Introduction
Ventilator-induced lung injury (VILI) is caused by
the interaction between the mode in which the ventilator delivers
to the lung parenchyma and the mode in which the lung parenchyma
accepts (1). VILI is the most
serious complication of mechanical ventilation (MV) and increases
the morbidity and mortality of patients receiving ventilator
therapy (1). Currently, the main
measures to alleviate VILI include high frequency oscillatory
ventilation, low tidal volume and inspiratory pressure ventilation,
prone position ventilation and positive end-expiratory pressure
(2). Although several studies
have focused on VILI (3–6), the effectiveness of current
treatment regimens is still limited. Thus, it is important to
identify more effective therapeutic targets to alleviate the pain
of patients with VILI.
Aggregation of inflammatory cells and overproduction
of inflammatory cytokines are two key processes in the pathogenesis
of VILI (7,8). Cytokines released by
pro-inflammatory factors can increase the permeability of the
alveolar capillary barrier, resulting in pulmonary dysfunction
(9). Macrophages are the most
abundant immune cells in several organs, including the lungs, and
play an important role in restoring tissue homeostasis after
triggering inflammatory signals (10,11). Previous studies have reported that
macrophages effectively respond to environmental signals by
changing their phenotype, thus altering their physiological effect
to immune responses (12,13). Macrophages are classified as M1
and M2 phenotypes (14),
according to their immunomodulatory function. M1 macrophages
(classically activated) can enhance inflammatory response, whereas
M2 macrophages (alternatively activated) reduce inflammatory
response and promote tissue regeneration (15,16). Transition of macrophages can occur
between these two phenotypes under certain conditions (17,18). Safavian et al (13) demonstrated that a reduction in the
M2 phenotype of alveolar macrophages (AMs) increased
lipopolysaccharide (LPS)-induced lung injury. Therefore, the
phenotypic transformation of AMs may inhibit the progression of
VILI.
Increasing evidence suggest that long non-coding
RNAs (lncRNAs) act as competing endogenous RNAs (ceRNAs) and
interact with microRNAs (miRNAs) to regulate gene expression
(19–21). Previous research has indicated
that MALAT1 knockdown alleviates neuronal cell death by
upregulating miR-30a expression in cerebral ischemic stroke
(22). Wang et al
(23) demonstrated that GAS5
knockdown inhibits renal tubular epithelial fibrosis by
competitively binding to miR-96-5p. Notably, nuclear enriched
abundant transcript 1 (NEAT1) knockdown inhibits the fibrogenesis
and epithelial-to-mesenchymal transition in diabetic nephropathy
(DN) by targeting miR-27b-3p and ZEB1 expression (24). Zhou et al (25) reported that NEAT1 expression is
upregulated in LPS-induced A549 cells, and inhibition of lncRNA
NEAT1 inhibits the production of inflammatory cytokines. In hepatic
ischemia/reperfusion (I/R) injury, lncRNA HOTAIR also regulates
autophagy via the miR-20b-5p/ATG7 axis (26). However, whether lncRNA NEAT1 in
AMs participates in the VILI process by regulating miR-20b
expression remains unclear.
The present study aimed to investigate the effect of
lncRNA NEAT1 in AMs on VILI. For this purpose, mouse and cell
models of VILI were established to detect the effect of NEAT1 on
pathological changes, apoptosis and levels of inflammatory markers.
Furthermore, the downstream miRNA and gene of NEAT1 were predicted
to explore the underlying molecular mechanism of NEAT1 in VILI.
Materials and methods
VILI mouse model
A total of 40 adult male C57BL/6 mice (age, 10±2
weeks; weight, 25±2 g) were purchased from Nanjing Biomedical
Research Institute of Nanjing University. The animals were fed with
common feed and tap water in a specific pathogen-free laboratory
with a 12 h light/dark cycle at 22±1°C and 45–55% relative
humidity. The mice were randomly divided into three groups
(n=8/group), as follows: i) Control (mice without any treatment);
ii) Sham (mice that underwent tracheotomy but were still breathing
spontaneously); and iii) VILI (mice that underwent tracheotomy and
MV for 4 h) groups. To construct the VILI mouse model, mice were
anaesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) by
intraperitoneal injection and tracheotomy was performed. The
ventilator (Harvard Apparatus) was connected to the mice and the
animals were ventilated with 30 ml/kg air at a rate of 70
breaths/min for 4 h.
RNA interference
NEAT1 short hairpin (sh)RNA (sh-NEAT1,
5′-GCTTATAAGTTTGTTGTGTTG-3′) and negative control (sh-NC,
5′-GGTTCGGTTTATTGAGTTTAT-3′) were purchased from Shanghai
GenePharma Co., Ltd. The second-generation lentivirus encoding
green fluorescent protein (GFP)-expressing NEAT1 shRNA (GFP-NEAT1
lentivirus) was generated for the present study. The pCMVdr-8.91
and pMD2G plasmids (Thermo Fisher Scientific, Inc.) were used as
package systems. The recombinant lentivirus was generated by
co-transfection of 293FT cells (Thermo Fisher Scientific, Inc.)
with lentiCRISPR V2 (10 µg; Addgene, Inc.), pCMVdr-8.91 (7.5 µg),
and pMD2G plasmids (5 µg), using Lipofectamine® 3000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) at room
temperature. The medium was collected and ultracentrifuged at
12,000 × g for 3 h at 4°C and filtered using 0.22-µm filters, 48 h
post-transfection. The resulting pellets were resuspended in PBS,
cooled and stored at −80°C. For transduction of target cells, the
lentivirus vector (MOI=20) was directly added to the complete
medium, followed by transfection at 37°C for 72 h. Stably
transfected cells were selected by adding puromycin (6 g/ml) in
medium and the maintenance concentration of puromycin in
transfected cells was 3 g/ml. The infection efficiency of AMs was
observed under a fluorescence microscope (magnification, ×100).
Subsequent experiments were performed 72 h post-transfection.
A total of 16 mice were randomly divided into two
groups (eight mice/group), VILI + sh-NC and VILI + sh-NEAT1 groups.
Mice in the VILI + sh-NEAT1 group received sh-NEAT1 by intravenous
injection prior to VILI treatment, while mice in the VILI + sh-NC
group received sh-NC by intravenous injection prior to VILI
treatment. All mice were sacrificed via cervical dislocation after
anesthesia with 50 mg/kg pentobarbital sodium. All animal
experiments were approved by the Ethics Committee of The Affiliated
Huaian No.1 People's Hospital of Nanjing Medical University
(Huaian, China; approval no. HASDY20201009).
Sample collection
After establishing the VILI model or performing
spontaneous respiration, the mice were sacrificed by
intraperitoneal injection of 100 mg/kg sodium pentobarbital. The
right lung was collected for bronchoalveolar lavage fluid (BALF)
collection, and the left lung was collected for histopathology and
reverse transcription-quantitative PCR (RT-qPCR) detection of
NEAT1. BALF and the lung tissues were collected and stored at −80°C
until subsequent experimentation. The total protein concentration
of BALF was determined using the BCA method (Beyotime Institute of
Biotechnology).
Histopathology
Lung tissues were fixed in 10% formaldehyde at 4°C
overnight and embedded in paraffin. Paraffin-embedded tissue
samples were cut into 3-µm-thick sections for hematoxylin and eosin
(H&E) staining. Samples were subsequently stained with
hematoxylin for 10 min and eosin for 10 sec at room temperature.
The sections were observed under a light microscope (magnification,
×200).
ELISA
The levels of interleukin (IL)-1β (cat. no.
SEA563Mu; Wuhan USCN Business Co., Ltd.), IL-6 (cat. no. SEA079Mu;
Wuhan USCN Business Co., Ltd.), tumor necrosis factor-α (TNF-α;
cat. no. SCA133Mu; Wuhan USCN Business Co., Ltd.) and inducible
nitric oxide synthase (iNOS; cat. no. ml057773; Shanghai
Enzyme-linked Biotechnology Co., Ltd.) in BALF and AMs were
determined using respective ELISA kits (Thermo Fisher Scientific,
Inc.), according to the manufacturer's instructions.
Isolation, collection and counting of
AMs
BALF was collected by injecting PBS intratracheally
three times. BALF was centrifuged at 500 × g for 10 min at 4°C and
the supernatant was discarded. The red blood cells were lysed using
ACK lysis buffer (Beyotime Institute of Biotechnology) for 5 min.
This was followed by centrifugation at 500 × g for 10 min at 4°C,
after which the supernatant was discarded and the pelleted cells
were resuspended in RPMI-1640 medium (Gibco; Thermo Fisher
Scientific, Inc.). The single cell suspension made of the culture
solution was incubated overnight at 37°C, the adherent cells were
macrophages. A hemocytometer was used to count the number of
AMs.
In vitro cell stretch (CS)
experiments
In vitro CS experiments were performed using
the FX-5000T Flexercell Tension Plus system (Flexcell International
Corp.) to stretch isolated AMs and simulate the stretching of MV.
Briefly, AMs were loaded in a Flexercell FX-4000T strain unit; the
cells were stretched for 4 h at a frequency of 30 cycles/min (0.5
Hz) and a 20% range with a stretch-to-relaxation ratio of 1:1
(27). The control cells were
also placed in the Flexercell FX-4000T strain unit without
mechanical stretching.
Cell transfection
Sh-NEAT1 and sh-NC, miR-20b mimics (50 nM; forward,
5′-ACUGCAGUGUGAGCACUUCUAG-3′ and reverse,
5′-AGAAGUGCUCACACUGCAGUUU-3′) and mimics NC (50 nM; forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′), miR-20b inhibitor (100 nM;
5′-GTGCTCATAGTGCAGGTAGTT-3′) and inhibitor NC (100 nM;
5′-GGGTCTGACGAGGTACTATTT-3′), STAT3 overexpression plasmid pcDNA3.1
(pc-STAT3, 5 µg) and the parental negative control (pc-NC, 5 µg)
were all purchased from Shanghai GenePharma, Co., Ltd., and
transfected into AMs using Lipofectamine® 3000 reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 48 h,
according to the manufacturer's instructions. Cells were harvested
for subsequent experimentation 48 h post-transfection.
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The cell viability of AMs was detected using an MTT
assay kit (Nanjing KeyGen Biotech Co., Ltd.). Cells
(1×104/well) were cultured in a 96-well plate at 37°C
for 48 h. The MTT solution was added and incubated for 2 h at 37°C.
Following incubation, the supernatant was removed carefully and the
insoluble formazan was dissolved in 150 µl DMSO. The absorbance was
measured at 570 nm to assess cell viability.
Apoptosis analysis
Apoptosis of AMs was detected using the Annexin
V-FITC/propidium iodide (PI) apoptosis detection kit (Vazyme
Biotech Co., Ltd.). Briefly, AMs were resuspended in 500 µl binding
buffer (Vazyme Biotech Co., Ltd.) at a density of 1×105
cells/ml. AMs were subsequently stained with Annexin V and PI for
10 min at room temperature, in the dark. Apoptotic cells were
detected using a BD Accuri C6 Plus flow cytometer (BD Biosciences)
and FACSDiva software (version 6.13; BD Biosciences).
RT-qPCR
Total RNA was extracted from lung tissues and AMs
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and reverse transcribed into cDNA using the
TaqMan MicroRNA Reverse Transcription kit (Thermo Fisher
Scientific, Inc.) and PrimeScript RT Master Mix (Takara
Biotechnology, Co., Ltd.). The cDNA synthesis was performed at 37°C
for 15 min and 85°C for 5 sec. qPCR was subsequently performed
using TaqMan 2X Universal PCR Master Mix (Thermo Fisher Scientific,
Inc.). The following thermocycling conditions were used for qPCR:
Initial denaturation at 95°C for 10 min; 40 cycles of 95°C for 1
min, 63°C for 2 min, 72°C for 1 min; final 72°C for 10 min. The
following primer sequences were used for qPCR: NEAT1 forward,
5′-TGGCTAGCTCAGGGCTTCAG-3′ and reverse,
5′-TCTCCTTGCCAAGCTTCCTTC-3′; STAT3 forward,
5′-CCTTCCTCACCGTGTACTGG-3′ and reverse,
5′-AGCGTAGGGTAAGGTTCTTGC-3′; CD68 forward,
5′-GCTACATGGCGGTGGAGTACAA-3′ and reverse,
5′-ATGATGAGAGGCAGCAAGATGG-3′; CD206 forward,
5′-TTCGACACCCATCGGAATT-3′ and reverse, 5′-CACAAGCGCTGCGTGGAT-3′;
GAPDH forward, 5′-AGTCAGCTCTCTCCTTTCAGG-3′ and reverse,
5′-TCCACCACCCTGTTGCTGTA-3′; miR-20b forward,
5′-GCTCATAGTGCAGGTAGAA-3′ and reverse, 5′-TGTCAACGATACGCTACG-3′;
and U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′. Relative expression levels were
calculated using the 2−ΔΔCq method (28) and normalized to U6 or GAPDH.
Dual-luciferase reporter assay
The dual-luciferase reporter assay was performed to
validate the binding site between NEAT1 and miR-20b, and between
miR-20b and STAT3, in AMs. The luciferase reporter plasmids (pGL3
vectors; Promega Corporation) containing NEAT1-wild-type (WT),
NEAT1-mutant (MUT), STAT3-WT and STAT3-MUT were synthesized by
Shanghai GenePharma Co., Ltd. The constructed plasmids were
co-transfected into AM cells with miR-20b mimics or mimics NC using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.). After transfection for 48 h, relative luciferase
activities were detected using the Dual-Luciferase Reporter Assay
kit (Promega Corporation). Relative luciferase activity was
normalized to Renilla luciferase activity.
RNA immunoprecipitation (RIP)
assay
The Magna RNA-binding protein immunoprecipitation
kit (Millipore, Sigma) was used to perform the RIP assay, according
to the manufacturer's instructions. Briefly, cells
(1×107) were centrifugated at 3,000 × g at 4°C for 10
min and resuspended with RIPA buffer (100 µl; Sigma-Aldrich; Merck
KGaA) containing protease and RNase inhibitors. Cell lysates (100
µl) were incubated with human anti-Ago2 antibody (5 µg; Cell
Signaling Technology, Inc.) or negative control IgG (5 µg;
ProteinTech Group, Inc.) coated magnetic beads (50 µl; Thermo
Fisher Scientific, Inc.) at 4°C overnight, while shaking to aid
digestion of proteins and the precipitated RNA was isolated. Beads
were washed twice using PBS buffer (Sangon Biotech Co., Ltd.), and
the mixture was centrifuged at 2,500 × g for 10 min at 4°C. RNA was
treated with proteinase K for 30 min at 55°C and extracted using
TRIzol (Thermo Fisher Scientific, Inc.). The relative expression
levels of NEAT1, miR-20b and STAT3 were determined via RT-qPCR
analysis.
Western blotting
Total protein was extracted from lung tissues or AMs
using RIPA lysate containing protease inhibitors (Thermo Fisher
Scientific, Inc.). The enhanced BCA protein assay kit (Beyotime
Institute of Biotechnology) was used to quantify the protein
concentration. Protein (50 µg/per lane) were separated via 10%
SDS-PAGE, transferred onto PVDF membranes and blocked with non-fat
milk for 1 h at room temperature. The membranes were incubated with
primary antibodies against STAT3 (cat. no. ab68153; 1:1,000; Abcam)
and GAPDH (cat. no. ab8245; 1:1,000; Abcam) overnight at 4°C.
Following the primary incubation, membranes were incubated with
HRP-conjugated anti-rabbit IgG secondary antibody (cat. no. ab6721;
1:5,000; Abcam) at room temperature for 1 h. Protein bands were
visualized with ECL detection reagents (Cytiva), and blots were
semi-quantitated using ImageJ software (version V1.8.0; National
Institutes of Health).
Bioinformatics analysis
The DIANA tools database (http://carolina.imis.athena-innovation.gr/diana_tools/web/index.php?r=lncbasev2%2Findex-predicted)
and differentially expressed genes (DEGs; GEO accession no.
GSE148649; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148649)
were used to screen the targets of lncRNA NEAT1. The binding sites
between lncRNA NEAT1 and miR-20b were predicted using StarBase3.0
(http://starbase.sysu.edu.cn/index.php), while the
binding sites between miR-20b and STAT3 were predicted using
TargetScan 7.2 (http://www.targetscan.org/vert_72).
Statistical analysis
Statistical analysis was performed using SPSS 23.0
software (IBM Corp.) and GraphPad Prism 7.0 software (GraphPad
Software, Inc.). All experiments were repeated three times and data
are presented as the mean ± SD. The comparisons among multiple
groups were analyzed using one-way ANOVA followed by Tukey's post
hoc test. The comparisons between two groups were analyzed using an
unpaired Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
NEAT1 knockdown alleviates lung injury
caused by VILI
Abnormal NEAT1 expression was observed in VILI mice.
The results demonstrated that NEAT1 expression was significantly
higher in the lung tissues and BALF of the VILI group compared with
the Sham group (P<0.001; Fig. 1A
and B). Furthermore, total protein in the BALF was
significantly higher in the VILI group compared with the Sham group
(P<0.001; Fig. 1C).
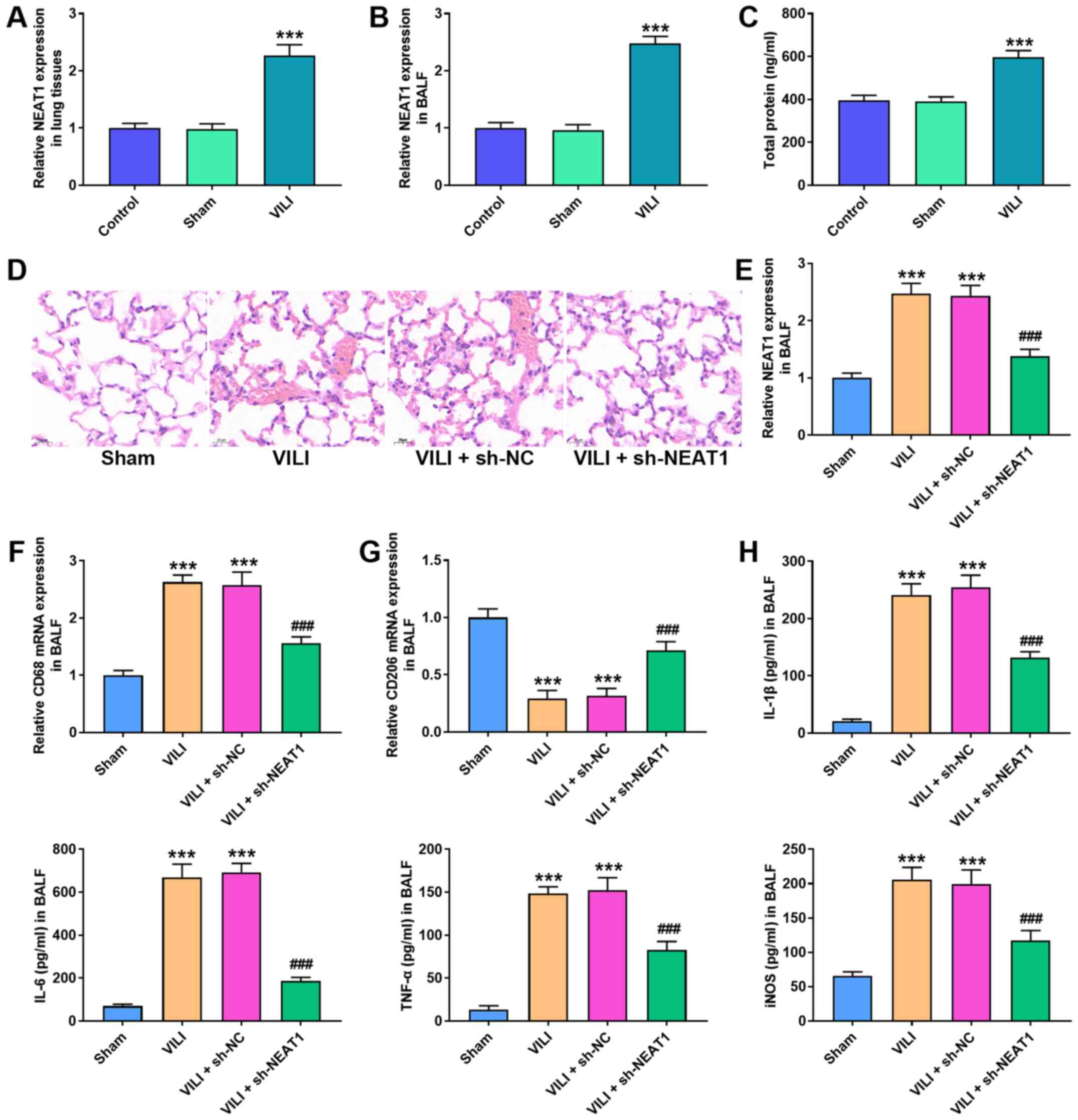 | Figure 1.NEAT1 knockdown alleviates lung
injury caused by VILI. RT-qPCR analysis was performed to detect
NEAT1 expression in (A) lung tissues and (B) BALF. (C) The total
protein level of alveolar macrophages was calculated via the BCA
method. (D) The histopathological changes of lung tissues were
measured via H&E staining. (E) RT-qPCR analysis was performed
to detect NEAT1 expression in BALF. RT-qPCR analysis was performed
to detect the expression of the (F) M1 phenotype marker, CD86 and
the (G) M2 phenotype marker, CD206 in BALF. (H) The ELISA assay was
performed to detect the expression levels of IL-1β, IL-6, TNF-α and
iNOS. ***P<0.05 vs. the control and sham groups;
###P<0.05 vs. the VILI + sh-NC group. NEAT1, nuclear
enriched abundant transcript 1; VILI, ventilator-induced lung
injury; RT-qPCR, reverse transcription-quantitative PCR; BALF,
broncho-alveolar lavage fluid; sh, short hairpin; NC, negative
control; IL, interleukin, TNF, tumor necrosis factor; iNOS,
inducible nitric oxide synthase. |
To investigate the role of NEAT1 in VILI, sh-NEAT1
was transfected into mice prior to VILI treatment. H&E staining
demonstrated that VILI destroyed lung injury of mice, and NEAT1
knockdown suppressed the destructive effect of VILI on lung tissues
(Fig. 1D). RT-qPCR analysis
demonstrated that NEAT1 expression decreased in the VILI + sh-NEAT1
group compared with the VILI + sh-NC group (P<0.001; Fig. 1E).
The activation of AMs is classified as classically
activated M1 and alternatively activated M2 (15,16). RT-qPCR analysis was performed to
detect the mRNA expression levels of the M1 phenotype marker, CD68
and M2 phenotype marker, CD206. The results demonstrated that VILI
increased CD68 mRNA expression but decreased CD206 mRNA expression,
the effects of which were reversed following NEAT1 knockdown
(P<0.001; Fig. 1F and G). The
ELISA assay was performed to assess the effect of NEAT1 on the
expression levels of IL-1β, IL-6, TNF-α and iNOS. The results
demonstrated that NEAT1 knockdown decreased the expression levels
of IL-1β, IL-6, TNF-α and iNOS, the effects of which were reversed
following VILI treatment (P<0.001; Fig. 1H).
NEAT1 knockdown decreases the cell
injury of CS treatment on AMs
NEAT1 shRNA was transfected into AMs to detect the
effect of NEAT1 in VILI in vitro. NEAT1 expression
significantly decreased following transfection with NEAT1 shRNA
(P<0.001; Fig. 2A). CS
experiments were performed to imitate the stretching of MV. Similar
to the results of the VILI mouse model, CS significantly increased
NEAT1 expression (P<0.001; Fig.
2B), inhibited cell viability (P<0.05; Fig. 2C) and induced the apoptosis of AMs
(P<0.001; Fig. 2D). Notably,
CS treatment increased CD68 mRNA expression (P<0.001; Fig. 2E) and decreased CD206 mRNA
expression in AMs (P<0.001; Fig.
2F) compared with the control group. The effect of NEAT1 on
inflammation was assessed via the ELISA assay. The results
demonstrated that CS treatment significantly increased the
expression levels of IL-1β, IL-6, TNF-α and iNOS in AMs compared
with the control group (all P<0.001; Fig. 2G).
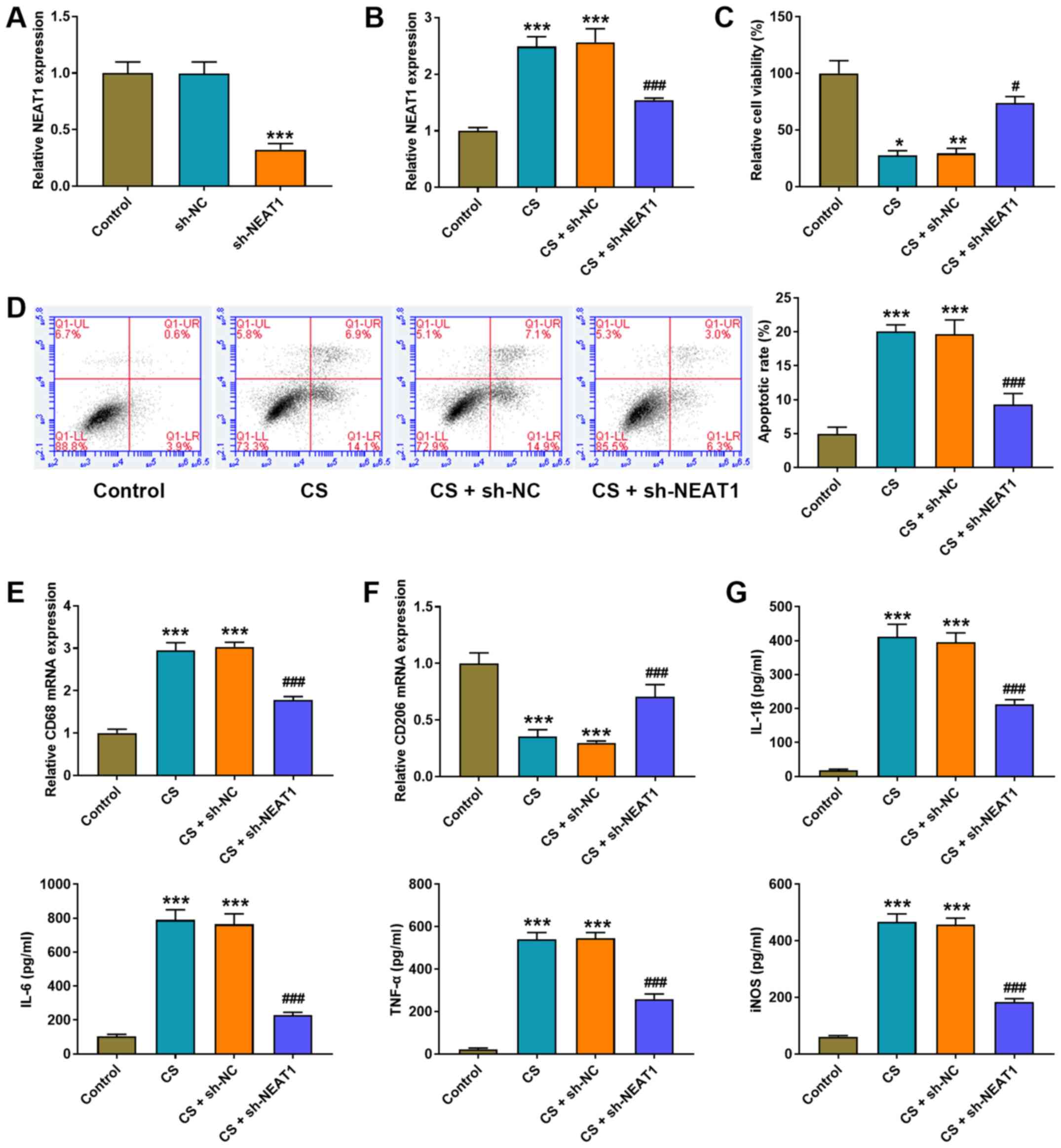 | Figure 2.NEAT1 knockdown decreases the cell
injury of CS treatment in AMs. (A and B) RT-qPCR analysis was
performed to detect NEAT1 expression in AMs. (C) The MTT assay was
performed to detect the viability of AMs. (D) Flow cytometric
analysis was performed to detect the apoptosis of AMs. RT-qPCR
analysis was performed to detect the expression of the (E) M1
phenotype marker, CD86 and the (F) M2 phenotype marker, CD206 in
AMs. (G) The ELISA assay was performed to detect the expression
levels of IL-1β, IL-6, TNF-α and iNOS in AMs. *P<0.05,
**P<0.01, ***P<0.001 vs. the control group;
#P<0.05, ###P<0.001 vs. the CS + sh-NC
group. NEAT1, nuclear enriched abundant transcript 1; CS, cell
stretch; AMs, alveolar macrophages; RT-qPCR, reverse
transcription-quantitative PCR; IL, interleukin, TNF, tumor
necrosis factor; iNOS, inducible nitric oxide synthase; sh, short
hairpin; NC, negative control. |
NEAT1 shRNA was transfected into AMs to confirm the
effect of NEAT1 on VILI. As presented in Fig. 2A and B, NEAT1 expression
significantly decreased following transfection with NEAT1 shRNA
(P<0.001). Notably, NEAT1 knockdown significantly increased cell
viability (P<0.05; Fig. 2C),
inhibited cell apoptosis (P<0.001; Fig. 2D), decreased CD68 mRNA expression
(P<0.001; Fig. 2E), increased
CD206 mRNA expression (P<0.001; Fig. 2F) and decreased the expression
levels of IL-1β, IL-6, TNF-α and iNOS in AMs (P<0.001; Fig. 2G).
NEAT1 sponges miR-20b expression in
AMs
The underlying molecular mechanism of NEAT1 in VILI
was further investigated. A total of 10 miRNAs were predicted to
directly interact with NEAT1 via bioinformatics analysis (Fig. 3A). RT-qPCR analysis was performed
to detect the expression levels of the miRNAs. The results
demonstrated that the expression levels of the screened miRNAs
increased following transfection with sh-NEAT1 in AMs, and the most
significant increase was observed in miR-20b (P<0.001; Fig. 3B). The StarBase 3.0 database was
used to predict the potential binding site between NEAT1 and
miR-20b (Fig. 3C), and the
dual-luciferase reporter assay was performed to confirm the
interaction between NEAT1 and miR-20b. The results demonstrated
that luciferase activity significantly decreased in cells
co-transfected with NEAT1-WT and miR-20b mimic (P<0.01), while
no significant changes were observed in cells co-transfected with
NEAT1-MUT and miR-20b mimics (Fig.
3D). The results of the RIP assay proved that both NEAT1 and
miR-20b were immunoprecipitated by anti-Ago2 (P<0.001; Fig. 3E). In addition, RT-qPCR analysis
demonstrated that NEAT1 knockdown in both VILI mice (P<0.001;
Fig. 3F) and CS-treated AMs
(P<0.001; Fig. 3G) increased
miR-20b expression compared with the VILI + sh-NC or CS + sh-NC
groups. As expected, miR-20b expression significantly increased
following transfection with miR-20b mimics and significantly
decreased following transfection with miR-20b inhibitor (both
P<0.001; Fig. 3H).
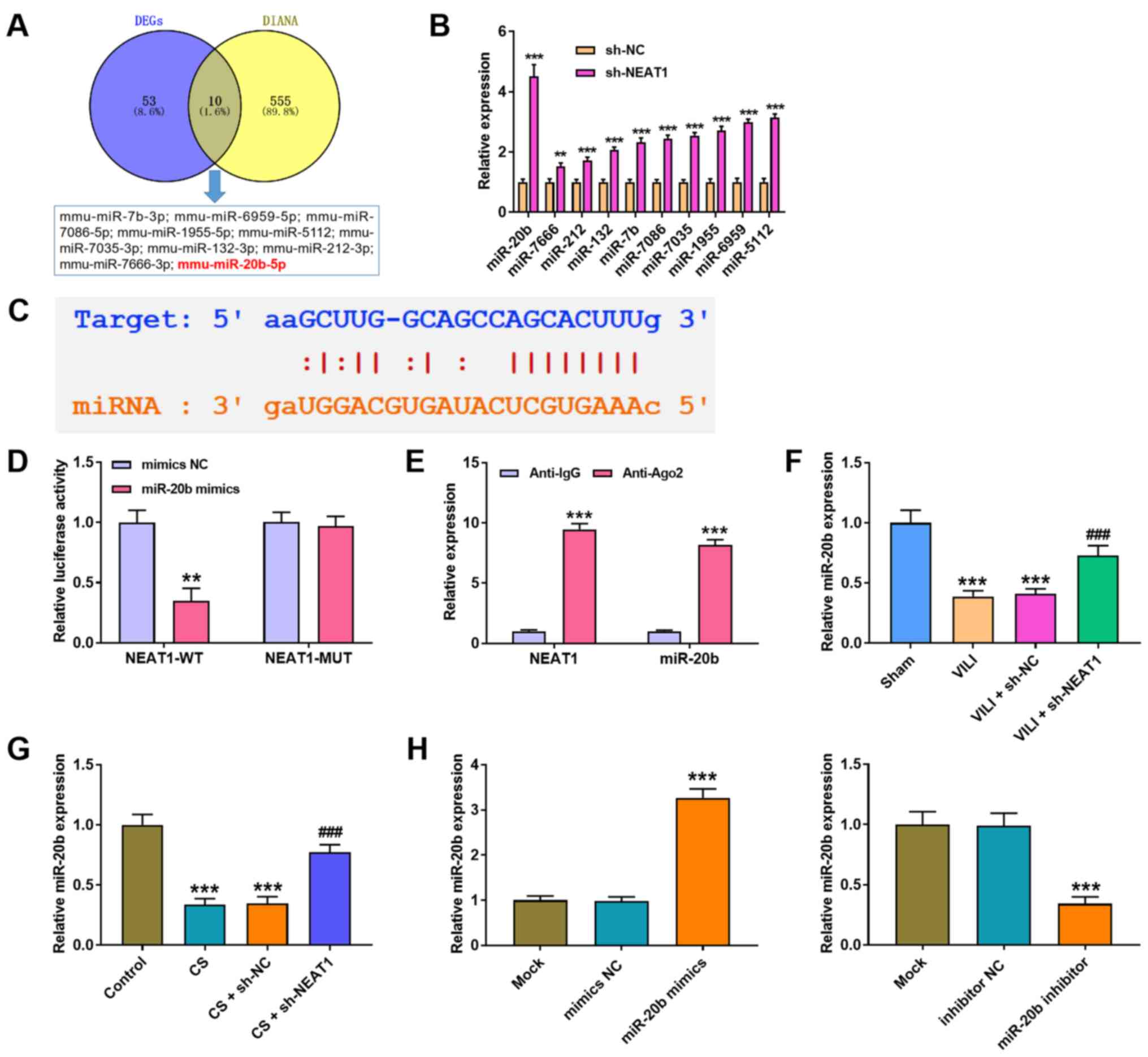 | Figure 3.NEAT1 sponges miR-20b expression in
AMs. (A) A total of 10 miRNAs were identified to potentially bind
to NEAT1 through the intersection of DEGs and DIANA-LncBase. (B)
RT-qPCR analysis was performed to detect the expression levels of
the 10 miRNAs in AMs. **P<0.01, ***P<0.001 vs. the sh-NC
group. (C) The binding site between NEAT1 and miR-20b was predicted
using the StarBase 3.0 database. (D) Luciferase activity was
determined via the dual-luciferase reporter gene assay. **P<0.01
vs. the mimics NC group. (E) The RIP assay was performed to assess
the interaction between NEAT1 and miR-20b. ***P<0.001 vs. the
anti-lgG group. RT-qPCR analysis was performed to detect miR-20b
expression in the lung tissues of (F) VILI mice and (G) CS treated
AMs. ***P<0.001 vs. the sham and control groups;
###P<0.001 vs. the VILI + sh-NC group and CS + sh-NC
groups. (H) RT-qPCR analysis was performed to detect miR-20b
expression in AMs transfected with miR-20b mimics or miR-20b
inhibitor. ***P<0.001 vs. the mimics NC and the inhibitor NC
groups. NEAT1, nuclear enriched abundant transcript 1; miR/miRNA,
microRNA; AMs, alveolar macrophages; DEGs. differentially expressed
genes; RT-qPCR, reverse transcription-quantitative PCR; VILI,
ventilator-induced lung injury; CS, cell stretch; sh, short
hairpin; NC, negative control; WT, wild-type; MUT, mutant. |
NEAT1 modulates miR-20b expression to
regulate STAT3 in AMs
To determine the regulatory mechanism of miR-20b in
VILI, the potential target genes of miR-20b were predicted using
TargetScan. The results revealed that STAT3 has a binding site for
miR-20b (Fig. 4A). The results of
the dual-luciferase reporter assay (Fig. 4B) showed that the luciferase
activity was significantly decreased in cells co-transfected with
STAT3-WT and miR-20b mimic (P<0.001), while no change in cells
co-transfected with STAT3-MUT and miR-20b mimics were observed. The
RIP assay (Fig. 4C) also
demonstrated that both STAT3 and miR-20b were immunoprecipitated by
anti-Ago2 (P<0.001). Notably, STAT3 protein expression
significantly increased following transfection with pc-STAT3
compared with the pc-NC group (P<0.001; Fig. 4D). Western blot analysis was
performed to detect STAT3 protein expression in AMs following
transfection with miR-20b mimics. The results demonstrated that
STAT3 protein expression significantly decreased in the miR-20b
mimics group compared with the mimics NC group (P<0.001;
Fig. 4E). Furthermore, western
blot analysis was performed to detect STAT3 protein expression in
the lung tissues of VILI mice and CS-treated AMs. As presented in
Fig. 4F and G, both VILI and CS
treatment significantly increased STAT3 protein expression, the
effects of which were reversed following NEAT1 knockdown
(P<0.001).
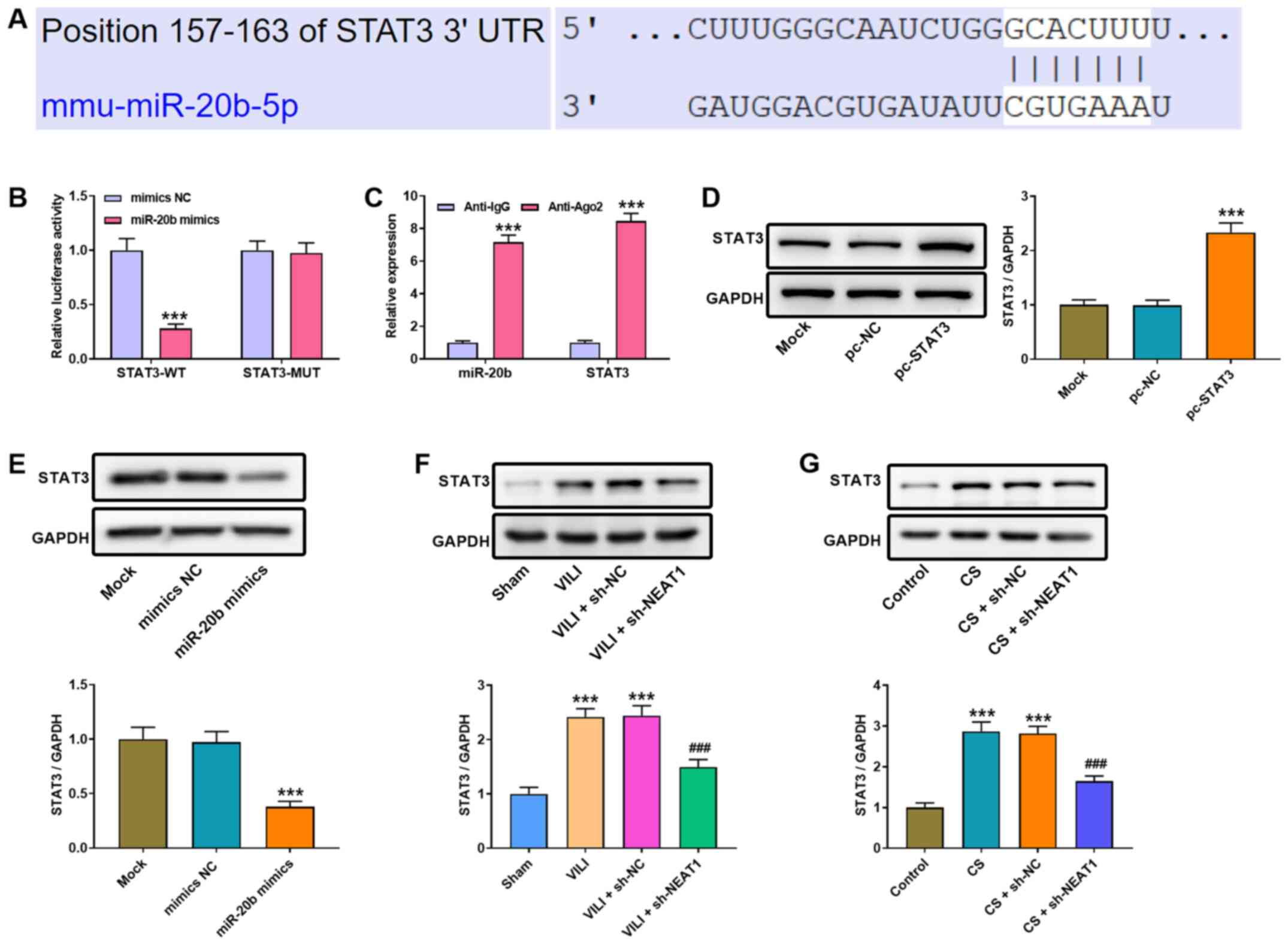 | Figure 4.NEAT1 modulates miR-20b expression to
regulate STAT3 expression in AMs. (A) The binding site between
miR-20b and STAT3 was predicted using TargetScan. (B) Luciferase
activities were determined via the dual-luciferase reporter assay.
***P<0.001 vs. the mimics NC group. (C) The RIP assay was
performed to assess the interaction between NEAT1 and miR-20b.
***P<0.001 vs. the anti-lgG group. (D) Western blot analysis was
performed to detect STAT3 protein expression in AMs. ***P<0.001
vs. the pc-NC group. (E) Western blot analysis was performed to
detect STAT3 protein expression following transfection with miR-20b
mimics or NC mimics. ***P<0.001 vs. the mimics NC group. Western
blot analysis was performed to detect STAT3 protein expression in
the lung tissues of (F) VILI mice or (G) CS-treated AMs.
***P<0.001 vs. the sham and control groups;
###P<0.001 vs. the VILI + sh-NC and CS + sh-NC
groups. NEAT1, nuclear enriched abundant transcript 1; miR,
microRNA; AMs, alveolar macrophages; NC, negative control; VILI,
ventilator-induced lung injury; CS, cell stretch; sh, short
hairpin; UTR, untranslated region; WT, wild-type; MUT, mutant. |
NEAT1 knockdown alleviates CS-induced
AMs injury by increasing miR-20b expression
To determine whether NEAT1 exerts its function via
miR-20b, CS-treated AMs were co-transfected with NEAT1 shRNA and
miR-20b inhibitor. RT-qPCR analysis demonstrated that miR-20b
expression significantly increased following transfection with
NEAT1 shRNA, the effects of which were reversed following
transfection with miR-20b inhibitor (P<0.001; Fig. 5A). The results of the MTT assay
demonstrated that transfection with NEAT1 shRNA significantly
increased the viability (Fig. 5B)
and inhibited the apoptosis of AMs (Fig. 5C and D) (both P<0.001).
Notably, these effects were reversed following transfection with
miR-20b inhibitor (P<0.001; Fig.
5B-D). RT-qPCR analysis was performed to detect the expression
levels of CD68 (M1 phenotype marker) and CD206 (M2 phenotype
marker) following co-transfection with NEAT1 shRNA and miR-20b
inhibitor. The results demonstrated that transfection with miR-20b
inhibitor increased CD68 mRNA expression (Fig. 5E) and decreased CD206 mRNA
expression (Fig. 5F) compared
with the sh-NEAT1 group (both P<0.001), which suggests that low
miR-20b expression enhances polarization into M1 and inhibits
polarization into M2. The results of the ELISA assay demonstrated
that transfection with miR-20b inhibitor increased the expression
levels of IL-1β, IL-6, TNF-α and iNOS, the effects of which were
reversed following NEAT1 knockdown (P<0.001; Fig. 5G).
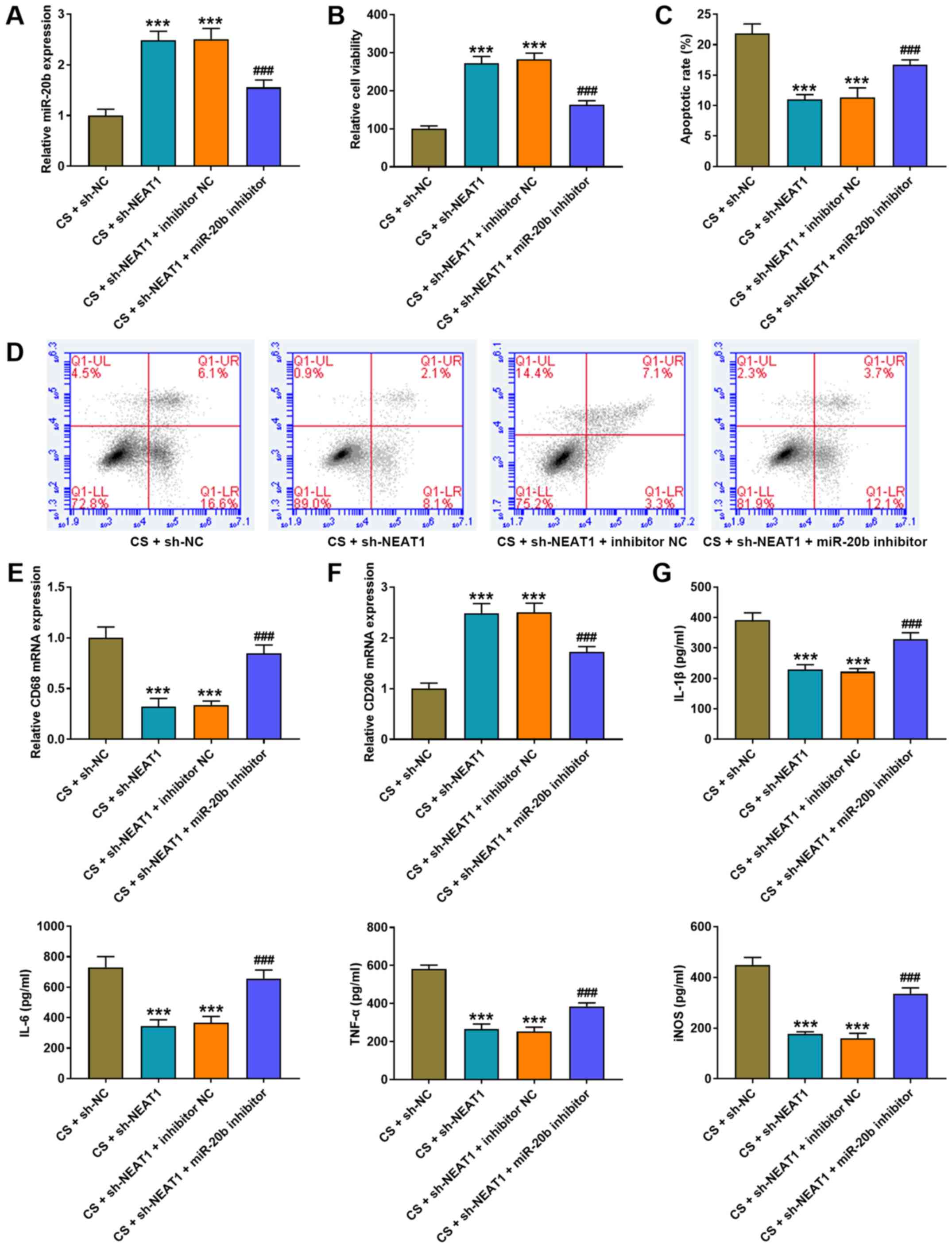 | Figure 5.NEAT1 knockdown alleviates CS-induced
AMs injury by increasing miR-20b expression. (A) RT-qPCR analysis
was performed to detect miR-20b expression in AMs following
co-transfection with NEAT1 shRNA and miR-20b inhibitor. (B) The MTT
assay was performed to assess the viability of AMs following
co-transfection with NEAT1 shRNA and miR-20b inhibitor. (C and D)
Flow cytometric analysis was performed to detect the apoptosis of
AMs following co-transfection with NEAT1 shRNA and miR-20b
inhibitor. RT-qPCR analysis was performed to detect the expression
of (E) the M1 phenotype marker, CD86 and (F) the M2 phenotype
marker, CD206 in AMs following co-transfection with NEAT1 shRNA and
miR-20b inhibitor. (G) The ELISA assay was performed to detect the
expression levels of IL-1β, IL-6, TNF-α and iNOS in AM following
co-transfection with NEAT1 shRNA and miR-20b inhibitor.
***P<0.001 vs. the CS + sh-NC group; ###P<0.001
vs. the CS + sh-NEAT1 + inhibitor NC group. NEAT1, nuclear enriched
abundant transcript 1; CS, cell stretch; AMs, alveolar macrophages;
miR, microRNA; RT-qPCR, reverse transcription-quantitative PCR; sh,
short hairpin; NC, negative control; IL, interleukin; TNF, tumor
necrosis factor; iNOS, inducible nitric oxide synthase. |
Overexpression of STAT3 eliminates the
protective effect of miR-20b on cell injury of CS-induced AMs
To verify whether miR-20b exerts its function via
STAT3, CS-treated AMs were co-transfected with miR-20b mimics and
STAT3 overexpressed plasmid (pcDNA3.1 STAT3). Western blot analysis
demonstrated that STAT3 protein expression significantly decreased
following transfection with miR-20b mimics, the effects of which
were reversed following overexpression of STAT3 (P<0.001;
Fig. 6A). In addition,
transfection with miR-20b mimics significantly increased the
viability (Fig. 6B) and inhibited
the apoptosis (Fig. 6C) of AMs
(both P<0.001). Conversely, overexpression of STAT3
significantly inhibited the viability (Fig. 6B) and induced the apoptosis
(Fig. 6C) of AMs (both
P<0.001). Notably, transfection with miR-20b mimics
significantly decreased CD68 mRNA expression (Fig. 6D) and increased CD206 mRNA
expression (Fig. 6E) in
CS-treated AMs, the effects of which were reversed following
overexpression of STAT3 (P<0.001). The results of the ELISA
assay demonstrated that transfection with miR-20b mimics decreased
the expression levels of IL-1β, IL-6, TNF-α and iNOS in AMs, the
effects of which were reversed following overexpression of STAT3
(all P<0.001; Fig. 6F). The
aforementioned results indicated that lncRNA NEAT1 regulated the
expression of STAT3 by targeting miR-20b, thereby affecting the
polarization of alveolar macrophages in VILI mice and increasing
inflammation and lung injury (Fig.
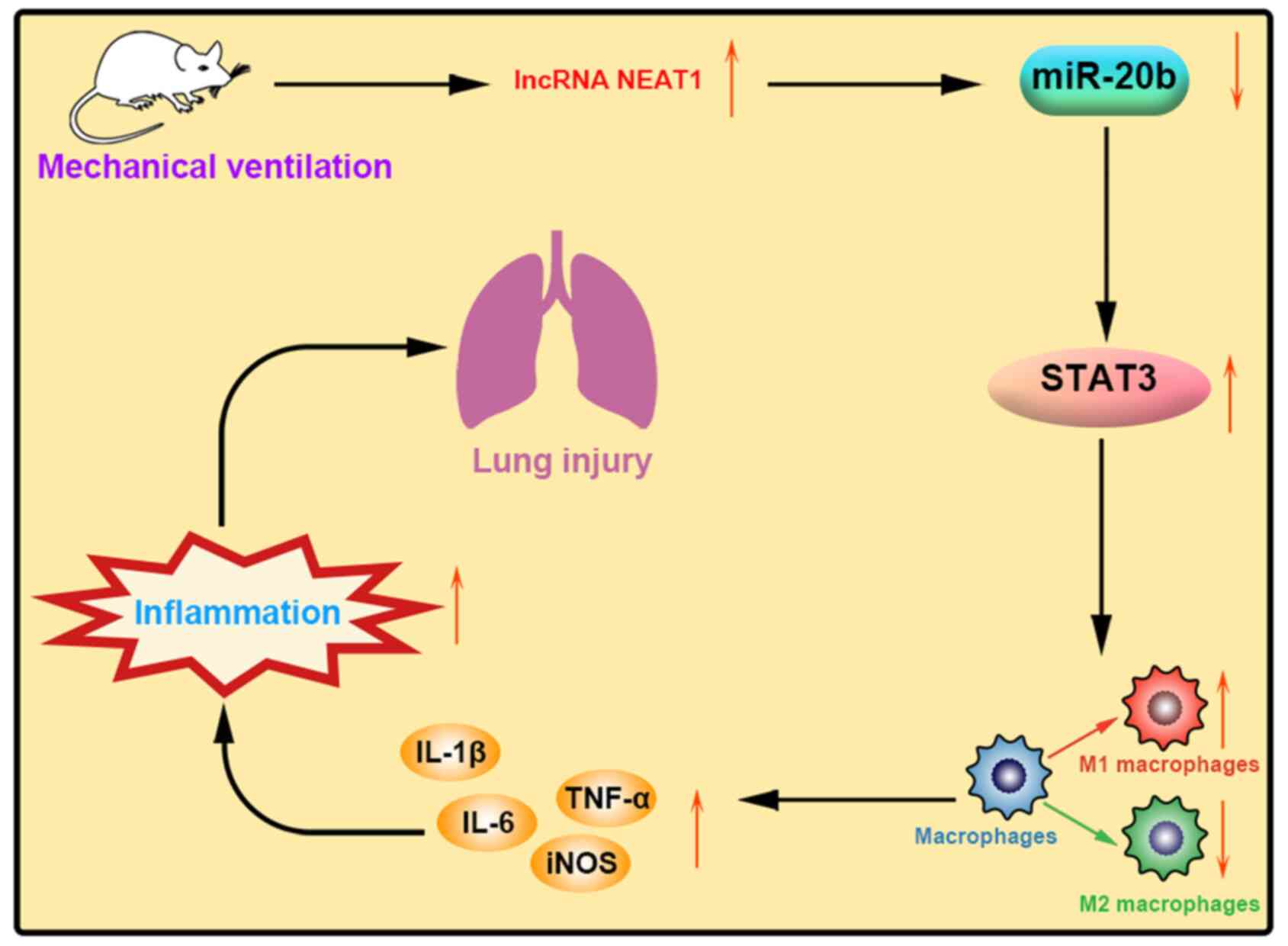
7).
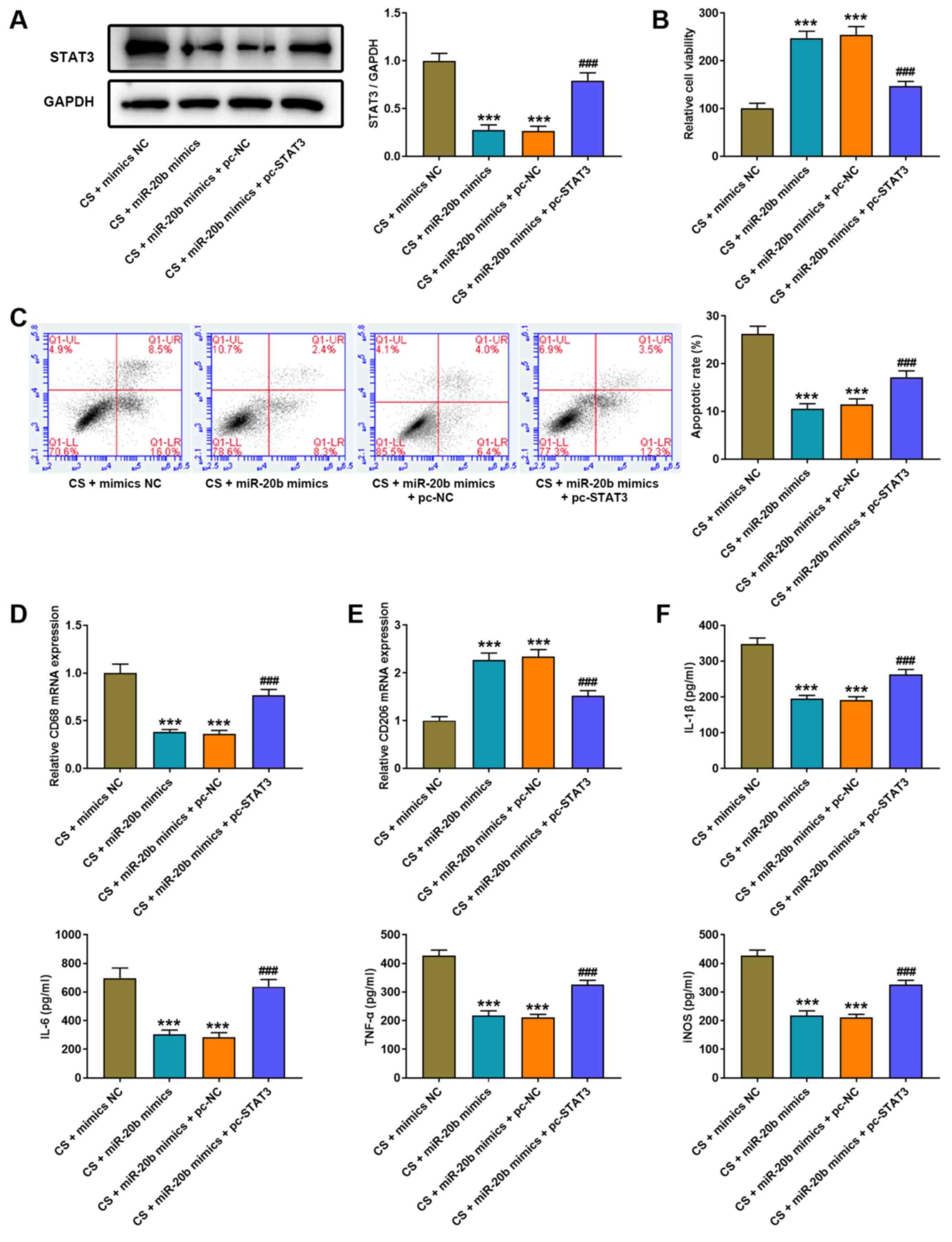 | Figure 6.Overexpression of STAT3 eliminates
the protected effect of miR-20b on cell injury of CS-induced AMs.
(A) Western blot analysis was performed to detect STAT3 protein
expression in AMs following co-transfection with miR-20b mimics and
pcDNA3.1 STAT3. (B) The MTT assay was performed to detect the
viability of AMs following co-transfection with miR-20b mimics and
pcDNA3.1 STAT3. (C) Flow cytometric analysis was performed to
assess the apoptosis of AMs following co-transfection with miR-20b
mimics and pcDNA3.1 STAT3. Reverse transcription-quantitative PCR
analysis was performed to detect the expression of (D) the M1
phenotype marker, CD86 and (E) the M2 phenotype marker, CD206 in
AMs following co-transfection with miR-20b mimics and pcDNA3.1
STAT3. (F) The ELISA assay was performed to detect the expression
levels of IL-1β, IL-6, TNF-α and iNOS in AMs following
co-transfection with miR-20b mimics and pcDNA3.1 STAT3.
***P<0.001 vs. the CS + mimics NC group;
###P<0.001 vs. the CS + miR-20b mimics + pc-NC group.
miR, microRNA; CS, cell stretch; AMs, alveolar macrophages; IL,
interleukin; TNF, tumor necrosis factor; iNOS, inducible nitric
oxide synthase; NC, negative control. |
Discussion
MV is an essential therapeutic tool for patients
with acute respiratory distress syndrome (1,29).
However, MV can also induce or exacerbate lung injury (29). The present study aimed to
investigate the protective effect of NEAT1 knockdown in the in
vitro and in vivo model of VILI. The results
demonstrated that NEAT1 knockdown promoted the phenotypic
transformation of AMs from the pro-inflammatory (M1) to the
anti-inflammatory (M2) phenotype, and reduced inflammation by
regulating miR-20b and STAT3 expression.
Previous studies have demonstrated that regulating
the expression of lncRNAs can alleviate lung injury (30–32). Wang et al (33) reported that CASC9 is significantly
downregulated in human small airway epithelial cells (HSAECs)
treated with LPS, and in the lung tissues of rats with sepsis,
whereas overexpression of CASC9 significantly promotes the
viability of HSAECs. MALAT1 is highly expressed in lung transplant
ischemia-reperfusion (LTIR), and MALAT1 knockdown ameliorates
injury following LTIR (34). The
results of the present study demonstrated that NEAT1 expression was
significantly upregulated in the lung tissues of VILI mice and
CS-treated AMs. NEAT1 knockdown weakened the histopathological
injury of the lung tissues, decreased apoptosis of AMs and
decreased the number of macrophages. Furthermore, NEAT1 knockdown
alleviated lung injury induced by VILI and suppressed apoptosis in
CS-treated AMs.
The interaction between miRNAs and lncRNAs is
indispensable in the development of diseases (32,35,36). Tang et al (26) reported that as the ceRNA for
miR-20b-5p, HOTAIR knockdown attenuates autophagy in hepatic I/R
injury by improving the inhibitory effect of miR-20b-5p on ATG7.
Furthermore, You et al (37) demonstrated that miR-20b expression
was downregulated in a CCI rat model, and miR-20b-5p mimics
alleviates neuropathic pain by inhibiting Akt3 expression in CCI
rats, which supports the results of the present study. In the
present study, miR-20b expression was downregulated in both the
lung tissues of VILI mice and CS-treated AMs. The dual-luciferase
reporter and RIP assays confirmed that miR-20b is the target miRNA
of NEAT1. Notably, the inhibitory effect of sh-NEAT1 on apoptosis,
CD68 expression and the expression levels of IL-1β, IL-6, TNF-α and
iNOS was eliminated following transfection with miR-20b inhibitor.
Taken together, these results suggest that NEAT1 may inhibit the
progression of VILI by upregulating miR-20b expression.
Previous studies have reported that STAT3 is
implicated in the pathogenesis of alcoholic hepatitis (AH) and lung
injury (38,39). Facilitation of STAT3
phosphorylation in AMs exacerbates pulmonary inflammation in acute
lung injury (ALI) and aggravates lung injury in ALI rats (39). STAT3 is activated in mice with AH,
inhibition of which alleviates liver injury (38). The results of the present study
demonstrated that STAT3 was highly expressed in the lung tissues of
VILI mice and CS-treated AMs. The results confirmed that STAT3 is
the direct target of miR-20b, and was positively regulated by
NEAT1. STAT3 expression decreased following transfection with both
miR-20b mimics and sh-NEAT1, the effects of which were reversed
following transfection with miR-20b inhibitor. Collectively, these
results suggest that NEAT1 knockdown may inhibit the progression of
VILI by regulating miR-20b-mediated STAT3.
Recent studies have reported that activated
macrophages play a vital role in the progression of several
inflammatory diseases (30,40–42). The M1 and M2 phenotypes play
opposing roles in the regulation of inflammation. M1 macrophages
play important roles in immune inflammatory effects, whereas M2
macrophages mainly exert anti-inflammatory effects (43,44). In acute spinal cord injury (SCI)
mice, lncGBP9 knockdown promotes M2 polarization of the
macrophages, which reduces the inflammatory levels and facilitates
repair following SCI (41). Ji
et al (45) demonstrated
that promoting the transformation of M2 macrophages can protect the
podocytes from high-glucose-induced injury in DN, which supports
the results of the present study. CD68 mRNA expression (M1
phenotype marker) increased and CD206 mRNA expression (M2 phenotype
marker) decreased in the lung tissues of VILI mice, and NEAT1
knockdown inhibited CD68 expression and enhanced CD206 expression.
Similar results were observed in CS-treated AMs. Notably, the
expression levels of IL-1β, IL-6, TNF-α and iNOS were decreased in
VILI mice and CS-treated AMs concomitant with the increasing
transformation of M2 macrophages. Taken together, these results
suggest that NEAT1 knockdown may remit injury of VILI by promoting
the phenotypic transformation of AMs from M1 to M2.
In conclusion, the results of the present study
demonstrated that NEAT1 expression was upregulated in the lung
tissues of VILI mice and CS-treated AMs, and played an important
role in accelerating the progress of VILI. NEAT1 knockdown promoted
the phenotypic transformation of AMs from M1 to M2, thereby
suppressing inflammatory levels and reducing histopathological
injury and apoptosis of VILI. The regulatory effect of NEAT1 on
VILI may be achieved by regulating miR-20b and STAT3 expression.
However, the present study did not explore whether there are other
targets that interact with NEAT1 in VILI, and investigation into
its underlying mechanisms require further research in the
future.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL designed the present study and performed the
literature review. YL and GT performed the experiments. GT and JL
analyzed the data and drafted the initial manuscript. GT and JL
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Affiliated Huaian No.1 People's Hospital of Nanjing Medical
University (Huaian, China; approval no. HASDY20201009) and
performed in accordance with the Guide for the Care and Use of
Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gattinoni L, Tonetti T, Cressoni M,
Cadringher P, Herrmann P, Moerer O, Protti A, Gotti M, Chiurazzi C,
Carlesso E, et al: Ventilator-related causes of lung injury: The
mechanical power. Intensive Care Med. 42:1567–1575. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fan E, Brodie D and Slutsky AS: Acute
Respiratory Distress Syndrome: Advances in Diagnosis and Treatment.
JAMA. 319:698–710. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu B, Wang Y, Li X, Mao Y and Deng X: RNA
sequencing analysis of aberrantly expressed long non coding RNAs
and mRNAs in a mouse model of ventilator induced lung injury. Mol
Med Rep. 18:882–892. 2018.PubMed/NCBI
|
|
4
|
Zhang B, Zhang X, Li Q, Ma F, Sun L and
Wang M: Dexmedetomidine attenuates ventilator-induced lung injury
in rats by up-regulating NLRC3. Ann Palliat Med. 9:2474–2484. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun Z, Wang F, Yang Y, Wang J, Sun S, Xia
H and Yao S: Resolvin D1 attenuates ventilator-induced lung injury
by reducing HMGB1 release in a HO-1-dependent pathway. Int
Immunopharmacol. 75:1058252019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Veskemaa L, Graw JA, Pickerodt PA, Taher
M, Boemke W, González-López A and Francis RC:
Tert-butylhydroquinone augments Nrf2-dependent resilience against
oxidative stress and improves survival of ventilator-induced lung
injury in mice. Am J Physiol Lung Cell Mol Physiol. 320:L17–L28.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin JY, Jing R, Lin F, Ge WY, Dai HJ and
Pan L: High tidal volume induces mitochondria damage and releases
mitochondrial dna to aggravate the ventilator-induced lung injury.
Front Immunol. 9:14772018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dai H, Zhang S, Du X, Zhang W, Jing R,
Wang X and Pan L: RhoA inhibitor suppresses the production of
microvesicles and rescues high ventilation induced lung injury. Int
Immunopharmacol. 72:74–81. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matthay MA and Zimmerman GA: Acute lung
injury and the acute respiratory distress syndrome: Four decades of
inquiry into pathogenesis and rational management. Am J Respir Cell
Mol Biol. 33:319–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gautier EL, Shay T, Miller J, Greter M,
Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, et al
Immunological Genome Consortium, : Gene-expression profiles and
transcriptional regulatory pathways that underlie the identity and
diversity of mouse tissue macrophages. Nat Immunol. 13:1118–1128.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wynn TA, Chawla A and Pollard JW:
Macrophage biology in development, homeostasis and disease. Nature.
496:445–455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mosser D and Edwards J: Exploring the full
spectrum of macrophage activation. Nat Rev Immunol 8:958-969. Nat
Rev Immunol. 8:958–969. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Safavian D, Leung CH, Kapus A, Ailenberg
M, Szaszi K, Shani R, Di Ciano-Oliveira C, Ghazarian M and Rotstein
O: Hemorrhagic shock/resuscitation reduces the M2 phenotype of
alveolar macrophages: A potential mechanism contributing to
increased LPS-induced lung injury. Shock. 51:213–220. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Genin M, Clement F, Fattaccioli A, Raes M
and Michiels C: M1 and M2 macrophages derived from THP-1 cells
differentially modulate the response of cancer cells to etoposide.
BMC Cancer. 15:5772015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brown BN, Ratner BD, Goodman SB, Amar S
and Badylak SF: Macrophage polarization: An opportunity for
improved outcomes in biomaterials and regenerative medicine.
Biomaterials. 33:3792–3802. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Biswas SK and Mantovani A: Macrophage
plasticity and interaction with lymphocyte subsets: Cancer as a
paradigm. Nat Immunol. 11:889–896. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chistiakov DA, Bobryshev YV, Nikiforov NG,
Elizova NV, Sobenin IA and Orekhov AN: Macrophage phenotypic
plasticity in atherosclerosis: The associated features and the
peculiarities of the expression of inflammatory genes. Int J
Cardiol. 184:436–445. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao Z, Jia X, Megger DA, Chen J, Liu Y, Li
J, Sitek B and Yuan Z: Label-free proteomic analysis of exosomes
secreted from THP-1-derived macrophages treated with IFN-α
identifies antiviral proteins enriched in exosomes. J Proteome Res.
18:855–864. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qiao Y, Peng C, Li J, Wu D and Wang X:
LncRNA MALAT1 is neuroprotective in a rat model of spinal cord
Ischemia-Reperfusion injury through miR-204 regulation. Curr
Neurovasc Res. 15:211–219. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fang Y, Hu J, Wang Z, Zong H, Zhang R and
Sun L: LncRNA H19 functions as an Aquaporin 1 competitive
endogenous RNA to regulate microRNA-874 expression in LPS sepsis.
Biomed Pharmacother. 105:1183–1191. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yamamura S, Imai-Sumida M, Tanaka Y and
Dahiya R: Interaction and cross-talk between non-coding RNAs. Cell
Mol Life Sci. 75:467–484. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo D, Ma J, Yan L, Li T, Li Z, Han X and
Shui S: Down-regulation of Lncrna MALAT1 attenuates neuronal cell
death through suppressing Beclin1-dependent autophagy by regulating
Mir-30a in cerebral ischemic stroke. Cell Physiol Biochem.
43:182–194. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang W, Jia YJ, Yang YL, Xue M, Zheng ZJ,
Wang L and Xue YM: LncRNA GAS5 exacerbates renal tubular epithelial
fibrosis by acting as a competing endogenous RNA of miR-96-5p.
Biomed Pharmacother. 121:1094112020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Xu Y, Zhu YC, Wang YK, Li J, Li
XY, Ji T and Bai SJ: LncRNA NEAT1 promotes extracellular matrix
accumulation and epithelial-to-mesenchymal transition by targeting
miR-27b-3p and ZEB1 in diabetic nephropathy. J Cell Physiol.
234:12926–12933. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou H, Wang X and Zhang B: Depression of
lncRNA NEAT1 antagonizes LPS-evoked acute injury and inflammatory
response in alveolar epithelial cells via HMGB1-RAGE signaling.
Mediators Inflamm. 2020:80194672020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang B, Bao N, He G and Wang J: Long
noncoding RNA HOTAIR regulates autophagy via the miR-20b-5p/ATG7
axis in hepatic ischemia/reperfusion injury. Gene. 686:56–62. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Minshall RD, Schwartz DE and Hu G:
Cyclic stretch induces alveolar epithelial barrier dysfunction via
calpain-mediated degradation of p120-catenin. Am J Physiol Lung
Cell Mol Physiol. 301:L197–L206. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ding H, Wang Y, Dong W, Ren R, Mao Y and
Deng X: Proteomic lung analysis of mice with ventilator-induced
lung injury (VILI) using iTRAQ-based quantitative proteomics. Chem
Pharm Bull (Tokyo). 66:691–700. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cui H, Banerjee S, Guo S, Xie N, Ge J,
Jiang D, Zörnig M, Thannickal VJ and Liu G: Long noncoding RNA
Malat1 regulates differential activation of macrophages and
response to lung injury. JCI Insight. 4:e1245222019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Song X, Li L, Zhao Y and Song Y:
Down-regulation of long non-coding RNA XIST aggravates
sepsis-induced lung injury by regulating miR-16-5p. Hum Cell.
34:1335–1345. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li X, Mo J, Li J and Chen Y: lncRNA CASC2
inhibits lipopolysaccharide induced acute lung injury via miR
27b/TAB2 axis. Mol Med Rep. 22:5181–5190. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang HR, Guo XY, Liu XY and Song X:
Down-regulation of lncRNA CASC9 aggravates sepsis-induced acute
lung injury by regulating miR-195-5p/PDK4 axis. Inflamm Res.
69:559–568. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wei L, Li J, Han Z, Chen Z and Zhang Q:
Silencing of lncRNA MALAT1 prevents inflammatory injury after lung
transplant ischemia-reperfusion by downregulation of IL-8 via p300.
Mol Ther Nucleic Acids. 18:285–297. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Liu M, Li W, Song F, Zhang L and Sun X:
Silencing of lncRNA MIAT alleviates LPS-induced pneumonia via
regulating miR-147a/NKAP/NF-κB axis. Aging (Albany NY).
13:2506–2518. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang M, Cheng K, Chen H, Tu J, Shen Y,
Pang L, Wu W and Yu Z: LncRNA AK020546 protects against cardiac
ischemia-reperfusion injury by sponging miR-350-3p. Aging (Albany
NY). 13:14219–14233. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
You H, Zhang L, Chen Z, Liu W, Wang H and
He H: MiR-20b-5p relieves neuropathic pain by targeting Akt3 in a
chronic constriction injury rat model. Synapse. 73:e221252019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wan Y-M, Li Z-Q, Zhou Q, Liu C, Wang MJ,
Wu HX, Mu YZ, He YF, Zhang Y, Wu XN, et al: Mesenchymal stem cells
alleviate liver injury induced by chronic-binge ethanol feeding in
mice via release of TSG6 and suppression of STAT3 activation. Stem
Cell Res Ther. 11:24. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wu J, Yan X and Jin G: Ulinastatin
protects rats from sepsis-induced acute lung injury by suppressing
the JAK-STAT3 pathway. J Cell Biochem. 120:2554–2559. 2018.
View Article : Google Scholar
|
|
40
|
Chow F, Ozols E, Nikolic-Paterson DJ,
Atkins RC and Tesch GH: Macrophages in mouse type 2 diabetic
nephropathy: Correlation with diabetic state and progressive renal
injury. Kidney Int. 65:116–128. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhou J, Li Z, Wu T, Zhao Q and Cao Y:
LncGBP9/miR-34a axis drives macrophages toward a phenotype
conducive for spinal cord injury repair via STAT1/STAT6 and SOCS3.
J Neuroinflammation. 17:020–01805. 2020. View Article : Google Scholar
|
|
42
|
Lee JW, Chun W, Lee HJ, Min JH, Kim SM,
Seo JY, Ahn KS and Oh SR: The role of macrophages in the
development of acute and chronic inflammatory lung diseases. Cells.
10:8972021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gordon S and Taylor PR: Monocyte and
macrophage heterogeneity. Nat Rev Immunol. 5:953–964. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gordon S: Alternative activation of
macrophages. Nat Rev Immunol. 3:23–35. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ji L, Chen Y, Wang H, Zhang W, He L, Wu J
and Liu Y: Overexpression of Sirt6 promotes M2 macrophage
transformation, alleviating renal injury in diabetic nephropathy.
Int J Oncol. 55:103–115. 2019.PubMed/NCBI
|