Introduction
Mitochondrial trifunctional protein (MTP) deficiency
(MTPD) is a disease caused by blocked fatty acid metabolism due to
defective MTP activity, and the incidence of MTPD is 1/140,000
worldwide (1). MTP is an enzyme
complex that catalyzes the last three steps in long-chain fatty
acid metabolism, and is located in the mitochondrial inner membrane
(2). MTP is an octamer composed
of two distinct subunits of four α and four β subunits, including
three enzymes, long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD),
long-chain 2,3-enoyl-CoA hydratase (LCEH) and
long-chain-3-ketothiolase (LCKAT) (3). HADHA (OMIM 600890) encodes
LCHAD and LCEH, whereas HADHB (OMIM 143450) encodes LCKAT.
LCHAD catalyzes the reduced dehydrogenation reaction between the
β-hydroxyl group and the β-ketone group of C12 to C18 fatty acids,
and NAD is used as the hydrogen carrier. Hydrogenases and thiolases
are also specific for targeting the β-oxidation of long chain fatty
acids. Enzymatic activity defects are associated with long-chain
fatty acid metabolism disorders, in which long-chain fatty acids
cannot be oxidized for energy supply, consequently accumulating in
the cell, and causing toxic effects on the cardiac muscle, skeletal
muscle and liver. The clinical manifestations of MTPD are
hypoketotic hypoglycemia, rhabdomyolysis, decreased muscle tone,
cardiomyopathy and liver disease, and the acute onset period can
lead to death due to metabolic crisis. In routine laboratory
examinations, patients show low ketotic hypoglycemia, increased
creatine kinase (CK) and elevated uric acid levels. In
acyl-carnitine detection, blood samples show significant increases
in multiple acyl carnitines, particularly long-chain acyl carnitine
and 3-hydroxyl long-chain acyl carnitine (C12, C14, C16, C18,
C14-OH, C16-OH, C18-OH and C18:1-OH), accompanied by a decrease in
free carnitine (1,4,5).
MTPD can involve three enzymatic defects; however,
most patients have only LCHAD defects. LCHAD deficiency (LCHADD) is
caused by mutations in the HADHA gene encoding LCHAD, thus
causing fatty acid oxidation defects. It is divided into two types
according to the gene mutation: i) Isolation LCHADD, patients carry
the hotspot mutation 1528G>C, LCHAD has significantly reduced
activity, whereas the integrity of MTP is not affected, and the
activity of the other two enzymes is >60%; ii) LCHADD with MTPD,
the coding genes of α and β subunits of MTPD are mutated, and the
folding stability of the multicomplex is affected, thus resulting
in a decrease in activity of the three enzymes (6). The incidence of LCHADD is higher in
Europe compared with in other regions, particularly in the Baltic
region. For example, the incidence in Poland is 1/115,450 (7).
HADHA mutations are common in the Caucasian
population, and HADHB mutations mainly exist in the East
Asian population, such as in Japan, South Korea and China (8–12).
Since the initial discovery of pathogenic mutations of HADHA
in 1990, most of the 59 HADHA mutations reported have been
frameshift mutations, nonsense mutations or alternative splicing
mutations, which determine the clinical manifestations of MTPD,
ranging from the life-threatening phenotype in newborns to mild
rhabdomyolysis in adolescence (6,8).
The most common mutation of HADHA is c.1528G>C, which
results in the inactivation of LCHAD. The typical manifestations
are hypoketosis, hypoglycemia, cardiomyopathy, liver disease and
rhabdomyolysis. The homozygous c.1528G>C mutation often causes
neonatal death (5,13,14).
The present study reported on a Chinese family with
MTPD caused by HADHA gene mutation. On the basis of clinical
symptoms, biochemical testing, filter paper blood carnitine
testing, autopsy and molecular detection, to the best of our
knowledge, the present study was the first to identify the compound
heterozygous c.703C>T (p.R235W) and c.2107G>A (p.G703R)
mutations in HADHA associated with MTPD in China.
Materials and methods
Human participants
One boy and one girl from a southern Chinese family
died suddenly for unknown reasons. The core family members were
called to the Genetics Department of Liuzhou Maternal and Child
Health Hospital (Liuzhou, China) in July 2019. The clinical history
of the two patients was collected and evaluated. II2 and II3
routine examinations, including blood biochemical indexes and
filter paper blood spot acyl-carnitine testing through liquid
chromatography-tandem mass spectrometry analysis, were performed.
The neonatal filter paper blood spots, which had been collected at
birth, were used to test acyl-carnitine and extract DNA. Collection
of the two patients' urine for organic acid detection by gas
chromatography-mass spectrometry was not possible because of the
unexplained death of the two patients. After approval was granted
by the family members, the Judicial Appraisal Center of Southern
Medical University (Guangzhou, China) performed systematic
anatomical and forensic pathological examinations on the body of
the male patient to determine the cause of death. At that time, the
mother was pregnant and expected to have a prenatal diagnosis. The
present study was approved by the Ethical Committee of Liuzhou
Maternity and Child Health Hospital (approval no. 2021-007) and the
written informed consent was obtained from the parents.
Exome sequencing (ES) analysis
Peripheral blood anticoagulated with EDTA was
collected from the parents, II1 and proband II2, and genomic DNA
was extracted from peripheral blood lymphocytes. Only exome
sequencing of proband II2 was performed and analyzed by AmCare
Genomics Lab, and the variant detected was validated by Sanger
sequencing in the whole pedigree. According to the manufacturer's
instructions, genomic DNA was extracted using the SolPure Blood DNA
kit (Magen Biotechnology Co., Ltd.) followed by DNA fragmentation
using Q800R Sonicator (Qsonica LLC). Based on the paired-end
libraries, custom designed NimbleGen SeqCap solution-based exome
capture reagent (Roche NimbleGen, Inc.) was used for fragment
enrichment prior to sequencing on a NextSeq500 sequencer (Illumina,
Inc.). The DNA of II3, was extracted from the neonatal filter paper
blood spots, which had been collected at birth, and was used for
verification via Sanger sequencing. Amniotic DNA of II:4 was
extracted through amniocentesis. The parents of the patients
provided written informed consent for the test, and the samples of
the patients were sequenced and analyzed. The genetic analysis
included 1,739 genes and 25,978 coding regions containing 4,262,587
bases, with an average coverage depth of 343/-206X, with >10X
coverage at an interval >99.9% and >20X coverage at an
interval of 99.8%. All variants were filtered and annotated as
follows: i) A frequency <0.01 in the 1000 Genomes Project
(https://pubmed.ncbi.nlm.nih.gov/26432245/), ExAc
(https://pubmed.ncbi.nlm.nih.gov/27899611/) and gnomAD
(https://pubmed.ncbi.nlm.nih.gov/32461654/) was
required; ii) mutations beyond exons and splicing sites were
filtered and excluded; iii) bioinformatics prediction software was
used for annotation, including the Mendelian Clinically Applicable
Pathogenicity Score (https://pubmed.ncbi.nlm.nih.gov/27776117/), Combined
Annotation Dependent Deletion (https://pubmed.ncbi.nlm.nih.gov/30371827/) and
Polymorphism Phenotyping v2 (https://pubmed.ncbi.nlm.nih.gov/20354512/). The data
from the present study have been deposited into the Sequence Read
Archive (SRR16871898; http://www.ncbi.nlm.nih.gov/sra/?term=PRJNA778796).
Sanger sequencing
The detected mutations were confirmed by Sanger
sequencing in all members of the family. Total DNA of whole blood
leukocytes or amniotic fluid was isolated using the QIAamp DNA
Blood Mini Kit (Qiagen GmbH). Total DNA of dry blood spots was
isolated by standard chloroform extraction. The primers were as
follows: Forward (F) 5′-AGACCGCGTTTTCTACCTAGA-3′ and reverse (R)
5′-AGGCTGACTTTATGCTTTGAGT-3′ for the c.703C>T mutation; F
5′-AGATTCTCCTTGGCCCCTTC-3′ and R 5′-GTGGCTTCAGATGGCTCTTG-3′ for the
c.2107G>A mutation. Amplification was performed using the
EmeraldAmp MAX PCR Master Mix (Takara Bio USA, Inc.). PCR amplicons
were sequenced on an ABI PRISM 3130 Genetic Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The following
thermocycling conditions were used: 95°C for 5 min; followed by 35
cycles of 95°C for 30 sec, 58°C for 30 sec and 72°C for 60 sec; and
finally 72°C for 7 min.
Molecular dynamics simulation of the
mutant protein
To study the effects of key amino acid mutations in
HADHA on its structure, molecular dynamics simulation was used to
generate virtual mutations and for structural optimization. The
three-dimensional structure of wild-type HADHA (PDB ID: 5zqz) was
downloaded from the RCSB Protein Data Bank (www.rcsb.org). PyMOL 2.30 (https://www.pymol.org) was used to mutate Arg235 to
Trp and Gly703 to Arg.
The structure of the mutant protein was optimized by
molecular dynamics simulation in AMBER16 (https://ambermd.org). The simulation temperature was
set to 300 K, and the GROMACS all-atom position and SPC water model
were selected. The water molecules added around the protein formed
a water box simulation system as a periodic boundary for dynamic
simulation. In the simulation process, the PME algorithm was used
to calculate the long-range electrostatic interactions. The
integration step was set to 2 fs. Under the NVT ensemble (with the
atomic number, volume and temperature kept constant), the system
was balanced, and the water was optimized at 500 ps; then under the
NPT ensemble (with the atomic number, pressure and temperature kept
constant), the system was balanced at 500 ps. Finally, a 100 ns
molecular dynamics simulation was performed.
Routine examination and blood
acyl-carnitine analysis
Blood glucose, the myocardial enzyme spectrum and
liver function of II2 and II3 were analyzed during hospitalization.
Neonatal blood samples of II2 and II3 had been collected at birth,
and dried blood spots of the parents and II1 were collected at the
clinic visit. Acyl-carnitine content in dried blood spots was
determined using the liquid chromatography tandem mass spectrometer
API3200 LC-MS/MS system (Shanghai AB SCIEX Analytical Instrument
Trading Co.). An API3200 triple quadrupole mass spectrometer
equipped with an electrospray ionization (ESI) interface operated
in positive ionization mode was used for the mass spectrometric
detection. A multiple reaction monitoring mode was operated to
detect specific precursor and product ions. The ESI parameters were
curtain gas, 25 psi; collision gas, 6 psi; lonspray voltage, 5,500
V; temperature, 450°C; ion source gas1, 45 psi; ion source gas2, 55
psi; interface heater, on. According to the ratio of index ions and
the internal standard ion intensity in the mass, the relative
response factor value and known internal standard concentration
were used to calculate the index concentration. ChemoView™(1.4.2)
software enabled screening of large sets of flow injection triple
quadrupole mass spectrometry data for various analytes.
Pathological autopsy examination
The examination was carried out in accordance with
the public safety standards of the People's Republic of China,
Forensic Autopsy (GA/T147-1996), Forensic Medical Corpse
Examination (GA/T149-1996), Methods of Extraction, Fixation,
Packaging and Examination of Forensic Pathological Materials
(GA/T148-1996) and (NYSJ-JS-BL02) of the Forensic Pathological
Corpse Examination of Southern Medical University, the Instructions
for the Operation of Forensic Pathological Autopsy (NYSJ-JS-BL03)
of Southern Medical University, and the Instructions for the
Extraction and Fixation of Forensic Pathological Materials
(NYSJ-JS-BL04) of Forensic Pathology.
Through autopsy, the pathological changes observed
in the organs of the patient II3 prior to death were determined,
and scientific analysis and inference were performed. Subsequently,
a pathological and anatomical diagnosis was reached, thus providing
a theoretical basis for disease diagnosis.
Results
Clinical manifestation
The 3-year-old proband II2 was delivered at 39 weeks
+ 2 days with a weight of 3,990 g. She was the second child of this
family, and the father I1 and mother I2 denied consanguineous
marriage. The proband died in the hospital after fever for half a
day; sleepiness, diminished speech and progressive disturbance of
consciousness for 1 day; and respiratory weakness for 30 min. The
child had shown weakness in both lower limbs and poor swallowing
function. The child had been unable to walk at 1 year old and
underwent surgery. The parents I1 and I2, and sister II1 had no
significant clinical manifestations (Table I). Patient II3 was delivered at 38
weeks + 3 days with a body weight of 3,070 g; this child died
suddenly of fever and diarrhea at 7 months old, and was examined by
autopsy (Table I). Routine
examination analysis revealed that lactate dehydrogenase (LDH), CK,
CK-MB and liver enzymes were abnormal in the past hospital
examinations of proband II2 and their younger brother II3, and the
biochemical test results prior to death were the same (Table II).
 | Table I.Clinical phenotypic and genotypic
information of the affected family. |
Table I.
Clinical phenotypic and genotypic
information of the affected family.
| ID | Sex | Age of death | MTPD state | Inheritance testing
method | Genotype | Long chain tandem
mass spectrometry results | Pathological results
of autopsy |
|---|
| I1 | M | – | U | ES, Sanger | c.703C>T/WT | N | – |
| I2 | F | – | U | ES, Sanger |
c.2107G>A/WT | N | – |
| II1 | F | – | U | Sanger | c.703C>T/WT | N | – |
| II2 | F | 3 years | A | ES, Sanger |
c.703C>T/c.2107G>A | P | – |
| II3 | M | 7 months | A | Sanger |
c.703C>T/c.2107G>A | P | Upper respiratory
tract infection, liver and cardiac cell fat modification |
| II4 | – | – | – | Sanger |
c.2107G>A/WT | – | – |
| Table II.Laboratory biochemistry findings. |
Table II.
Laboratory biochemistry findings.
| Variable | Proband II2 | Patient II3 | Reference
range |
|---|
| Blood
biochemistry |
|
|
|
| ALT
(IU/l) | 822 | 237.4 | 0–49 |
| AST
(IU/l) | 1,103 | 595.4 | 0–49 |
| LDH
(IU/l) | 4,732 | 1,513.6 | 109–245 |
| CK
(IU/l) | 16,448 | 5,987 | 0–200 |
| CK-MB
(IU/l) | 3,965 | 427.5 | 0–25 |
| Glu
(mmol/l) | – | 2.84 | 3.86–6.11 |
|
NT-proBNP (pg/ml) | – | >9,000 | 0–150 |
| Blood spot
analysis |
|
|
|
| C0
(µmol/l) | 28.55 | 17.47 | 9–55 |
| C14:1
(µmol/l) | 0.389 | 0.63 | 0.01–0.25 |
| C14:2
(µmol/l) | 0.036 | 0.08 | 0001-0.05 |
| C16-OH
(µmol/l) | 0.249 | 0.33 | 0-0.06 |
|
C18:1-OH | 0.114 | 0.15 | 0-0.09 |
Family members II2 and II3 died because of unknown
causes after symptoms including fever and diarrhea, with a high
possibility of genetic disease. Proband II2 had poor walking
function and poor swallowing function; II2 and II3 had findings
including abnormal LDH, CK, CK-MB and liver enzyme levels, as well
as hypoglycemia and liver disease. Other diseases, such as Reye
syndrome, have appeared in similar clinical cases (15). Results of biochemical tests of
these diseases are similar; therefore, it is important to make a
differential diagnosis between them. During the last clinic visit,
the physicians suspected that II2 had a muscle-related disease, and
consequently suggested a muscle biopsy and related genetic tests,
which were rejected by the parents. Therefore, the reason of the
disease in II2 could not be identified.
After a systematic autopsy and forensic pathological
examination of II3, death due to physical violence was excluded.
Allergy-related indexes of cadaver blood from II3 were normal.
These findings combined with systematic dissection and forensic
pathological examination indicated no typical acute allergic
pathological changes, such as clear laryngeal edema, eosinophil
infiltration in the larynx and trachea submucosa, and spleen
parenchyma, and the possibility of death due to acute allergy was
excluded. The dissection and forensic pathology showed that II3 had
extensive neutrophil infiltration in the bronchi of both lungs,
diffuse inflammation of neutrophil monocytes in some alveolar
cavities and edema of multiple organs. The aforementioned lesions
were consistent with the pathological changes associated with
typical bronchopneumonia and were serious; consequently, they were
considered the cause of death for II3. Simultaneously, diffuse
hepatocyte steatosis and some cardiomyocyte steatosis was detected,
thus suggesting the presence of abnormal lipid metabolism or gene
defects. From these results combined with the medical history
analysis, it was concluded that II3 died as a cause of
bronchopneumonia due to abnormal lipid metabolism.
Pathological examination showed diffuse hepatocyte
steatosis and some cardiomyocyte steatosis, thus suggesting
abnormal lipid metabolism or the possibility of genetic defects.
Acyl-carnitine assays in past blood samples from II2 and II3 were
performed and considered together with the results of the autopsy
performed on II3. Acyl-carnitine analysis of the two patients
revealed that the levels of C0 were normal, whereas those of C14:1,
C14:2, C16-OH and C18:1-OH were elevated (Table II). The increase in long-chain
3-OH-acyl-carnitine (C16-OH and C18:1-OH) indicated the lack of
very-long-chain acyl-CoA dehydrogenase deficiency (VLCADD) or
MTP.
Mutation analysis
ES of proband II2 was performed and analyzed. The
compound heterozygous mutations of the HADHA gene
c.703C>T (p.R235W) and c.2107G>A (p.G703R) were detected, and
the verification of ES used Sanger sequencing (Fig. 1A). The c.2107G>A variant was
inherited from the father and the c.2107G>A variant was
inherited from the mother. To date, the frequency of these two
mutations is relatively low in the reference population genome
database. The regions of the two mutations were confirmed to affect
important components of the protein, and the amino acid sequences
among species are highly conserved (Fig. 1C). The p.R235W mutation affected
the second step of hydration in the four-step cycle of LCEH.
Furthermore, the p.G703R mutation affected the third step of the
four-step cycle of LCHAD dehydrogenation. Computer-aided analysis
predicted that these two variants were highly likely to affect the
structure/function of the protein. In conclusion, combined with the
clinical manifestations and pedigree analysis of the patients,
according to the ACMG-guideline-based variant classification
(16), p.R235W and p.G703R are
‘likely pathogenic.’ Genetic testing of II3 also revealed the
HADHA gene compound heterozygous variants c.703C>T
(p.R235W) and c.2107G>A (p.G703R); genetic testing of the sister
II1 revealed a c.703C>T (p.R235W) gene mutation; and genetic
testing of the fetus II4 via amniotic fluid gene testing revealed a
c.2107G>A (p.G703R) gene mutation (Table I; Fig. 1B). The results confirmed that the
parents I1 and I2, and sister II1 were asymptomatic, and that their
long chain tandem mass spectrometry results were normal. Therefore,
the clinical phenotypes of the two heterozygous variants of the
HADHA gene are non-lethal.
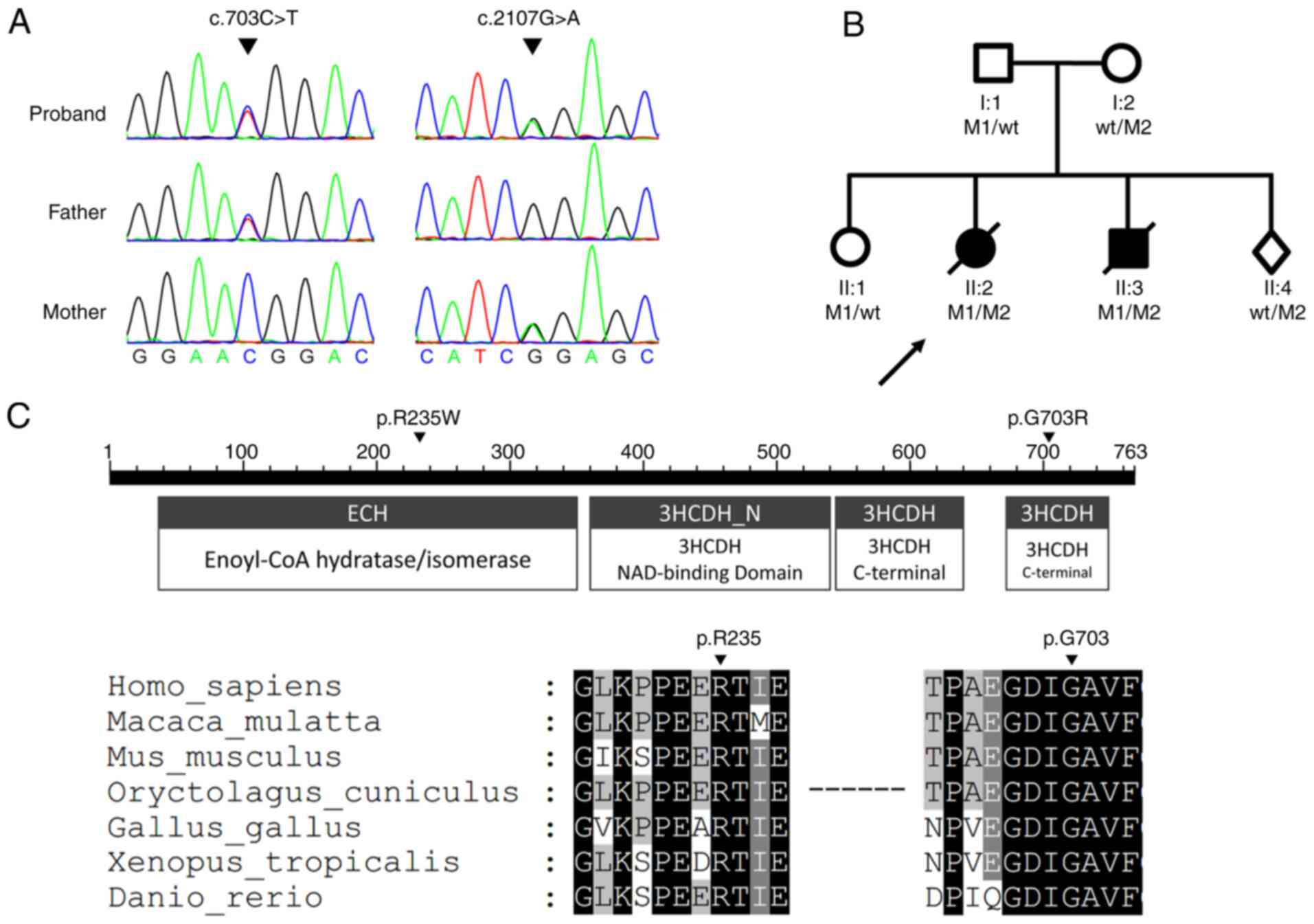 | Figure 1.Genomic DNA sequencing of the
pedigree. (A) HADHA compound heterozygous variants analyzed
by sequencing. ES of proband II2 was performed and analyzed, and
the verification of ES used Sanger sequencing. The proband II2 had
compound heterozygous c.703C>T and c.2107G>A mutations. The
father had a heterozygous c.703C>T mutation. The mother had a
heterozygous c.2107G>A mutation. (B) Pedigree chart. Square,
male; circle, female; dark symbol, affected; arrow, proband; slash,
deceased; diamond, fetus; M1, c.703C>T; M2, c.2107G>A; WT,
wild type. Genotypes of II2 and II3 are both
c.703C>T/c.2107G>A. (C) Conservation analysis of HADHA
protein. The p.R235W mutation occurred in the enoyl-CoA
hydratase/isomerase domain, and the p.G703R mutation occurred in
the 3-hydroxyacyl CoA-dehydrogenase C-terminal domain. Comparison
of the conservation of arginine 235 and glycine 703 among species.
Black indicates 100% identity, dark grey indicates 80% identity,
and gray indicates 60% identity. ES, exome sequencing. |
Molecular simulation and molecular
mechanics
In terms of molecular characteristics, the present
study demonstrated the two mutated residues were located in an
essential domain of the protein that is highly conserved among
species. In the mutant protein, Gly703 was changed to Arg703 in the
mutated protein, residue Arg703 formed hydrogen bond interactions
with nearby Gly700, and also formed a π-cation interaction with
Tyr546; Arg235 was changed to Trp235 in the mutated protein,
residue Trp235 formed hydrogen bond interactions with nearby
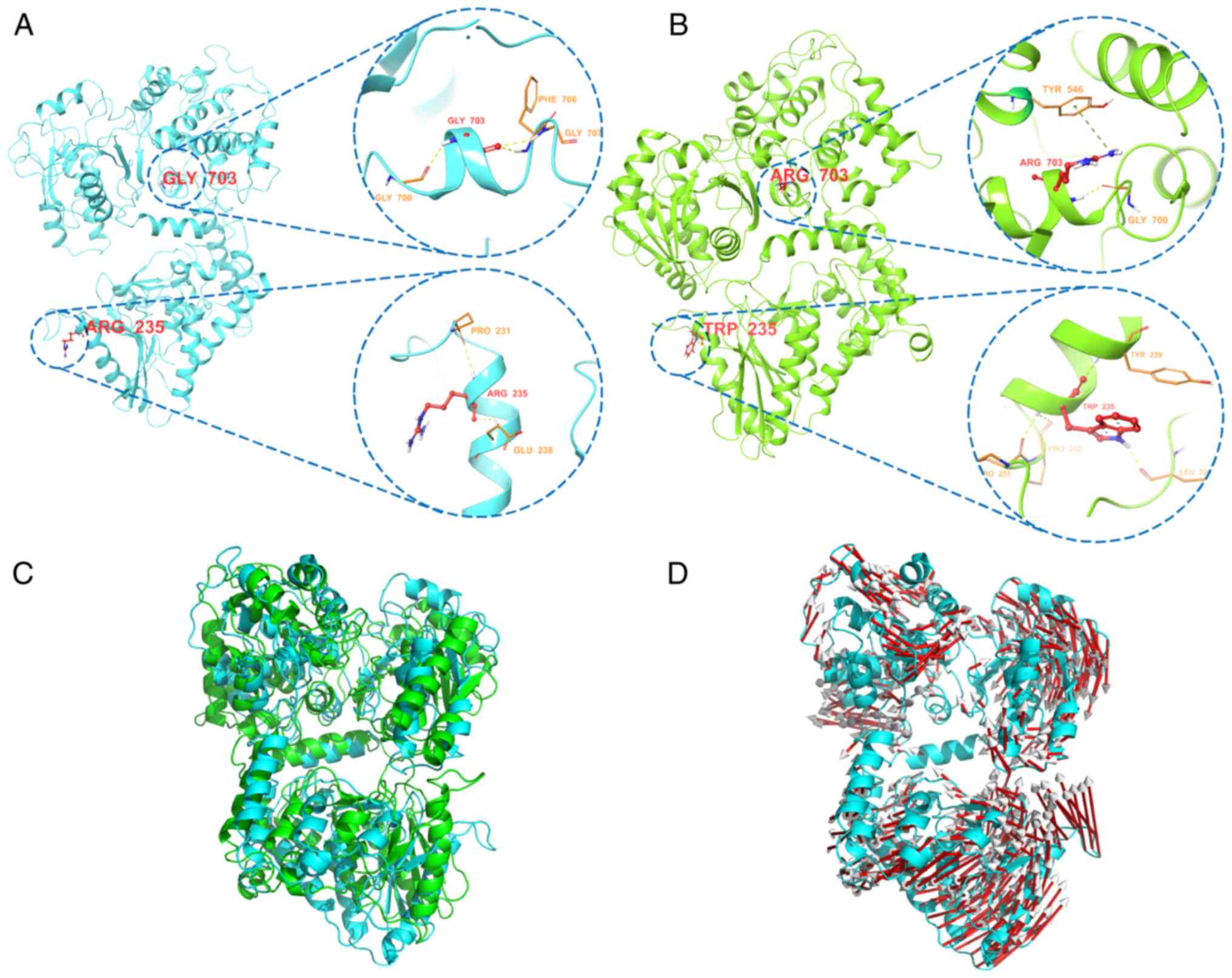
Pro231, Pro232, Tyr239 and Leu225 residues (Fig. 2A and B). The two mutations may
affect the activity of the encoded enoyl-CoA hydratase/isomerase
and the C-terminus of 3-hydroxyacyl-CoA-dehydrogenase, thus causing
a conformational shift of other amino acids (Fig. 2C and D). The conformation of the α
subunit of the MTPase complex could thus be destroyed, thereby
affecting the stability and function of the enzyme complex.
Consequently, long-chain fatty acids could not be metabolized and
converted into energy.
Discussion
In the present study, ES was performed on samples
obtained from a child who died of an infectious disease along with
weakness in both lower limbs and swallowing dysfunction. After
bioinformatics filtering and Sanger sequencing, the compound
heterozygous mutations c.703C>T and c.2107G>A were identified
in HADHA, and the first molecular diagnosis of familial MTPD
caused by HADHA gene mutations in China was determined.
In the present study, the II2 and II3 had compound
heterozygous c.703C>T and c.2107G>A mutations, which are not
common mutation sites in HADHA. The p.R235W mutation
affected the enoyl-CoA hydratase/isomerase domain, thus hindering
the second step of hydration in the four-step cycle of LCEH.
Furthermore, the p.G703R mutation affected the
3-hydroxyacyl-CoA-dehydrogenase C-terminal domain, thereby
hindering the third step of the four-step cycle of LCHAD
dehydrogenation. Any mutation in HADHB or HADHA may
interfere with the stability of the entire MTP complex, thus
increasing MTP degradation and decreasing the activity of all three
enzymes (3). Among the clinical
cases of mitochondrial DNA deficiency, an Israeli woman has
previously been reported to be homozygous for c.703C>T
(p.R235W). The clinical manifestations were fatigue, fever,
hypotonia, rhabdomyolysis, and a significant increase in LDH, CK
and liver enzymes, in agreement with the clinical manifestations
and biochemical test results in the two patients of the present
study (17). The c.2107G>A
variant has been reported in a clinical case of LCHADD; the patient
was a compound heterozygote for c.1528G>C and c.2107G>A, and
died suddenly at the age of 7 (7). In the present study, the patients
showed typical myasthenia, as well as significantly increased LDH,
CK and liver enzymes, which are typical clinical features of
myasthenia.
To assess the effects of p.R235W and p.G703R
mutations on the structure and stability of the α subunit in
HADHA, molecular dynamics simulation was performed. Before
mutation, Gly703 formed hydrogen bond interactions with the three
nearby residues Gly700, Phe706 and Gly707. After mutation, Arg703
formed hydrogen bond interactions with the nearby residue Gly700
and also formed a π-cation interaction with Tyr546. The previous
hydrogen bond interaction with the residues Gly700, Phe706 and
Gly707 was destroyed, and the π-cation interaction formed by Tyr546
was weaker than the hydrogen bond. In wild-type HADHA, Arg235
formed hydrogen bond interactions with only two nearby residues,
Pro231 and Glu238, whereas in the mutant, Trp235 formed hydrogen
bond interactions with four nearby residues, Pro231, Pro232, Tyr239
and Leu225, but was unable to form a hydrogen bond with Glu238. The
two mutations led to changes in hydrogen bonding, and eventually
led to shifts in the positional conformation of other residues. The
structure and stability of the α subunit was affected, and the
function of the MTPase complex was destroyed, thus preventing
metabolism of long-chain fatty acids and conversion into energy.
Any mutation in HADHB or HADHA has been reported to
potentially interfere with the stability of the entire MTP complex,
thereby increasing MTP degradation and decreasing the activity of
all three enzymes (3). To the
best of our knowledge, no previous study has determined
abnormalities in the function of specific enzymes involved in
heterozygous individuals for HADHA mutations. LCHAD and LCKAT
enzymatic activity in cultured fibroblasts can be assessed by skin
biopsy to verify MTPase dysfunction; however, because both patients
in the present study died, fibroblasts could not be obtained for
validation. Designing molecular experiments to validate the
mechanisms of these two variants in regulating the stability and
function of the MTP enzyme complex will be a focus of future
studies. Although our institute did not perform such assessments,
we intend to cooperate with conditional laboratories to improve
this deficiency.
Tandem mass spectrometry technical analysis is a
technology used to detect the acyl-carnitine content in dry blood
spots and blood samples. It can simultaneously detect a variety of
carnitines with high sensitivity. Fatty acid β-oxidation disorder
can occur in each link of fatty acids in the mitochondria and the
fatty acid oxidation process. Acyl-carnitine is an intermediate of
fatty acid and organic acid metabolism, and different fatty acid
metabolism diseases are characterized by different acyl-carnitine
profiles. Fatty acid metabolic diseases can be screened and
diagnosed on the basis of changes in acyl-carnitine levels with
different lengths of carbon chains (18). Tandem mass spectrometry is the
gold standard analysis for the determination of carnitine
concentration. The metabolite 3-OH-acyl-carnitine is usually
increased during the acute phase of MTPD; however, MTPD cannot be
distinguished from other fatty acid metabolic disorders (19). In the present study, neonatal
screening for carnitine was not performed in the two cases (II2 and
II3) at birth; however, neonatal blood samples had been collected
at birth and were used after the patients' death to assess
acyl-carnitine levels. Two non-acute neonatal filter paper blood
samples were collected for carnitine detection. Numerous types of
acyl-carnitine were revealed to be increased, particularly the
C16-OH contents of II2 and II3 were 0.249 and 0.33 µmol/l,
respectively (the normal reference range of C16-OH is 0–0.06
µmol/l). Non-acute phase tandem mass spectrometry of acyl-carnitine
can also indicate MTPD (20);
therefore, despite the possibility of missed detection, tandem mass
spectrometry remains an important first-line method for neonatal
screening (1). However, it has
been reported that analysis of acyl-carnitine in the blood and
organic acids in the urine alone are insufficient for final
diagnosis, and molecular or functional analysis is essential for
the diagnosis of MTPD/LCHA (21).
Notably, the enzymatic analysis of fibroblasts can be performed
only in several special laboratories and cannot be performed
routinely; therefore, molecular detection is the first choice for
the diagnosis of patients with suspected genetic metabolic disease
(22,23).
In the present study, detection and analysis of ES
were performed, and Sanger sequencing was used to confirm the
mutation sites. In addition, pathological results of autopsies can
help diagnose metabolic diseases. In the present study, diffuse
steatosis of hepatocytes and partial steatosis of the myocardium
were detected in the autopsy of II3, thus potentially suggesting
abnormal lipid metabolism. II2 and II3 were not referred to the
center of Genetic Metabolism for further diagnosis despite
indications from clinical symptoms and biochemical laboratory
results. The present study emphasizes the importance of confirming
or eliminating the diagnosis of suspicious biochemical results and
clinical symptoms in a professional center in in a timely manner.
The results of the study suggested that, according to clinical
symptoms, routine biochemical tests and the detection of
acyl-carnitine in filter paper blood spots, patients with suspected
metabolic disorders should undergo ES or functional analysis to
confirm or eliminate MTPD, and rapid professional metabolic
consultations are very important and helpful for early diagnosis,
treatment and prevention of metabolic crisis (7).
Acute fatty liver of pregnancy (AFLP) is a rare
liver disease with an incidence rate of 1/13,000 worldwide, and is
associated with significant perinatal morbidity and mortality
(24). In clinical practice,
women with AFLP often have elevated liver enzymes and bilirubin,
elevated blood ammonia, hypoglycemia, coagulation dysfunction,
acute renal failure and hepatic encephalopathy. Pregnant women and
their fetuses are at risk of death even if birth occurs rapidly
(25). In 1991, Schoeman et
al (26) first reported the
relationship between AFLP and LCHAD defects, indicating that the
affected women may have a genetic deficiency in a β-oxidation
enzyme, thus making them prone to AFLP. In addition, Kobayashi
et al (27) reported that
fetal homozygous mutations in exon 13 of the HADHB gene
resulted in the development of acute fatty liver in pregnant woman.
In the present study, the mother was asked about any history of
AFLP. All of the children were born at full-term with normal
weight. It was hypothesized that although the fetus had compound
heterozygous c.703C>T (p.R235W) and c.2107G>A (p.G703R)
mutations, the possibility of AFLP in the mother was low. After
identifying the pathogenic gene in the patients through familial
and genetic analysis, when the mother of the child became pregnant
again, amniotic fluid was collected for relevant testing. The
results revealed that the fetal genotype was the same as that of
the mother, who was a carrier.
In conclusion, the present study analyzed a Chinese
family with MTPD caused by a HADHA gene mutation. On the
basis of combined clinical symptoms, biochemical testing, filter
paper blood carnitine testing, autopsy and molecular detection, to
the best of our knowledge, the present study identified the first
compound heterozygous c.703C>T (p.R235W), c.2107G>A (p.G703R)
mutations in HADHA associated with MTPD in China.
Acknowledgements
Not applicable.
Funding
The present study was sponsored by a grant from the Liuzhou
Science and Technology Major Special Project (grant no.
2018AF10501).
Availability of data and materials
The sequencing data from the present study have been
deposited into the Sequence Read Archive (SRR16871898; http://www.ncbi.nlm.nih.gov/sra/?term=PRJNA778796).
The data that support the findings of this study are available from
AmCare Genomics Lab but restrictions apply to the availability of
these data, which were used under license for the current study,
and so are not publicly available. Data are however available from
the authors upon reasonable request and with permission of AmCare
Genomics Lab.
Authors' contributions
JY analyzed the data and wrote the initial draft. TY
revised the draft and study design. RC and JT are pediatricians who
were responsible for diagnosing and treating the children. RC and
DY were in charge of collecting the clinical data and following up
with the children. XT, YZ, NT, DC and JH performed screening by
mass spectrometry and collected the data. JY and DY confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to participate
statements
The present study was approved by the Ethical
Committee of Liuzhou Maternity and Child Health Hospital (approval
no. 2021-007) and written informed consent was obtained from the
parents.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Lotz-Havla AS, Röschinger W, Schiergens K,
Singer K, Karall D, Konstantopoulou V, Wortmann SB and Maier EM:
Fatal pitfalls in newborn screening for mitochondrial trifunctional
protein (MTP)/long-chain 3-Hydroxyacyl-CoA dehydrogenase (LCHAD)
deficiency. Orphanet J Rare Dis. 13:1222018. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Van Vliet P, Berden AE, van Schie MK,
Bakker JA, Heringhaus C, de Coo IF, Langeveld M, Schroijen MA and
Arbous MS: Peripheral neuropathy, episodic rhabdomyolysis, and
hypoparathyroidism in a patient with mitochondrial trifunctional
protein deficiency. JIMD Rep. 38:101–105. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Uchida Y, Izai K, Orii T and Hashimoto T:
Novel fatty acid beta-oxidation enzymes in rat liver mitochondria.
II. Purification and properties of enoyl-coenzyme A (CoA)
hydratase/3-hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase
trifunctional protein. J Biol Chem. 267:1034–1041. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
den Boer ME, Ijlst L, Wijburg FA, Oostheim
W, van Werkhoven MA, van Pampus MG, Heymans HS and Wanders RJ:
Heterozygosity for the common LCHAD mutation (1528g>c) is not a
major cause of HELLP syndrome and the prevalence of the mutation in
the dutch population is low. Pediatr Res. 48:151–154. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Karall D, Brunner-Krainz M, Kogelnig K,
Konstantopoulou V, Maier EM, Möslinger D, Plecko B, Sperl W,
Volkmar B and Scholl-Bürgi S: Clinical outcome, biochemical and
therapeutic follow-up in 14 Austrian patients with long-chain
3-Hydroxy Acyl CoA dehydrogenase deficiency (LCHADD). Orphanet J
Rare Dis. 10:212015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wanders RJ, IJlst L, van Gennip AH, Jakobs
C, de Jager JP, Dorland L, van Sprang FJ and Duran M: Long-chain
3-hydroxyacyl-CoA dehydrogenase deficiency: Identification of a new
inborn error of mitochondrial fatty acid beta-oxidation. J Inherit
Metab Dis. 13:311–314. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sykut-Cegielska J, Gradowska W,
Piekutowska-Abramczuk D, Andresen BS, Olsen RK, Ołtarzewski M,
Pronicki M, Pajdowska M, Bogdańska A, Jabłońska E, et al: Urgent
metabolic service improves survival in long-chain 3-hydroxyacyl-CoA
dehydrogenase (LCHAD) deficiency detected by symptomatic
identification and pilot newborn screening. J Inherit Metab Dis.
34:185–195. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boutron A, Acquaviva C, Vianey-Saban C, de
Lonlay P, de Baulny HO, Guffon N, Dobbelaere D, Feillet F, Labarthe
F, Lamireau D, et al: Comprehensive cDNA study and quantitative
analysis of mutant HADHA and HADHB transcripts in a French cohort
of 52 patients with mitochondrial trifunctional protein deficiency.
Mol Genet Metab. 103:341–348. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hong YB, Lee JH, Park JM, Choi YR, Hyun
YS, Yoon BR, Yoo JH, Koo H, Jung SC, Chung KW and Choi BO: A
compound heterozygous mutation in HADHB gene causes an axonal
Charcot-Marie-tooth disease. BMC Med Genet. 14:1252013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fu X, Zheng F, Zhang Y, Bao X, Wang S,
Yang Y and Xiong H: Mitochondrial trifunctional protein deficiency
due to HADHB gene mutation in a Chinese family. Mol Genet Metab
Rep. 5:80–84. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bo R, Yamada K, Kobayashi H, Jamiyan P,
Hasegawa Y, Taketani T, Fukuda S, Hata I, Niida Y, Shigematsu Y, et
al: Clinical and molecular investigation of 14 Japanese patients
with complete TFP deficiency: A comparison with caucasian cases. J
Hum Genet. 62:809–814. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu ZR, Dong HL, Ma Y and Wu ZY:
Identification and functional characterization of mutations within
HADHB associated with mitochondrial trifunctional protein
deficiency. Mitochondrion. 49:200–205. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tyni T, Palotie A, Viinikka L, Valanne L,
Salo MK, von Döbeln U, Jackson S, Wanders R, Venizelos N and Pihko
H: Long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency
with the G1528C mutation: Clinical presentation of thirteen
patients. J Pediatr. 130:67–76. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Barvinska O, Olkhovych N and Gorovenko N:
High prevalence of c.1528G>C rearrangement in patients with long
chain 3-Hydroxyacyl-CoA dehydrogenase deficiency from Ukraine.
Cytol Genet. 52:198–203. 2018. View Article : Google Scholar
|
|
15
|
Glasgow JF and Middleton B: Reye
syndrome-insights on causation and prognosis. Arch Dis Child.
85:351–353. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Scheuerman O, Wanders RJ, Waterham HR,
Dubnov-Raz G and Garty BZ: Mitochondrial trifunctional protein
deficiency with recurrent rhabdomyolysis. Pediatr Neurol.
40:465–467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zheng Y, Chen Y, Qiu X, Chen W, Lin Q,
Zeng Y, Zhao H and Zhu W: A verification of the application of the
non-derivatized mass spectrometry method in newborns screening of
metabolic disorders. Medicine (Baltimore). 98:e155002019.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Terrone G, Ruoppolo M, Brunetti-Pierri N,
Cozzolino C, Scolamiero E, Parenti G, Romano A, Andria G, Salvatore
F and Frisso G: Child Neurology: Recurrent rhabdomyolysis due to a
fatty acid oxidation disorder. Neurology. 82:e1–e4. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Landau YE, Waisbren SE, Chan LM and Levy
HL: Long-term outcome of expanded newborn screening at Boston
children's hospital: Benefits and challenges in defining true
disease. J Inherit Metab Dis. 40:209–218. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Diebold I, Schön U, Horvath R, Schwartz O,
Holinski-Feder E, Kölbel H and Abicht A: HADHA and HADHB gene
associated phenotypes-identification of rare variants in a patient
cohort by next generation sequencing. Mol Cell Probes. 44:14–20.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pena LD, van Calcar SC, Hansen J, Edick
MJ, Vockley CW, Leslie N, Cameron C, Mohsen AW, Berry SA, Arnold
GL, et al: Outcomes and genotype-phenotype correlations in 52
individuals with VLCAD deficiency diagnosed by NBS and enrolled in
the IBEM-IS database. Mol Genet Metab. 118:272–281. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang E, Kim YM, Kang M, Heo SH, Kim GH,
Choi IH, Choi JH, Yoo HW and Lee BH: Clinical and genetic
characteristics of patients with fatty acid oxidation disorders
identified by newborn screening. BMC Pediatr. 18:1032018.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ibdah JA: Acute fatty liver of pregnancy:
An update on pathogenesis and clinical implications. World J
Gastroenterol. 12:7397–7404. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ronen J, Shaheen S, Steinberg D and Justus
KR: Acute fatty liver of pregnancy:A thorough examination of a
harmful obstetrical syndrome and its counterparts. Cureus.
10:e21642018.PubMed/NCBI
|
|
26
|
Schoeman MN, Batey RG and Wilcken B:
Recurrent acute fatty liver of pregnancy associated with a
fatty-acid oxidation defect in the offspring. Gastroenterology.
100:544–548. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kobayashi T, Minami S, Mitani A, Tanizaki
Y, Booka M, Okutani T, Yamaguchi S and Ino K: Acute fatty liver of
pregnancy associated with fetal mitochondrial trifunctional protein
deficiency. J Obstet Gynaecol Res. 41:799–802. 2015. View Article : Google Scholar : PubMed/NCBI
|