Introduction
Diabetes, a widespread metabolic disorder, primarily
manifests as chronic hyperglycemia and is caused by insulin
dysfunction or impaired insulin secretion, or a combination of both
(1). Patients with diabetes are
further predisposed to a greater risk of acquiring ischemic heart
disease and myocardial ischemia-reperfusion injury (MI/RI)
(2). In lieu of the increasing
incidence rate of diabetes complicated with myocardial ischemia, it
is important to explore new avenues to relieve MI/RI in diabetic
patients (3,4). Currently, myocardial ischemia caused
by the blockage of heart blood vessels is treated via reperfusion,
restoring blood flow, however, the process of reperfusion is known
to lead to ischemic myocardial injury (2). MI/RI contributes to the morbidity and
mortality of a large range of pathologies, including ischemic
stroke, myocardial infarction, acute kidney damage and trauma
(5). Therefore, the present study
aimed to investigate the cellular mechanisms between diabetic MI/RI
and cardioprotective mechanisms.
Propofol, a widely-used intravenous anesthetic, is
known to interact with numerous pathophysiological processes in
different types of cancer (6).
Furthermore, propofol can reduce myocardial cell apoptosis and thus
exhibits a cardio-protective effect (7). Similarly, propofol is effective in
alleviating hemodynamic instability in diabetic patients treated
with glibenclamide (8). As a result
of its anti-oxidant properties, propofol protects against MI/RI
(9). It has also been shown that
propofol may protect against MI/RI by reducing oxidative stress and
inhibiting cardiomyocyte apoptosis (10). Nevertheless, the role of propofol in
diabetic MI/RI remains unclear.
Propofol has previously been reported to protect
against hepatic I/R via upregulation of microRNAs (miRNAs)
(11). Inherently, miRNAs are small
endogenous RNAs, which possess the ability to
post-transcriptionally regulate gene expression and are regarded as
crucial promising biomarkers for disease development (12). miRNAs have also been associated with
diabetic MI/RI (2). For example,
propofol is suggested to serve as a therapeutic target for MI/RI
via interactions with miRNA (13).
One of the most important regulatory factors in diabetic MI/RI is
the interacting miRNA family. For example, miR-144, miR-34a,
miR-200 and miR-155 are identified as being pro-apoptotic, among
which miR-200c may be related to MI/RI in diabetes (14). Moreover, inhibition of miR-200c is
shown to diminish reactive oxygen species (ROS) production, lipid
peroxidation and malondialdehyde (MDA) peroxidation and upregulate
anti-oxidant enzyme superoxide dismutase (SOD) activity in
cardiomyocytes (15). However, at
present there is no evidence on whether propofol postconditioning
can attenuate diabetic MI/RI through miR-200c-3p. Therefore, the
present study aimed to investigate the protective mechanism of
propofol postconditioning on diabetic MI/RI, hoping to provide a
new theoretical basis for propofol postconditioning to attenuate
diabetic MI/RI.
Materials and methods
Ethics statement
The present study was approved by the Academic
Ethics Committee of The Affiliated People's Hospital of Ningbo
University (approval number: 2019-010; Ningbo, China). Animal
experimentation was carried out in strict accordance with the
Guidelines for the Management and Use of Laboratory Animals issued
by the Laboratory Association of China. Extensive efforts were made
to minimize the suffering of the animals.
Diabetic MI/RI model establishment and
grouping
A total of 54 healthy male Sprague-Dawley (SD) rats
(age, 8–10 weeks; weight, 250–300 g) were purchased from Beijing
Vital River Laboratory Animal Technology Co., Ltd. The rats were
housed at 22–24°C with 12-h light/dark cycles, at 50–60% humidity
and had free access to food and water.
The obtained SD rats were randomly assigned into a
high-glucose (HG) group (fed with high fat and HG feed; n=54).
After four weeks all rats were fasted for 12 h and the rats were
intraperitoneally injected with 65 mg/kg streptozotocin
(Sigma-Aldrich; Merck KGaA) (16).
Blood samples were collected from the tail vein after 72 h to
assess fasting blood glucose levels. Diabetic rat models were
regarded as successfully established when blood glucose levels were
≥16.7 mmol/l and the animals presented with diabetic symptoms,
including excessive drinking, eating, polyuria and weight loss.
Subsequently, the diabetic rats were fed with high-fat and HG feed
and had their blood glucose monitored for 4 weeks. The rats with
unqualified blood glucose were excluded and the qualified ones were
used for I/R experiments.
Diabetic rats were intraperitoneally anesthetized
with 2% pentobarbital sodium (50 mg/kg; Sigma-Aldrich; Merck KGaA).
Following tracheal intubation, mechanical ventilation was performed
with a ventilator. The ventilator was set to the following
conditions: tidal volume, 6 ml/kg; respiratory ratio, 1:1.5; and
respiratory rate, 80 times/min. An 8-channel physiological signal
recorder (PowerLab/8s; ADInstruments Pty Ltd.) was used to
continuously monitor a 3-lead electrocardiogram (ECG). The femoral
vein was then punctured, catheterized and connected with a
micro-perfusion pump for infusion of propofol or normal saline. The
left anterior descending coronary artery was isolated through
thoracotomy. A 6/0 thread was threaded under the artery and a rigid
polyethylene tube with a diameter of 2 mm and a length of 0.5 cm
was placed between the myocardium and the ligation thread. When the
ligation thread was tightened the color of the myocardium at the
ligation site became dark and the ECG displayed significant
elevation of the ST segment, indicating successful ischemia. After
30 min of ischemia the ligation thread was released, the color of
heart turned back to red, the ST elevation dropped, and reperfusion
was performed for 120 min to establish the MI/RI rat model
(17).
Diabetic rats were randomly assigned into the
following: the sham group (DS group), the MI/RI group (DI group)
and the MI/RI + propofol group (DP group), with 18 rats in each
group, and with 6 rats used for TTC staining, 6 rats for
homogenization treatment, and 6 rats for section staining. Only
chest opening and threading were performed in DS rats, equal
volumes of normal saline were infused in DI rats and other
operations were the same as those in the DP group. Propofol was
infused at constant velocity into the femoral vein 3 min prior to
perfusion in the DP group for 5 min (propofol was infused 3 min
before the start of perfusion until 2 min after the start of
perfusion) (20 mg/kg/h) (18).
Following I/R treatment, rats were euthanized by intraperitoneal
injection of 2% pentobarbital sodium (200 mg/kg).
2,3,5-triphenyl-2H-tetrazolium
chloride (TTC) staining
The hearts of rats in each group were removed and
sliced into 2 mm coronal sections following animal sacrifice.
Sections were stained with 1% TTC solution (Sigma-Aldrich; Merck
KGaA) at 37°C and incubated in the dark for 30 min. After staining,
the sections were fixed with 10% formalin at 4°C for 24 h. Healthy
myocardial tissues were stained brick-red and the infarcted
myocardial tissues were white. A digital camera was used to capture
images. Image J software (version 1.8.0; National Institutes of
Health) was used to calculate myocardial infarction size.
Hematoxylin and eosin (HE)
staining
Ischemic myocardial tissues from each group were
collected and fixed using 4% paraformaldehyde at 4°C for 24 h,
embedded in paraffin, and sliced into 4 µm sections. Sections were
then dewaxed with xylene, washed twice with anhydrous ethanol,
dehydrated using an ethanol gradient, stained with hematoxylin for
5 min and washed with double-distilled water. Subsequently, at room
temperature, the sections were differentiated with 0.5% alcohol
hydrochloride for 30 sec, stained with eosin for 2 min, washed with
xylene for 2 min and sealed with neutral resin. Finally, the
sections were observed under a light microscope.
Cell culture
The rat cardiomyocyte H9C2 and 293T cell lines
(American Type Culture Collection) was cultured in Dulbecco's
modified Eagle's medium (Gibco; Thermo Fisher Scientific, Inc.)
containing 10% fetal bovine serum (Gibco; Thermo Fisher Scientific,
Inc.), 100 mg/ml penicillin and 100 mg/ml streptomycin in a
humidified incubator at 37°C containing 5% CO2 and 95%
O2. Subsequent experimentation was carried out at 50%
confluency.
Cell Counting Kit-8 (CCK-8)
H9C2 cells were collected and treated with various
concentrations of propofol (0, 6.25, 12.5, 25, 50, 100 and 200 µM)
at 37°C for 6 h to determine propofol cytotoxicity. The cells were
seeded into 96-well plates (3,000 cells/well) and cultured at 37°C.
After culture for 0, 24, 48 and 72 h, samples were incubated with
10 µl CCK-8 reagent (Beyotime Institute of Biotechnology).
Following incubation at 37°C for 4 h, the optical density was
measured at 450 nm and the cell proliferation curve was drawn.
Establishment of the HG
hypoxia/reperfusion (H/R) model
For hypoxia, H9C2 cells were incubated in 94%
N2, 5% CO2 and 1% O2 at 37°C for
12 h, and then reoxygenated for 6 h (9,19).
Cells were treated with 0, 6.25, 12.5, 25, 50, 100 and 200 µM
propofol following 12 h of hypoxia and then propofol treatment and
reoxygenation were simultaneously performed for 6 h at 37°C.
Cells were assigned into the following groups: the
blank group (H9C2 cells without any treatment), the HG group
(cultured in 4.5 g/l HG medium without H/R treatment), the HG + H/R
group (H/R treatment was performed following 48 h of HG culture),
the P25 group (H/R treatment + propofol postconditioning was
performed following HG culture), the P25 + mimic negative control
(NC) group (mimic NC + H/R + 25 µM propofol postconditioning was
performed following HG culture), the P25 + miR-200c-3p mimic
(miR-200C-3p mimic + H/R + 25 µM propofol postconditioning was
performed following HG culture), the P25 + small interfering RNA
(si)-NC group (si-NC + H/R + 25 µM propofol postconditioning was
performed following HG culture) and the P25 + si-AdipoR2 group
(si-Adipor2 + H/R + 25 µM propofol postconditioning was performed
following HG culture). miR-200c-3p mimic, mimic NC, si-Adipor2, and
si-NC were designed by Shanghai GenePharma Co., Ltd. The 293T cells
(American Type Culture Collection) were transfected with mimic NC,
miR-200c-3p mimic, si-NC, or si-AdipoR2 (Shanghai Genechem Co.,
Ltd.; 30 nM miRNA-mimic and 40–100 nM siRNA) using Lipofectamine
2000® (Invitrogen; Thermo Fisher Scientific, Inc.). The
sequence of the miR-200c-3p mimic was 5′-UAAUACUGCCGGGUAAUGAUG−3′
and the sequence of si-AdipoR2 was 5′-GCATCAAGCTGTACATATA-3′, the
sequence of mimic NC was 5′-AUGUCCGUAAGGAUACUGGUA−3′ and the
sequence of si-NC was 5′-TCGCACTAATAAGGTACAT-3′. miR-200c-3p mimic,
si-AdipoR2 and their controls were transfected into the target
cells using the Lipofectamine RNAiMAX Transfection Kit (Invitrogen;
Thermo Fisher Scientific, Inc.) at 37°C for 48 h according to the
manufacturer's instructions. Prior to transfection, the cells were
seeded into 6-well plates at 1×106 cells/well. On the
day of transfection, Lipofectamine RNAiMAX transfection reagent and
opti-MEM culture medium (Thermo Fisher Scientific, Inc.) were at
room temperature for 10 min and added into the 6-well plates. After
24 h, the transfection efficiency in H/R cells was verified by
reverse transcription-quantitative PCR (Fig. S1) and other experiments were
performed.
Dual-luciferase reporter assay
The binding sites of miR-200c-3p and AdipoR2 were
predicted using the TargetScan bioinformatics database v. 7.1
(http://www.targetscan.org/vert71/)
(20). Subsequently, the wild-type
(Wt) and mutant (Mut) 3′untranslated region of the AdipoR2 and
miR-200c-3p binding site sequences were amplified and cloned into
the pmirGLO dual-luciferase miRNA target expression vector (Promega
Corp.). The Wt plasmid AdipoR2-Wt and the corresponding Mut plasmid
AdipoR2-Mut were constructed and co-transfected with mimic NC or
miR-200c-3p mimic into 293T cells at a density of 2×104
using Lipofectamine 2000 reagent according to the manufacturer's
protocol. After 48 h of the transfection at room temperature, the
luciferase activity was detected using the Dual-Luciferase Reporter
Assay System (Promega Corp.) according to the manufacturer's
instructions, with Renilla luciferase used for normalization. The
fluorescence intensity was normalized to the mimic NC plasmid
group.
ROS detection
The ROS Assay kit (Nanjing Jiancheng Bioengineering
Institute) was used for ROS detection according to the
manufacturer's protocol. Briefly, myocardial tissue homogenate or
cells were collected and suspended in diluted
2,7-dichlorodi-hydrofluorescein diacetate, incubated at 37°C for 30
min and centrifuged at 1,000 × g at 37°C for 5 min to obtain the
precipitates. Precipitates were suspended in phosphate buffer
saline (PBS) and observed under a confocal microscope (LSM 510 Meta
microscope; Zeiss GmbH) using an emission wavelength of 525 nm.
Detection of related biochemical
indexes
Following the reperfusion procedure, after
euthanasia by intraperitoneal injection of 2% pentobarbital sodium
(200 mg/kg), 1 ml femoral arterial blood was collected and
centrifuged at 300 × g at 4°C for 15 min and the supernatant was
extracted. The H9C2 cell line was also used in this experiment.
Creatinine kinase-myocardial band levels (CK-MB) were subsequently
detected using the Rat CK-MB Isoenzyme ELISA kit (cat. no.
E02C0085; BlueGene Biotech) according to the manufacturer's
protocol. Lactate dehydrogenase (LHD) levels were determined using
a LDH Assay Kit (cat. no. A020-2-2; Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer's
instructions. Similarly, SOD levels were quantified using a Total
SOD Assay kit (water-soluble tetrazolium-8 method; cat. no. S0101S;
Beyotime Institute of Biotechnology), and MDA levels were detected
using a Lipid Peroxidation MDA Assay kit (cat. no. S0131S; Beyotime
Institute of Biotechnology) according to the manufacturer's
protocol. Bax and B cell lymphoma-2 (BCL-2) levels were detected
using a Rat Bax ELISA kit (cat. no. E-EL-R0098; Elabscience
Biotechnology, Inc.) or a Rat Bcl-2 ELISA kit (E-EL-R0096;
Elabscience Biotechnology, Inc.) according to the manufacturer's
protocol. Absorbance values were measured at the corresponding
wavelengths.
TdT-mediated dUTP Nick-End Labeling (TUNEL) assay.
Myocardial tissues were fixed with 4% paraformaldehyde at room
temperature for 24 h. Cardiomyocyte apoptosis was detected using
the One-Step TUNEL Cell Apoptosis Detection kit (cat. no. C1088;
Beyotime Institute of Biotechnology) on 4 µm-thick cardiac sections
or H9C2 cells. Briefly, the treated sections or cells were
incubated with 50 µl TUNEL reagent at 37°C for 1 h, rinsed three
times with PBS and counterstained with
4′,6-diamidino-2-phenylindole (DAPI; 1 mg/ml) at room temperature
in the dark for 5 min. The cells (1×106) were collected
and sealed with anti-fluorescence quenching sealing solution as
mounting medium (Beyotime Institute of Biotechnology). Following
another three PBS washes, the sections or cells were observed under
a fluorescence microscope (Leica Microsystems GmbH). The percentage
of apoptotic cells was calculated using five randomly selected
fields. The cell apoptotic rate=the number of apoptotic cells
(green)/total cells (blue) ×100.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from myocardial tissues or
H9C2 cells using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. Total RNA was reverse transcribed using the Taqman
Micro-RNA Reverse Transcription Reagents kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
qPCR was performed using the SYBR Green Real-Time PCR Master mix
(Thermo Fisher Scientific, Inc.) according to the manufacturer's
protocol. The following thermocycling conditions were used for
qPCR: initial denaturation at 95°C for 5 min; 40 cycles of
denaturation at 95°C for 15 sec, annealing at 58°C for 35 sec,
elongation at 72°C for 30 sec; and final extension at 72°C for 10
min. Expression levels were quantified using the 2−∆∆Cq
method (21) and normalized to the
internal reference genes, GAPDH and U6. Primer sequences are shown
in Table I.
 | Table I.Sequences of primers used for reverse
transcription-quantitative PCR. |
Table I.
Sequences of primers used for reverse
transcription-quantitative PCR.
| Gene | Sequence
(5′→3′) |
|---|
| miR-200c-3p | F:
TAATACTGCCGGGTAAT |
|
| R:
TGTCGTGGAGTCGGC |
| AdipoR2 | F:
ATGAACGAGCCAACCGAACA |
|
| R:
TCAGAAGAGCAATACCAGAG |
| U6 | F:
CGCTTCGGCAGCACATATAC |
|
| R:
AAATATGGAACGCTTCACGA |
| GAPDH | F:
ATGGTGAAGGTCGGTGTG |
|
| R:
TCACCCCATTTGATGTTA |
Western blotting
Total protein was extracted from myocardial tissues
or H9C2 cells using radio-immunoprecipitation assay (RIPA) buffer
(Beyotime Institute of Biotechnology). Total protein was quantified
using a Bicinchoninic Acid (BCA) Protein Concentration
Determination kit (Beyotime Institute of Biotechnology).
Subsequently, proteins (20 µg) were bathed in boiling water for 5
min. Proteins were separated by SDS-PAGE on a 15% gel following
denaturation. Proteins were transferred onto polyvinylidene
fluoride membranes using the wet-transfer method, blocked with 5%
skimmed milk-TBS-Tween 20 (TBST; containing 0.05% Tween-20) at room
temperature for 60 min. The samples were developed using ECL
working fluid (MilliporeSigma). Membranes were then washed using
TBST and incubated with the primary antibodies phosphorylated-STAT3
(88 kDa; 1:20,000; cat. no. ab76315; Abcam), STAT3 (88 kDa;
1:1,000; cat. no. ab68153; Abcam) and GAPDH (36 kDa; 1:1,000; cat.
no. ab8245; Abcam) overnight at 4°C. Membranes were washed with
TBST. Following the primary incubation, membranes were incubated
with the secondary goat anti-rabbit IgG (heavy chain and light
chain; HRP; 1:1,000; cat. no. ab205718; Abcam) at 37°C for 2 h. A
chemiluminescence method was used for visualization and Image J
software (version 1.52a; National Institutes of Health) was used
for gray value analysis. GAPDH was used as the loading control.
Statistical analysis
SPSS 21.0 statistical software (IBM Corp.) and
GraphPad Prism 8.0.1 (GraphPad Software, Inc.) were used for data
analysis. Cell experiments were repeated three times. The
Shapiro-Wilk test demonstrated that all data were normal
distributed. All data are expressed as the mean ± standard
deviation. Unpaired student's t-test was used to analyze
comparisons between two groups, whereas one-way analysis of
variance was used to analyze comparisons between more than two
groups followed by Tukey's post-hoc test. P<0.05 was considered
to indicated a statistically significant difference.
Results
Propofol postconditioning has a
protective effect on diabetic MI/RI
To verify the effect of propofol postconditioning on
diabetic MI/RI, diabetic MI/RI rat models were established and the
infarct size was detected using TTC staining. There was no infarct
in the myocardium in the DS group. The results demonstrated that
the infarct size of the DI group was significantly increased
compared with the DP group (Fig.
1A; P<0.01). The results of HE staining demonstrated that in
the DS group, rat cardiac structure and cell morphology were
normal, the myocardial cells were organized, the capsule was intact
and no inflammatory cell or red blood cell infiltration was
observed in the stroma. However, in the DI group there was clear
myocardial infarction, disordered arrangement of myocardial cells,
widened myocardial fiber gap and inflammatory cell infiltration in
the stroma. All these conditions were markedly improved in the DP
group (Fig. 1B). Furthermore, it
has been reported that propofol can relieve I/R injury by reducing
oxidative stress and suppressing apoptosis (10). Therefore, oxidative stress indexes
and apoptosis in the various groups were detected. In the DI group,
ROS, CK-MB, MDA and LDH levels were significantly upregulated,
whereas SOD levels were significantly reduced, all compared with
the DP group (Fig. 1C-G; all
P<0.01). Moreover, the results of TUNEL staining demonstrated
that compared with the DI group, the apoptosis of myocardial tissue
was significantly downregulated in the DP group (Fig. 1H; P<0.01). Bax and Bcl-2 levels
in serum were also detected using ELISA. Compared with the DI
group, Bax was significantly downregulated and Bcl-2 was
significantly upregulated in the DP group (Fig. 1I; P<0.01). Therefore, these
findings suggested that propofol postconditioning may inhibit
oxidative stress and apoptosis, thus improving MI/RI.
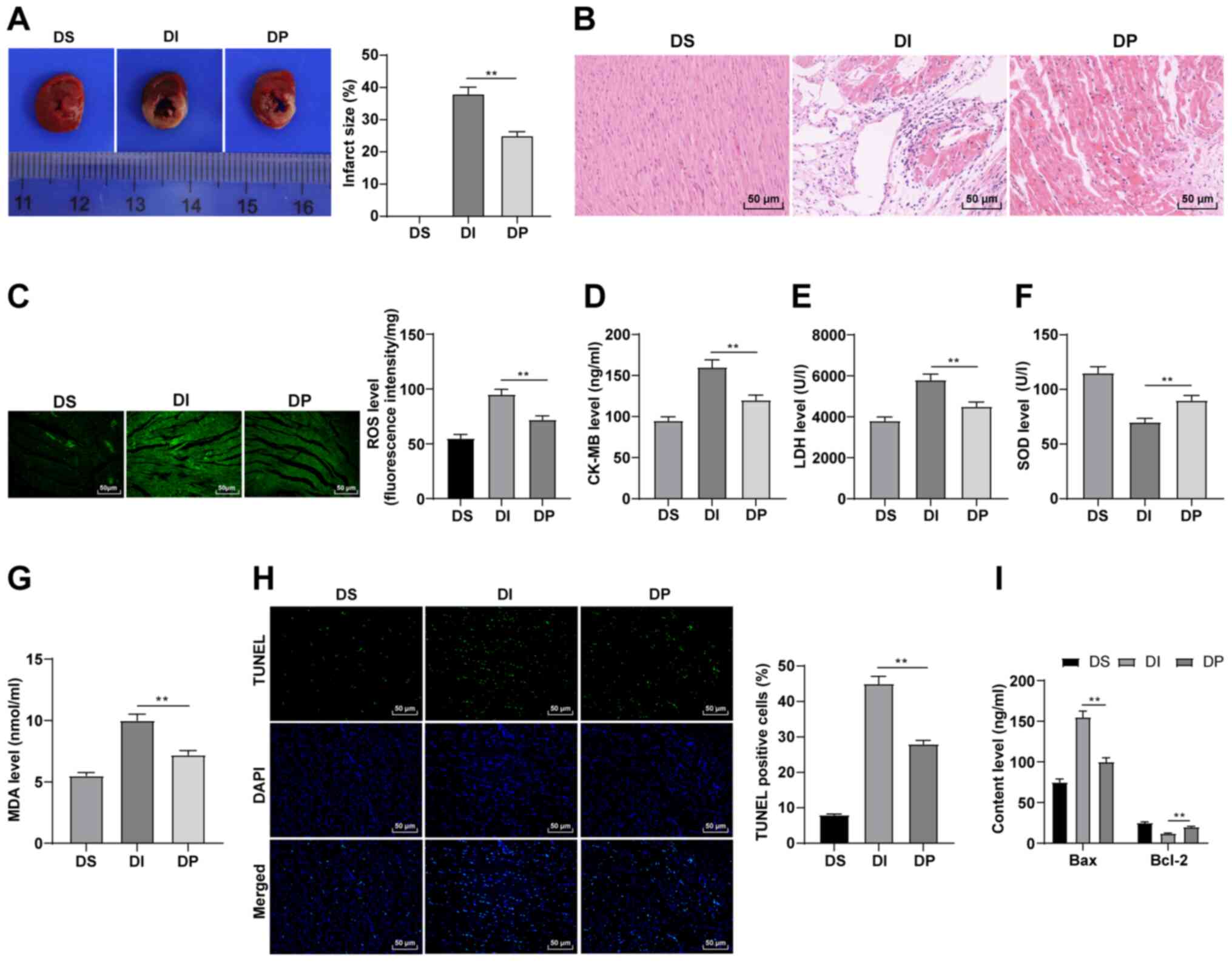 | Figure 1.Propofol postconditioning has
protective effect on diabetic MI/RI. (A) Area of myocardial
infarction was detected using 2,3,5-triphenyl-2H-tetrazolium
chloride staining. (B) Pathological changes of MI/RI tissue were
detected using HE staining. Oxidative stress index levels were
detected using ELISA: (C) ROS, (D) CK-MB, (E) LDH, (F) SOD and (G)
MDA levels. (H) Apoptosis was detected using TUNEL staining. (I)
Bax and Bcl-2 serum levels were detected using ELISA. n=6. Three
independent cell tests were performed. Data are presented as the
mean ± standard deviation. One-way ANOVA was used for comparisons
between groups followed by Tukey's post hoc test. **P<0.01.
MI/RI, myocardial ischemia reperfusion injury; ROS, reactive oxygen
species; CK-MB, creatinine kinase-myocardial band; LDH, lactate
dehydrogenase; SOD, superoxide dismutase; MDA, malondialdehyde; DS,
sham group; DI, MI/RI group; DP, MI/RI + propofol group. |
Propofol postconditioning has a
protective effect on H9C2 cells induced by HG and H/R
The effect of propofol postconditioning using in
vitro models was also investigated. The CCK-8 assay was
performed to detect the effect of propofol on H9C2 cells. The
results demonstrated that H9C2 cells treated with 25 µM propofol
had a significantly higher cell viability compared with 0 µM
concentrations (Fig. 2A;
P<0.05). Therefore, a propofol concentration of 25 µM was used
to perform postconditioning on H9C2 cells induced by HG and H/R.
Following propofol postconditioning, ROS, CK-MB, MDA and LDH levels
were significantly reduced and SOD levels were significantly
increased compared with the HG + H/R group (Fig. 2B-F; P<0.01). Furthermore, the
results of the TUNEL assay demonstrated that propofol
postconditioning significantly suppressed apoptosis compared with
the HG + H/R group (Fig. 2G;
P<0.01). Compared with the HG + H/R group, it was also observed
that Bax levels were significantly reduced and Bcl-2 levels were
significantly upregulated as a result of propofol postconditioning
(Fig. 2H; P<0.05). These
findings suggested that propofol postconditioning may protect H9C2
cells from HG and H/R.
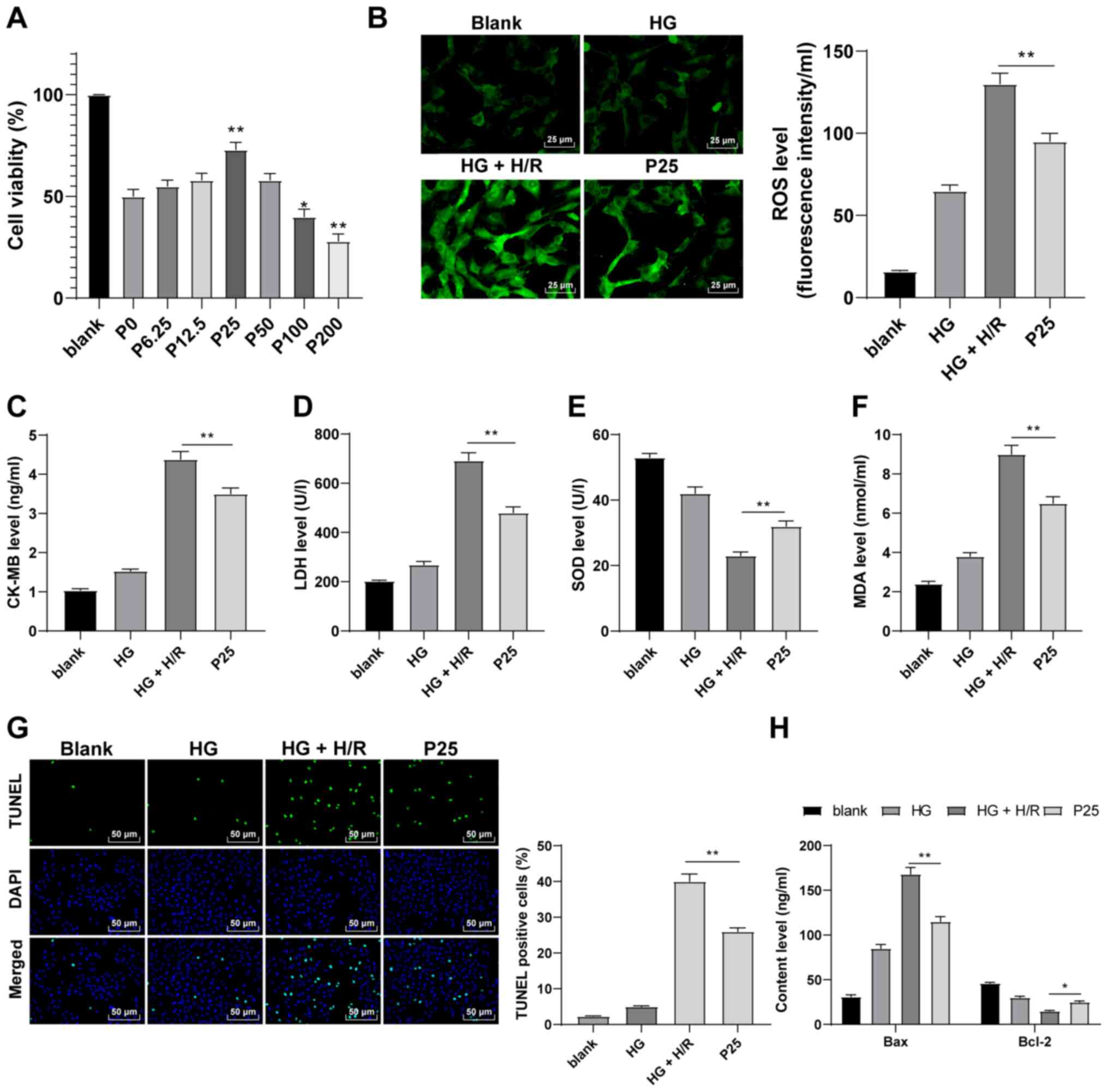 | Figure 2.Propofol postconditioning has a
protective effect on HG and H/R-induced H9C2 cells. (A) Cell
Counting Kit-8 assay was used to detect the toxicity of H9C2 cells.
The results determined that cell activity was significantly higher
at a propofol concentration of 25 µM compared with the P0 group.
Therefore, 25 µM propofol was used in subsequent experiments.
Oxidative stress index levels were detected using ELISA: (B) ROS,
(C) CK-MB, (D) LDH, (E) SOD and (F) MDA. (G) H9C2 cell apoptosis
was detected using TUNEL staining. (H) Bax and Bcl-2 levels in H9C2
cells were detected using ELISA. Three independent cell tests were
performed. Data are presented as the mean ± standard deviation.
One-way ANOVA was used for comparisons between groups followed by
Tukey's post hoc test. *P<0.05 and **P<0.01. HG, high
glucose; H/R, hypoxia/reperfusion; P, µM propofol; ROS, reactive
oxygen species; CK-MB, creatinine kinase-myocardial band; LDH,
lactate dehydrogenase; SOD, superoxide dismutase; MDA,
malondialdehyde. |
Propofol postconditioning demonstrates
antioxidant and antiapoptotic effects on H9C2 cells by inhibiting
miR-200c-3p
Previous studies indicate that propofol exerts its
protective effects on I/R injury via miRNAs (11,13).
Among them, miR-200c is known to promote an oxidative reaction and
is associated with diabetic MI/RI (14). Subsequently, the expression patterns
of miR-200c-3p were detected using RT-qPCR, which demonstrated that
miR-200c-3p expression levels were significantly upregulated in
in vivo and in vitro models, compared with the
propofol postconditioning groups which exhibited significantly
downregulated miR-200c-3p expression levels (Fig. 3A-B; all P<0.01). Moreover,
miR-200c-3p expression levels were significantly increased using
miR-200c-3p mimic to further investigate the effect of propofol on
miR-200c-3p compared with the P25 + mimics NC group (P<0.01).
ROS, CK-MB, MDA and LDH levels were significantly upregulated and
SOD levels were significantly downregulated as a result of
miR-200c-3p overexpression, compared with P25 + mimics NC (Fig. 3C-G; all P<0.01). Furthermore,
TUNEL assay results demonstrated that overexpression of miR-200c-3p
significantly increased cell apoptosis compared with P25 + mimics
NC (Fig. 3H and I; all P<0.05).
Overall, these findings indicated that overexpression of
miR-200c-3p may partially reverse the antioxidant and antiapoptotic
effects of propofol postconditioning.
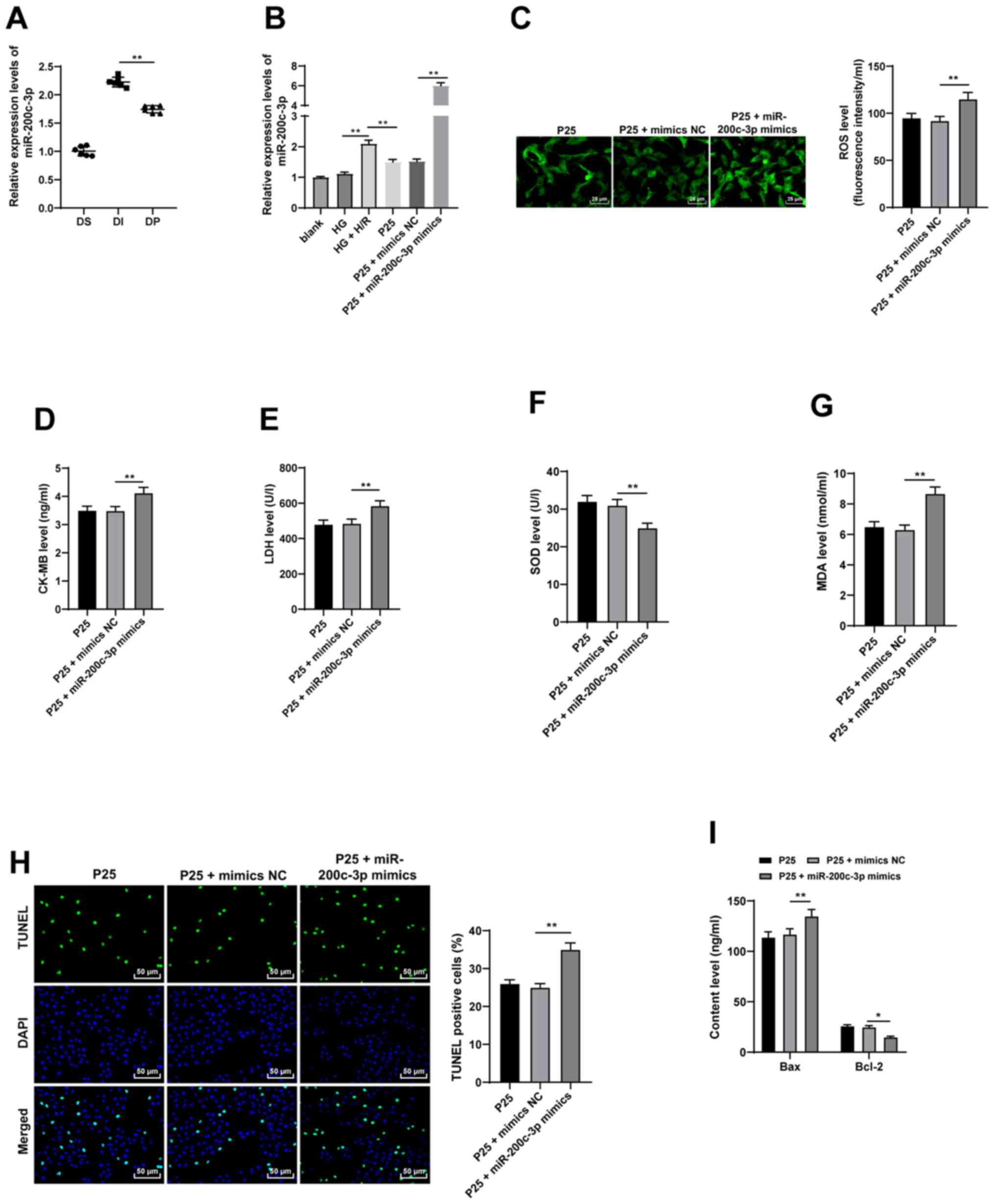 | Figure 3.Propofol postconditioning has
antioxidant and antiapoptotic effects on H9C2 cells via miR-200c-3p
inhibition. miR-200c-3p mimic was transfected into P25-treated
cells and mimics NC was used as control. (A) miR-200c-3p expression
levels in the diabetic MI/RI rat model were detected by RT-qPCR,
n=6. (B) miR-200c-3p expression levels in H9C2 cells induced by HG
and H/R were detected using RT-qPCR. Oxidative stress index levels
were detected using ELISA: (C) ROS, (D) CK-MB, (E) LDH, (F) SOD and
(G) MDA. (H) H9C2 cell apoptosis was detected using TUNEL staining.
(I) Bax and Bcl-2 levels in H9C2 cells were detected using ELISA.
Three independent cell tests were performed. Data are presented as
the mean ± standard deviation. One-way ANOVA was used for
comparisons between groups followed by Tukey's post hoc test.
*P<0.05 and **P<0.01. miR, microRNA; P25, 25 µM propofol; NC,
negative control; MI/RI, myocardial ischemic reperfusion; RT-qPCR,
reverse transcription-quantitative PCR; ROS, reactive oxygen
species; CK-MB, creatinine kinase-myocardial band; LDH, lactate
dehydrogenase; SOD, superoxide dismutase; MDA, malondialdehyde; DS,
sham group; DI, MI/RI group; DP, MI/RI + propofol group; HG, high
glucose; H/R, hypoxia/reperfusion. |
miR-200c-3p targets AdipoR2
The targeted mRNA of miR-200c-3p was predicted and
analyzed using the TargetScan database (20) to explore the downstream molecular
mechanism of miR-200c-3p. Among the predicted targets, AdipoR2 was
previously associated with the recovery of diabetic MI/RI (22). Therefore, the binding sites between
AdipoR2 and miR-200c-3p were predicted using the TargetScan
database (Fig. 4A). Subsequently,
the binding relationship was verified using the dual-luciferase
reporter assay. Compared with the control group, following
transfection with miR-200c-3p mimic in the AdipoR2-WT group, the
relative luciferase activity was significantly decreased, while
that of the AdipoR2-MUT group did not change (Fig. 4B; P<0.01). AdipoR2 mRNA
expression levels were detected by RT-qPCR. The results
demonstrated that propofol postconditioning significantly
upregulated the expression of AdipoR2 in vivo and in
vitro compared with the DI or HG + H/R group, respectively
(Fig. 4C and D; all P<0.01).
Furthermore, miR-200c-3p overexpression significantly decreased
AdipoR2 mRNA expression levels compared with the P25 + mimics NC
group. Overall, these results suggested that propofol
postconditioning might manipulate MI/RI by downregulating
miR-200c-3p expression and upregulating AdipoR2 expression.
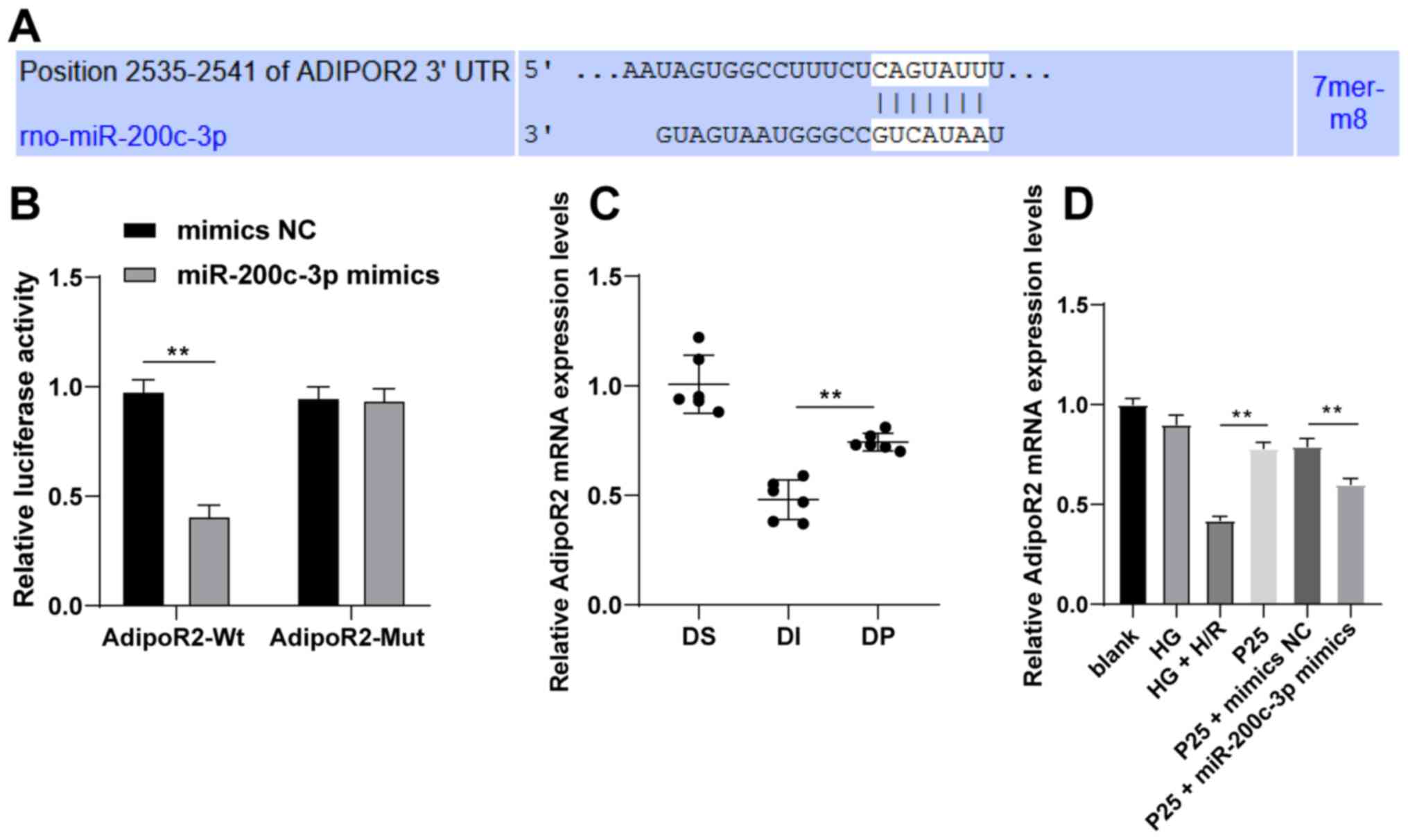 | Figure 4.Propofol postconditioning upregulates
AdipoR2 and downregulates miR-200c-3p. (A) Binding sites of
miR-200c-3p and AdipoR2 were predicted using TargetScan. (B)
Dual-luciferase reporter assay was used to detect the targeting
relationship between miR-200c-3p and AdipoR2. (C) AdipoR2 mRNA
expression levels in diabetic myocardial ischemic reperfusion rat
model following propofol postconditioning were detected by RT-qPCR,
n=6. (D) AdipoR2 mRNA expression levels in H9C2 cells following
propofol postconditioning were detected by RT-qPCR. Three
independent cell tests were performed. Data are presented as the
mean ± standard deviation. Unpaired Student's t-test was used for
comparisons between two groups in panel B, and one-way ANOVA
followed by Tukey's post hoc test was used for comparisons between
multiple groups in panels C and D. **P<0.01. AdipoR2,
adiponectin receptor 2; miR, microRNA; MI/RI; RT-qPCR, reverse
transcription-quantitative PCR; UTR, untranslated region; NC,
negative control; P25, 25 µM propofol; Wt, wild-type; Mut, mutant;
DS, sham group; DI, MI/RI group; DP, MI/RI + propofol group; HG,
high glucose; H/R, hypoxia/reperfusion; rno, Rattus norvegicus. |
AdipoR2 downregulation reverses the
antioxidative and antiapoptotic effects of propofol on H9C2
cells
To further investigate the function of AdipoR2,
AdipoR2 expression levels were silenced in P25-treated cells using
si-AdipoR2. Compared with the si-NC group, the mRNA expression
levels of AdipoR2 were significantly reduced in the P25 +
si-AdipoR2 group (Fig. 5A;
P<0.01). The results demonstrated that ROS, CK-MB, MDA and LDH
levels were significantly enhanced and SOD levels were
significantly downregulated as a result of AdipoR2 knockdown,
compared with P25 + si-NC (Fig.
5B-F; all P<0.01). Furthermore, the results of the TUNEL
assay and Bax and Bcl-2 ELISAs demonstrated that AdipoR2 silencing
significantly increased apoptosis in H9C2 cells compared with P25 +
si-NC (Fig. 5G and H; all
P<0.05). Overall, these results indicated that silencing AdipoR2
may partially reverse the antioxidant and antiapoptotic effects of
propofol postconditioning on MI/RI.
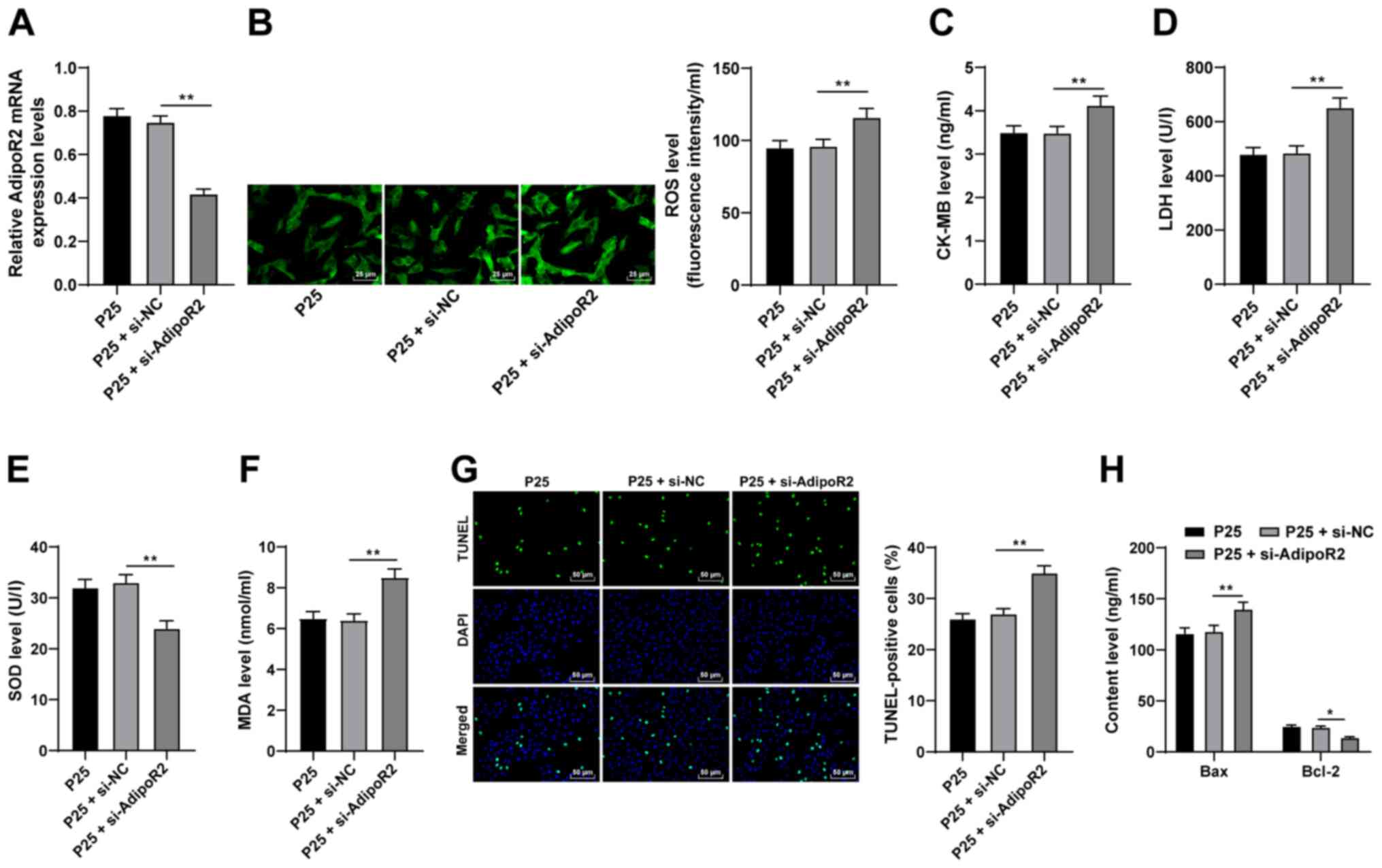 | Figure 5.Silencing AdipoR2 partially reverses
the antioxidant and antiapoptotic effects of propofol
postconditioning on myocardial ischemic reperfusion injury.
si-AdipoR2 was transfected into P25-treated cells and si-NC was
used as control. (A) AdipoR2 mRNA expression levels in H9C2 cells
were detected using reverse transcription-quantitative PCR.
Oxidative stress index levels were detected using ELISA: (B) ROS,
(C) CK-MB, (D) LDH, (E) SOD and (F) MDA. (G) H9C2 cell apoptosis
was detected using TUNEL staining. (H) Bax and Bcl-2 levels in H9C2
cells were detected using ELISA. Three independent cell tests were
performed. Data are presented as the mean ± standard deviation.
One-way ANOVA was used for comparisons between groups followed by
Tukey's post hoc test. *P<0.05 and **P<0.01. AdipoR2,
adiponectin receptor 2; si, small interfering RNA; P25, 25 µM
propofol; NC, negative control; ROS, reactive oxygen species;
CK-MB, creatinine kinase-myocardial band; LDH, lactate
dehydrogenase; SOD, superoxide dismutase; MDA, malondialdehyde. |
Propofol postconditioning activated
the STAT3 pathway via the miR-200c-3p/AdipoR2 axis
Previous studies have reported that adiponectin
(APN) can restore STAT3 activity and suppress apoptosis through
AdipoR2 following anoxia of cardiomyocytes (22,23).
Therefore, in order to validate whether the STAT3 pathway was
involved in the protective effect of propofol postconditioning on
diabetic MI/RI, STAT3 protein expression levels were detected via
western blotting. The results demonstrated that propofol
postconditioning significantly upregulated the phosphorylation
levels of STAT3 compared with the DI group. Furthermore,
miR-200c-3p overexpression or AdipoR2 knockdown of AdiopR2
significantly reduced STAT3 phosphorylation levels compared with
P25 + mimics NC and P25 + si-NC, respectively (Fig. 6A and B; all P<0.01). These
results demonstrated that propofol postconditioning may activate
the STAT3 pathway and alleviate MI/RI via the miR-200c-3p/AdipoR2
axis.
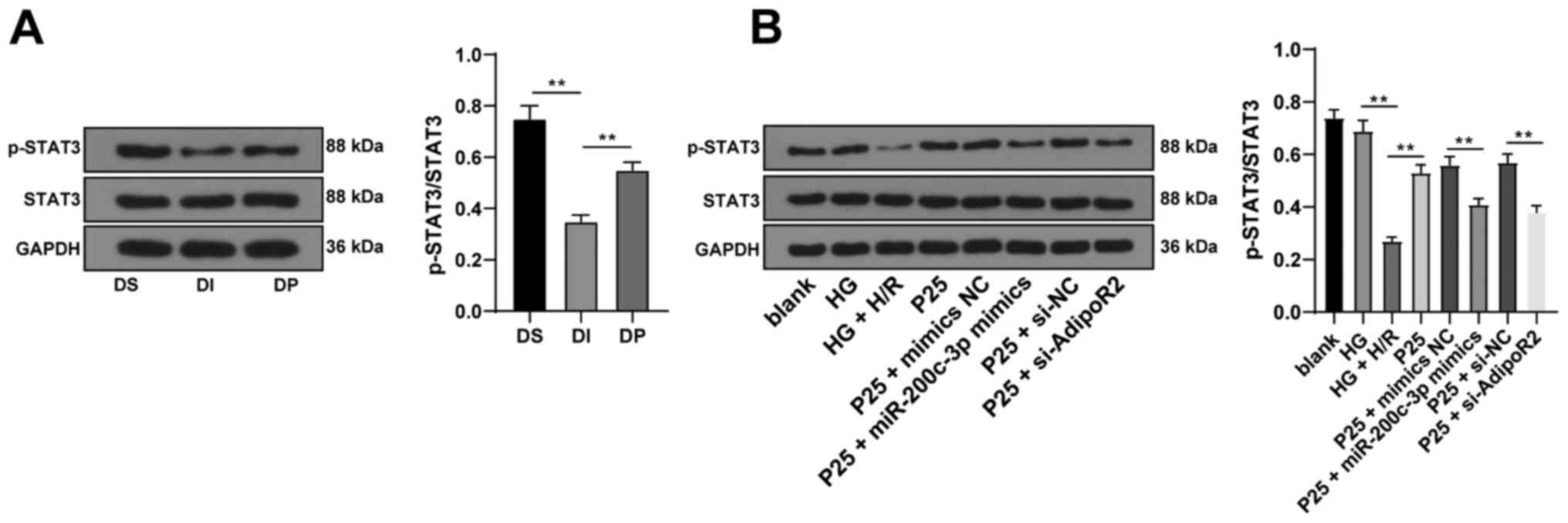 | Figure 6.Propofol postconditioning activates
the STAT3 pathway via the miR-200c-3p/AdipoR2 axis. (A) STAT3
protein expression levels were detected via western blotting, n=6.
(B) STAT3 protein expression levels in the H9C2 cell high sugar H/R
model were detected via western blotting. Three independent cell
tests were performed. Data are presented as the mean ± standard
deviation. One-way ANOVA was used for comparisons between groups
followed by Tukey's post hoc test. **P<0.01. miR, microRNA;
AdipoR2, adiponectin receptor 2; H/R, hypoxia/reperfusion; DS, sham
group; DI, MI/RI group; DP, MI/RI + propofol group; HG, high
glucose; p, phosphorylated; P25, 25 µM propofol; NC, negative
control; si, small interfering RNA. |
Discussion
Patients with diabetes are predisposed to MI/RI,
which is one of the leading causes of mortality and disability
(24). Preconditioning with
propofol during the process of reperfusion is known to confer
protection against MI/RI (25).
Therefore, the present study aimed to elucidate the mechanism of
propofol postconditioning on diabetic MI/RI and demonstrated that
propofol postconditioning attenuated diabetic MI/RI via the
miR-200c-3p/AdipoR2/STAT3 pathway (Fig. S2).
Oxidative stress and apoptosis serve a vital role in
the pathogenesis of I/R injury (26). ROS, CK-MB, LDH and SOD are regarded
as important indexes of oxidative stress (27,28).
Notably, propofol pretreatment was previously indicated to inhibit
oxidative stress and further improve nerve function in rats with
I/R injury (29). Preconditioning
occurs before ischemia. Although propofol preconditioning has been
reported to have a protective effect on ischemia-reperfusion in the
myocardium and brain, it is often unable to predict the occurrence
of ischemia clinically, which limits the effect of preconditioning
(30). Postconditioning is applied
for the ischemic tissues. It is to establish a more effective
reperfusion method to treat and save the ischemic tissues and to
minimize reperfusion injury (30).
Therefore, postconditioning of propofol may have a greater
application as a treatment. Shedding a new light on the effect of
propofol postconditioning on oxidative stress in diabetic MI/RI,
the present study demonstrated that infarct size was significantly
reduced and adverse changes in myocardial cells were markedly
reduced following I/R treatment, as a result of propofol
postconditioning. Moreover, significantly decreased levels of ROS,
CK-MB, MDA and LDH and a significant increase in SOD levels were
documented upon propofol postconditioning, which suggested reduced
oxidative stress. Moreover, Bax and Bcl-2 are widely-used
indicators of apoptosis (28). Bax
levels were significantly downregulated, whereas Bcl-2 levels were
significantly upregulated as a result of propofol postconditioning.
Furthermore, propofol has been previously indicated to exert an
inhibitory effect on I/R injury in a variety of experimental models
by suppressing apoptosis and reducing oxidative stress (10). These results demonstrated that
propofol postconditioning may protect against diabetic MI/RI by
attenuating oxidative stress and apoptosis. The present study also
revealed that propofol postconditioning conferred protective
effects on H9C2 cardiomyocytes induced by HG and H/R injury in
vitro. A recent study also reported that the cardioprotective
effects of propofol against MI/RI are dependent on miRNAs (25). miR-200c was previously shown to
exhibit proapoptotic cytotoxicity in tumors and endothelial cells
(31). Overexpression of miR-200c
is also known to inhibit the protective effect of sevoflurane
preconditioning against I/R injury (32). miR-200c is further associated with
diabetic MI/RI, possibly through the upsurge of ROS (14). The present study reported that
propofol postconditioning downregulated miR-200c-3p expression
levels. The present study also demonstrated that in addition to
significantly increased cell apoptosis, ROS, CK-MB, MDA and LDH
levels were all significantly enhanced, and SOD levels was
significantly reduced as a result of miR-200c-3p overexpression in
P25-treated H9C2 cells. A previous study demonstrated the rapid
upregulation of miR-200c following transient ischemia, leading to
transient ischemic injury (31).
Upregulation of miR-200c has also been previously associated with
oxidative stress (33,34). In view of these findings, it would
be plausible to suggest that overexpression of miR-200c-3p
partially reversed the antioxidant and antiapoptotic effects of
propofol postconditioning. Therefore, propofol postconditioning may
serve an antioxidant and antiapoptotic role in H9C2 cells by
inhibiting miR-200c-3p.
Prediction analyses performed using the TargetScan
database indicated AdipoR2 as a target gene of miR-200c. AdipoR2 is
a type of APN, a cardioprotective molecule derived from adipocytes,
and its reduction is known to exacerbate diabetic MI/RI (35). The dual-luciferase reporter assay
verified the existence of a targeted binding relationship between
miR-200c-3p and AdipoR2. Furthermore, this indicated that propofol
postconditioning regulated MI/RI by significantly downregulating
miR-200c-3p expression levels and significantly upregulating
AdipoR2 mRNA expression levels. Moreover, AdipoR2 silencing in
P25-treated cells via si-AdipoR2, resulted in ROS, CK-MB, MDA and
LDH levels all being significantly elevated and SOD levels being
significantly downregulated. Cardiomyocyte apoptosis was also
significantly promoted as a result of AdipoR2 silencing. APN is
further known to exert protective effects through antiapoptotic
activities and antioxidative stress and has been previously
indicated as a potential therapy for HI/RI (36). Furthermore, another study
illustrated that AdipoR knockdown contributes to aggravating
diabetic MI/RI, wherein AdipoR activation represents an effective
intervention against diabetic MI/RI (37). Collectively, these findings indicate
that silencing AdipoR2 partially reversed the antioxidant and
antiapoptotic effects of propofol postconditioning on MI/RI.
APN possesses the ability to restore STAT3
activation and reduce apoptosis, thus AdipoR2 can function as a
mediator to prevent diabetic MI/RI through STAT3 (22). Moreover, the present study
demonstrated that propofol postconditioning significantly
upregulated STAT3 phosphorylation levels, whereas miR-200c-3p
overexpression or AdipoR2 downregulation of AdiopR2 brought about
the opposite effect. A recent study documented significant
downregulation of STAT3 in I/R injury, which may serve a role in
myocardial protection (38).
Propofol was also previously shown to inhibit cardiomyocyte
apoptosis in MI/RI by repressing the STAT3 pathway (39). Collectively, the results of the
present study suggested that propofol postconditioning might have
activated the STAT3 pathway and alleviated diabetic MI/RI via the
miR-200c-3p/AdipoR2 axis.
In conclusion, results obtained in the present study
indicated that propofol postconditioning could protect against
MI/RI by inhibiting miR-200c-3p and activating the AdipoR2/STAT3
pathway. However, the protective effect of propofol
postconditioning cannot be realized by a single and simple
molecular mechanism. Whether propofol postconditioning can exert an
effect by regulating other miRNAs is largely unknown. For example,
miR-29b has been shown to protect against MI/RI (40). Furthermore, whether the protective
effect of propofol postconditioning depends on miR-29b or other
miRNAs remains to be studied. The regulation mechanism of propofol
postconditioning on AdipoR2/STAT3 axis and whether miR-200c-3p
could be a target of MI/RI in diabetes remains to be elucidated.
The anti-oxidative and anti-apoptotic properties of propofol
postconditioning could be attributed to complex processes of
multiple pathways and receptors and the AdipoR2/STAT3 axis is only
one of these mechanisms. The protective effect of propofol on MI/RI
is not dose-dependent and the mechanism remains unclear. It may be
possible that the internal lipid components of propofol can reduce
the antioxidant effect of propofol during metabolism, however, this
also needs further validation. The results of the present study
have provided new ideas for the clinical treatment of diabetic
MI/RI.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
LH designed the study. LD, SY, XH and QR carried out
the experiments and wrote the manuscript. LH and QR were
responsible for reviewing and editing the manuscript. LH and LD
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
All procedures were authorized by the Academic
Ethics Committee of The Affiliated People's Hospital of Ningbo
University (Approval number: 2019-010). Experiments was carried out
in strict accordance with the Guidelines for the Management and Use
of Laboratory Animals issued by the Laboratory Association of
China.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kerner W and Bruckel J; German Diabetes
Association, : Definition, classification and diagnosis of diabetes
mellitus. Exp Clin Endocrinol Diabetes. 122:384–386. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu SY, Dong B, Fang ZF, Hu XQ, Tang L and
Zhou SH: Knockdown of lncRNA AK139328 alleviates myocardial
Ischaemia/reperfusion injury in diabetic mice via modulating
miR-204-3p and inhibiting autophagy. J Cell Mol Med. 22:4886–4898.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tao A, Xu X, Kvietys P, Kao R, Martin C
and Rui T: Experimental diabetes mellitus exacerbates
ischemia/reperfusion-induced myocardial injury by promoting
mitochondrial fission: Role of down-regulation of myocardial Sirt1
and subsequent Akt/Drp1 interaction. Int J Biochem Cell Biol.
105:94–103. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li J, Zhao Y, Zhou N, Li L and Li K:
Dexmedetomidine attenuates myocardial ischemia-reperfusion injury
in diabetes mellitus by inhibiting endoplasmic reticulum stress. J
Diabetes Res. 2019:78693182019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu Y, Pan S, Jiang W, Xue F and Zhu X:
Effects of propofol on the development of cancer in humans. Cell
Prolif. 53:e128672020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yan HJ, Qi GQ and Ma Y: Effect of propofol
on myocardial ischemia-reperfusion injury through MAPK/ERK pathway.
Eur Rev Med Pharmacol Sci. 23:11051–11061. 2019.PubMed/NCBI
|
|
8
|
Facey J, Young L and Nwokocha C:
Relaxation responses of ketamine and propofol to vasoactive agents
in Streptozotocin-induced diabetic rats. Niger J Physiol Sci.
35:33–39. 2020.PubMed/NCBI
|
|
9
|
Deng F, Wang S, Zhang L, Xie X, Cai S, Li
H, Xie GL, Miao HL, Yang C, Liu X and Xia Z: Propofol through
upregulating Caveolin-3 attenuates Post-hypoxic mitochondrial
damage and cell death in H9C2 cardiomyocytes during hyperglycemia.
Cell Physiol Biochem. 44:279–292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin C, Sui H, Gu J, Yang X, Deng L, Li W,
Ding W, Li D and Yang Y: Effect and mechanism of propofol on
myocardial ischemia reperfusion injury in type 2 diabetic rats.
Microvasc Res. 90:162–168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hao W, Zhao ZH, Meng QT, Tie ME, Lei SQ
and Xia ZY: Propofol protects against hepatic ischemia/reperfusion
injury via miR-133a-5p regulating the expression of MAPK6. Cell
Biol Int. 41:495–504. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu TX and Rothenberg ME: MicroRNA. J
Allergy Clin Immunol. 141:1202–1207. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhen W, Hui D, Wenying S and Yulong S:
MicroRNA-20b-5p regulates propofol-preconditioning-induced
inhibition of autophagy in hypoxia-and-reoxygenation-stimulated
endothelial cells. J Biosci. 45:352020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dehaini H, Awada H, El-Yazbi A, Zouein FA,
Issa K, Eid AA, Ibrahim M, Badran A, Baydoun E, Pintus G and Eid
AH: MicroRNAs as potential pharmaco-targets in ischemia-reperfusion
injury compounded by diabetes. Cells. 8:1522019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Climent M, Viggiani G, Chen YW, Coulis G
and Castaldi A: MicroRNA and ROS crosstalk in cardiac and pulmonary
diseases. Int J Mol Sci. 21:43702020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao B, Gao WW, Liu YJ, Jiang M, Liu L,
Yuan Q, Hou JB and Xia ZY: The role of glycogen synthase kinase 3
beta in brain injury induced by myocardial ischemia/reperfusion
injury in a rat model of diabetes mellitus. Neural Regen Res.
12:1632–1639. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H, Yao W, Liu Z, Xu A, Huang Y, Ma XL,
Irwin MG and Xia Z: Hyperglycemia abrogates ischemic
postconditioning cardioprotection by impairing
AdipoR1/Caveolin-3/STAT3 signaling in diabetic rats. Diabetes.
65:942–955. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang HY, Wang GL, Yu YH and Wang Y: The
role of phosphoinositide-3-kinase/Akt pathway in propofol-induced
postconditioning against focal cerebral ischemia-reperfusion injury
in rats. Brain Res. 1297:177–184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhu HJ, Wang DG, Yan J and Xu J:
Up-regulation of microRNA-135a protects against myocardial
ischemia/reperfusion injury by decreasing TXNIP expression in
diabetic mice. Am J Transl Res. 7:2661–2671. 2015.PubMed/NCBI
|
|
20
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:e050052015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang T, Mao X, Li H, Qiao S, Xu A, Wang J,
Lei S, Liu Z, Ng KF, Wong GT, et al: N-Acetylcysteine and
allopurinol up-regulated the Jak/STAT3 and PI3K/Akt pathways via
adiponectin and attenuated myocardial postischemic injury in
diabetes. Free Radic Biol Med. 63:291–303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang T, Qiao S, Lei S, Liu Y, Ng KF, Xu A,
Lam KS, Irwin MG and Xia Z: N-acetylcysteine and allopurinol
synergistically enhance cardiac adiponectin content and reduce
myocardial reperfusion injury in diabetic rats. PLoS One.
6:e239672011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhan L, Zhang Y, Su W, Zhang Q, Chen R,
Zhao B, Li W, Xue R, Xia Z and Lei S: The roles of autophagy in
acute lung injury induced by myocardial ischemia reperfusion in
diabetic rats. J Diabetes Res. 2018:50475262018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li YM, Sun JG, Hu LH, Ma XC, Zhou G and
Huang XZ: Propofol-mediated cardioprotection dependent of
microRNA-451/HMGB1 against myocardial ischemia-reperfusion injury.
J Cell Physiol. 234:23289–23301. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang Y, Che J, Zhao H, Tang J and Shi G:
Platycodin D inhibits oxidative stress and apoptosis in H9c2
cardiomyocytes following hypoxia/reoxygenation injury. Biochem
Biophys Res Commun. 503:3219–3224. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rahimi R, Karimi J, Khodadadi I, Tayebinia
H, Kheiripour N, Hashemnia M and Goli F: Silymarin ameliorates
expression of urotensin II (U-II) and its receptor (UTR) and
attenuates toxic oxidative stress in the heart of rats with type 2
diabetes. Biomed Pharmacother. 101:244–250. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hu C, Zhang X, Wei W, Zhang N, Wu H, Ma Z,
Li L, Deng W and Tang Q: Matrine attenuates oxidative stress and
cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via
maintaining AMPKα/UCP2 pathway. Acta Pharm Sin B. 9:690–701. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen Y and Li Z: Protective effects of
propofol on rats with cerebral ischemia-reperfusion injury via the
PI3K/Akt Pathway. J Mol Neurosci. 71:810–820. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li H, Zhang X, Tan J, Sun L, Xu LH, Jiang
YG, Lou JS, Shi XY and Mi WD: Propofol postconditioning protects
H9c2 cells from hypoxia/reoxygenation injury by inducing autophagy
via the SAPK/JNK pathway. Mol Med Rep. 17:4573–4580.
2018.PubMed/NCBI
|
|
31
|
Tang M, Liu P, Li X, Wang JW, Zhu XC and
He FP: Protective action of B1R antagonist against cerebral
ischemia-reperfusion injury through suppressing miR-200c expression
of Microglia-derived microvesicles. Neurol Res. 39:612–620. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu Y, Gu C and Huang X: Sevoflurane
protects against hepatic ischemia/reperfusion injury by modulating
microRNA-200c regulation in mice. Biomed Pharmacother.
84:1126–1136. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi D, Guo L, Sun X, Shang M, Meng D, Zhou
X, Liu X, Zhao Y and Li J: UTMD inhibit EMT of breast cancer
through the ROS/miR-200c/ZEB1 axis. Sci Rep. 10:66572020.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nunomura A and Perry G: RNA and oxidative
stress in Alzheimer's disease: Focus on microRNAs. Oxid Med Cell
Longev. 2020:26381302020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang Y, Zhao J, Li R, Lau WB, Yuan YX,
Liang B, Li R, Gao EH, Koch WJ, Ma XL and Wang YJ: AdipoRon, the
first orally active adiponectin receptor activator, attenuates
postischemic myocardial apoptosis through both AMPK-mediated and
AMPK-independent signalings. Am J Physiol Endocrinol Metab.
309:E275–E282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li D, Song LL, Wang J, Meng C and Cui XG:
Adiponectin protects against lung ischemia-reperfusion injury in
rats with type 2 diabetes mellitus. Mol Med Rep. 17:7191–7201.
2018.PubMed/NCBI
|
|
37
|
Wang Y, Liang B, Lau WB, Du Y, Guo R, Yan
Z, Gan L, Yan W, Zhao J, Gao E, et al: Restoring diabetes-induced
autophagic flux arrest in ischemic/reperfused heart by ADIPOR
(adiponectin receptor) activation involves both AMPK-dependent and
AMPK-independent signaling. Autophagy. 13:1855–1869. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Y, Zheng LM, Wang CX, Gu JM and Xue
S: SENP3 protects H9C2 cells from apoptosis triggered by H/R via
STAT3 pathway. Eur Rev Med Pharmacol Sci. 22:2778–2786.
2018.PubMed/NCBI
|
|
39
|
Chen X, Wang Y, Xiao ZY, Hou DN, Li DB and
Zhang XP: Effect of propofol on myocardial ischemia/reperfusion
injury in rats through JAK/STAT signaling pathway. Eur Rev Med
Pharmacol Sci. 23:6330–6338. 2019.PubMed/NCBI
|
|
40
|
Li K, Zhou P, Li S, Zheng S and Wang D:
MicroRNA-29b reduces myocardial ischemia-reperfusion injury in rats
via down-regulating PTEN and activating the Akt/eNOS signaling
pathway. J Thromb Thrombolysis. 53:123–135. 2021. View Article : Google Scholar : PubMed/NCBI
|














