Introduction
Osteoarthritis (OA), characterized by enhanced
cartilage degradation and cartilage cell death, is a degenerative
joint disease caused by the progressive erosion of articular
cartilage (1,2). It has been suggested that oxidative
stress, characterized by excessive production of reactive oxygen
species (ROS), serves a significant role in OA pathology (3). ROS can disturb cartilage homeostasis
via promoting chondrocyte apoptosis and matrix catabolism (1). Furthermore, lipopolysaccharide (LPS)
primes the proinflammatory innate immune response via Toll-like
receptor 4 and that progresses to a full-blown inflammatory
response and structural damage of the joint, thus it is considered
as a key pro-inflammatory factor that serves a crucial role in the
pathogenesis of OA (4–7). LPS-induced chondrocytes are used to
establish in vitro chondrocyte injury models, which are
characterized by increased inflammation and apoptosis (8).
FXYD domain containing ion transport regulator 5
(Fxyd5) is a widely expressed single-pass transmembrane protein
that increases the apparent affinity for intracellular
Na+/K+-ATPase and serves several roles in
different cell types (9).
Previous studies demonstrate that Fxyd5 overexpression is
associated with tumor progression, including breast cancer and
renal cell carcinoma (10,11).
In addition, Fxyd5 promotes inflammation in epithelial cells
(12). Increased expression of
Fxyd5 may be associated with chronic inflammatory responses and the
excessive activity of the immune system during human brain aging
(13). Another study shows that
Fxyd5 is upregulated in the epithelium of patients with cystic
fibrosis, accompanied by excessive airway inflammation (14). Fxyd5 overexpression in mice
promotes alveolar epithelial cell injury via activating NF-κB
signaling and promoting cytokine secretion in response to treatment
with LPS (15). However, the
effect of Fxdy5 upregulation in chondrocytes on promoting the
development of OA has not been previously investigated to the best
of the authors' knowledge.
The significant role of NF-κB signaling in the
occurrence and progression of OA has been extensively studied
(16–19). Emerging evidence suggests that the
NF-κB signaling pathway can be activated in response to mechanical
stress, pro-inflammatory factors and extracellular matrix (ECM)
degradation products, thereby affecting cartilage matrix remodeling
and promoting chondrocyte apoptosis and synovial inflammation
(20). Therefore, the current
treatment approach for OA, specifically targeting NF-κB signaling,
is gaining increased attention. The current study aimed to
investigate whether Fxyd5 could promote chondrocyte inflammation
via activating NF-κB signaling. Therefore, to explore the
expression profile of Fxdy5 in OA and its potential regulatory
mechanism, murine ATDC5 chondrocytes were treated with LPS to
establish an in vitro chondrocyte injury model.
Materials and methods
Cell culture and treatment
The murine ATDC5 chondrocytes were obtained from the
American Type Culture Collection and cells were maintained in
DMEM/F12 (MilliporeSigma) supplemented with 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin solution
(Beyotime Institute of Biotechnology) at 37°C in a humidified
CO2 (5%) incubator. To construct the OA cell model in
vitro, ATDC5 cells were incubated with 5 µg/ml LPS (Shanghai
Yuanye Bio. Tech. Co., Ltd.) for 5 h at 37°C (21,22), co-treated or not with 10 nM
betulinic acid (BA; MedChemExpress), an NF-κB activator, for 48 h
at 37°C.
Cell transfection
Short hairpin RNAs (shRNAs) against mouse Fxyd5
(accession no. NM_001111073; shFxyd5-1 and shFxyd5-2) were designed
using the online design software (https://portals.broadinstitute.org/gpp/public/seq/search)
and then cloned into the pLKO.1 vector (MilliporeSigma; cat. no.
SHC001) to silence Fxyd5 expression. A non-targeting shRNA clone
was used as the negative control (shNC). Briefly, ATDC5 cells were
seeded into 6-well plates at a density of 1×106
cells/well and cultured overnight at 37°C. Cells were then
transfected with 50 pmol shRNAs using Lipofectamine®
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C for 24 h
according to the manufacturer's recommendation. Transfection
efficiency was detected via western blot at 48 h after
transfection. The sequences for shRNAs were as follows: shFxyd5-1,
forward
5′-CCGGCGCCTGTGTCTCCTCACTATTCTCGAGAATAGTGAGGAGACACAGGCGTTTTTG-3′,
reverse
5′-AATTCAAAAACGCCTGTGTCTCCTCACTATTCTCGAGAATAGTGAGGAGACACAGGCG-3′;
shFxyd5-2, forward
5′-CCGGGCTGTTCATCACGGGAATTATCTCGAGATAATTCCCGTGATGAACAGCTTTTTG-3′,
reverse
5′-AATTCAAAAAGCTGTTCATCACGGGAATTATCTCGAGATAATTCCCGTGATGAACAGC-3′;
shNC, forward
5′-CACCGCAATTTTTTTTTTTGATTCACGAATGAATCAAAAAAAAAAAAATGC-3′, reverse
5′-AAAAGCAATTTTTTTTTTTGATTCATTCGTGAATCAAAAAAAAAAAAATGC-3′.
Cell Counting Kit-8 (CCK-8) assay
Following cell treatment or transfection, a total of
2×104 ATDC5 cells/well were seeded into a 96-well and
were then incubated with CCK-8 regent for 2 h at 37°C. The
absorbance at a wavelength of 450 nm was measured in each well
using a microplate reader (Multiskan SkyHigh Microplate
Spectrophotometer; Thermo Fisher Scientific, Inc.). Each sample was
assessed five times.
Western blot analysis
A total of 1×106 ATDC5 cells were seeded
into 6-well plates, washed with PBS and lysed using RIPA Cell lysis
buffer (Beyotime Institute of Biotechnology). Following
centrifugation at 12,000 × g for 20 min at 4°C, the supernatant was
collected to perform western blot analysis. Briefly, total protein
concentration was quantified using a BCA kit (Invitrogen; Thermo
Fisher Scientific, Inc.), then equal amount of protein (30 µg) was
separated through the 10% SDS-PAGE, followed by transfer onto PVDF
membranes. After blocking with 5% non-fat milk for 1 h at room
temperature, the PVDF membranes were incubated with primary
antibodies overnight at 4°C. The primary antibodies were: Rabbit
anti-Fxyd5 (1:1,000; cat. no. ABIN2780210), rabbit anti-aggrecan
(1:500; cat. no. ABIN6996681; both from antibodies-online), rabbit
anti-GAPDH (1:10,000; cat. no. ab181602), rabbit anti-Bcl2
(1:2,000; cat. no. ab182858), rabbit anti-Bax (1:8,000; cat. no.
ab32503), rabbit anti-cleaved caspase 3 (1:5,000; cat. no.
ab214430), rabbit anti-caspase 3 (1:2,000; cat. no. ab184787),
rabbit anti-MMP3 (1:20,000; cat. no. ab52915; all from Abcam),
rabbit anti-MMP13 (1:2,000; cat. no. NBP2-66954), rabbit
anti-phosphorylated (p)-IκBα (1:800; cat. no. NB100-81987), rabbit
anti-IκBα (1:5,000; cat. no. NBP2-67369), rabbit anti-collagen II
(1:10,000; cat. no. NBP1-77795; all from Novus Biologicals, Ltd.),
rabbit anti-p-p65 (1:1,000; cat. no. 3033S) and rabbit anti-p65
(1:1,000; cat. no. 8242S; Cell Signaling Technology, Inc.).
Subsequently, membranes were treated with the goat anti-rabbit IgG
(HRP) secondary antibody (1:10,000; cat. no. ab6721; Abcam) at room
temperature for 1 h. Then, enhanced chemiluminescence
(MilliporeSigma) was used to detect the protein expression levels.
Band semi-quantification was performed using Image J software
(version 1.54; National Institutes of Health).
Determination of intracellular ROS,
superoxide dismutase (SOD) and malondialdehyde (MDA)
Following cell treatment or transfection, ATDC5
cells were seeded into 6-well plates at a density of
1×106 cells/well and the levels of intracellular ROS,
SOD and lipid peroxidation product MDA were determined using ROS
Assay kit (cat. no. S0033M; Beyotime Institute of Biotechnology),
SOD Assay Kit (cat. no. 7500-100-K; R&D systems, Inc.) and
TBARS Parameter Assay Kit (cat. no. KGE013; R&D systems, Inc.),
respectively, according to the manufacturer's recommendations.
ELISA
Control or transfected ATDC5 cells were seeded into
96-well plates at a density of 2×104 cells/well,
following indicated treatment, the secretion levels of IL-1β, IL-6
and IL-18 in the culture supernatants were measured using the Mouse
IL-1β (cat. no. ab197742), IL-6 (cat. no. ab222503) and IL-18 (cat.
no. ab216165) SimpleStep ELISA kits (Abcam) according to the
manufacturer's instructions.
Statistical analysis
All statistical analyses were performed using
GraphPad Prism 8.0.1 (GraphPad Software, Inc.). Data were expressed
as the mean ± standard deviation from three independent
experiments. One-way ANOVA followed by Tukey's multiple comparison
test was applied to compare the differences among multiple groups,
while those between two groups were compared using an unpaired
two-tailed Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Fxyd5 knockdown enhances cell
viability and inhibits apoptosis and ECM degradation in LPS-induced
ATDC5 cells
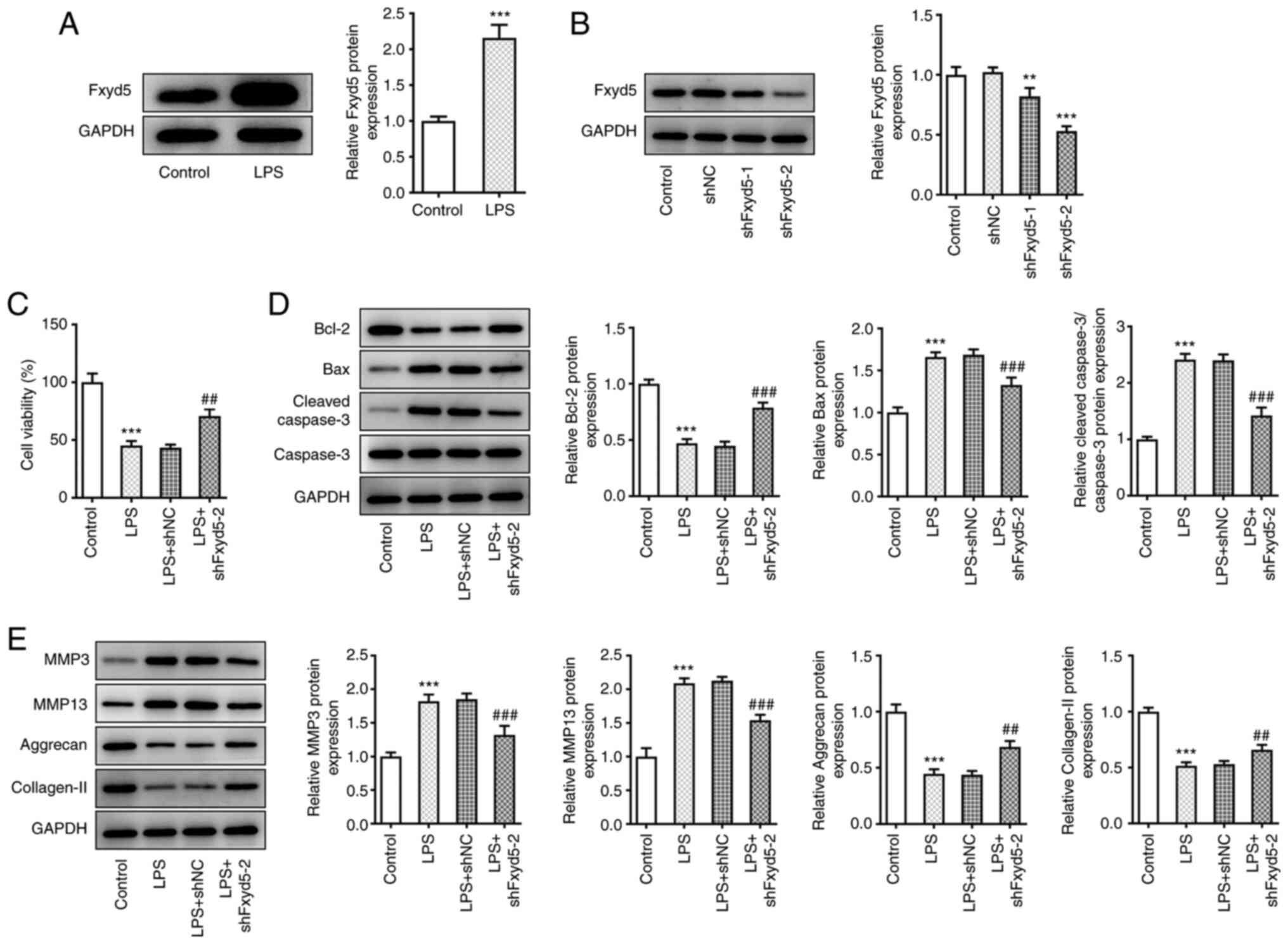
To explore the expression profile of Fxyd5 in the
LPS-induced chondrocyte injury model (21), total proteins were extracted from
ATDC5 cells treated with 5 µg/ml LPS for 5 h followed by western
blot analysis. The results showed that the expression levels of
Fxyd5 were significantly increased in LPS-treated cells compared
with untreated ones (Fig. 1A).
Furthermore, to investigate the cell-specific effect of Fxyd5,
ATDC5 cells were transfected with shRNA clones targeting Fxyd5
(shFxyd5-1 and shFxyd5-2) to knockdown its expression. The results
showed that shFxyd5-2 clone exhibited a more potent effect on
silencing Fxyd5 expression compared with shFxyd5-1, since the
protein expression levels of Fxyd5 in ATDC5 cells transfected with
shFxyd5-2 were decreased to ~50% compared with cells transfected
with shNC (Fig. 1B). Therefore,
the shFxyd5-2 clone was used in subsequent experiments.
Additionally, cell viability was assessed by CCK-8 assays.
Following cell treatment with LPS, cell viability was notably
higher in Fxyd5-depleted ATDC5 cells compared with those
transfected with shNC (Fig. 1C).
Western blot analysis also revealed that the protein expression
levels of the apoptosis-related markers (23) Bax and cleaved caspase 3/caspase 3
were markedly reduced, while those of Bcl-2 were increased in
Fxyd5-depleted LPS-induced ATDC5 cells compared with cells
transfected with shNC (Fig. 1D).
Subsequently, the role of Fxyd5 in chondrocyte ECM homeostasis was
also evaluated by western blot analysis. The results demonstrated
that cell treatment with LPS inhibited ECM-factors production, and
promoted ECM degradation, as verified by Aggrecan and collagen II
downregulation accompanied by MMP3 and MMP13 upregulation when
compared with control cells (Fig.
1E). Fxyd5 silencing reversed these effect caused by LPS
(Fig. 1E). Taken together, the
aforementioned findings indicated that Fxyd5 knockdown could
protect ATDC5 cells against LPS-mediated cell injury via enhancing
cell viability and inhibiting cell apoptosis and ECM
degradation.
Fxyd5 silencing attenuates
LPS-mediated inflammation and oxidative stress in ATDC5 cells
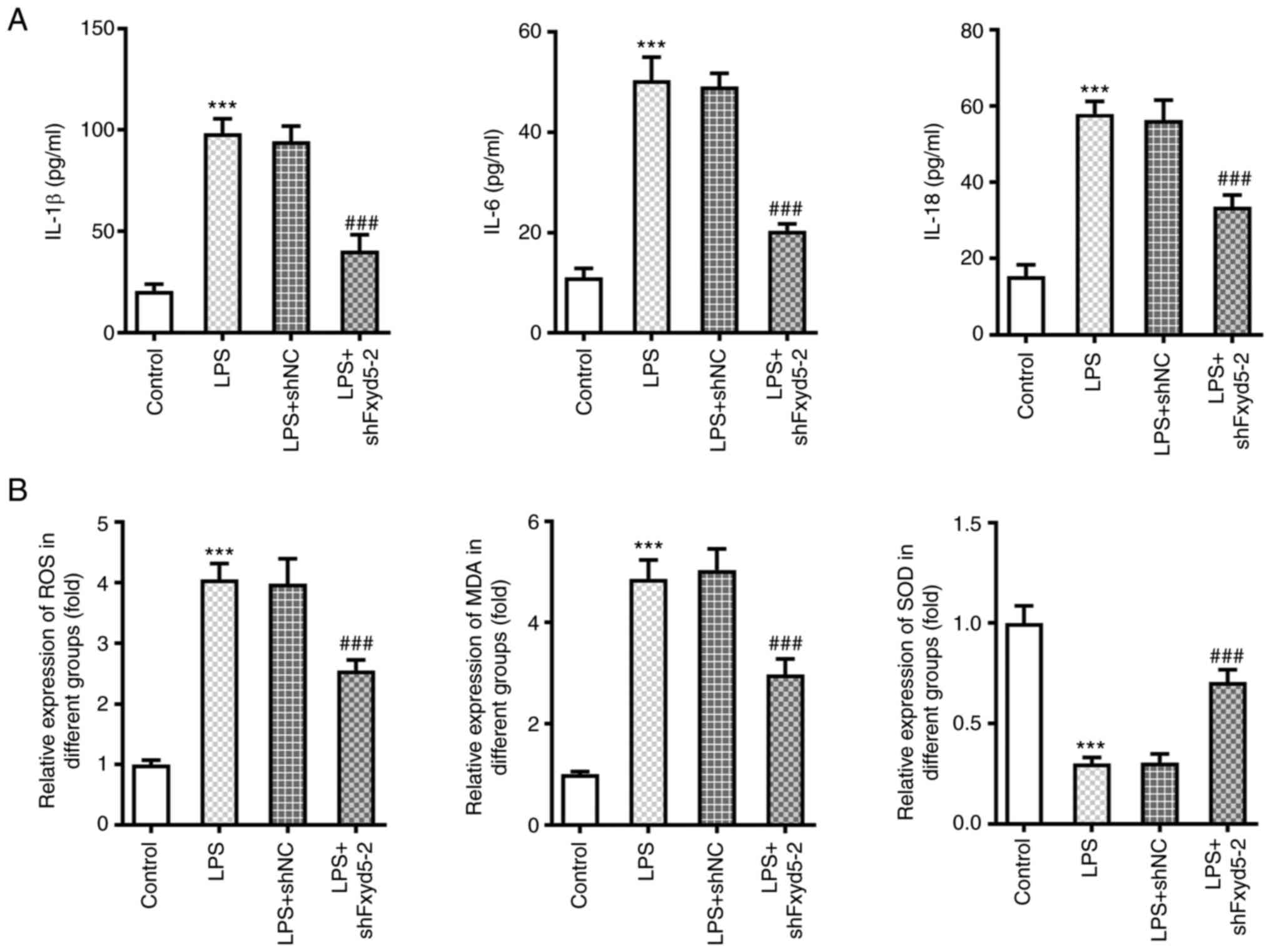
To further explore the effect of Fxyd5 on
LPS-induced chondrocyte inflammation and oxidative stress, ELISA
was carried out to determine the secretion levels of
pro-inflammatory cytokines and the content of ROS, MDA and SOD in
LPS-induced ATDC5 cells. The results revealed that Fxyd5 was
involved in LPS-induced cellular inflammation. Therefore, the
secretion levels of the inflammatory factors IL-1β, IL-6 and IL-18
were significantly decreased in Fxyd5-depleted ATDC5 cells treated
or not with LPS compared with the shNC group (Fig. 2A). Similar results were observed
in the levels of ROS, MDA and SOD, suggesting that the increased
expression of Fxyd5 could be involved in LPS-induced oxidative
stress in ATDC5 cells. Therefore, the levels of ROS and MDA were
significantly lower and those of SOD were notably higher in
Fxyd5-depleted ATDC5 cells compared with the shNC group (Fig. 2B). These results suggested that
Fxyd5 knockdown could alleviate LPS-induced cellular inflammatory
responses and oxidative stress in ATDC5 cells.
Fxyd5 activates NF-κB signaling in
LPS-induced ATDC5 cells
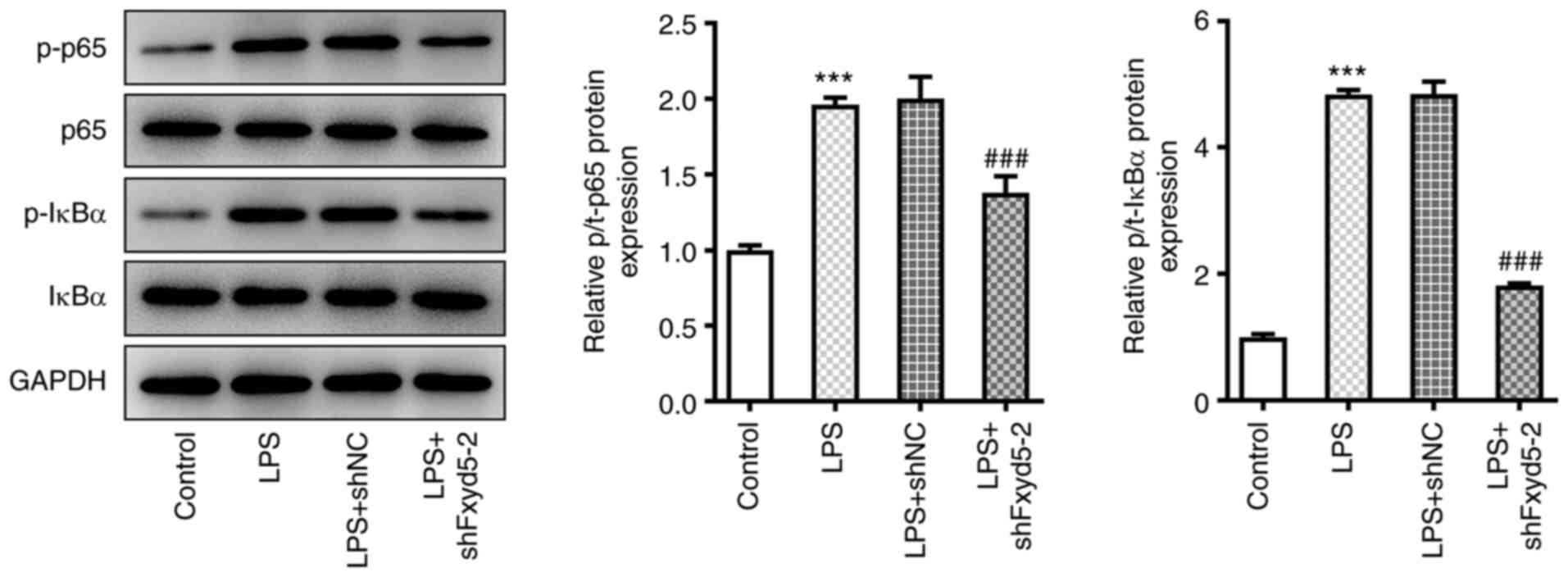
To investigate whether Fxyd5 was involved in the
regulation of NF-κB pathway in the LPS-induced chondrocyte injury
model (8), western blot analysis
was performed. The results showed that Fxyd5 knockdown in ATDC5
cells restored the increased LPS-mediated p65 and IκBα
phosphorylation (Fig. 3).
Therefore, the increased expression of Fxyd5 could be involved in
LPS-induced chondrocyte injury via activating the NF-κB signaling
pathway.
Fxyd5 promotes LPS-induced chondrocyte
injury via activating the NF-κB signaling pathway
Since Fxyd5 knockdown in ADTC5 cells could inhibit
the activation of the NF-κB pathway (Fig. 3), the present study aimed to
verify that Fxyd5 could promote LPS-mediated chondrocyte injury via
activating NF-κB signaling. Therefore, LPS-stimulated
Fxyd5-depleted ATDC5 cells were treated with 10 nM BA, a NF-κB
activator. Western blot analysis showed that the phosphorylation
levels of p65 and IκBα were significantly increased in BA-treated
Fxyd5-depleted ATDC5 cells compared with BA untreated
Fxyd5-depleted ATDC5 cells (Fig.
4A). Additionally, treatment with BA attenuated cell viability
and enhanced cell apoptosis and ECM degradation in LPS-induced
Fxyd5-depleted ATDC5 cells compared with untreated cells (Fig. 4B-D). To this end, the present
study investigated whether Fxyd5 could be also involved in
LPS-induced inflammatory responses and oxidative stress in ATDC5
cells via activating the NF-κB signaling pathway. The results
demonstrated that cell treatment with BA restored the levels of
IL-1β, IL-6, IL-18, ROS and MDA, but not those of SOD, compared
with untreated cells (Fig. 5).
The above findings indicated that Fxyd5 upregulation could promote
LPS-mediated chondrocyte injury via activating NF-κB signaling.
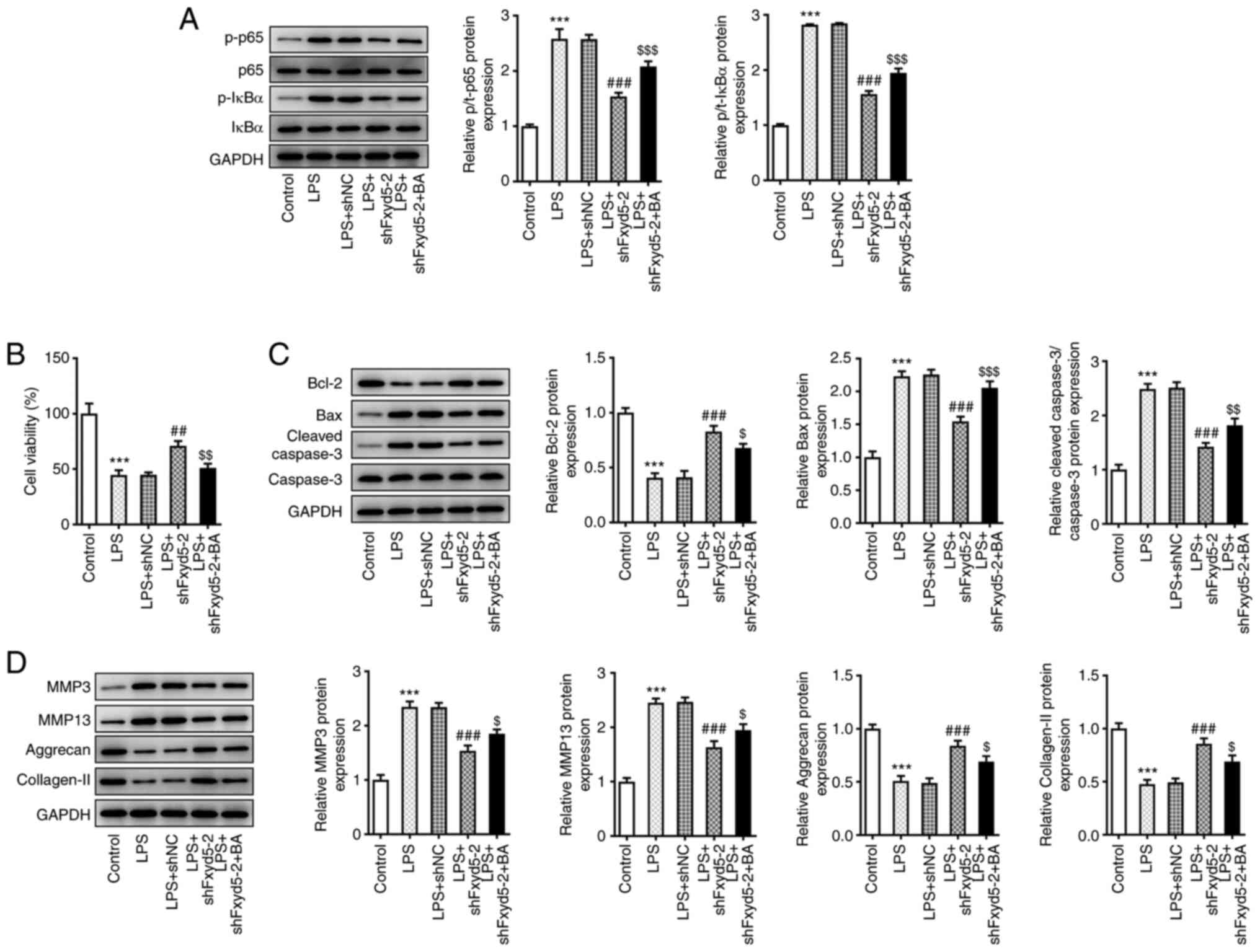 | Figure 4.Fxyd5 regulates cell viability,
apoptosis and ECM degradation via activating the NF-κB signaling
pathway in ATDC5 cells. Following cell treatment with 10 nM BA, a
NF-κB activator, for an additional 48 h (A) the phosphorylation
levels of p65 and IκBα were increased, as demonstrated by western
blot analysis. (B) Cell Counting Kit 8 assay showed that cell
viability was increased in Fxyd5-depleted ATDC5 cells co-treated
with LPS and BA compared with cells treated only with LPS. (C and
D) Western blotting was used to evaluate the expression of proteins
involved in cell apoptosis and ECM degradation. Untreated and
untransfected ATDC5 cells served as the control group. GAPDH served
as the loading control. ***P<0.001 vs. the control group;
##P<0.01 and ###P<0.001 vs. the LPS +
shNC group; $P<0.05, $$P<0.01 and
$$$P<0.001 vs. the LPS + shFxyd5-2 + BA group. Fxyd5,
FXYD domain containing ion transport regulator 5; ECM,
extracellular matrix; BA, betulinic acid; LPS, lipopolysaccharide;
shRNA, short hairpin RNA; NC, negative control. |
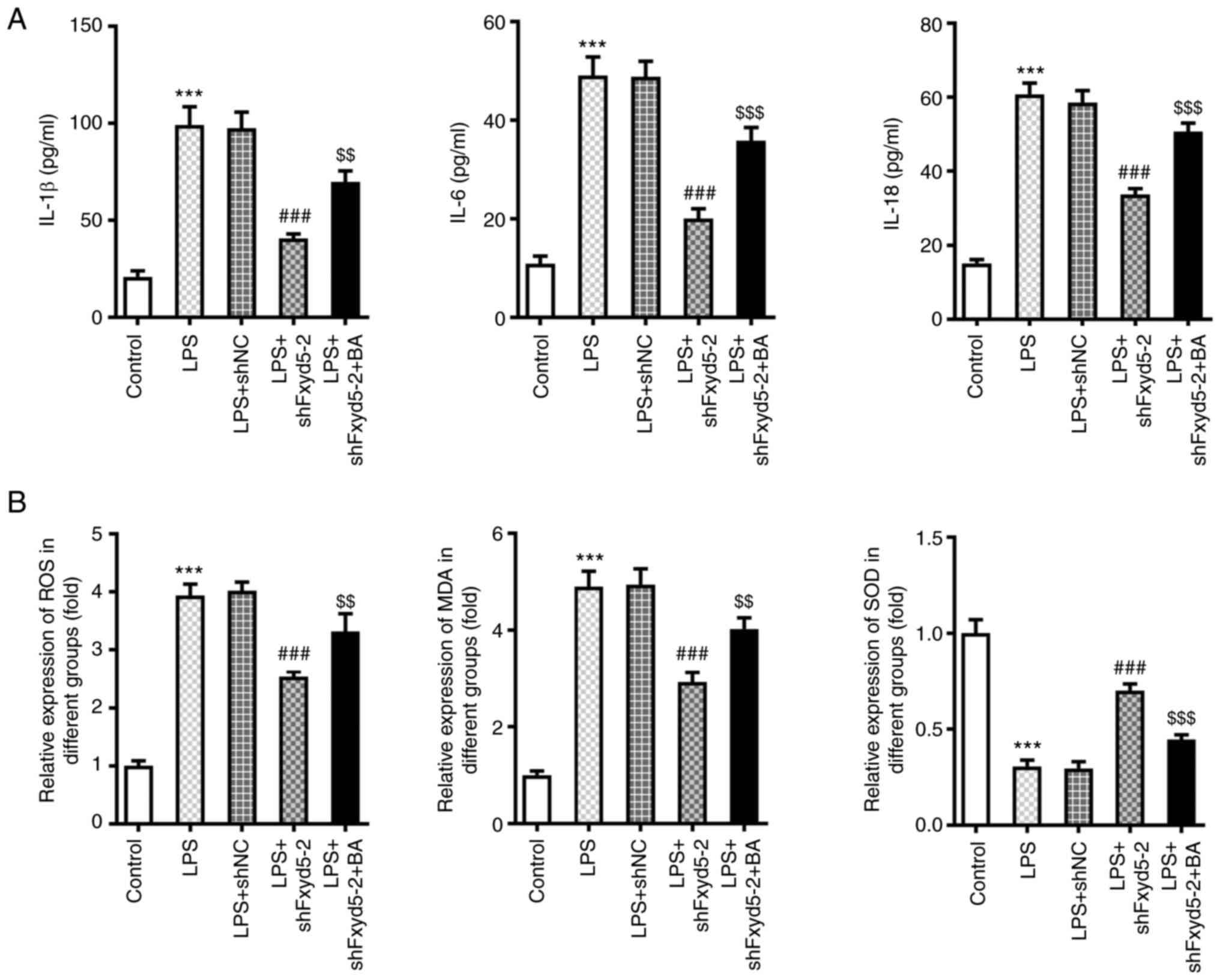 | Figure 5.Fxyd5 regulates cellular inflammation
and oxidative stress in ATDC5 cells via activating NF-κB signaling.
Following cell treatment with 10 nM BA for additional 48 h, the
secretion levels of the (A) pro-inflammatory factors IL-1β, IL-6
and IL-18 were increased in ELISA. (B) The content of ROS and MDA
was increased and that of SOD was decreased in Fxyd5-depleted ATDC5
cells co-treated with LPS and BA compared with cells treated only
with LPS. Untreated and untransfected ATDC5 cells served as the
control group. GAPDH served as the loading control. ***P<0.001
vs. the control group; ###P<0.001 vs. the LPS + shNC
group; $$P<0.01 and $$$P<0.001 vs. the
LPS + shFxyd5-2 + BA group. Fxyd5, FXYD domain containing ion
transport regulator 5; BA, betulinic acid; LPS, lipopolysaccharide;
shRNA, short hairpin RNA; NC, negative control; ROS, reactive
oxygen species MDA, malondialdehyde; SOD, superoxide dismutase. |
Discussion
Articular cartilage is a non-self-renewing avascular
tissue that lacks inherent repair capability (1). Although, cartilage-derived
stem/progenitor cells are involved in the maintenance of tissue
homeostasis (2), the gradual loss
of chondrocytes still leads to cartilage homeostasis disorders and
eventually to OA (24). It has
been reported that non-medicinal and non-surgical approaches such
as exercise mode and light equipment-assisted training can prevent
and delay the deterioration of early OA to a certain extent
(25). However, for the treatment
of severe OA, symptomatic treatment and joint replacement surgery
should be performed (24).
Nevertheless, the quality of life remains poor over time.
LPS is used to establish chondrocyte injury models
by inducing cell apoptosis, oxidative stress and inflammation
(21). In the present study,
ATDC5 cell treatment with LPS increased matrix catabolism via
upregulating the expression of matrix degradation-related enzymes,
including MMP3 and MMP13, and reducing the content of collagen and
proteoglycans. Using fast 3D confocal cartilage imaging technology
combined with standard histological examination, Zhang et al
(26) demonstrate that cartilage
cell apoptosis by itself cannot cause articular cartilage injury,
which could be mainly triggered by cartilage cell catabolism.
The current study aimed to investigate the effect of
transport regulator Fxyd5 on LPS-induced ATDC5 cell injury. Unlike
other members of the FXYD family, Fxyd5 exhibits several biological
functions in different cell types (12). A previous study revealed that
during lung injury, Fxyd5 upregulation is associated with the
excessive secretion of cytokines and chemokines by alveolar
epithelial cells in response to IFN-α and TNF-α via activating
NF-κB signaling (15). Consistent
with the aforementioned results, the present study showed that
Fxyd5 was upregulated in LPS-induced ATDC5 cells. The increased
expression of Fxyd5 further responded to LPS stimulation via
promoting the secretion of the inflammatory cytokines IL-1β, IL-6
and IL-18. However, the detection of the mRNA level of these
pro-inflammatory cytokines could further support the above
findings.
The current study also demonstrated that Fxyd5
silencing could relieve LPS-induced oxidative stress in ATDC5 cells
as evidenced by reduced ROS and MDA, while increased SOD levels
upon Fxyd5 silencing. Excessive production of ROS contributes to
the occurrence of oxidative stress; MDA is a production of lipid
peroxidation and SOD belongs to a member of the enzyme antioxidant
system (27). Although the
combination of the changes in ROS, SOD and MDA levels can
illustrate the changes in oxidative stress (28,29), detection of other molecules
involved in oxidative stress such as catalase, glutathione
peroxidase, 8-hydroxyldeoxyguanosine and nicotinamide adenine
dinucleotide phosphate, may further support the findings of the
present study. Emerging evidence suggests that ROS-mediated
oxidative stress in chondrocytes can affect intracellular signal
transduction, chondrocyte life cycle and cartilage matrix
metabolism, which may lead to synovial inflammation and subchondral
bone dysfunction (1,16). In addition to ECM degradation and
cellular inflammation, the increased expression of Fxyd5 could also
promote ATDC5 cell apoptosis, possibly via a series of complex and
comprehensive reactions. Cellular inflammation can undoubtedly
induce cell death, while chondrocyte-specific ECM proteins can also
regulate cell proliferation and survival via transmitting cell
signals by combining with integrins (30).
It has been reported that LPS binds to and alters
the configuration of its associated receptors, promotes IκBα
phosphorylation and activates the classic NF-κB pathway (31). In addition, the post-translational
modification of p65 can also regulate the activity of NF-kB
signaling (31), which is a
classic pathway associated with the occurrence and development of
OA (20). In the present study,
the phosphorylation levels of p65 and IκBα were enhanced in
LPS-induced Fxyd5-depleted ATDC5 cells. Combined with the results
obtained by the co-treatment of ATDC5 cells with the NF-kB
activator, BA, the elevated expression of Fxyd5 promoted
chondrocyte injury partially via activating the NF-κB pathway.
However, other signaling pathways such as Wnt/β-catenin and
MAPK/ERK signaling pathways, which have been reported to regulate
OA (32,33), may also be involved in the above
process, thus future studies should be performed to investigate.
Additionally, further studies should focus on the effect of Fxyd5
on chondrocyte differentiation, as chondrocytes show
differentiation plasticity during the healing process (34). Moreover, in vivo studies
using IL-1β-induced cell model and animal OA model need to be
performed to validate the in vitro findings of the present
study.
The results of the current study revealed that Fxyd5
was significantly upregulated in the LPS-induced chondrocyte injury
model. Furthermore, the results demonstrated that the increased
expression levels of Fxyd5 could promote inflammation, oxidative
stress and ECM degradation in ATDC cells via activating the NF-κB
signaling pathway. Overall, these findings could provide novel
insights to increase our understanding in the role of NF-κB
signaling in the pathogenesis of OA.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LS guided the project and wrote the manuscript. LS
and XL conceived the technical details and designed the
experiments. LS, XL and YZ performed the experiments. QS analyzed
the data. All authors read and approved the final manuscript. LS
and XL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hosseinzadeh A, Kamrava SK, Joghataei MT,
Darabi R, Shakeri-Zadeh A, Shahriari M, Reiter RJ, Ghaznavi H and
Mehrzadi S: Apoptosis signaling pathways in osteoarthritis and
possible protective role of melatonin. J Pineal Res. 61:411–425.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiang Y and Tuan RS: Origin and function
of cartilage stem/progenitor cells in osteoarthritis. Nat Rev
Rheumatol. 11:206–212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blanco FJ, Rego I and Ruiz-Romero C: The
role of mitochondria in osteoarthritis. Nat Rev Rheumatol.
7:161–169. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang ZY, Stabler T, Pei FX and Kraus VB:
Both systemic and local lipopolysaccharide (LPS) burden are
associated with knee OA severity and inflammation. Osteoarthritis
Cartilage. 24:1769–1775. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhao C, Wang Y, Jin H and Yu T: Knockdown
of microRNA-203 alleviates LPS-induced injury by targeting MCL-1 in
C28/I2 chondrocytes. Exp Cell Res. 359:171–178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang Z and Kraus VB: Does
lipopolysaccharide-mediated inflammation have a role in OA? Nat Rev
Rheumatol. 12:123–129. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen G, Liu T, Yu B, Wang B and Peng Q:
CircRNA-UBE2G1 regulates LPS-induced osteoarthritis through
miR-373/HIF-1a axis. Cell Cycle. 19:1696–1705. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ding Y, Wang L, Zhao Q, Wu Z and Kong L:
MicroRNA-93 inhibits chondrocyte apoptosis and inflammation in
osteoarthritis by targeting the TLR4/NF-κB signaling pathway. Int J
Mol Med. 43:779–790. 2019.PubMed/NCBI
|
|
9
|
Geering K: Function of FXYD proteins,
regulators of Na, K-ATPase. J Bioenerg Biomembr. 37:387–392. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nam JS, Kang MJ, Suchar AM, Shimamura T,
Kohn EA, Michalowska AM, Jordan VC, Hirohashi S and Wakefield LM:
Chemokine (C-C motif) ligand 2 mediates the prometastatic effect of
dysadherin in human breast cancer cells. Cancer Res. 66:7176–7184.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schüler Y, Lee-Thedieck C, Geiger K,
Kaiser T, Ino Y, Aicher WK and Klein G: Osteoblast-secreted factors
enhance the expression of dysadherin and CCL2-dependent migration
of renal carcinoma cells. Int J Cancer. 130:288–299. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lubarski-Gotliv I, Asher C, Dada LA and
Garty H: FXYD5 Protein Has a Pro-inflammatory role in epithelial
cells. J Biol Chem. 291:11072–11082. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nikas JB: Inflammation and immune system
activation in aging: A mathematical approach. Sci Rep. 3:32542013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Miller TJ and Davis PB: FXYD5 modulates
Na+ absorption and is increased in cystic fibrosis airway
epithelia. Am J Physiol Lung Cell Mol Physiol. 294:L654–L664. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brazee PL, Soni PN, Tokhtaeva E, Magnani
N, Yemelyanov A, Perlman HR, Ridge KM, Sznajder JI, Vagin O and
Dada LA: FXYD5 is an essential mediator of the inflammatory
response during lung injury. Front Immunol. 8:6232017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lepetsos P, Papavassiliou KA and
Papavassiliou AG: Redox and NF-κB signaling in osteoarthritis. Free
Radic Biol Med. 132:90–100. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiang RH, Xu JJ, Zhu DC, Li JF, Zhang CX,
Lin N and Gao WY: Glycyrrhizin inhibits osteoarthritis development
through suppressing the PI3K/AKT/NF-κB signaling pathway in vivo
and in vitro. Food Funct. 11:2126–2136. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi MC, Jo J, Park J, Kang HK and Park Y:
NF-κB signaling pathways in osteoarthritic cartilage destruction.
Cells. 8:7342019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Saito T and Tanaka S: Molecular mechanisms
underlying osteoarthritis development: Notch and NF-kappaB.
Arthritis Res. 19:942017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rigoglou S and Papavassiliou AG: The NF-κB
signalling pathway in osteoarthritis. Int J Biochem Cell Biol.
45:2580–2584. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mei X, Tong J, Zhu W and Zhu Y:
lncRNA-NR024118 overexpression reverses LPS-induced inflammatory
injury and apoptosis via NF-κB/Nrf2 signaling in ATDC5
chondrocytes. Mol Med Rep. 20:3867–3873. 2019.PubMed/NCBI
|
|
22
|
Chang Q, Ji M, Li C and Geng R:
Downregulation of miR-486-5p alleviates LPS-induced inflammatory
injury, oxidative stress and apoptosis in Chondrogenic cell ATDC5
by targeting NRF1. Mol Med Rep. 22:2123–2131. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheng EH, Kirsch DG, Clem RJ, Ravi R,
Kastan MB, Bedi A, Ueno K and Hardwick JM: Conversion of Bcl-2 to a
Bax-like death effector by caspases. Science. 278:1966–1968. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hashimoto M, Nakasa T, Hikata T and
Asahara H: Molecular network of cartilage homeostasis and
osteoarthritis. Med Res Rev. 28:464–481. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bierma-Zeinstra S, van Middelkoop M,
Runhaar J and Schiphof D: Nonpharmacological and nonsurgical
approaches in OA. Best Pract Res Clin Rheumatol. 34:1015642020.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang M, Mani SB, He Y, Hall AM, Xu L, Li
Y, Zurakowski D, Jay GD and Warman ML: Induced superficial
chondrocyte death reduces catabolic cartilage damage in murine
posttraumatic osteoarthritis. J Clin Invest. 126:2893–2902. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sies H: Oxidative stress: A concept in
redox biology and medicine. Redox Biol. 4:180–183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ansari MY, Ahmad N and Haqqi TM: Oxidative
stress and inflammation in osteoarthritis pathogenesis: Role of
polyphenols. Biomed Pharmacother. 129:1104522020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Feng K, Chen Z, Pengcheng L, Zhang S and
Wang X: Quercetin attenuates oxidative stress-induced apoptosis via
SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and
prevents the progression of osteoarthritis in a rat model. J Cell
Physiol. 234:18192–18205. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Loeser RF: Integrins and
chondrocyte-matrix interactions in articular cartilage. Matrix
Biol. 39:11–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lawrence T: The nuclear factor NF-kappaB
pathway in inflammation. Cold Spring Harb Perspect Biol.
1:a0016512009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou Y, Wang T, Hamilton JL and Chen D:
Wnt/β-catenin signaling in osteoarthritis and in other forms of
arthritis. Curr Rheumatol Rep. 19:532017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
He Z, Li H, Han X, Zhou F, Du J, Yang Y,
Xu Q, Zhang S, Zhang S, Zhao N, et al: Irisin inhibits osteocyte
apoptosis by activating the Erk signaling pathway in vitro and
attenuates ALCT-induced osteoarthritis in mice. Bone.
141:1155732020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Varela-Eirin M, Loureiro J, Fonseca E,
Corrochano S, Caeiro JR, Collado M and Mayan MD: Cartilage
regeneration and ageing: Targeting cellular plasticity in
osteoarthritis. Ageing Res Rev. 42:56–71. 2018. View Article : Google Scholar : PubMed/NCBI
|