Introduction
Cervical cancer is one of the most common
malignancies in females worldwide (1). Surgery and concurrent
chemoradiotherapy are the two primary treatment options for
patients with cervical cancer (2). Traditional chemotherapy drugs cause
serious adverse reactions and thus, the development of more
effective therapeutics will provide a marked benefit for patients
with cervical cancer (3).
Ginsenoside Rh2 (G-Rh2) is a major bioactive
component of ginseng (4) that has
been indicated to significantly inhibit the proliferation of cancer
cells in cell culture and in tumor models of various human cancers
(5–8). Previous studies have indicated that
apoptosis of cancer cells induced by G-Rh2 is caused by an
excessive accumulation of mitochondrial reactive oxygen species
(mtROS), accompanied by the occurrence of numerous downstream
apoptotic events, such as mitochondrial depolarization, cytochrome
c release and activation of caspase enzymes (9–11).
However, the upstream mechanisms through which G-Rh2 induces
mtROS-mediated apoptosis have remained elusive.
Recently, numerous studies have demonstrated that
mitochondrial dysfunction resulting in mtROS overproduction is a
potential cause of cancer cell apoptosis (12–14). Mitochondria perform oxidative
phosphorylation (OXPHOS) to synthesize ATP through the
mitochondrial electron transport chain (ETC), which comprises
complexes I–V. The leakage of electrons from the gradient formed by
the ETC complexes in the electron transfer process is the main
source of mtROS (15–17). As an unavoidable byproduct of
metabolism, the mtROS concentration is normally maintained at a low
level by the corresponding scavenger system and thereby a steady
state is maintained, which is important because mtROS are a
significant signal transducer for various molecular pathways
(18). However, if an imbalance
or decompensation of mtROS production/scavenging occurs, cells
become damaged (19,20) with reduced mitochondrial function
and they subsequently undergo apoptosis (21–23). In addition, a recent study by our
group reported that G-Rh2 induces apoptosis in HeLa cervical cancer
cells and that the process was related to ROS accumulation and
mitochondrial dysfunction (24).
Based on these previous findings, it was hypothesized that G-Rh2
targets ETC complexes, which mediates the generation of mtROS and
induces apoptosis of cervical cancer cells. The aim of the present
study was to further investigate the novel upstream direct target
of mtROS production and apoptosis induction by G-Rh2. The present
study provided novel insight into how G-Rh2 induces apoptosis in
cervical cancer cells.
Materials and methods
Cell culture
The human cervical cancer cell lines HeLa [human
papillomavirus (HPV)-positive] and C33A (HPV-negative) were
obtained from the Type Culture Collection of the Chinese Academy of
Sciences and cultured in DMEM supplemented with 10% fetal bovine
serum, 100 U/ml penicillin and 100 µg/ml streptomycin (all from
Hyclone; Cytiva). The nontumorigenic (control) End1/e6e7 cell line
was purchased from the American Type Culture Collection and
cultured in KSFM medium (Gibco; Thermo Fisher Scientific, Inc.)
with bovine pituitary extract (0.05 mg/ml), human recombinant
epidermal growth factor (0.1 ng/ml), calcium chloride (44.1 mg/l)
(all from Shanghai Absin Biotechnology Co., Ltd) and 1% antibiotics
(100 U/ml penicillin and 100 µg/ml streptomycin; Hyclone; Cytiva).
All cell lines were cultured in a humidified normoxic chamber
(Thermo Fisher Scientific, Inc.) with 5% CO2 at
37°C.
Assessment of cell viability
HeLa, C33A and End1/e6e7 cells (2×104
cells/well) were inoculated onto 96-well plates and incubated for
24 h. To determine the 50% growth inhibition concentration
(IC50) (25,26), cells were treated with 35, 45, 55
and 65 µM G-Rh2 (Yuanye Biotechnology Co., Ltd) for 24 h. Cell
viability was measured using the Cell Counting Kit-8 (CCK-8) assay
(Wuhan Boster Biological Technology, Ltd.) according to the
manufacturer's protocol. Evaluation was performed using an Infinite
M200 PRO plate reader (Tecan Group, Ltd.) at 450 nm.
Flow cytometric analysis of cell death
mode
The mode of cell death was assessed using an FITC
Annexin V Apoptosis Detection Kit I (BD Biosciences). Cells were
inoculated onto 6-well plates and incubated for 24 h. HeLa cells
were treated with 35 and 45 µM G-Rh2 for 24 h and C33A cells were
treated with 45 and 55 µM G-Rh2 for 24 h. After treatment, the
cells were collected by centrifugation, washed twice with ice-cold
PBS, suspended in 1X Binding Buffer and then incubated with 5 µl
each of propidium iodide (PI) and Annexin V-FITC solution. Probes
were incubated at 37°C in the dark for 15 min. Samples were
examined with a flow cytometer (Amnis Corporation) and quantified
using IDEAS software v6.1 (Amnis Corporation).
Apoptosis morphology detection with
Hoechst 33342 staining
The mode of cell death was also demonstrated by
Hoechst 33342 staining. After treating cells as above, the culture
medium was removed and cells were washed twice with PBS and then
submerged in 1 ml Hoechst 33342 staining solution (Beyotime
Institute of Biotechnology). The cells were washed twice again
after labelling for 30 min at 37°C in the dark and then observed
with the EVOS FL Auto Imaging System (Thermo Fisher Scientific,
Inc.).
Determination of mitochondrial
membrane potential (MMP)
JC-1 (Beijing Solarbio Science and Technology, Co.,
Ltd.) and rhodamine 123 (Beyotime Institute of Biotechnology) were
used to evaluate the MMP. HeLa cells were treated with G-Rh2 at 35
and 45 µM for 24 h and C33A cells were treated with G-Rh2 at 45 and
55 µM for 24 h. After removing the medium and washing the cells
twice with cold PBS, 1 ml of cell culture medium supplemented with
1 ml JC-1 dye working solution was added, followed by incubation at
37°C in the dark for 20 min. Cells were then washed twice with JC-1
staining buffer prior to imaging with the EVOS FL Auto Imaging
System (Thermo Fisher Scientific, Inc.). Rhodamine 123 was also
used to estimate changes in MMP. In brief, cells were stained with
rhodamine 123 in the dark at 37°C for 30 min. Cells were washed
twice with PBS prior to loading and evaluation by flow cytometry
(Amnis Corporation).
Analysis of intracellular ATP
levels
An ATP analysis kit (Beyotime Institute of
Biotechnology) was used to detect intracellular ATP levels. In
brief, cells inoculated onto 6-well cell culture plates were
incubated for 24 h and subsequently, HeLa cells were treated with
G-Rh2 at 35 and 45 µM for 24 h and C33A cells were treated with
G-Rh2 at 45 and 55 µM for 24 h. The culture medium was then removed
and replaced with 200 µl ice-cold ATP lysis buffer and the lysate
was then centrifuged at 12,000 × g for 10 min at 4°C. The
supernatant was then transferred to a new tube. The reaction was
initiated at 37°C for 3 min to consume background ATP with 100 µl
ATP detection buffer prior to adding 10 µl supernatant for
detection. Optical density values were recorded using an Infinite
M200 PRO plate reader (Tecan Group, Ltd.) and the ATP content was
converted according to the standard curve.
Measurement of oxygen consumption rate
(OCR)
The OCR was tested using the Seahorse Bioscience XFp
Extracellular Flux Analyzer (Agilent Technologies, Inc.) according
to the manufacturer's protocol. The Seahorse XFp Mito Stress Test
Kit (Agilent Technologies, Inc.) was applied to measure the OCR.
After treatment with G-Rh2 (35 and 45 µM) for 24 h, HeLa cells
(1×104/well) were inoculated onto XFp cell culture
plates for 24 h. The cells were then washed with analysis medium
(supplemented with 10 mM glucose, 2 mM glutamine and 1 mM sodium
pyruvate). Subsequently, the plates were placed in a
non-CO2 incubator at 37°C for at least 1 h for
degassing. After baseline measurement, 1 µM oligomycin (ATP
synthase inhibitor), 1 µM carbonyl cyanide-4-(trifluoromethoxy)
phenylhydrazone (FCCP; uncoupler) and 0.5 µM rotenone
(rot)/antimycin A (AA) mixture (inhibitors of ETC complexes I/III)
were used to perform real-time OCR quantifications. Data were
evaluated using Wave software version 2.6.3 (Agilent Technologies,
Inc.) and OCR was expressed in pmol/min.
Measurement of extracellular
acidification rate (ECAR)
The ECAR was detected using the Seahorse XF
Glycolysis Stress Test Kit (Agilent Technologies, Inc.) on the
Seahorse XFp Extracellular Flux Analyzer (Agilent Technologies,
Inc.) according to the manufacturer's protocol. In brief, HeLa
cells were treated with 35 or 45 µM G-Rh2 for 24 h and then
inoculated onto Seahorse XFp cell culture microtiter plates for 24
h (1×104 cells/well). The cells were then washed with
assay medium (supplemented with 2 mM glutamine) prior to degassing
treatment at 37°C in a non-CO2 incubator for 1 h. After
the ECAR baseline was calibrated, 10 mM glucose, 1 µM oligomycin
(OXPHOS inhibitor) and 50 mM 2-deoxy-d-glucose (glycolysis
inhibitor) were injected into the detection wells at the specified
time points. Data were evaluated using Wave software version 2.6.3
(Agilent Technologies, Inc.) and ECAR was expressed in mpH/min.
Cell fractionation
The Cell Mitochondrial Isolation Kit (Beyotime
Institute of Biotechnology) was used to extract mitochondrial
protein. HeLa cells were collected and rinsed with cold PBS after
treatment with 35 and 45 µM G-Rh2 for 24 h, and then treated with
ice-cold mitochondrial separation reagent containing 1 mM PMSF for
10 min. Next, the cells were homogenized and the supernatant was
centrifuged at 11,000 × g for 10 min at 4°C. The mitochondrial
protein precipitate was lysed with mitochondrial lysate for
subsequent western blotting analysis. Finally, total protein was
obtained by lysing HeLa cells with RIPA buffer (Beyotime Institute
of Biotechnology). The protein concentration of the samples was
detected using a BCA Protein Assay Kit.
Western blot analysis
Western blot was applied to analyze protein
expression. In brief, denatured protein samples (30 µg) were
separated by 12% SDS-PAGE, transferred to PVDF membranes (EMD
Millipore), blocked in 5% skimmed milk (Beijing Solarbio Science
& Technology, Co., Ltd.) for 2 h at room temperature, washed
with PBS containing Tween-20 (PBST) and incubated with primary
antibodies [total OXPHOS human WB antibody cocktail (cat no.
ab110411); anti-myc tag (cat no. ab206486); anti-Flag (cat no.
ab205606); and anti-6×His tag (cat no. ab18184); all 1:1,000
dilution; Abcam] at 4°C overnight on an orbital shaker. PVDF
membranes were then washed with PBST and incubated with horseradish
peroxidase-conjugated anti-mouse IgG (cat no. sc-2005; 1:1,000
dilution; Santa Cruz Biotechnology, Inc.) at room temperature for 1
h prior to washing with PBST and adding a working solution of
Enhanced Chemiluminescence Reagent Kit (Beyotime Institute of
Biotechnology). Membranes were imaged using the iBright FL1000
Imaging System (Invitrogen; Thermo Fisher Scientific, Inc.). Image
J software 1.53a (National Institutes of Health) was used to
quantify the gray value of western blot bands and the protein
expression level was normalized as the gray value of the target
protein/the loading control protein.
ETC complex activity assay
ETC complex activity detection kits (cat no. BC0515,
BC3235, BC3245, BC0945 and BC1445; Beijing Solarbio Science &
Technology, Co., Ltd.) were used to measure the enzymatic activity
of ETC complexes I–V with an Infinite M200 PRO Plate Reader (Tecan
Group, Ltd.) according to the manufacturer's protocol. The
oxidation rate of NADH was recorded at 340 nm to assess the
activity of ETC complex I. The rate of 2,6-dichloroindolephenol
reduction was used to measure the activity of ETC complex II at 605
nm. The activity of ETC complex III was detected by measuring the
increase rate of the light absorption of cytochrome c-reduced at
550 nm. The activity of ETC complex IV was monitored by measuring
the rate of decrease in the absorbance of reduced cytochrome c at
550 nm. The activity of ETC complex V was quantified by calculating
the rate of addition of inorganic phosphate (Pi) at 660 nm.
Molecular docking
Molecular docking analyses were performed for G-Rh2
and the ETC complexes. Data on the structures of ETC complexes were
obtained from the RCSB Protein Data Bank (PDB; http://www.rcsb.org/pdb), including ETC complex I (PDB
ID: 4G73) (27), ETC complex III
(PDB ID: 4PD4) (28) and ETC
complex V (1BMF) (29,30). Molecular docking was performed
with AutoDock tools (version 4.2.6) and visualized with the
Discovery Studio 4.0 Visualizer (BIOVIA).
Measurement of mtROS
The mitoSOX probe (Beyotime Institute of
Biotechnology) was used to determine mtROS production. First, cells
were seeded onto 6-well cell culture plates. After 24 h of
incubation, G-Rh2 specificity was determined by pretreating HeLa
cells with mitoQ (5 µM) for 1 h prior to treatment with G-Rh2 (35
and 45 µM) for 24 h. The cells were collected by centrifugation and
washed with cold PBS prior to adding 5 µM mitoSOX probe (cat. no.
M36008; Invitrogen; Thermo Fisher Scientific, Inc.) and incubating
the mixture at 37°C for 20 min in the dark. The cells were then
washed twice with PBS, 200 µl cell suspension (1×106
cells/ml) was added to the 96-well black plate and fluorescence
units were detected with an Infinite M200 PRO plate reader (Tecan
Group, Ltd.) at 510/580 nm excitation/emission wavelengths.
Construction of the overexpression
vectors and transfection
According to the PDB ID of ETC complexes I, III and
V in molecular docking, the representative subunits of
corresponding human genes were selected as NADH-ubiquinone
oxidoreductase core subunit S1(NDUFS1), biquinol-cytochrome
c reductase core protein 1 (UQCRC1) and ATP synthase F1
subunit beta (ATP5F1B), respectively. The NDUFS1, UQCRC1 and
ATP5F1B cDNA fragments were amplified with primers using a cDNA
library prepared from HeLa cells as the template. The PCR program
was as follows: 98°C for 10 sec, 58°C for 10 sec and then 72°C for
1 min per kb for a total of 35 cycles. The target fragments were
digested with double restriction enzymes and then ligated to the
corresponding plasmids (Guangzhou RiboBio Co., Ltd.) with T4 Ligase
(Takara Bio, Inc.) and then transformed into E. coli turbo
(New England Biolabs). Positive recombinant plasmids were
identified by restriction digestion and sequencing. All plasmids,
primers and enzymes used are provided in Table SI. For transfection, HeLa cells
were seeded in 6-well plates and cultured for 24 h. Cells were
transfected with 2 µg of the plasmids using
Lipofectamine® 2000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) for 6 h. The culture medium was then replaced
with fresh opti-DMEM (Gibco; Thermo Fisher Scientific, Inc.) and
the cells were continued to be grown in culture for 48 h. For G-Rh2
treatment, HeLa cells were treated with 45 µM G-Rh2 for 24 h after
transfection.
Statistical analysis
Statistical analyses were performed with GraphPad
Prism software version 6 (GraphPad Software Inc.). For quantitative
analyses, data were collected from three independent experiments
and presented as the mean ± standard deviation. One-way ANOVA
followed by Dunnett's post-hoc test or Tukey's post-hoc test as
indicated within the figure legends were utilized to compare among
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
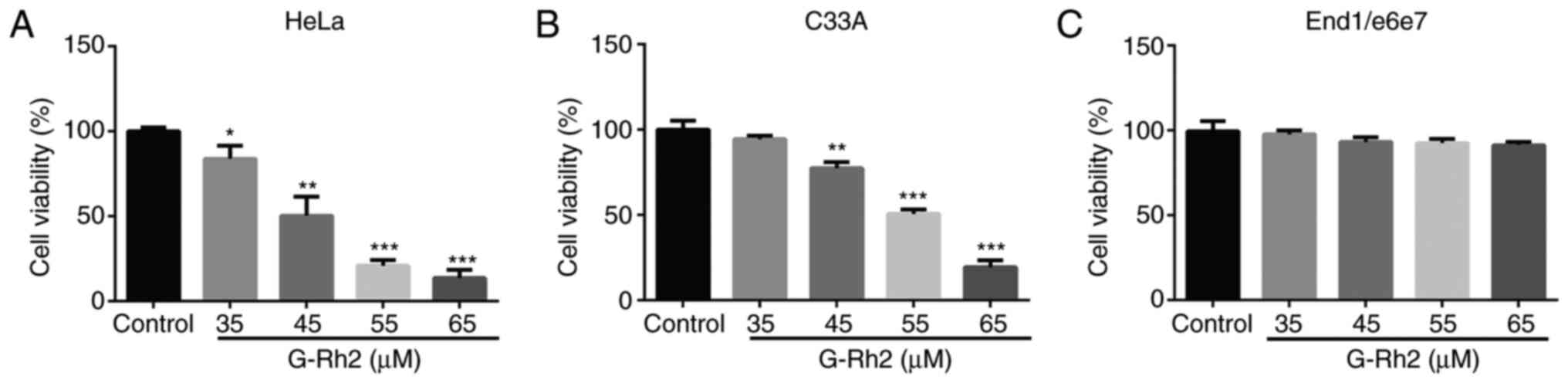
G-Rh2 inhibits the viability of
cervical cancer cells but is not cytotoxic to End1/e6e7 cells
To study the impact of G-Rh2 on the viability of
cancer and normal cells, two cervical cancer cell lines, including
HeLa (integrated HPV18) and C33A (without HPV) and human cervical
epithelial cells (End1/e6e7) as a control were exposed to 35, 45,
55 and 65 µM G-Rh2 for 24 h. As presented in Fig. 1A and B, G-Rh2 was cytotoxic to all
cervical cancer cell lines assessed, with IC50 values of 45 µM for
HeLa and 55 µM for C33A cells at 24 h. However, compared with the
effect on the two cervical cancer cell lines, the effect of
treatments at all concentrations on the viability of End1/e6e7
cells was negligible, indicating that G-Rh2 was highly selective
for cancer cells (Fig. 1C).
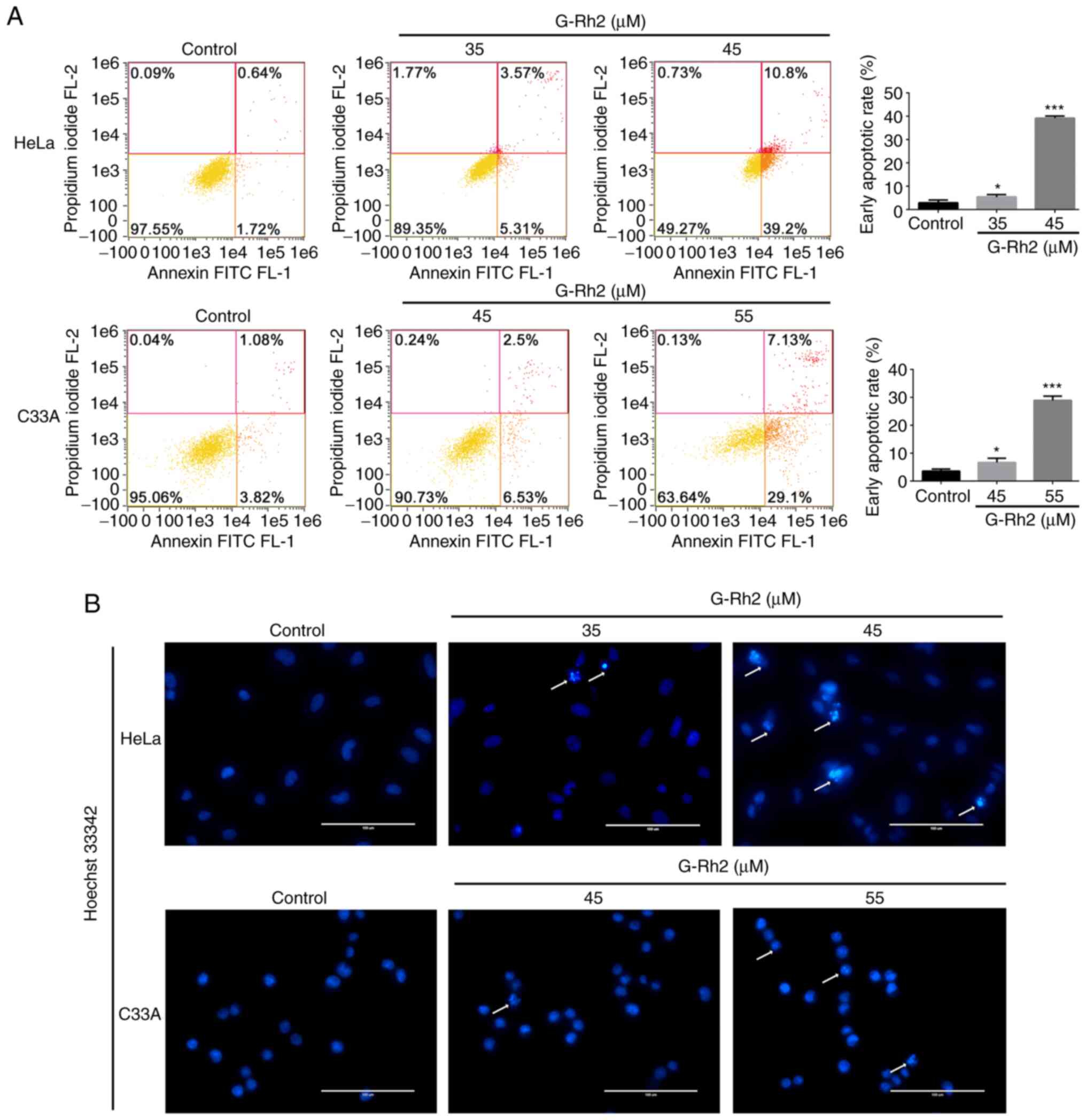
G-Rh2 induces apoptosis in cervical
cancer cells
Next, Annexin V-FITC/PI and Hoechst 33342 staining
assays were used to analyze the number of apoptotic cells following
exposure to different concentrations of G-Rh2. As indicated in
Fig. 2A, after 24 h of treatment,
the rate of early apoptosis was considerably increased in
G-Rh2-treated cervical cancer cells (HeLa and C33A) compared with
that in the control group. Furthermore, more intense blue staining
and more irregular chromatin were observed in G-Rh2-treated HeLa
and C33A cells (Fig. 2B) compared
with that in the control group, indicating that G-Rh2 treatment
induced apoptosis in cervical cancer cells. Of note, G-Rh2
significantly induced early apoptosis in both cancer cell lines in
a dose-dependent manner. However, treatment with high-dose G-Rh2
(55 and 65 µM) slightly increased the rate of cell death in
End1/e6e7 cells compared with that in the control group (Fig. S1) and demonstrated that the
effect on normal cells was not statistically significant as
determined for cancer cells.
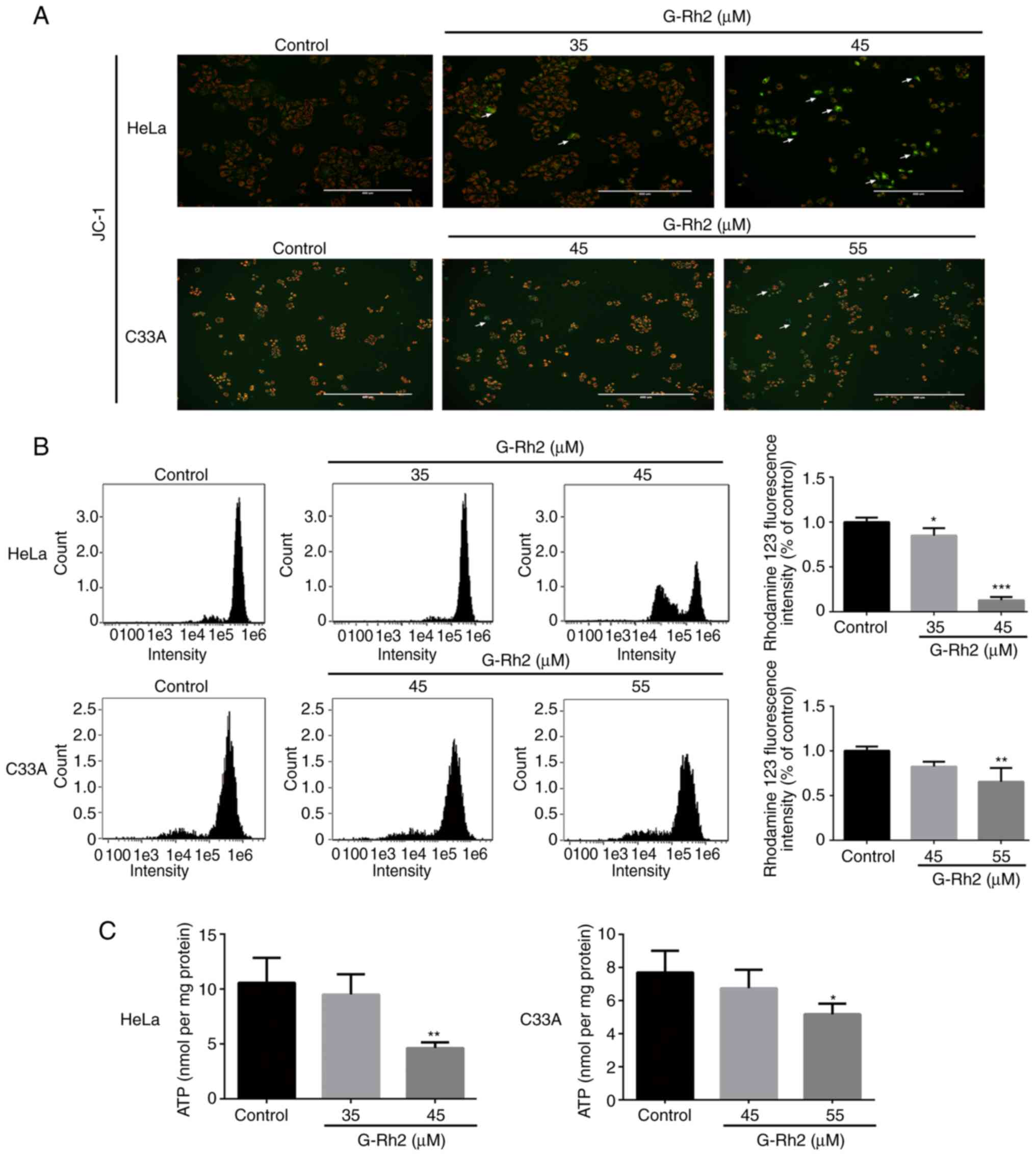
HeLa cells are more sensitive to G-Rh2
than C33A cells regarding MMP and ATP generation
According to a previous study by our group
suggesting that G-Rh2 possibly induced mitochondrial dysfunction in
cancer cells, the mitochondrial defects induced by G-Rh2 may
potentially result in apoptosis of cervical cancer cells (24). In the present study, to further
examine this, the JC-1 and rhodamine 123 probes were used to detect
changes in the MMP, an index of mitochondrial function. After
staining with the JC-1 fluorescent probe, HeLa cells had an obvious
response, displaying red to green transition with increasing G-Rh2
concentration. However, only a slight change in JC-1 probe
fluorescence was identified in C33A cells (Fig. 3A). In addition, flow cytometry was
employed to measure the fluorescence intensity of rhodamine 123;
G-Rh2 treatment significantly reduced the MMP in HeLa cells.
Although 55 µM G-Rh2 was able to significantly reduce the MMP in
C33A cells, the degree of reduction was significantly lower than
that in HeLa cells (Fig. 3B).
Next, the effect of G-Rh2 on cellular ATP levels was evaluated by a
chemiluminescence assay. Specifically, G-Rh2 treatment reduced
cellular ATP levels by 10.4% (35 µM) and 49.1% (45 µM) in HeLa
cells and by 7.5% (35 µM) and 35.2% (45 µM) in C33A cells compared
with that in in the control group (Fig. 3C). According to these
observations, HeLa cells were more sensitive to G-Rh2 than C33A
cells, at least with regard to MMP and ATP production. Therefore,
HeLa cells were selected for subsequent mechanistic examination of
these effects.
Inhibitory effect of G-Rh2 on
mitochondrial OXPHOS is greater than that on glycolysis in HeLa
cells
OXPHOS is essential for the ability of cells to hold
the MMP (7). To study the effect
of G-Rh2 on mitochondrial OXPHOS, a Seahorse XFp extracellular flux
analyzer was used to monitor mitochondrial aerobic respiration in
HeLa cells. As presented in Fig.
S2A, G-Rh2 treatment of HeLa cells resulted in a rapid decline
of OXPHOS. G-Rh2 treatment also significantly inhibited the
cellular response to typical ETC complex inhibitors [e.g.,
oligomycin (ATP synthase inhibitor), FCCP (mitochondrial uncoupling
agent), AA (ETC complex III inhibitor) and rot (ETC complex I
inhibitor)]. In addition, G-Rh2 significantly inhibited the basal
and maximum mitochondrial OXPHOS capacity. Furthermore, in HeLa
cells, ATP-linked proton leak as well as reserve capacity were also
more significantly inhibited by G-Rh2 compared with those in the
control group (Fig. S2B).
Cancer cells provide energy and produce lactic acid
through the glycolysis pathway, via a process termed the ‘Warburg
effect’. Therefore, it was investigated whether G-Rh2 was involved
in blocking glycolysis. The results indicated that G-Rh2 reduced
ECAR, which reflects glycolytic capacity. Glycolysis, glycolytic
capacity and glycolytic reserve were all significantly inhibited by
G-Rh2 (Fig. S2C and D). Of note,
G-Rh2 inhibited both OXPHOS and glycolysis in HeLa cells and had a
stronger effect on OXPHOS.
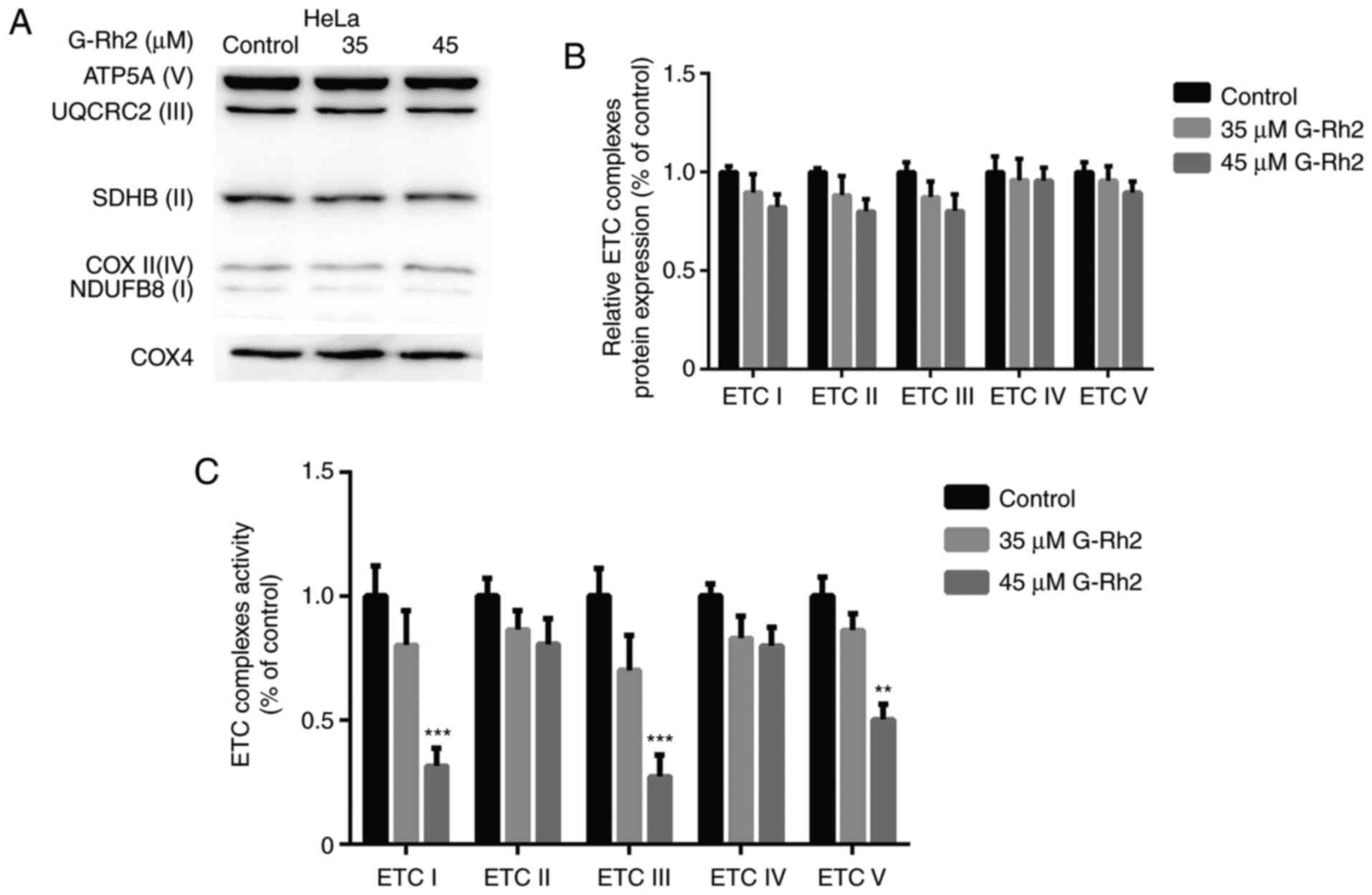
Impairment of OXPHOS by G-Rh2 occurs
via direct inhibition of ETC complexes activity, not by
downregulation of ETC proteins
Decreasing OXPHOS may contribute to the
downregulation of ETC proteins or to the inhibition of ETC complex
enzymes (31). Therefore, in the
present study, the expression levels of five ETC subunits were
detected in G-Rh2-treated cells. As presented in Fig. 4A and B, ETC protein expression was
not significantly altered after G-Rh2 treatment, indicating that
G-Rh2 damages OXPHOS not by downregulating ETC protein expression
but by directly inhibiting the activity of ETC complexes. As
expected, treatment with 45 µM G-Rh2 significantly inhibited the
activity of ETC complexes I, III and V in HeLa cells but did not
affect ETC complexes II and IV (Fig.
4C). These results suggested that the impairment of G-Rh2 on
OXPHOS was not caused by downregulation of ETC protein expression
but by directly inhibiting the activity of ETC complexes.
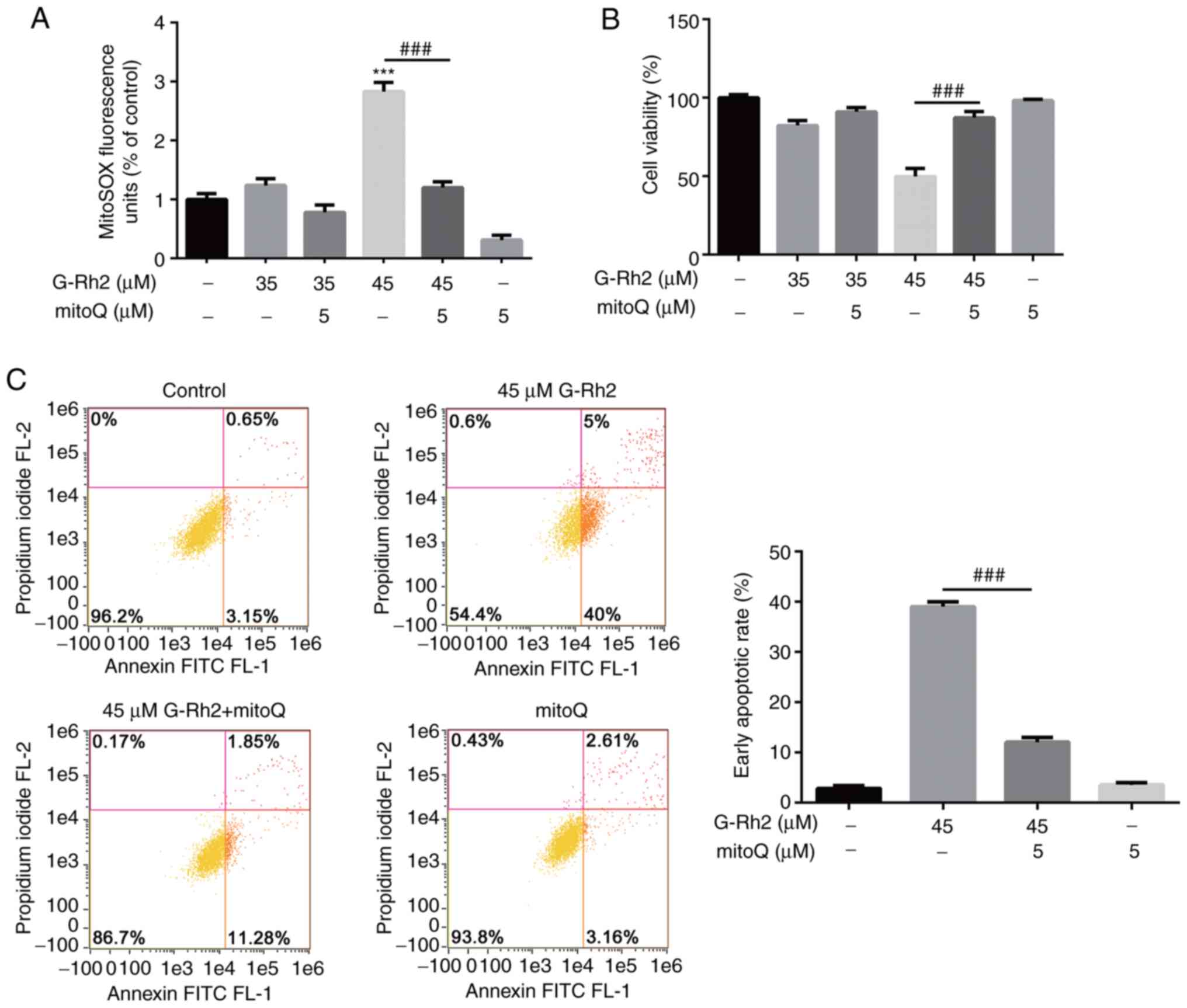
Increased ROS levels in G-Rh2-mediated
apoptosis
MtROS is an outgrowth of OXPHOS that is primarily
generated by the ETC complexes. As G-Rh2 significantly inhibited
the activity of complexes I, III and V, its impact on mtROS
production was examined. As presented in Fig. 5A, treatment of HeLa cells with 45
µM G-Rh2 led to overproduction of mtROS, with mtROS levels
exhibiting a significant increase of 2.8-fold compared with that in
the control group. This burst of mtROS production was partially
rescued by the antioxidant mitoQ.
To study whether mtROS was involved in G-Rh2-induced
apoptosis, the protective effect of mitoQ on G-Rh2-induced
cytotoxicity was detected using CCK-8 assays. As indicated in
Fig. 5B, the effect of G-Rh2 on
HeLa cells was partially attenuated by mitoQ pretreatment.
Furthermore, quantitative measurements of Annexin V/PI-positive
cells by flow cytometry (Fig. 5C)
verified that mitoQ partially rescued G-Rh2-induced early
apoptosis. These results demonstrated that G-Rh2-induced mtROS
generation mediated the subsequent apoptosis.
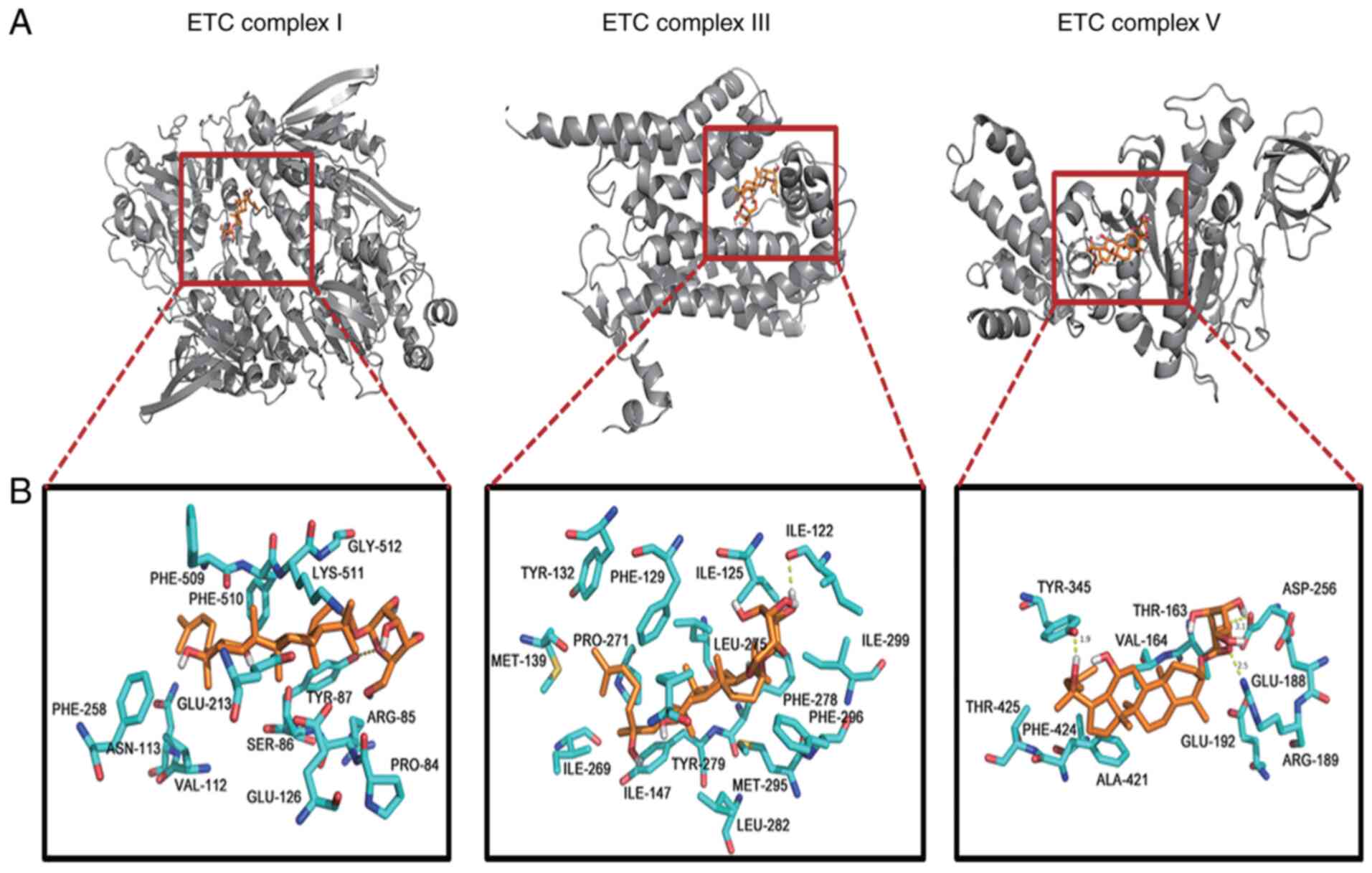
Molecular docking reveals a putative
G-Rh2 binding site on ETC complexes I, III and V
Although the above results confirmed that G-Rh2
induced mtROS, triggering apoptosis via suppressing ETC complexes
I, III and V, the detailed binding mechanism remained to be further
elucidated. Therefore, protein-ligand docking was performed to
understand the mode of G-Rh2 binding and to locate the ETC complex
residues that may have crucial roles in G-Rh2 binding.
The molecular docking results revealed that the
optimal docking conformation binding energy of G-Rh2 and ETC
complex III was −10.6 kcal/mol, which was lower than that of ETC
complex I (−7.73 kcal/mol) and ETC complex V (−6.48 kcal/mol). The
hydroxyl oxygen atom of the six-membered ring of G-Rh2 was
indicated to be able to form hydrogen bonds with Thr87 in ETC
complex I, Ile122 in ETC complex III and Thr163 and Asp256 in ETC
complex V, with hydrogen bond lengths of 3.3, 2.1, 1.9 and 3.1 Å,
respectively. The oxygen atom of the six-membered ring of G-Rh2 was
also able to form a 2.5 Å hydrogen bond with Arg189 in ETC complex
V. In addition, the oxygen of Tyr345 in ETC complex V formed a
hydrogen bond with the six-membered ring hydrogen of G-Rh2, the
length of which was 1.9 Å (Fig. 6A
and B; Table I). The
formation of these hydrogen bonds enhanced the ability of G-Rh2 to
target mitochondrial ETC complexes by better inhibiting protein
activity.
 | Table I.Binding affinities (free energies of
binding) and the types of interactions of the ligand ginsenoside
Rh2 with its receptors ETC complex I, III and V. |
Table I.
Binding affinities (free energies of
binding) and the types of interactions of the ligand ginsenoside
Rh2 with its receptors ETC complex I, III and V.
| ETC complex | Docking score
(kcal/M) | Residues involved
in hydrogen bonding | Residues involved
in weak interactions |
|---|
| I | −7.73 | THR87 | PRO84, ASP85,
SER86, THR87, VAL112, ASN113, GLU126, GLU213, PHE258, PHE509,
PHE510, LYS511, GLY512 |
| III | −10.60 | ILE122 | ILE125, PHE129,
TYR132, MET139, ILE147, PRO271, LEU275, TYR279, PHE278, LEU282,
MET295, PHE9, ILE299 |
| V | −6.48 | THR163, ASP256,
ARG189, TYR345 | THR163, VAL164,
ARG189, GLU192, ASP256, TYR345, PHE424, ALA421, THR425 |
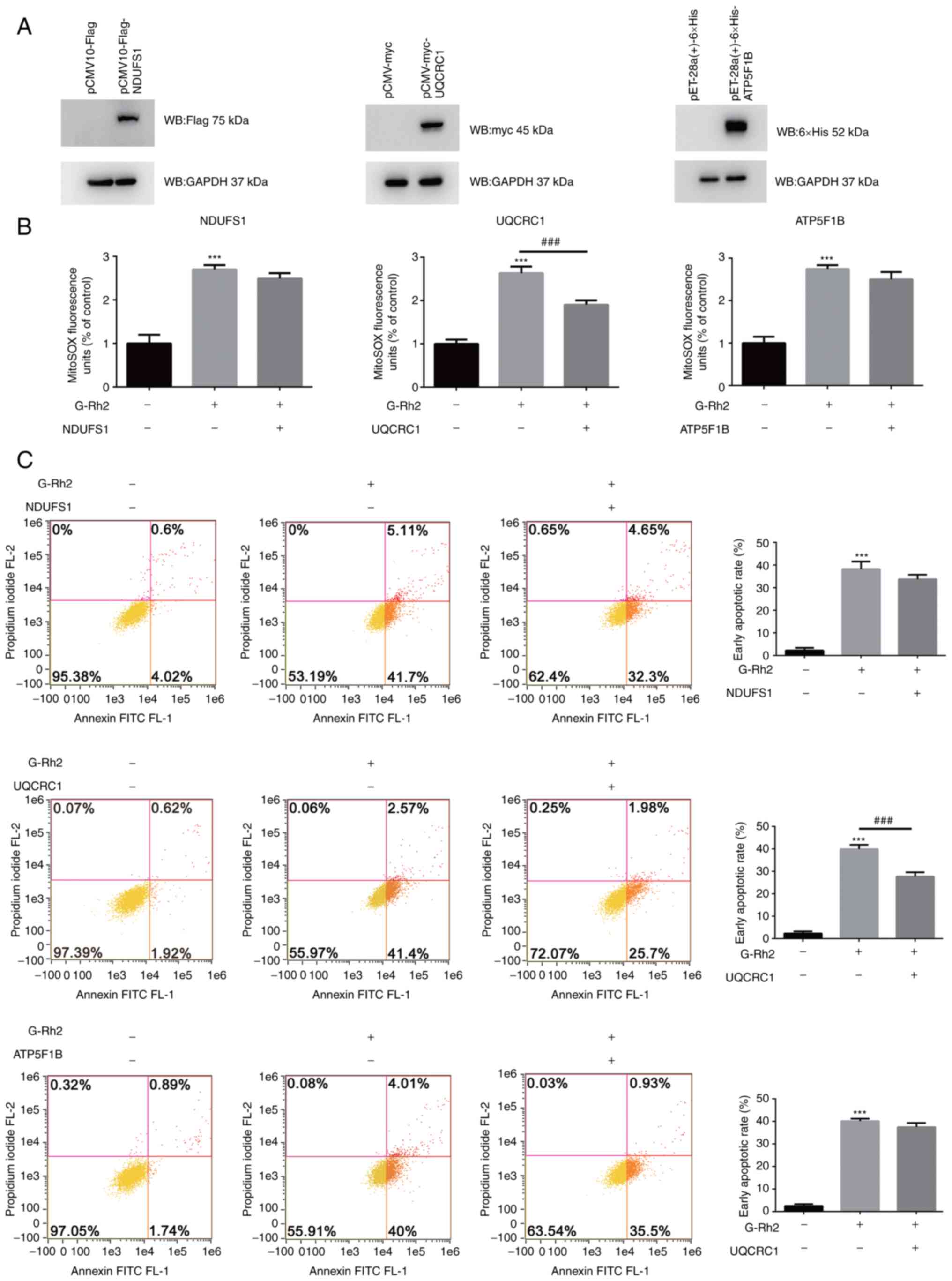
Overexpression of UQCRC1 partially
rescued G-Rh2-induced mtROS production and apoptosis
In order to verify that the effect of ETC complexes
I, III and V in G-Rh2 induced cervical cancer cell apoptosis,
overexpression vectors for the representative subunits of ETC
complex I (NDUFS1), ETC complex III (UQCRC1) and ETC complex V
(ATP5F1B) were constructed. HeLa cells transfected with these
overexpression vectors exhibited a significant increase in the
expression levels of NDUFS1, UQCRC1 and ATP5F1B protein compared to
mock-transfected cells (Fig. 7A).
G-Rh2 treatment increased mtROS and apoptosis in HeLa cells
compared with that in the control group, which was partially
attenuated by overexpression of NDUFS1, UQCRC1 and ATP5F1B and the
overexpression of UQCRC1 had a significant effect (Fig. 7B and C).
Discussion
In the present study, the cytotoxicity of G-Rh2 in
the HeLa (HPV18-positive) and C33A (HPV-negative) cervical cancer
cell lines compared with that of the non-tumorigenic spontaneous
End1/e6e7 cell line was assessed. The results demonstrated that
G-Rh2 exposure had specific dose-dependent cytotoxicity on both
cervical cancer cell lines but not on End1/e6e7 cells. It was
further reported that G-Rh2-induced apoptosis was closely related
to the specific activity inhibition of ETC complexes I, III and V,
which led to reduced OXPHOS, decreased oxygen consumption and
increased mtROS production; these processes are summarized in
Fig. 8.
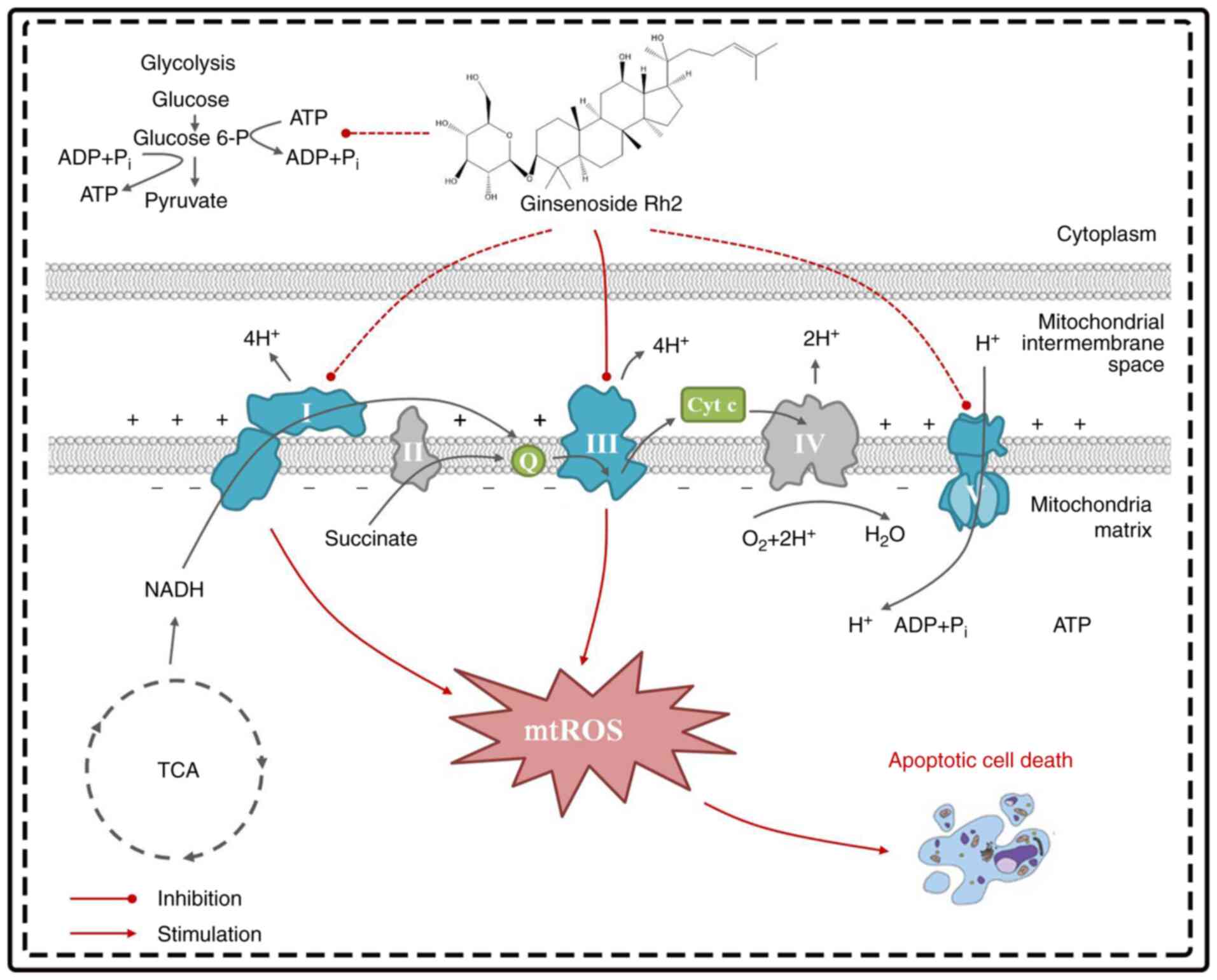 | Figure 8.Schematic diagram of G-Rh2 targeting
mitochondria. In the process of reducing mitochondrial metabolism,
G-Rh2 primarily targets ETC complexes III and indirectly affects I
and V, thereby impairing mitochondrial oxidative phosphorylation,
blocking electron transfer related to the TCA cycle and stimulating
excess mtROS production, leading to apoptosis. G-Rh2, ginsenoside
Rh2; ETC, electron transport chain; mtROS, mitochondrial reactive
oxygen species; Cyt c, cytochrome c; Q, ubiquinone; TCA,
tricarboxylic acid. |
The vast majority of G-Rh2-based anti-cervical
cancer studies have been performed in HeLa cells (32,33). The present results are consistent
with these reports; in addition, it was indicated that G-Rh2 was
cytotoxic to both HeLa and C33A cells, with IC50 values of 45 and
55 µM, respectively. Compared with the effect in C33A cells, HeLa
cells had a more markedly decreased viability following treatment
with a relatively low concentration of G-Rh2. This may indicate
that cervical cancer caused by HPV infection is more sensitive to
G-Rh2. By contrast, G-Rh2 did not inhibit the viability of
End1/e6e7 cells to a marked degree, although their viability was
slightly decreased at higher concentrations tested. Similar to
reports on other human cancer cell lines (34,35), the specific effect of G-Rh2 on
cervical cancer cells determined in the present study suggests its
low intrinsic toxicity and its differential effects on normal cells
vs. cancer cells.
One of the major apoptotic pathways in cancer cells
involves the mitochondria. The presence of mitochondrial
dysfunction was considered as a possible mechanism of G-Rh2-induced
cytotoxicity (36). A previous
study by our group suggested that G-Rh2 upregulated
voltage-dependent anion channel 1 to trigger the mitochondrial
translocation of BAX, promoting cytochrome c release and initiating
mitochondrial-dependent apoptosis in HeLa cells (24). In the present study, it was
revealed that the effect of G-Rh2 on the dissipation of the MMP and
the reduction in ATP generation in HeLa cells was much stronger
than that in C33A cells, consistent with the cell viability and
apoptosis results. It is worth noting that p53 is mutated in C33A
cells, while p53 is wild-type in HeLa (HPV-positive cervical cancer
cell line) and p53 is involved in maintaining mitochondrial
homeostasis. The response of wild-type p53 to mitochondrial stress
in HeLa cells may be responsible for the higher sensitivity to
decreased MMP and ATP levels (37,38). In summary, mitochondria may be the
primary intracellular target of G-Rh2.
Mitochondria primarily synthesize ATP via OXPHOS,
which involves the ETC complex along with several carriers that
localize to the inner mitochondrial membrane (39). The next goal of the present study
was to investigate whether G-Rh2 affected mitochondrial OXPHOS and
the underlying mechanism. In these experiments, it was demonstrated
that G-Rh2 inhibited mitochondrial respiration (basal OCR, maximal
OCR and reserve capacity). Mitochondrial defects may lead to
reduced expression of ETC complexes I–V (40). The results suggested that G-Rh2
primarily suppressed ETC by inhibiting the activity of ETC complex
enzymes rather than by affecting their expression. It was further
demonstrated that G-Rh2 exhibited potent inhibitory effects on ETC
complexes I, III and V, but had no impact on ETC complexes II and
IV. The suppression of ETC complexes I, III and V by G-Rh2 caused
significantly decreased MMP and cellular ATP levels. However, the
inhibition of the MMP by G-Rh2 was not consistent with that of ATP,
with the inhibitory effect on MMP being stronger. This was probably
due to the ATP produced in compensation by the glycolytic pathway.
More active glycolysis takes place to compensate for the decreased
ATP production due to impaired mitochondrial OXPHOS (41). During homeostasis, ~70% of ATP is
synthesized in the mitochondria; however, this percentage declines
to 30% under a hypoxic environment (42). In the present study, it was also
demonstrated that G-Rh2 does not completely inhibit glycolysis,
exerting a stronger inhibitory effect of G-Rh2 on OXPHOS than on
glycolysis in HeLa cells.
The overproduction of mtROS is a result of the
events that inhibit the activity of ETC complexes (43). In fact, in the process of electron
transfer from NADH to coenzyme Q in complex I, mtROS may be
generated at the flavin mononucleotide site and the coenzyme Q
binding site; the hemiquinone radical (QH-) of complex III produces
mtROS by leaking electrons to O2 through a non-enzymatic reaction
(17). Although ETC complex V
(ATP synthase) is not a direct source of mtROS, it participates in
the regulation of energy metabolism, which in turn affects mtROS
generation (44). In the present
study, a suppressive effect of G-Rh2 on the activity of ETC
complexes I, III and V was observed. To this end, the necessity of
mtROS was further validated, which was stimulated by the inhibition
of ETC complexes I, III and V in the induction of apoptosis. The
results indicated that the overproduction of mtROS mediated by
G-Rh2 was primarily responsible for the cytotoxicity caused by
G-Rh2. After G-Rh2 treatment, cellular mtROS levels increased by
~2.8-fold and this was partially reversed by the mitochondrial
target antioxidant Mito Q. Furthermore, Mito Q also inhibits
apoptosis induced by G-Rh2. These results are in line with previous
reports that suggested G-Rh2 induces apoptosis by promoting mtROS
accumulation in human leukemia Jurkat, HepG2 and Hep3B cells
(34,36,45).
The molecular docking experiments indicated that due
to powerful hydrogen bonding and/or hydrophobic interactions, G-Rh2
exhibited strong affinities towards ETC complex III (−10.6
kcal/mol), ETC complex I (−7.73 kcal/mol) and ETC complex V (−6.48
kcal/mol). Enzymatic activity experiments suggested that G-Rh2
significantly inhibited the activity of ETC complexes I, III and V
and the degree of inhibition was basically the same. Conversely,
the molecular docking results indicated that G-Rh2 has a higher
affinity for ETC complex III. This implied that G-Rh2 may directly
bind to ETC complex III and indirectly interfere with ETC complexes
I and V. The binding sites of the molecular docking results
suggested that post-translational modification of the protein is
likely responsible for the ability of G-Rh2 to reduce the activity
of the ETC complex enzymes under the condition that the protein
expression of the representative subunits of complex I–V did not
change. G-Rh2 may catalyze the phosphorylation of corresponding
amino acids through hydrogen bonding.
To gain additional insight into the mechanism of
action of ETC complex I, III and V in G-Rh2-induced mtROS-mediated
apoptosis, overexpression vectors for NDUFS1, UQCRC1 and ATP5F1B,
which are representative subunits of ETC complex I, III and V,
respectively, were constructed. The results suggested that
overexpression of UQCRC1 significantly attenuated G-Rh2-induced
mtROS-mediated apoptosis in HeLa cells. Consistent with the
molecular docking results, G-Rh2 mainly targets ETC complex III,
which promotes the production of mtROS and ultimately induces
apoptosis. It has been reported that ETC complex I and ETC complex
III are closely connected through the interaction between protein
subunits (46), which may be the
cause of the decrease in ETC complex I activity. In addition, ETC
complexes I–IV not only transfer electrons, but also establish the
proton gradient of ETC complex V to synthesize ATP (17). The inhibition of ETC complex III
hinders electron transfer and accumulated electrons may be
responsible for the decrease of ETC complex V enzyme activity.
The limitation of the present study on G-Rh2 is that
the experiments were all performed in cell lines cultured in
vitro. It has been reported that G-Rh2 exhibits a variety of
anti-tumor activities in vivo with no measurable toxicity
even at a dose of 120 mg/kg (47), which means that G-Rh2 possesses a
wide tolerance. However, due to the huge difference between in
vitro and in vivo experiments, the actual mechanisms of
G-Rh2 require to be further studied in vivo.
In conclusion, the present study indicated that
G-Rh2 is a major contributor to apoptosis in HeLa cells through the
mitochondrial pathway. In this process, G-Rh2 inhibits the activity
of ETC complex III and indirectly affects the activity of ETC
complexes I and V, leading to the overproduction of mtROS,
decreased ATP synthesis and bidirectional inhibition of OXPHOS and
glycolysis. To the best of our knowledge, the present study was the
first to elucidate that the mechanism through which mtROS
overproduction induces apoptosis in HPV-positive cervical cancer
cells following G-Rh2 treatment was through inhibiting the activity
of ETC complex III. This artificial regulation of mtROS at ETC
complex III may be of great significance for cancer treatment.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The research was supported by key projects at the
central government level: The Ability Establishment of Sustainable
use for Valuable Chinese Medicine Resources (grant no. 2060302),
the National Key Research and Development Program of China (grant
no. 2017YFC1702104) and the National Natural Foundation of China
(grant no. 81703663).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
ML and JW conceived and designed the study. YL, SY,
XX and JQ performed experiments. YY helped with the collection and
assembly of data. YY, WZ and JQ analyzed the data and prepared the
figures. ML, YL and WZ drafted and revised the manuscript. All
authors read and approved the final manuscript. ML and WZ confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
G-Rh2
|
ginsenoside Rh2
|
|
MMP
|
mitochondrial membrane potential
|
|
ETC
|
electron transport chain
|
|
mtROS
|
mitochondrial reactive oxygen
species
|
|
CCK-8
|
Cell Counting Kit-8
|
|
OCR
|
oxygen consumption rate
|
|
FCCP
|
carbonyl cyanide-4-(trifluoromethoxy)
phenylhydrazone
|
|
ECAR
|
extracellular acidification rate
|
|
OXPHOS
|
oxidative phosphorylation
|
|
NDUFS1
|
NADH: ubiquinone oxidoreductase core
subunit S1
|
|
UQCRC1
|
biquinol-cytochrome c reductase core
protein 1
|
|
ATP5F1B
|
ATP synthase F1 subunit beta
|
References
|
1
|
Wang R, Pan W, Jin L, Huang W, Li Y, Wu D,
Gao C, Ma D and Liao S: Human papillomavirus vaccine against
cervical cancer: Opportunity and challenge. Cancer Lett.
471:88–102. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yang K, Park W, Huh SJ, Bae DS, Kim BG and
Lee JW: Clinical outcomes in patients treated with radiotherapy
after surgery for cervical cancer. Radiat Oncol J. 35:39–47. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nokihara H, Lu S, Mok TSK, Nakagawa K,
Yamamoto N, Shi YK, Zhang L, Soo RA, Yang JC, Sugawara S, et al:
Randomized controlled trial of S-1 versus docetaxel in patients
with non-small-cell lung cancer previously treated with
platinum-based chemotherapy (East Asia S-1 Trial in Lung Cancer).
Ann Oncol. 28:2698–2706. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhuang J, Yin J, Xu C, Mu Y and Lv S:
20(S)-Ginsenoside Rh2 induce the apoptosis and autophagy in U937
and K562 cells. Nutrients. 10:3282018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li X, Chu S, Lin M, Gao Y, Liu Y, Yang S,
Zhou X, Zhang Y, Hu Y, Wang H and Chen N: Anticancer property of
ginsenoside Rh2 from ginseng. Eur J Med Chem. 203:1126272020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
He XL, Xu XH, Shi JJ, Huang M, Wang Y,
Chen X and Lu JJ: Anticancer effects of ginsenoside Rh2: A
systematic review. Curr Mol Pharmacol. Mar 8–2021.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu Y, Han M, Li X, Wang H, Ma M, Zhang S,
Guo Y, Wang S, Wang Y, Duan N, et al: Age-related changes in the
mitochondria of human mural granulosa cells. Hum Reprod.
32:2465–2473. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang YS, Lin Y, Li H, Li Y, Song Z and Jin
YH: The identification of molecular target of (20S) ginsenoside Rh2
for its anti-cancer activity. Sci Rep. 7:124082017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li B, Zhao J, Wang CZ, Searle J, He TC,
Yuan CS and Du W: Ginsenoside Rh2 induces apoptosis and
paraptosis-like cell death in colorectal cancer cells through
activation of p53. Cancer Lett. 301:185–192. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhu C, Liu F, Qian W, Zhang T and Li F:
Combined effect of sodium selenite and ginsenoside Rh2 on HCT116
human colorectal carcinoma cells. Arch Iran Med. 19:23–29.
2016.PubMed/NCBI
|
|
11
|
Xia T, Wang JC, Xu W, Xu LH, Lao CH, Ye QX
and Fang JP: 20S-Ginsenoside Rh2 induces apoptosis in human
Leukaemia Reh cells through mitochondrial signaling pathways. Biol
Pharm Bull. 37:248–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng MH, Pan CY, Chen NF, Yang SN, Hsieh
S, Wen ZH, Chen WF, Wang JW, Lu WH and Kuo HM: Piscidin-1 induces
apoptosis via mitochondrial reactive oxygen species-regulated
mitochondrial dysfunction in human osteosarcoma cells. Sci Rep.
10:50452020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Payen VL, Zampieri LX, Porporato PE and
Sonveaux P: Pro- and antitumor effects of mitochondrial reactive
oxygen species. Cancer Metastasis Rev. 38:189–203. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Du X, Zhang P, Fu H, Ahsan HM, Gao J and
Chen Q: Smart mitochondrial-targeted cancer therapy: Subcellular
distribution, selective TrxR2 inhibition accompany with declined
antioxidant capacity. Int J Pharm. 555:346–355. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Forman HJ and Kennedy JA: Role of
superoxide radical in mitochondrial dehydrogenase reactions.
Biochem Biophys Res Commun. 60:1044–1050. 1974. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baldissera MD, Souza CF, Grings M,
Parmeggiani BS, Leipnitz G, Moreira KLS, da Rocha MIUM, da Veiga
ML, Santos RCV, Stefani LM and Baldisserotto B: Inhibition of the
mitochondrial respiratory chain in gills of Rhamdia quelen
experimentally infected by Pseudomonas aeruginosa: Interplay with
reactive oxygen species. Microb Pathog. 107:349–353. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao RZ, Jiang S, Zhang L and Yu ZB:
Mitochondrial electron transport chain, ROS generation and
uncoupling (Review). Int J Mol Med. 44:3–15. 2019.PubMed/NCBI
|
|
18
|
Milkovic L, Cipak Gasparovic A, Cindric M,
Mouthuy PA and Zarkovic N: Short Overview of ROS as cell function
regulators and their implications in therapy concepts. Cells.
8:7932019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL
and Lesnefsky EJ: Production of reactive oxygen species by
mitochondria: Central role of complex III. J Biol Chem.
278:36027–36031. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brand MD: The sites and topology of
mitochondrial superoxide production. Exp Gerontol. 45:466–472.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fan W, Shen T, Ding Q, Lv Y, Li L, Huang
K, Yan L and Song S: Zearalenone induces ROS-mediated mitochondrial
damage in porcine IPEC-J2 cells. J Biochem Mol Toxicol. 312017.doi:
10.1002/jbt.21944.
|
|
22
|
Wang HW, Zhang Y, Tan PP, Jia LS, Chen Y
and Zhou BH: Mitochondrial respiratory chain dysfunction mediated
by ROS is a primary point of fluoride-induced damage in Hepa1-6
cells. Environ Pollut. 255:1133592019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li N, Ragheb K, Lawler G, Sturgis J, Rajwa
B, Melendez JA and Robinson JP: Mitochondrial complex I inhibitor
rotenone induces apoptosis through enhancing mitochondrial reactive
oxygen species production. J Biol Chem. 278:8516–8525. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Y, Wang J, Qiao J, Liu S, Wang S, Zhao
D, Bai X and Liu M: Ginsenoside Rh2 inhibits HeLa cell energy
metabolism and induces apoptosis by upregulating voltage-dependent
anion channel 1. Int J Mol Med. 46:1695–1706. 2020.PubMed/NCBI
|
|
25
|
Zhang H, Gong J and Kong D: Induction of
apoptosis and reversal of permeability glycoprotein-mediated
multidrug resistance of MCF-7/ADM by ginsenoside Rh2. Int J Clin
Exp Pathol. 8:4444–4456. 2015.PubMed/NCBI
|
|
26
|
Bannon JH, Fichtner I, O'Neill A,
Pampillón C, Sweeney NJ, Strohfeldt K, Watson RW, Tacke M and Mc
Gee MM: Substituted titanocenes induce caspase-dependent apoptosis
in human epidermoid carcinoma cells in vitro and exhibit antitumour
activity in vivo. Br J Cancer. 97:1234–1241. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mazumder MK, Paul R and Borah A:
β-phenethylamine-a phenylalanine derivative in brain-contributes to
oxidative stress by inhibiting mitochondrial complexes and
DT-diaphorase: An in silico study. CNS Neurosci Ther. 19:596–602.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kapur A, Beres T, Rathi K, Nayak AP,
Czarnecki A, Felder M, Gillette A, Ericksen SS, Sampene E, Skala
MC, et al: Oxidative stress via inhibition of the mitochondrial
electron transport and Nrf-2-mediated anti-oxidative response
regulate the cytotoxic activity of plumbagin. Sci Rep. 8:10732018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen LS, Nowak BJ, Ayres ML, Krett NL,
Rosen ST, Zhang S and Gandhi V: Inhibition of ATP synthase by
chlorinated adenosine analogue. Biochem Pharmacol. 78:583–591.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Emmanuel IA, Olotu FA, Agoni C and Soliman
MES: Deciphering the ‘Elixir of Life’: Dynamic perspectives into
the allosteric modulation of mitochondrial ATP synthase by J147, a
novel drug in the treatment of Alzheimer's disease. Chem Biodivers.
16:e19000852019.PubMed/NCBI
|
|
31
|
Larsen S, Nielsen J, Hansen CN, Nielsen
LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F
and Hey-Mogensen M: Biomarkers of mitochondrial content in skeletal
muscle of healthy young human subjects. J Physiol. 590:3349–3360.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim MJ, Yun H, Kim DH, Kang I, Choe W, Kim
SS and Ha J: AMP-activated protein kinase determines apoptotic
sensitivity of cancer cells to ginsenoside-Rh2. J Ginseng Res.
38:16–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo XX, Li Y, Sun C, Jiang D, Lin YJ, Jin
FX, Lee SK and Jin YH: p53-dependent Fas expression is critical for
Ginsenoside Rh2 triggered caspase-8 activation in HeLa cells.
Protein Cell. 5:224–234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xia T, Wang J, Wang Y, Cai J, Wang M, Chen
Q, Song J, Yu Z, Huang W and Fang J: Inhibition of autophagy
potentiates anticancer property of 20(S)-ginsenoside Rh2 by
promoting mitochondria-dependent apoptosis in human acute
lymphoblastic leukaemia cells. Oncotarget. 7:27336–27349. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zare-Zardini H, Taheri-Kafrani A, Amiri A
and Bordbar AK: New generation of drug delivery systems based on
ginsenoside Rh2-, Lysine- and Arginine-treated highly porous
graphene for improving anticancer activity. Sci Rep. 8:5862018.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chen F, Deng Z, Xiong Z, Zhang B, Yang J
and Hu J: A ROS-mediated lysosomal-mitochondrial pathway is induced
by ginsenoside Rh2 in hepatoma HepG2 cells. Food Funct.
6:3828–3837. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Charlot JF, Prétet JL, Haughey C and
Mougin C: Mitochondrial translocation of p53 and mitochondrial
membrane potential (Delta Psi m) dissipation are early events in
staurosporine-induced apoptosis of wild type and mutated p53
epithelial cells. Apoptosis. 9:333–343. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, Qi H, Li X, Hou X, Lu X and Xiao X:
A novel dithiocarbamate derivative induces cell apoptosis through
p53-dependent intrinsic pathway and suppresses the expression of
the E6 oncogene of human papillomavirus 18 in HeLa cells.
Apoptosis. 20:787–795. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Seyfried TN, Arismendi-Morillo G,
Mukherjee P and Chinopoulos C: On the origin of ATP synthesis in
cancer. iScience. 23:1017612020. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Solsona-Vilarrasa E, Fucho R, Torres S,
Nuñez S, Nuño-Lámbarri N, Enrich C, García-Ruiz C and
Fernández-Checa JC: Cholesterol enrichment in liver mitochondria
impairs oxidative phosphorylation and disrupts the assembly of
respiratory supercomplexes. Redox biology. 24:1012142019.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cappelli E, Cuccarolo P, Stroppiana G,
Miano M, Bottega R, Cossu V, Degan P and Ravera S: Defects in
mitochondrial energetic function compels Fanconi Anaemia cells to
glycolytic metabolism. Biochim Biophys Acta Mol Basis Dis.
1863:1214–1221. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jaña F, Faini F, Lapier M, Pavani M,
Kemmerling U, Morello A, Maya JD, Jara J, Parra E and Ferreira J:
Tumor cell death induced by the inhibition of mitochondrial
electron transport: The effect of 3-hydroxybakuchiol. Toxicol Appl
Pharmacol. 272:356–364. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lindsay DP, Camara AK, Stowe DF, Lubbe R
and Aldakkak M: Differential effects of buffer pH on Ca(2+)-induced
ROS emission with inhibited mitochondrial complexes I and III.
Front Physiol. 6:582015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kaludercic N and Giorgio V: The dual
function of reactive Oxygen/Nitrogen species in bioenergetics and
cell death: The role of ATP synthase. Oxid Med Cell Longev.
2016:38696102016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Park HM, Kim SJ, Kim JS and Kang HS:
Reactive oxygen species mediated ginsenoside Rg3- and Rh2-induced
apoptosis in hepatoma cells through mitochondrial signaling
pathways. Food Chem Toxico. 50:2736–2741. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gu J, Wu M, Guo R, Yan K, Lei J, Gao N and
Yang M: The architecture of the mammalian respirasome. Nature.
537:639–643. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Musende AG, Eberding A, Wood C, Adomat H,
Fazli L, Hurtado-Coll A, Jia W, Bally MB and Guns ET: Pre-clinical
evaluation of Rh2 in PC-3 human xenograft model for prostate cancer
in vivo: Formulation, pharmacokinetics, biodistribution and
efficacy. Cancer Chemother Pharmacol. 64:1085–1095. 2009.
View Article : Google Scholar : PubMed/NCBI
|