauvé OS, Beaudoin MS, Wright
DC and Connor MK: Proliferative endocrine effects of adipose tissue
from obese animals on MCF7 cells are ameliorated by resveratrol
supplementation. PLoS One. 12:e01838972017. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Tae CH, Kim SE, Jung SA, Joo YH, Shim KN,
Jung HK, Kim TH, Cho MS, Kim KH and Kim JS: Involvement of
adiponectin in early stage of colorectal carcinogenesis. BMC
Cancer. 14:8112014. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Li W, Saud SM, Young MR, Chen G and Hua B:
Targeting AMPK for cancer prevention and treatment. Oncotarget.
6:7365–7378. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Park SY, Kim D and Kee SH:
Metformin-activated AMPK regulates β-catenin to reduce cell
proliferation in colon carcinoma RKO cells. Oncol Lett.
17:2695–2702. 2019.PubMed/NCBI
|
|
127
|
Rutherford C, Speirs C, Williams JJ, Ewart
MA, Mancini SJ, Hawley SA, Delles C, Viollet B, Costa-Pereira AP,
Baillie GS, et al: Phosphorylation of Janus kinase 1 (JAK1) by
AMP-activated protein kinase (AMPK) links energy sensing to
anti-inflammatory signaling. Sci Signal. 9:ra1092016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Waldner MJ, Foersch S and Neurath MF:
Interleukin-6-a key regulator of colorectal cancer development. Int
J Biol Sci. 8:1248–1253. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Semaan J, Pinon A, Rioux B, Hassan L,
Limami Y, Pouget C, Fagnère C, Sol V, Diab-Assaf M, Simon A and
Liagre B: Resistance to 3-HTMC-induced apoptosis through activation
of PI3K/Akt, MEK/ERK, and p38/COX-2/PGE2 pathways in
human HT-29 and HCT116 colorectal cancer cells. J Cell Biochem.
117:2875–2885. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Yoshimura T: The chemokine MCP-1 (CCL2) in
the host interaction with cancer: A foe or ally? Cell Mol Immunol.
15:335–345. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Bonapace L, Coissieux MM, Wyckoff J, Mertz
KD, Varga Z, Junt T and Bentires-Alj M: Cessation of CCL2
inhibition accelerates breast cancer metastasis by promoting
angiogenesis. Nature. 515:130–133. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Tsuyada A, Chow A, Wu J, Somlo G, Chu P,
Loera S, Luu T, Li AX, Wu X, Ye W, et al: CCL2 mediates cross-talk
between cancer cells and stromal fibroblasts that regulates breast
cancer stem cells. Cancer Res. 72:2768–2779. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Saji H, Koike M, Yamori T, Saji S, Seiki
M, Matsushima K and Toi M: Significant correlation of monocyte
chemoattractant protein-1 expression with neovascularization and
progression of breast carcinoma. Cancer. 92:1085–1091. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Dommel S and Blüher M: Does C-C motif
chemokine ligand 2 (CCL2) link obesity to a pro-inflammatory state?
Int J Mol Sci. 22:15002021. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Faria SS, Corrêa LH, Heyn GS, de Sant'Ana
LP, Almeida RDN and Magalhães KG: Obesity and breast cancer: The
role of crown-like structures in breast adipose tissue in tumor
progression, prognosis, and therapy. J Breast Cancer. 23:233–245.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Hsieh CC and Huang YS: Aspirin breaks the
crosstalk between 3T3-L1 adipocytes and 4T1 breast cancer cells by
regulating cytokine production. PLoS One. 11:e01471612016.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Kuziel G, Thompson V, D'Amato JV and
Arendt LM: Stromal CCL2 signaling promotes mammary tumor fibrosis
through recruitment of myeloid-lineage cells. Cancers (Basel).
12:20832020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Kranjc MK, Novak M, Pestell RG and Lah TT:
Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme.
Radiol Oncol. 53:397–406. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Suárez-Nájera LE, Chanona-Pérez JJ,
Valdivia-Flores A, Marrero-Rodríguez D, Salcedo-Vargas M,
García-Ruiz DI and Castro-Reyes MA: Morphometric study of
adipocytes on breast cancer by means of photonic microscopy and
image analysis. Microsc Res Tech. 81:240–249. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Zazo S, González-Alonso P, Martín-Aparicio
E, Chamizo C, Luque M, Sanz-Álvarez M, Mínguez P, Gómez-López G,
Cristóbal I, Caramés C, et al: Autocrine CCL5 effect mediates
trastuzumab resistance by ERK pathway activation in HER2-positive
breast cancer. Mol Cancer Ther. 19:1696–1707. 2020.PubMed/NCBI
|
|
141
|
Song X, Zhou X, Qin Y, Yang J, Wang Y, Sun
Z, Yu K, Zhang S and Liu S: Emodin inhibits epithelial-mesenchymal
transition and metastasis of triple negative breast cancer via
antagonism of CC-chemokine ligand 5 secreted from adipocytes. Int J
Mol Med. 42:579–588. 2018.PubMed/NCBI
|
|
142
|
Gao D, Rahbar R and Fish EN: CCL5
activation of CCR5 regulates cell metabolism to enhance
proliferation of breast cancer cells. Open Biol. 6:1601222016.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Sax MJ, Gasch C, Athota VR, Freeman R,
Rasighaemi P, Westcott DE, Day CJ, Nikolic I, Elsworth B, Wei M, et
al: Cancer cell CCL5 mediates bone marrow independent angiogenesis
in breast cancer. Oncotarget. 7:85437–85449. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Velasco-Velazquez M and Pestell RG: The
CCL5/CCR5 axis promotes metastasis in basal breast cancer.
Oncoimmunology. 2:e236602013. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Velasco-Velázquez M, Jiao X, De La Fuente
M, Pestell TG, Ertel A, Lisanti MP and Pestell RG: CCR5 antagonist
blocks metastasis of basal breast cancer cells. Cancer Res.
72:3839–3850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Kim EJ, Kim YK, Kim S, Kim JE, Tian YD,
Doh EJ, Lee DH and Chung JH: Adipochemokines induced by ultraviolet
irradiation contribute to impaired fat metabolism in subcutaneous
fat cells. Br J Dermatol. 178:492–501. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Keophiphath M, Rouault C, Divoux A,
Clement K and Lacasa D: CCL5 promotes macrophage recruitment and
survival in human adipose tissue. Arterioscler Thromb Vasc Biol.
30:39–45. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Shao L, Feng B, Zhang Y, Zhou H, Ji W and
Min W: The role of adipose-derived inflammatory cytokines in type 1
diabetes. Adipocyte. 5:270–274. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Brett E, Sauter M, Timmins É, Azimzadeh O,
Rosemann M, Merl-Pham J, Hauck SM, Nelson PJ, Becker KF, Schunn I,
et al: Oncogenic linear collagen VI of invasive breast cancer is
induced by CCL5. J Clin Med. 9:9912020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Guo Y, Xu F, Lu T, Duan Z and Zhang Z:
Interleukin-6 signaling pathway in targeted therapy for cancer.
Cancer Treat Rev. 38:904–910. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Kim HS, Jung M, Choi SK, Woo J, Piao YJ,
Hwang EH, Kim H, Kim SJ and Moon WK: IL-6-mediated cross-talk
between human preadipocytes and ductal carcinoma in situ in breast
cancer progression. J Exp Clin Cancer Res. 37:2002018. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Lee J, Hong BS, Ryu HS, Lee HB, Lee M,
Park IA, Kim J, Han W, Noh DY and Moon HG: Transition into
inflammatory cancer-associated adipocytes in breast cancer
microenvironment requires microRNA regulatory mechanism. PLoS One.
12:e01741262017. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Bachelot T, Ray-Coquard I, Menetrier-Caux
C, Rastkha M, Duc A and Blay JY: Prognostic value of serum levels
of interleukin 6 and of serum and plasma levels of vascular
endothelial growth factor in hormone-refractory metastatic breast
cancer patients. Br J Cancer. 88:1721–1726. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Deng T, Lyon CJ, Bergin S, Caligiuri MA
and Hsueh WA: Obesity, inflammation, and cancer. Annu Rev Pathol.
11:421–449. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Liu S, Lee JS, Jie C, Park MH, Iwakura Y,
Patel Y, Soni M, Reisman D and Chen H: HER2 overexpression triggers
an IL1α proinflammatory circuit to drive tumorigenesis and promote
chemotherapy resistance. Cancer Res. 78:2040–2051. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Banerjee K and Resat H: Constitutive
activation of STAT3 in breast cancer cells: A review. Int J Cancer.
138:2570–2578. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Nickel A, Blücher C, Kadri OA, Schwagarus
N, Müller S, Schaab M, Thiery J, Burkhardt R and Stadler SC:
Adipocytes induce distinct gene expression profiles in mammary
tumor cells and enhance inflammatory signaling in invasive breast
cancer cells. Sci Rep. 8:94822018. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Gyamfi J, Lee YH, Eom M and Choi J:
Interleukin-6/STAT3 signalling regulates adipocyte induced
epithelial-mesenchymal transition in breast cancer cells. Sci Rep.
8:88592018. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Jin K, Pandey NB and Popel AS:
Simultaneous blockade of IL-6 and CCL5 signaling for synergistic
inhibition of triple-negative breast cancer growth and metastasis.
Breast Cancer Res. 20:542018. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Guo C, Chen Y, Gao W, Chang A, Ye Y, Shen
W, Luo Y, Yang S, Sun P, Xiang R and Li N: Liposomal nanoparticles
carrying anti-IL6R antibody to the tumour microenvironment inhibit
metastasis in two molecular subtypes of breast cancer mouse models.
Theranostics. 7:775–788. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Gyamfi J, Lee YH, Min BS and Choi J:
Niclosamide reverses adipocyte induced epithelial-mesenchymal
transition in breast cancer cells via suppression of the
interleukin-6/STAT3 signalling axis. Sci Rep. 9:113362019.
View Article : Google Scholar : PubMed/NCBI
|